ГОСТ Р ИСО 14971-2009
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИЗДЕЛИЯ МЕДИЦИНСКИЕ
Применение менеджмента риска к медицинским изделиям
Medical devices. Application of risk management to medical devices
ОКС 11.040.01
ОКП 94 4000
Дата введения 2011-01-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "МЕДИТЕСТ" на основе аутентичного перевода стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством и соответствующие аспекты для медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 2 декабря 2009 г. N 528-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14971:2007* "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" (ISO 14971:2007 "Medical devices - Application of risk management to medical devices")
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
5 ВЗАМЕН ГОСТ Р ИСО 14971-2006
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
Введение
Международный стандарт ИСО 14971:2007 разработан техническим комитетом ИСО/ТК 210 "Менеджмент качества и соответствующие общие аспекты медицинских изделий" и подкомитетом МЭК/ПК 62А "Общие аспекты электрических изделий, применяемых в медицинской практике". Приложение Н "Руководство по менеджменту риска медицинских изделий для диагностики in vitro" подготовлено техническим комитетом ИСО/ТК 212 "Клинические лабораторные исследования и диагностические тест-системы in vitro".
Настоящий стандарт рекомендуется рассматривать как основу для осуществления изготовителями результативного менеджмента всех рисков, связанных с применением медицинских изделий. Содержащиеся в настоящем стандарте требования являются для изготовителей той базой, в рамках которой практический опыт, интуитивное понимание и умение принять правильное решение используют для менеджмента вышеуказанных рисков.
Настоящий стандарт предназначен непосредственно для изготовителей медицинских изделий и систем, применяющих установленные в данном стандарте принципы менеджмента риска.
Виды деятельности, в которых участвуют отдельные лица, организации или органы государственного управления, могут стать для вышеуказанных или других участвующих сторон источником опасностей, способных причинить вред или нанести ущерб. Менеджмент рисков представляет собой сложный для рассмотрения предмет, т.к. каждая из участвующих сторон может по-разному оценивать вероятность причинения вреда и возможность нанесения ущерба в случае опасности.
Концепция риска включает два компонента:
a) вероятность причинения вреда;
b) последствия причиненного вреда, т.е. его тяжесть.
Рассматриваемая концепция особенно важна применительно к медицинским изделиям из-за большого числа участвующих сторон, включая практикующих врачей, учреждения здравоохранения, уполномоченные органы, промышленные предприятия, пациентов и других пользователей.
По сравнению с предыдущей редакцией основная часть настоящего стандарта расширена, но главное внимание уделено приложениям. Их число увеличилось за счет приложения F, содержащего план менеджмента риска, приложения I, посвященного биологическим опасностям, приложения по безопасности J и остаточному риску. Существенно увеличилось в объеме приложение, посвященное менеджменту риска медицинских изделий для диагностики in vitro.
1 Область применения
Настоящий стандарт устанавливает для изготовителя процесс определения опасностей, связанных с медицинскими изделиями, включая изделия для диагностики in vitro, и процедуры определения, оценивания, управления рисками и мониторинга результативности данного управления.
Требования настоящего стандарта применимы ко всем стадиям жизненного цикла медицинских изделий.
Настоящий стандарт не может быть использован для принятия клинических решений.
Настоящий стандарт не устанавливает уровни допустимого риска.
Настоящий стандарт не требует наличия у изготовителя системы менеджмента качества, однако менеджмент риска может быть составной частью системы менеджмента качества.
2 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
2.1 эксплуатационный документ (accompanying document): Документ, прилагаемый к медицинскому изделию и содержащий информацию для лиц, ответственных за сборку, применение и техническое обслуживание медицинского изделия, оператора или пользователя, особенно в отношении безопасности.
Примечание - Данное определение заимствовано из [1], 3.4.
2.2 вред (harm): Физическая травма или ущерб здоровью людей или имуществу, или окружающей среде (см. [2], 3.3).
2.3 опасность (hazard): Потенциальный источник вреда (см. [2], 3.5).
2.4 опасная ситуация (hazardous situation): Обстоятельства, при которых люди, имущество или окружающая среда подвержены одной или нескольким опасностям (см. [2], 3.6).
Примечание - См. приложение E для понимания взаимосвязи между терминами "опасность" и "опасная ситуация".
2.5 предусмотренное применение (intended use): Применение изделия, процесса или услуги по назначению в соответствии с техническими требованиями, инструкциями и информацией, предоставленными изготовителем.
2.6 медицинское изделие для диагностики in vitro (IVD) (in vitro diagnostic medical device) (IVD medical device): Медицинское изделие, предназначенное изготовителем для исследования проб, взятых из тела человека, в целях получения информации для диагностики, мониторинга и определения совместимости.
Пример - Такими изделиями могут быть реагенты, калибраторы, устройства для отбора и хранения проб, контрольные материалы и сопутствующие инструменты, аппараты или средства.
Примечание - Может быть использовано отдельно или в комбинации с принадлежностями или другими медицинскими изделиями.
2.7 жизненный цикл (life-cycle): Все стадии существования медицинского изделия, от первоначальной концепции до вывода из эксплуатации и утилизации.
2.8 изготовитель (manufacturer): Физическое или юридическое лицо, ответственное за проектирование, изготовление, упаковывание и/или маркировку медицинского изделия, установку/монтаж или модификацию медицинского изделия перед выпуском его в обращение или вводом в эксплуатацию независимо от того, выполняет ли эти операции вышеупомянутое лицо или третья сторона от его имени.
Примечания
1 В определении термина "изготовитель" следует учитывать положения национальных и региональных нормативных документов.
2 Определение термина "маркировка" см. [3], 3.6.
2.9 медицинское изделие (medical device): Любой инструмент, аппарат, прибор, устройство, оборудование, имплантат, in vitro реагент или калибратор, программное обеспечение, материал или иные подобные или связанные с ними изделия, предназначенные изготовителем для применения к человеку по отдельности или в сочетании друг с другом в целях:
- диагностики, профилактики, мониторинга, лечения или облегчения заболеваний;
- диагностики, мониторинга, лечения, облегчения или компенсации последствий травмы;
- исследования, замещения или изменения анатомического строения или физиологических процессов;
- поддержания или сохранения жизни;
- управления зачатием;
- дезинфекции медицинских изделий;
- получения информации для медицинских целей посредством исследования in vitro проб, взятых из тела человека, при условии, что их функциональное воздействие на человеческий организм не реализуется за счет фармакологических, иммунологических или метаболических средств, но может поддерживаться такими средствами.
Примечания
1 Данное определение разработано Целевой группой по глобальной гармонизации (Global Harmonization Task Force - GHTF) (см. [4]).
2 Следующие изделия могут рассматриваться в некоторых странах как медицинские, но в их отношении еще не выработан единый подход:
- вспомогательные средства для лиц с ограниченными возможностями или с физическими и умственными недостатками;
- изделия для лечения/диагностики заболеваний и травм у животных;
- принадлежности медицинских изделий (см. примечание 3);
- дезинфицирующие вещества;
- изделия, включающие ткани животных или человека.
3 Требования настоящего стандарта применяют также к принадлежностям, которые предназначены изготовителем для использования в комплекте с конкретным медицинским изделием в целях обеспечения его предусмотренного применения.
2.10 объективное свидетельство (objective evidence): Данные, подтверждающие наличие или истинность чего-либо.
Примечание - Объективное свидетельство может быть получено посредством наблюдения, измерения, испытания или другими способами (см. [5], 3.8.1).
2.11 постпроизводство (post-production): Часть жизненного цикла изделия после окончания его проектирования и изготовления.
Пример - К постпроизводству относят: транспортирование, хранение, монтаж, применение изделия, техническое обслуживание, ремонт, модификацию, снятие с эксплуатации и утилизацию.
2.12 процедура (procedure): Установленный способ осуществления деятельности или процесса (см. [5], 3.4.5).
2.13 процесс (process): Совокупность взаимосвязанных и взаимодействующих видов деятельности, преобразующая входы в выходы (см. [5], 3.4.1).
2.14 запись (record): Документ, содержащий достигнутые результаты или свидетельства осуществленной деятельности (см. [5], 3.7.6).
2.15 остаточный риск (residual risk): Риск, остающийся после выполнения мер по управлению риском.
Примечания
1 Заимствовано из [2], 3.9.
2 В [2], 3.9, использован термин "защитные меры" вместо термина "меры по управлению риском". Однако в контексте настоящего стандарта "защитные меры" являются только одной из возможностей управления риском, как описано в 6.2.
2.16 риск (risk): Сочетание вероятности причинения вреда и тяжести этого вреда (см. [2], 3.2).
2.17 анализ риска (risk analysis): Систематическое использование имеющейся информации для выявления опасностей и определения риска (см. [2], 3.10).
Примечание - Анализ риска включает изучение последовательностей событий, которые могут привести к опасным ситуациям и причинению вреда (см. приложение E).
2.18 оценка риска (risk assessment): Полный процесс анализа и оценивания риска (см. [2], 3.12).
2.19 управление риском (risk control): Процесс принятия решений и выполнения мер по уменьшению рисков до установленных уровней или поддержания рисков на установленных уровнях.
2.20 определение риска (risk estimation): Процесс, применяемый для присвоения значений вероятности наступления вреда и тяжести этого вреда.
2.21 оценивание риска (risk evaluation): Процесс сравнения риска, который уже определен, с установленными критериями риска для определения допустимости риска.
2.22 менеджмент риска (risk management): Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания, управления и мониторинга риска.
2.23 файл менеджмента риска (risk management file): Совокупность записей и других документов, создаваемых в процессе менеджмента риска.
2.24 безопасность (safety): Отсутствие недопустимого риска (см. [2], 3.1).
2.25 тяжесть (severity): Мера возможных последствий опасности.
2.26 высшее руководство (top management): Лицо или группа лиц, осуществляющих направление деятельности и управление организацией-изготовителем на высшем уровне.
Примечание - Заимствовано из [5], 3.2.7.
2.27 ошибка применения (use error): Выполнение или невыполнение действия, приводящее к функционированию медицинского изделия, отличающемуся от предусмотренного изготовителем или ожидаемого пользователем (см. [6], 2.12).
Примечания
1 К ошибкам применения относят промахи, упущения и заблуждения (см. [6], 2.12).
2 См. [6], а также приложения B и D.1.3.
3 Неадекватную физиологическую реакцию пациента саму по себе не относят к ошибке применения.
2.28 верификация (verification): Подтверждение на основе представления объективных свидетельств того, что установленные требования были выполнены (см. [5], 3.8.4).
Примечания
1 Термин "верифицировано" используют для обозначения соответствующего статуса.
2 Деятельность по подтверждению может включать:
- осуществление альтернативных расчетов;
- сравнение научной и технической документации по новому проекту с аналогичной документацией по апробированному проекту;
- проведение испытаний и демонстраций;
- анализ документов до их выпуска.
3 Общие требования к менеджменту риска
3.1 Процесс менеджмента риска
Изготовитель должен установить, документировать и поддерживать в рабочем состоянии непрерывный процесс идентификации опасностей, связанных с медицинским изделием, определения и оценивания сопутствующих рисков, управления данными рисками и мониторинга результативности такого управления на протяжении всего жизненного цикла медицинского изделия. Этот процесс должен включать следующие элементы:
- анализ риска;
- оценивание риска;
- управление риском;
- производственную и постпроизводственную информацию.
Документированные процессы жизненного цикла продукции (см. раздел 7 [3]) должны включать соответствующие элементы процесса менеджмента риска.
Примечания
1 Документированный процесс системы менеджмента качества может быть использован для обеспечения системного подхода к рассмотрению проблем безопасности, позволяющего, в частности, идентифицировать на ранних стадиях опасности и опасные ситуации, связанные со сложными медицинскими изделиями и системами.
2 Процесс менеджмента риска схематически представлен на рисунке 1.
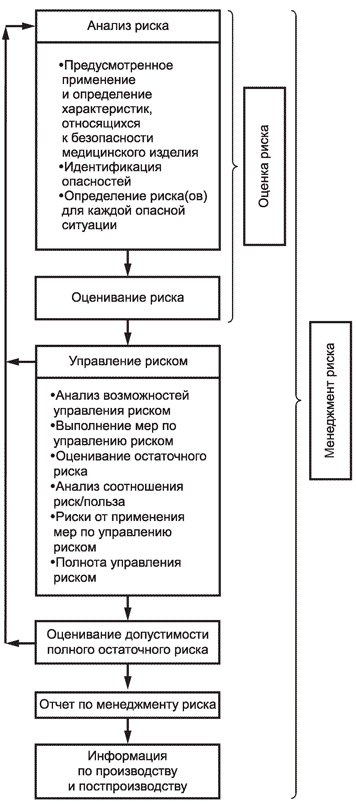
Рисунок 1 - Схематичное представление процесса менеджмента риска
В зависимости от конкретной стадии жизненного цикла медицинского изделия отдельным элементам менеджмента риска может быть уделено особое внимание. Деятельность по менеджменту риска может осуществляться итеративно или поэтапно в зависимости от рассматриваемого медицинского изделия. Приложение B содержит более подробный обзор этапов процесса менеджмента риска.
Соответствие требованиям данного подраздела проверяют путем экспертизы соответствующих документов.
3.2 Ответственность высшего руководства
Высшее руководство должно обеспечивать свидетельства своей приверженности процессу менеджмента риска посредством:
- обеспечения необходимыми ресурсами;
- назначения квалифицированного персонала (см. 3.3) для целей менеджмента риска.
Высшее руководство должно:
- разрабатывать и документировать политику установления критериев допустимости риска. Данная политика должна гарантировать, что установленные критерии основаны на применимых национальных и региональных нормативных документах и соответствующих международных стандартах, а также учитывают доступную информацию, такую как современный уровень научно-технического развития и потребности заинтересованных сторон;
- проводить анализ пригодности процесса менеджмента риска в запланированные промежутки времени для обеспечения постоянной результативности данного процесса и документировать все решения и предпринятые действия. Если изготовитель имеет действующую систему менеджмента качества, то данный анализ может быть частью анализа его системы менеджмента качества.
Примечание - Вышеуказанные документы могут быть включены в документы системы менеджмента качества изготовителя, и на них могут быть ссылки в файле менеджмента риска.
Соответствие требованиям данного подраздела проверяют путем экспертизы соответствующих документов.
3.3 Квалификация персонала
Персонал, выполняющий задачи по менеджменту риска, должен иметь знания и опыт, обеспечивающие возможность выполнения данных задач. Квалификация персонала должна включать знание рассматриваемых (или подобных) медицинских изделий, опыт их применения, а также владение применяемыми технологиями и методами менеджмента риска. Записи о необходимой квалификации персонала следует поддерживать в рабочем состоянии.
Примечание - Задачи по менеджменту риска могут решать представители разных функциональных подразделений с учетом имеющихся у них специальных знаний.
Соответствие требованиям данного подраздела проверяют путем экспертизы вышеуказанных записей.
3.4 План менеджмента риска
Деятельность по менеджменту риска необходимо планировать. Ввиду этого изготовитель должен составить и документировать план менеджмента риска для рассматриваемого медицинского изделия в соответствии с процессом менеджмента риска. План менеджмента риска должен быть частью файла менеджмента риска.
Данный план должен включать, как минимум:
a) объем применения запланированной деятельности по менеджменту риска, в том числе идентификацию и описание медицинского изделия и стадий его жизненного цикла, к которым применим каждый элемент плана;
b) распределение ответственности и полномочий;
c) требования к анализу деятельности по менеджменту риска;
d) критерии допустимости риска, основанные на политике изготовителя по установлению допустимого риска, включая случаи, когда вероятность причинения вреда не может быть определена;
e) действия по верификации;
f) действия по сбору и анализу информации, относящейся к менеджменту риска, на производственной и постпроизводственной стадиях.
Примечания
1 Руководящие указания по разработке плана менеджмента риска см. в приложении F.
2 Необязательно все элементы плана менеджмента риска разрабатывать одновременно. План или его элементы можно разрабатывать поэтапно.
3 Критерии допустимости риска имеют большое значение для определения конечной результативности процесса менеджмента риска. Для каждого плана менеджмента риска изготовителю следует выбирать надлежащие критерии допустимости риска.
Среди прочих можно рассматривать следующие варианты:
- построение матрицы [рисунки D.4 и D.5 (приложение D)], иллюстрирующей допустимые и недопустимые комбинации вероятности причинения вреда и тяжести вреда;
- дальнейшее подразделение области матрицы ("пренебрежимо малый риск", "допустимый риск при условии его минимизации" и т.д.) с требованием к рискам, чтобы они сначала были уменьшены, насколько это практически осуществимо, прежде чем признать их допустимыми [см. D.8 (приложение D)].
Любой из вариантов следует выбирать в соответствии с политикой изготовителя в отношении установления критериев допустимости риска и на основании применимых национальных или региональных нормативных документов, а также соответствующих международных стандартов с учетом доступной информации, такой как современный уровень научно-технического развития и интересы заинтересованных сторон (см. 3.2). Руководство по установлению данных критериев см. в D.4 (приложение D).
При внесении изменений в план менеджмента риска в течение жизненного цикла медицинского изделия необходимо сделать запись об изменениях в файле менеджмента риска.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
3.5 Файл менеджмента риска
Для рассматриваемого медицинского изделия изготовитель должен создать и поддерживать в рабочем состоянии файл менеджмента риска. В дополнение к требованиям других разделов настоящего стандарта файл менеджмента риска должен обеспечивать возможность прослеживания каждой идентифицированной опасности при:
- анализе риска;
- оценивании риска;
- выполнении и верификации мер по управлению риском;
- оценивании допустимости любого(ых) остаточного(ых) риска(ов).
Примечания
1 Записи и другие документы, составляющие файл менеджмента риска, могут быть частью других документов и файлов, требуемых, например, системой менеджмента качества изготовителя. Файл менеджмента риска необязательно должен непосредственно включать все записи и другие документы, относящиеся к настоящему стандарту. Однако он должен содержать, по меньшей мере, ссылки или указания на все требуемые документы. Изготовителю следует своевременно собрать ссылочную информацию в файле менеджмента риска.
2 Файл менеджмента риска может быть представлен в любой форме или на любом носителе информации.
4 Анализ риска
4.1 Процесс анализа риска
Анализ риска рассматриваемого медицинского изделия необходимо проводить в соответствии с 4.2-4.4. Деятельность по запланированному анализу риска, а также результаты анализа риска должны быть зарегистрированы в файле менеджмента риска.
Примечания
1 Если доступны результаты анализа риска или другая относящаяся к риску информация для подобного медицинского изделия, то их можно использовать в качестве отправной точки при новом анализе. Степень сопоставимости зависит от различий между изделиями и от того, могут ли данные различия стать источником новых опасностей или существенных различий в готовой продукции, характеристиках, функционировании или результатах применения. Возможность применения уже имеющегося анализа риска основана также на систематическом оценивании влияния изменений на развитие опасных ситуаций.
2 Некоторые методы анализа риска описаны в приложении G.
3 Дополнительные руководящие указания по методам анализа риска медицинских изделий для диагностики in vitro приведены в приложении H.
4 Дополнительные руководящие указания по методам анализа риска в отношении токсикологических опасностей приведены в приложении I.
В дополнение к записям, требуемым в 4.2-4.4, документы, относящиеся к проведению и результатам анализа риска, должны включать, как минимум:
a) описание и идентификацию рассматриваемого медицинского изделия;
b) идентификацию лица (лиц) и организации, выполнивших анализ риска;
c) область применения и дату проведения анализа риска.
5 Область применения анализа риска может быть очень широкой (например, разработка нового изделия, в отношении применения которого у изготовителя мало опыта или данный опыт вообще отсутствует) или ограниченной (например, анализ влияния привносимых изменений на выпускаемое изделие, о котором в файлах изготовителя имеется обширная информация).
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
4.2 Предусмотренное применение и определение характеристик, относящихся к безопасности медицинского изделия
Для рассматриваемого медицинского изделия изготовитель должен документировать все случаи предусмотренного применения и обоснованно прогнозируемого неправильного применения. Изготовитель должен также идентифицировать и документировать все качественные и количественные характеристики, которые могут повлиять на безопасность медицинского изделия, и при необходимости указать их предельно допустимые значения. Данные документы необходимо поддерживать в рабочем состоянии в файле менеджмента риска.
Примечания
1 В контексте настоящего стандарта неправильное применение означает непредусмотренное или ненадлежащее применение медицинского изделия.
2 Приложение С содержит вопросы, относящиеся к применению медицинского изделия, которые могут стать полезным ориентиром при идентификации характеристик данного изделия, способных повлиять на его безопасность.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
4.3 Идентификация опасностей
Изготовитель должен составить перечень известных или прогнозируемых опасностей, связанных с рассматриваемым медицинским изделием как для нормальных условий, так и для условий отказа.
Данный перечень необходимо поддерживать в рабочем состоянии в файле менеджмента риска.
Примечание - Примеры возможных опасностей, приведенные в E.2 (приложение Е) и H.2.4 (приложение Н), могут быть использованы как руководство для изготовителя, выполняющего идентификацию опасностей.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
4.4 Определение риска(ов) для каждой опасной ситуации
Необходимо рассматривать обоснованно прогнозируемые последовательности или комбинации событий, приводящие к возникновению опасной ситуации, и регистрировать возникающую(ие) опасную(ые) ситуацию(и).
Примечания
1 Для идентификации не выявленных ранее опасных ситуаций можно использовать системные методы, применимые в конкретной ситуации (см. приложение G).
2 Примеры опасных ситуаций приведены в H.2.4.5 (приложение Н) и E.4 (приложение Е).
3 Источником опасных ситуаций могут стать промахи, упущения и заблуждения.
Для каждой идентифицированной опасной ситуации необходимо определять связанный(е) с ней риск(и), используя для этого доступную информацию или данные. Для опасных ситуаций, в отношении которых не может быть определена вероятность причинения вреда, должен быть подготовлен перечень возможных последствий применения медицинского изделия, используемый при оценивании и управлении риском. Результаты этой деятельности должны быть зарегистрированы в файле менеджмента риска.
Любая система, используемая для качественной или количественной градации вероятности причинения вреда или тяжести вреда, также должна быть зарегистрирована в файле менеджмента риска.
Примечания
1 Определение риска медицинского изделия включает анализ вероятности возникновения риска и последствий применения данного изделия. В зависимости от применения медицинского изделия, возможно, достаточно рассмотреть лишь некоторые элементы процесса определения риска. Например, в отдельных случаях нет необходимости идти дальше анализа исходной опасности и последствий применения [см. также D.3 (приложение D)].
2 Определение риска может быть количественным или качественным. Методы определения риска, в том числе являющиеся следствием систематических отказов, описаны в приложении D. Приложение H содержит информацию, полезную при определении рисков медицинских изделий для диагностики in vitro.
3 Информацию или данные для определения рисков можно получить из:
a) опубликованных стандартов;
b) научно-технической информации;
c) данных о применении подобных медицинских изделий, включая опубликованные сведения об инцидентах;
d) данных испытаний эксплуатационной пригодности типичными пользователями;
e) клинических данных;
f) результатов соответствующих исследований;
g) экспертных заключений;
h) схем внешней оценки качества.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
5 Оценивание риска
Для каждой идентифицированной опасной ситуации изготовитель должен принять решение о необходимости уменьшения риска с учетом критериев, установленных в плане менеджмента риска. Если в уменьшении риска нет необходимости, то требования 6.2-6.6 для данной опасной ситуации не применяют (т.е. переходят к 6.7). Результаты такого оценивания риска должны быть зарегистрированы в файле менеджмента риска.
Примечания
1 Руководящие указания по принятию решения о допустимости риска приведены в D.4 (приложение D).
2 Применение соответствующих стандартов, как один из критериев проектирования медицинских изделий, может являться частью деятельности по управлению риском, что отвечает требованиям 6.3-6.6.
Соответствие требованиям данного раздела проверяют путем экспертизы файла менеджмента риска.
6 Управление риском
6.1 Уменьшение риска
При необходимости уменьшения риска следует осуществлять деятельность по управлению риском, как описано в 6.2-6.7.
6.2 Анализ возможностей управления риском
Изготовитель должен идентифицировать меру(ы) по управлению риском, необходимую(ые) для уменьшения риска(ов) до допустимого уровня.
Изготовитель должен применять один или несколько способов управления риском, перечисленных ниже в порядке приоритетов:
a) внутреннюю безопасность, обеспечиваемую проектом и конструкцией;
b) защитные меры, предусмотренные в самом медицинском изделии или в процессе его изготовления;
c) информацию по безопасности.
Примечания
1 При применении способов, приведенных в перечислениях b) и c), изготовитель может рассмотреть обоснованные и практически осуществимые меры по управлению риском и выбрать способ, обеспечивающий необходимое уменьшение риска, прежде чем будет установлено, является ли риск допустимым.
2 Меры по управлению риском могут уменьшить тяжесть вреда или вероятность причинения вреда, или и то и другое вместе.
3 Многие стандарты рассматривают вопросы внутренней безопасности, обеспечиваемой проектом и конструкцией, защитные меры и информацию по безопасности медицинских изделий. Кроме того, другие стандарты на медицинские изделия рассматривают те элементы процесса менеджмента риска (электромагнитную совместимость, эксплуатационную пригодность, биологическую совместимость), которые являются общими для медицинских изделий. При анализе возможностей управления риском следует применять соответствующие стандарты.
4 В отношении рисков, для которых не может быть определена вероятность причинения вреда, см. D.3.2.3 (приложение D).
5 Руководящие указания в отношении информации по безопасности приведены в приложении J.
Выбранные меры по управлению риском должны быть зарегистрированы в файле менеджмента риска.
Если в процессе анализа возможностей управления риском изготовитель устанавливает, что требуемое уменьшение риска практически неосуществимо, то он должен выполнить анализ соотношения риск/польза для остаточного риска (переход к 6.5).
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
6.3 Выполнение мер по управлению риском
Изготовитель должен выполнять меры по управлению риском, выбранные в соответствии с 6.2.
Выполнение каждой меры по управлению риском должно быть верифицировано. Данная верификация должна быть зарегистрирована в файле менеджмента риска.
Результативность мер по управлению риском должна быть верифицирована, а результаты верификации - зарегистрированы в файле менеджмента риска.
Примечание - Верификация результативности может включать действия по валидации.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
6.4 Оценивание остаточного риска
Любой остаточный риск, сохраняющийся после выполнения мер по управлению риском, необходимо оценивать в соответствии с критериями, установленными в плане менеджмента риска. Результаты данного оценивания должны быть зарегистрированы в файле менеджмента риска.
Если остаточный(е) риск(и) согласно установленным критериям оценен(ы) как недопустимый(е), то необходимо применить дополнительные меры по управлению риском (см. 6.2).
Если остаточные риски оценены как допустимые, то изготовитель должен принять решение, о каких остаточных рисках необходимо информировать и какую именно информацию необходимо включить в эксплуатационные документы в целях привлечения внимания к остаточным рискам.
Примечание - Руководящие указания по информированию об остаточном риске(ах) приведены в приложении J.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска и эксплуатационных документов.
6.5 Анализ соотношения риск/польза
Если остаточный риск по критериям, установленным в плане менеджмента риска, сочтен недопустимым, а дальнейшее управление риском - практически неосуществимым, то изготовитель может собрать и проанализировать имеющиеся данные и литературу, чтобы установить, превышает ли польза при предусмотренном применении медицинского изделия остаточный риск. Если собранные доказательства свидетельствуют о том, что польза от предусмотренного применения медицинского изделия не превышает остаточный риск, то риск считается недопустимым. Если польза от предусмотренного применения медицинского изделия превышает остаточный риск, то можно перейти к выполнению требований 6.6.
В отношении риска, где польза от применения медицинского изделия превышает риск, изготовитель должен решить, какую информацию по безопасности необходимо предоставить для информирования об остаточном риске.
Результаты оценивания должны быть зарегистрированы в файле менеджмента риска.
Примечание - См. также D.6 (приложение D).
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
6.6 Риски, возникающие вследствие выполнения мер по управлению риском
Эффективность выполнения мер по управлению риском необходимо анализировать с точки зрения:
a) возникновения новых опасностей и опасных ситуаций;
b) влияния выполненных мер по управлению риском на риски, определенные для ранее идентифицированных опасных ситуаций.
Любыми новыми или возросшими рисками необходимо управлять в соответствии с требованиями, приведенными в 4.4-6.5.
Результаты анализа мер по управлению риском следует зарегистрировать в файле менеджмента риска.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
6.7 Полнота управления риском
Изготовитель должен обеспечить рассмотрение риска(ов) для всех идентифицированных опасных ситуаций. Результаты данной деятельности должны быть зарегистрированы в файле менеджмента риска.
Соответствие требованиям данного подраздела проверяют путем экспертизы файла менеджмента риска.
7 Оценивание допустимости совокупного остаточного риска
После выполнения и верификации всех мер по управлению риском изготовитель должен принять решение о допустимости совокупного остаточного риска, создаваемого медицинским изделием, с точки зрения критериев, установленных в плане менеджмента риска.
Примечание - Руководящие указания по оцениванию совокупного остаточного риска приведены в D.7 (приложение D).
Если совокупный остаточный риск по критериям, установленным в плане менеджмента риска, оценен как недопустимый, то изготовитель может собрать и проанализировать имеющиеся данные и литературу, чтобы установить, превышает ли польза при предусмотренном применении медицинского изделия совокупный остаточный риск. Если собранные доказательства свидетельствуют о том, что польза от предусмотренного применения медицинского изделия превышает совокупный остаточный риск, то риск считается допустимым. В противном случае совокупный остаточный риск считается недопустимым.
В отношении совокупного остаточного риска, который оценен как допустимый, изготовитель должен решить, какую информацию необходимо включить в эксплуатационные документы для информирования о совокупном остаточном риске.
Примечание - Руководящие указания по информированию об остаточном риске(ах) приведены в приложении J.
Результаты оценивания совокупного остаточного риска должны быть зарегистрированы в файле менеджмента риска.
Соответствие требованиям данного раздела проверяют путем экспертизы файла менеджмента риска и эксплуатационных документов.
8 Отчет по менеджменту риска
Перед введением медицинского изделия в обращение изготовитель должен провести анализ процесса менеджмента риска. Данный анализ должен, по меньшей мере, свидетельствовать о том, что:
- менеджмент риска осуществлен в соответствии с планом;
- совокупный остаточный риск является допустимым;
- применяют надлежащие способы получения необходимой производственной и постпроизводственной информации.
Результаты анализа процесса менеджмента риска должны быть занесены в отчет по менеджменту риска и включены в файл менеджмента риска.
В плане менеджмента риска должны быть указаны лица, имеющие необходимые полномочия и ответственные за проведение анализа процесса менеджмента риска [см. 3.4, перечисление b)].
Соответствие данному разделу проверяют путем экспертизы файла менеджмента риска.
9 Производственная и постпроизводственная информация
Изготовитель должен разработать, документировать и поддерживать в рабочем состоянии систему сбора и анализа информации о рассматриваемом медицинском изделии или подобных изделиях на стадиях производства и постпроизводства.
При разработке системы сбора и анализа информации о рассматриваемом медицинском изделии изготовитель среди прочего должен учитывать:
a) механизмы, с помощью которых можно собирать и обрабатывать информацию, поступающую от операторов, пользователей или других лиц, ответственных за установку/монтаж, применение и поддержание в рабочем состоянии медицинского изделия;
b) новые или пересмотренные стандарты.
В рамках данной системы следует также собирать и анализировать общедоступную информацию, опубликованную о подобных медицинских изделиях, находящихся на рынке.
Данную информацию необходимо оценивать с точки зрения соответствия требованиям к безопасности, особенно с учетом того:
- существуют ли не выявленные ранее опасности или опасные ситуации;
- не стал ли недопустимым риск(и), определенный(ые) ранее для какой-либо опасной ситуации.
При положительном ответе на любой из данных вопросов необходимо:
- оценить влияние вышеуказанных факторов на ранее осуществленную деятельность по менеджменту риска и по результатам оценивания начать процесс менеджмента риска заново;
- провести анализ файла менеджмента риска по рассматриваемому медицинскому изделию; при наличии потенциальной возможности изменения остаточного риска(ов) или его (их) допустимости следует оценить воздействие вышеуказанных факторов на ранее выполненные меры по управлению риском.
Результаты оценивания должны быть зарегистрированы в файле менеджмента риска.
Примечания
1 Отдельные аспекты постпроизводственного мониторинга являются предметом национальных или региональных регламентов. В таких случаях могут быть задействованы дополнительные меры (например, перспективное постпроизводственное оценивание).
2 См. также [3], 8.2.
Соответствие требованиям данного раздела проверяют путем экспертизы файла менеджмента риска и другой соответствующей документации.
Приложение A
(справочное)
Обоснование требований
A.1 Общие положения
Объединенная рабочая группа 1 ИСО/ТК 210 - МЭК/ПК 62A "Применение менеджмента риска к медицинским изделиям" разработала данное приложение, для того чтобы документировать обоснованность требований, содержащихся в первой версии настоящего стандарта. В ходе разработки настоящей версии обоснование требований было обновлено с учетом изменений в нормативных документах. При дальнейшем пересмотре настоящего стандарта можно использовать данное приложение наряду с опытом, приобретенным в ходе применения настоящего стандарта, для того чтобы сделать настоящий стандарт еще более полезным для изготовителей, регулирующих органов и поставщиков медицинских услуг.
Важность разработки стандарта, рассматривающего менеджмент риска медицинских изделий, возросла благодаря осознанию регулирующими органами необходимости применения процесса менеджмента риска изготовителями медицинских изделий. Ранее не существовало ни одного стандарта по менеджменту риска медицинских изделий, и настоящий стандарт был призван заполнить пробел в данной области. Для разработки стандарта была сформирована рабочая группа 4 ИСО/ТК 210. Практически одновременно было запланировано включение в стандарт элементов, касающихся менеджмента риска. Была признана необходимость выделения деятельности по менеджменту риска и сформирована рабочая группа 15 МЭК/ПК 62A. Осознав, что интересы двух рабочих групп пересекаются, МЭК и ИСО сформировали объединенную рабочую группу 1 (JWG 1) по менеджменту риска. Совместная работа привела к изданию стандарта c двумя логотипами - ИСО и МЭК. Обе организации (как ИСО, так и МЭК) также признают стандарты друг друга, относящиеся к менеджменту риска, но опубликованные под каким-то одним логотипом, в качестве международных стандартов. Двойной логотип подчеркивает, что стандарт разработан усилиями двух сообществ - посредством участия государств - членов ИСО и национальных комитетов МЭК.
На первоначальном этапе обсуждения стандарта по менеджменту риска необходимо было решить ключевые вопросы менеджмента риска, такие как процесс оценивания риска, достижение баланса соотношения риск/польза для медицинских изделий. Изготовители, регулирующие органы и поставщики медицинских услуг пришли к осознанию того факта, что "абсолютная" безопасность в отношении медицинских изделий недостижима. Кроме того, риски, возникающие вследствие увеличения диверсификации медицинских изделий и расширения области их применения, невозможно всесторонне рассмотреть в рамках стандартов по безопасности продукции. Осознание данных фактов и вытекающая из них потребность в менеджменте риска медицинских изделий на протяжении всего их жизненного цикла способствовали принятию решения о разработке первой версии стандарта по менеджменту риска.
Первоначально стандарт планировали разработать в нескольких частях, каждая из которых была бы посвящена конкретному элементу менеджмента риска. Часть, посвященная анализу риска, должна была стать первой частью общего стандарта по менеджменту риска. Позже было принято решение о разработке единого документа, включающего все элементы менеджмента риска. Главной причиной стала очевидность того факта, что выполнение менеджмента риска является обязательным видом деятельности сразу в нескольких международных регулирующих системах. Поэтому было признано нецелесообразным иметь отдельный стандарт по анализу риска для каждой из этих систем. Кроме того, разработка единого стандарта по менеджменту риска вместо нескольких частей должна была лучше продемонстрировать связь между разными элементами менеджмента риска.
Настоящая версия разработана для того, чтобы внести дополнительные руководящие указания по применению настоящего стандарта. Незначительные изменения внесены в основную часть: добавлено требование планирования мониторинга постпроизводственной деятельности и из отчета по менеджменту риска удалено требование прослеживаемости.
В приложении E (ранее - приложение D) приведены новые руководящие указания в отношении взаимосвязи опасностей и опасных ситуаций. Все случаи использования данных терминов в настоящем стандарте проанализированы в целях обеспечения согласованности с данными руководящими указаниями.
Дальнейшая информация содержится в нижеприведенных разделах и подразделах настоящего стандарта.
A.2 Обоснование требований, содержащихся в конкретных разделах и подразделах настоящего стандарта
А.2.1 Область применения
Во введении к настоящему стандарту содержится объяснение необходимости разработки стандарта по менеджменту риска, применимого к проектированию и изготовлению всех медицинских изделий. Медицинские изделия для диагностики in vitro специально оговорены в области применения во избежание любого непонимания, которое в силу различий в регулирующих документах разных стран могло бы исключить данные изделия из области применения настоящего стандарта.
Риски существуют на протяжении всего жизненного цикла медицинского изделия. Рисками, выявленными на одной стадии жизненного цикла медицинского изделия, можно управлять с помощью действий, выполняемых на совершенно другой стадии жизненного цикла. По этой причине настоящий стандарт должен быть применим ко всем стадиям жизненного цикла медицинского изделия. Это означает, что стандарт ориентирует изготовителя применять принципы менеджмента риска к медицинскому изделию, начиная от первоначального замысла и до вывода из эксплуатации и утилизации.
Область применения настоящего стандарта не включает принятие решения относительно применения медицинского изделия. Принятие решения о проведении клинической процедуры с применением медицинского изделия требует достижения оптимального соотношения между остаточными рисками и ожидаемой пользой от проведения данной процедуры или альтернативных процедур. При вынесении таких решений необходимо учитывать предусмотренное применение, клиническую пользу и риски, связанные с конкретным медицинским изделием, а также риски и пользу, связанные с клинической процедурой или условиями применения медицинского изделия. Некоторые из этих решений могут быть приняты только квалифицированным медицинским работником, владеющим информацией о состоянии здоровья конкретного пациента или знающим субъективное мнение самого пациента по данному вопросу.
Несмотря на серьезную полемику по поводу уровней допустимости риска, настоящий стандарт не устанавливает данные уровни. Установление единого для всех случаев уровня допустимого риска было бы неуместным по следующим причинам:
- из-за большого разнообразия изделий и ситуаций, охватываемых областью применения настоящего стандарта, что лишает смысла выработку единого уровня допустимого риска;
- из-за наличия местных законов, обычаев, ценностей и особенностей восприятия риска, более подходящих для определения допустимого риска в конкретной культурной среде и конкретном регионе.
Поскольку не во всех странах требуется наличие системы менеджмента качества у изготовителей медицинских изделий, то система менеджмента качества не является требованием настоящего стандарта. Однако наличие системы менеджмента качества чрезвычайно важно для надлежащего менеджмента рисков. По этой причине, а также потому, что большинство изготовителей используют систему менеджмента качества, настоящий стандарт построен таким образом, что он легко может быть включен в применяемую изготовителем систему менеджмента качества.
A.2.2 Термины и определения
Во избежание изобретения множества новых и, возможно, непривычных терминов настоящий стандарт намеренно основан на большом количестве информации по менеджменту риска, содержащейся как в стандартах, так и в прочих изданиях. Там, где возможно, использованы уже имеющиеся термины с соответствующими определениями. Основными источниками терминов и определений являются [2], [3], [5].
Определения некоторых терминов в настоящем стандарте отличаются от общепринятых. Например, объединенная рабочая группа 1 (JWG 1) намеревалась включить в определение термина "вред" (2.2) упоминание о чрезмерном психологическом стрессе или нежелательной беременности как части "вреда, наносимого здоровью людей". Было известно, что выполнение менеджмента риска будет обязательным требованием, выраженным в явной или скрытой форме, во многих странах или регионах. В этой связи была сделана попытка использовать определения, широко распространенные в регулирующих документах. Например, определение термина "изготовитель" (2.8), содержащееся в [10], соответствует определению того же термина, используемому в США. Термин "медицинское изделие" и его определение (2.9) взяты из [3], где они заимствованы у Группы по глобальной гармонизации (GHTF) [4].
Определение термина "предусмотренное применение" (2.5) является комбинацией из определений терминов "предусмотренное применение" (США) и "предусмотренное назначение" (ЕС). Эти термины имеют практически одно и то же определение. Считается, что при рассмотрении предусмотренного применения медицинского изделия изготовитель принимает во внимание предполагаемых пользователей данного изделия.
Определения семи других терминов, приведенные в настоящем стандарте, не основаны на определениях, приведенных в других стандартах. Это определения таких терминов, как "жизненный цикл" (2.7), "постпроизводство" (2.11), "управление риском" (2.19), "оценивание риска" (2.21), "определение риска" (2.20), "менеджмент риска" (2.22) и "файл менеджмента риска" (2.23). Определение термина "жизненный цикл" необходимо для разъяснения того положения, что в контексте настоящего стандарта данный термин относится ко всем стадиям существования медицинского изделия. Определение термина "постпроизводство" введено, для того чтобы привлечь внимание к важности охвата всего жизненного цикла медицинского изделия для целей менеджмента риска. Определение термина "управление риском" согласовано с определением термина "анализ риска", приведенным в [2]. В первоначальной версии настоящего стандарта в определении термина "оценивание риска" была ссылка на "существующие общественные ценности". В данной версии эта ссылка отсутствует по двум причинам: 1) определение термина не должно содержать требование; 2) термин "существующие общественные ценности" не является достаточно точным. Удаление данного термина из определения компенсируется описанием концепции риска во введении к настоящему стандарту, обеспечением дополнительных нормативных требований к политике менеджмента риска, а также руководящими указаниями в отношении допустимости риска. В определении термина "менеджмент риска" сделан акцент на использовании системного подхода и необходимости контроля со стороны высшего руководства. Концепция "файла менеджмента риска" впервые была описана в [7], но в определение данного термина внесены изменения, поскольку определение из [7] относится к записям по качеству.
Определение термина "высшее руководство" (2.26) заимствовано из [5]. Оно относится к лицу или группе лиц, находящихся на высшей ступени организации.
А.2.3 Общие требования к менеджменту риска
А.2.3.1 Процесс менеджмента риска
Требование к изготовителю установить процесс менеджмента риска как часть проектирования и разработки медицинского изделия содержит 3.1. Это необходимо, для того чтобы изготовитель мог систематически обеспечивать наличие требуемых элементов в процессе менеджмента риска. Анализ риска, оценивание риска и управление риском являются общепризнанными основными элементами менеджмента риска. Однако в настоящем стандарте делается акцент на то, что процесс менеджмента риска не заканчивается на проектировании, разработке и изготовлении (включая при необходимости стерилизацию, упаковывание и маркирование) медицинского изделия, а продолжается на стадии постпроизводства. По этой причине сбор постпроизводственной информации был признан необходимым элементом процесса менеджмента риска. Кроме того, при наличии у изготовителя системы менеджмента качества процесс менеджмента риска должен быть полностью интегрирован в данную систему качества.
Несмотря на то, что деятельность по менеджменту риска во многом зависит от конкретного медицинского изделия, существуют основные элементы, которые необходимо включать в процесс менеджмента риска и которые рассмотрены в данном подразделе. В данном подразделе также отражена возможность различий в регулирующих документах по применению менеджмента риска к медицинским изделиям.
Требованиям стандартов по системе менеджмента качества строго следуют 3.2 и 3.3. В некоторых странах для введения изделия в обращение необходимо наличие системы менеджмента качества (кроме тех случаев, когда изделие специально исключено из области применения системы менеджмента качества). В других странах применение системы менеджмента качества является добровольным выбором изготовителя. Тем не менее выполнение требований 3.2 и 3.3 необходимо для обеспечения результативности менеджмента риска независимо от того, использует ли изготовитель все прочие элементы системы менеджмента качества.
A.2.3.2 Ответственность высшего руководства
Обязательства высшего руководства играют решающую роль в достижении результативности процесса менеджмента риска. Высшее руководство должно нести ответственность за процесс менеджмента риска в целом, и в данном подразделе уделено особое внимание роли высшего руководства, особенно следующим моментам:
a) при отсутствии необходимых ресурсов деятельность по менеджменту риска будет менее результативной, даже если она отвечает всем другим требованиям настоящего стандарта;
b) менеджмент риска является специализированной дисциплиной и требует вовлечения в процесс профессионалов, специально обученных методам менеджмента риска (см. А.2.3.3);
c) поскольку настоящий стандарт не устанавливает уровни допустимого риска, от высшего руководства требуется разработать политику установления допустимости рисков;
d) менеджмент риска представляет собой эволюционный процесс, и поэтому необходимо проводить периодический анализ деятельности по менеджменту риска, для того чтобы удостовериться в ее правильности, исправить недостатки, внедрить усовершенствования и адаптироваться к изменениям.
А.2.3.3 Квалификация персонала
Для выполнения задач по менеджменту риска важно иметь квалифицированный персонал. Процесс менеджмента риска требует привлечения персонала, имеющего практический опыт в том, как:
- устроено медицинское изделие;
- функционирует медицинское изделие;
- изготовлено медицинское изделие;
- применять медицинское изделие;
- применять процесс менеджмента риска.
К выполнению менеджмента риска необходимо привлекать представителей разных функциональных подразделений и дисциплин - специалистов в своей области. При этом следует взвешенно подходить к вопросу взаимоотношений между сотрудниками, выполняющими задачи менеджмента риска.
Для обеспечения объективности свидетельств необходимо вести записи о квалификации персонала. Настоящий стандарт не содержит требования хранить эти записи в файле менеджмента риска во избежание дублирования и из соображений конфиденциальности и защиты информации.
А.2.3.4 План менеджмента риска
План менеджмента риска необходим по следующим соображениям:
a) для надлежащего менеджмента риска важен организованный подход;
b) план является оперативной схемой для менеджмента риска;
c) план способствует объективности менеджмента риска и помогает не забывать о его основных элементах.
Перечисления a)-f) (см. 3.4) обязательны по следующим соображениям:
- область применения плана включает две отличные друг от друга составляющие. Первая из них идентифицирует рассматриваемое медицинское изделие, вторая - стадии жизненного цикла данного изделия и соответствующие им элементы плана. Определив область применения плана менеджмента риска, изготовитель создает базу, на которой строится вся деятельность по менеджменту риска;
- распределение ответственности и полномочий необходимо для гарантии того, что нет упущенной ответственности;
- анализ такого вида деятельности, как менеджмент риска, является обязанностью высшего руководства;
- критерии допустимости риска являются основополагающими для менеджмента риска, их следует устанавливать до начала анализа риска. Это поможет сохранить объективность процесса, описанного в разделе 5;
- верификация является одним из основных видов деятельности, ее проведение является требованием 6.3. Планирование данного вида деятельности помогает обеспечить наличие необходимых ресурсов. Если верификация не запланирована, ее важные составляющие могут быть упущены;
- необходимо разработать специальные методы получения производственной и постпроизводственной информации об изделии, для того чтобы существовала официальная и надлежащим образом организованная обратная связь, обеспечивающая включение производственной и постпроизводственной информации в процесс менеджмента риска.
Для облегчения аудита и анализа процесса менеджмента риска включено требование хранить записи об изменениях.
А.2.3.5 Файл менеджмента риска
В настоящем стандарте термин "файл менеджмента риска" применяют для обозначения намерения изготовителя разместить в конкретном месте или обозначить местоположение всех записей, относящихся к менеджменту риска. Это облегчает процесс менеджмента риска и способствует более эффективному проведению аудита на соответствие требованиям настоящего стандарта. Выполнение требования о прослеживаемости необходимо для демонстрации того, что процесс менеджмента риска применен к каждой идентифицированной опасности.
Важную роль в процессе менеджмента риска играет его завершенность. Незавершенная задача может означать, что идентифицированной опасностью не управляют, следствием чего может стать причинение вреда. Проблема может быть следствием незавершенности действий на любой стадии процесса менеджмента риска. Это неидентифицированные опасности, неоцененные риски, неустановленные меры по управлению риском, невыполненные меры по управлению риском или меры по управлению риском, признанные нерезультативными. Для обеспечения уверенности в завершенности процесса менеджмента риска необходимо осуществление прослеживаемости.
А.2.4 Анализ риска
А.2.4.1 Процесс анализа риска
В 4.1 описано, как применять доступную полезную информацию, используя результаты анализа риска подобного медицинского изделия. Примечание 1 (см. 4.1) информирует пользователей настоящего стандарта о том, что при наличии необходимой информации о подобном изделии ее можно и нужно использовать для экономии времени, усилий и других ресурсов. Однако пользователям настоящего стандарта необходимо внимательно и систематически оценивать данную информацию с точки зрения ее применимости к текущему анализу риска.
Следует отметить, что перечисления a), b) и c) (см. 4.1, примечание 4) содержат требования к основному минимальному набору данных, необходимому для обеспечения прослеживаемости и представляющему важность для анализа со стороны высшего руководства и последующих аудитов. Требования, содержащиеся в перечислении c) (см. 4.1, примечание 4), помогают также уточнить область применения анализа риска и верифицировать его завершенность и полноту.
А.2.4.2 Предусмотренное применение и идентификация характеристик, относящихся к безопасности медицинского изделия
На данном этапе процесса менеджмента риска изготовитель должен проанализировать характеристики, влияющие на безопасность медицинского изделия. Изготовитель также должен принимать во внимание предполагаемого пользователя(ей) медицинского изделия, т.е. учитывать, будет ли это необученный пользователь или квалифицированный медицинский работник. При проведении анализа риска необходимо также учитывать, что медицинские изделия могут быть применены в отличных от предусмотренных изготовителем ситуациях, а также в ситуациях, не предусмотренных на этапах проектирования и разработки медицинского изделия. Важно, чтобы изготовитель попытался заглянуть в будущее и предусмотреть опасности, возможные при применении медицинского изделия.
Приложение С предназначено для оказания помощи в описании характеристик конкретного медицинского изделия и условий его применения. Приведенный в нем перечень вопросов, на которые следует дать ответы, не является исчерпывающим. Каждый изготовитель должен творчески отнестись к определению характеристик, относящихся к безопасности конкретного медицинского изделия. Список, приведенный в приложении С, взят из первоначальной версии настоящего стандарта и имеет дополнения, появившиеся в результате изучения комментариев к проектам стандарта. Список должен побуждать изготовителя к проведению анализа в отношении того, "когда и что может пойти неправильно". Приложение H в отношении медицинских изделий для диагностики in vitro разработано ИСО/TК 212 "Клинические лабораторные исследования и диагностические тест-системы in vitro" специально для настоящего стандарта. Текст приложения I в отношении токсикологических опасностей заимствован из приложения B первоначальной версии настоящего стандарта с незначительными изменениями.
А.2.4.3 Идентификация опасностей
На данном этапе изготовителю необходимо систематически идентифицировать прогнозируемые опасности, связанные с конкретным медицинским изделием, как для нормальных условий, так и для условий отказа. Идентификация должна быть основана на характеристиках, относящихся к безопасности медицинского изделия, идентифицированных в 4.2.
А.2.4.4 Определение риска(ов) для каждой опасной ситуации
Оценивать риск и осуществлять менеджмент риска можно только после того, как опасная ситуация идентифицирована. Необходимо систематически документировать обоснованно прогнозируемые последовательности событий, которые могут преобразовать опасность в опасную ситуацию.
Приложение E включает перечень типичных опасностей и примеры, демонстрирующие взаимосвязь между опасностями, прогнозируемыми последовательностями событий, опасными ситуациями и возможным вредом, что позволяет помочь изготовителю в идентификации опасностей и опасных ситуаций. Это особенно важно при наличии последовательности событий, которая может привести к опасной ситуации и в итоге - к причинению вреда. Изготовителю следует распознавать и идентифицировать такие последовательности событий, для того чтобы должным образом определить возможный(ые) риск(и) [см. рисунок E.1 (приложение Е)].
Перечень, приведенный в приложении E, не является исчерпывающим и не должен рассматриваться как контрольный перечень; он призван стимулировать творческое мышление изготовителя.
Это последний этап анализа риска. Главной трудностью на данном этапе является то, что деятельность по определению риска будет разной для каждой исследуемой опасной ситуации и для каждого рассматриваемого медицинского изделия. Поэтому было принято решение изложить содержание данного подраздела в общих чертах. Поскольку опасности могут возникнуть как при нормальном, так и при неправильном применении изделия, следует внимательно рассмотреть обе ситуации. На практике обе составляющие риска - вероятность причинения и тяжесть вреда - рекомендуют анализировать отдельно. Если изготовитель систематически применяет способ разделения на категории в зависимости от уровней тяжести или вероятности причинения вреда, то ему следует разработать свою схему разделения на категории и зарегистрировать ее в файле менеджмента риска. Это позволит изготовителю правильно реагировать на повторное появление эквивалентных рисков и послужит свидетельством того, как он ранее поступал в подобном случае.
Некоторые опасные ситуации являются следствием систематических ошибок или последовательностей событий. До сих пор нет единого мнения в отношении того, как рассчитать вероятность систематической ошибки. В случае, когда вероятность причинения вреда не может быть рассчитана, возможные опасности все равно необходимо принимать во внимание, а составление перечня связанных с ними опасных ситуаций позволит изготовителю сконцентрироваться на уменьшении рисков вследствие развития этих опасных ситуаций.
Часто нелегко получить достаточные количественные данные при определении риска(ов), поэтому предложение осуществлять определение риска(ов) только количественным способом было отвергнуто.
Приложение D содержит полезные руководящие указания по анализу риска. Информация взята из нескольких источников, включая [8]. Настоящий стандарт признает практическую пользу [8], при этом область его применения распространена на все медицинские изделия и все этапы процесса менеджмента риска. Несмотря на широкое использование в качестве примеров в приложении D графиков и матриц по риску, настоящий стандарт не требует обязательного их применения.
A.2.5 Оценивание риска
Изготовитель должен принимать решения в отношении допустимости риска. Он может использовать информацию о рисках, которые уже определены, и оценить эти риски с помощью критериев допустимости, установленных в плане менеджмента риска. Он может также рассмотреть риски в целях определения тех из них, которые требуется уменьшить. Раздел 5 написан таким образом, чтобы помочь пользователю избежать ненужной работы.
A.2.6 Управление риском
А.2.6.1 Уменьшение риска
Этапы 6.2-6.7 образуют логическую последовательность. Такой систематический подход важен для обеспечения доступа к необходимой информации.
A.2.6.2 Анализ возможностей управления риском
Часто существует несколько способов уменьшения риска. В настоящем стандарте приведены три способа:
a) внутренняя безопасность, обеспечиваемая проектом и конструкцией;
b) защитные меры, предусмотренные в самом медицинском изделии или в процессе его изготовления;
c) информация по безопасности.
Таковы стандартные меры по уменьшению риска, приведенные в [2] и перечисленные в порядке приоритета. Данный принцип описан в нескольких документах, включая [9], в том числе в местных и региональных документах (например, в [10]). Когда это практически осуществимо, безопасность изделия рекомендуется закладывать в его конструкции. В противном случае необходимо предусмотреть защитные меры, например ограждения или системы сигнализации. Наименее предпочтительной защитной мерой являются письменные предупреждения или ограничения/противопоказания.
Общепризнано, что одним из результатов анализа возможностей управления риском может стать отсутствие практически возможного способа уменьшения риска до допустимого уровня согласно предварительно установленным критериям допустимости риска. Например, может быть практически неосуществимым проектирование изделия для поддержания жизни с оцененным допустимым остаточным риском. В этом случае следует выполнить анализ соотношения риск/польза, как это описано в 6.5, чтобы определить, перевешивает ли польза от применения изделия остаточный риск. Такая возможность описана в соответствующем подразделе настоящего стандарта и служит для обеспечения уверенности в том, что предприняты все усилия для уменьшения рисков до предварительно установленного допустимого уровня.
A.2.6.3 Выполнение мер по управлению риском
Выполнение мер по управлению риском включает проведение двух разных верификаций. Первая верификация необходима для обеспечения того, что в окончательном проекте приняты соответствующие меры по управлению риском, вторая верификация - что принятые меры по управлению риском действительно приводят к его уменьшению. В некоторых случаях для подтверждения результативности мер по управлению риском можно выполнить действия по валидации.
А.2.6.4 Оценивание остаточного риска
На данном этапе определяют достаточность принятых мер, для того чтобы риск стал допустимым. Если остаточный риск не удовлетворяет критериям, установленным в плане менеджмента риска, то изготовитель должен принять дополнительные меры по управлению риском. Данный итеративный процесс рекомендуется осуществлять до тех пор, пока остаточный риск не будет уменьшен до допустимого уровня, установленного в плане менеджмента риска.
Пользователя медицинского изделия следует обеспечить необходимой информацией об остаточных рисках, для того чтобы дать ему возможность принимать информированное решение. Однако изготовитель сам принимает решение о том, какую информацию об остаточном риске и в каком объеме следует предоставить пользователю. Это требование отражает подход, принятый во многих странах и регионах.
A.2.6.5 Анализ соотношения риск/польза
Возможны случаи, когда риск применения медицинского изделия превышает критерии допустимости риска, установленные изготовителем. В 6.5 предусмотрена возможность введения в обращение медицинского изделия с высокой степенью потенциального риска применения при условии, что изготовитель провел в отношении этого изделия тщательное оценивание и может продемонстрировать, что польза от применения данного медицинского изделия превышает риск. Важно довести до пользователя сведения о значимых остаточных рисках и конечной пользе, чтобы он мог принять информированное решение (см. приложение J).
A.2.6.6 Риски, возникающие вследствие выполнения мер по управлению риском
В 6.6 содержится информация о том, что меры по управлению риском, предпринятые отдельно или в сочетании друг с другом, могут стать источником новой опасности, отличной от уже известных, а меры, предпринятые для уменьшения одного риска, могут привести к увеличению другого риска.
A.2.6.7 Полнота управления риском
На данном этапе следует провести оценивание рисков всех известных опасностей. Такое оценивание необходимо для обеспечения уверенности в том, что ни одна из опасностей не осталась за пределами полного анализа риска.
А.2.7 Оценивание допустимости совокупного остаточного риска
При осуществлении процесса, описанного в разделах 4-6, изготовитель идентифицирует опасности, оценивает риски и принимает необходимые меры по управлению риском в рамках своего проекта. На данном этапе изготовитель должен остановиться, рассмотреть совокупное влияние отдельных остаточных рисков и принять решение о возможности дальнейшей работы над медицинским изделием. Совокупный остаточный риск может превышать установленные изготовителем критерии допустимости риска, даже если составляющие его отдельные остаточные риски будут допустимы. Это касается прежде всего сложных систем и изделий с большим числом возможных рисков. Даже если совокупный остаточный риск не удовлетворяет критериям плана менеджмента риска, изготовитель имеет возможность осуществить всестороннее оценивание соотношения риск/польза, чтобы определить, можно ли вводить в обращение медицинское изделие с высокой степенью потенциального риска применения, но приносящее при этом большую пользу. Очень важно проинформировать пользователя о значимых совокупных остаточных рисках, поэтому изготовителям предписано включать соответствующую информацию в эксплуатационные документы.
А.2.8 Отчет по менеджменту риска
Отчет по менеджменту риска является важнейшей частью файла менеджмента риска. Предполагается, что он должен представлять собой сводную информацию по анализу окончательных результатов менеджмента риска. Отчет является документом высшего уровня и служит доказательством обеспечения изготовителем удовлетворительного выполнения плана менеджмента риска, а также того, что полученные им результаты подтверждают достижение поставленной цели. Первоначальная версия содержала требование, чтобы прослеживаемость была частью отчета по менеджменту риска. Это требование было отменено, так как анализ прослеживаемости в отношении сложных изделий значительно усложняет форму отчета по менеджменту риска по сравнению с первоначально предусмотренной объединенной рабочей группой 1 (JWG 1). Однако выполнение прослеживаемости остается частью файла менеджмента риска, и 3.5 был изменен с учетом данного требования.
А.2.9 Производственная и постпроизводственная информация
Необходимо подчеркнуть, что менеджмент риска не заканчивается выпуском готового медицинского изделия. Менеджмент риска часто начинается еще на стадии идеи, когда медицинское изделие отсутствует физически. Оценочные показатели риска могут быть уточнены на стадии проектирования и разработки, особенно в случае изготовления функционирующего опытного образца. Информация для менеджмента риска может поступать из любых источников, в том числе из производственных записей и записей по качеству. Однако никакой опытный образец не может заменить реальное медицинское изделие в процессе его применения реальными пользователями. Поэтому изготовитель должен осуществлять мониторинг производственной и постпроизводственной информации, которая может повлиять на определение им рисков и, следовательно, на принятие решений по менеджменту риска. Изготовителю также необходимо учитывать современный уровень научно-технического развития и практическую применимость медицинского изделия. Данную информацию следует использовать для улучшения процесса менеджмента риска. При наличии постпроизводственной информации процесс менеджмента риска становится действительно итеративным процессом с обратной связью.
В настоящей версии стандарта наименование данного раздела изменено с "Постпроизводственная информация" на "Производственная и постпроизводственная информация" по той причине, что важная информация по менеджменту риска может быть получена еще на стадии изготовления медицинского изделия. Требования раздела 9 также были пересмотрены, для того чтобы подчеркнуть значение последовательности действий, ожидаемых от изготовителя.
Приложение B
(справочное)
Обзор процесса менеджмента риска медицинских изделий
Рисунок B.1 предназначен для того, чтобы дать пользователям настоящего стандарта краткое представление о процессе менеджмента риска медицинских изделий. Как показано на рисунке B.1, данный процесс является итеративным, каждый риск рассматривают последовательно, возвращаясь на более ранние стадии в случае, если меры по управлению риском приводят к возникновению новых опасностей или если становится доступной не известная ранее информация.
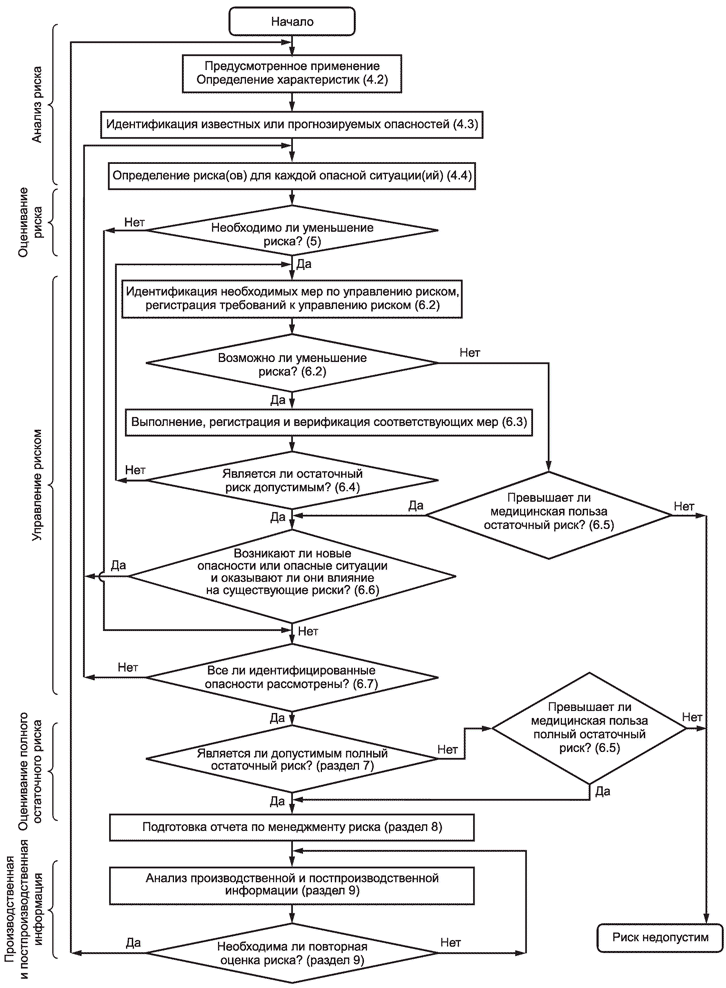
Рисунок В.1 - Обзор видов деятельности по менеджменту риска медицинских изделий
Приложение C
(справочное)
Вопросы, на которые необходимо ответить для определения характеристик медицинского изделия, влияющих на безопасность его применения
С.1 Общие положения
Требование к изготовителю идентифицировать характеристики медицинского изделия, влияющие на безопасность его применения, содержится в 4.2. Рассмотрение этих характеристик является основным требованием в идентификации опасностей, связанных с медицинским изделием (см. 4.3). Одним из способов выполнения данного требования является составление перечня вопросов, относящихся к изготовлению, предусмотренному применению, предполагаемым пользователям, обоснованно прогнозируемому неправильному применению и утилизации медицинского изделия. Если задавать эти вопросы от имени всех заинтересованных лиц (например, пользователей, специалистов по техническому обслуживанию, пациентов и т.д.), то можно получить более полную картину о возможных опасностях. Приведенные ниже вопросы могут помочь в идентификации характеристик медицинского изделия, влияющих на безопасность его применения. Вопросы, помогающие определить риск для пациента от применения медицинских изделий для диагностики in vitro, содержит H.2.5.4 (приложение Н).
Данный перечень не является исчерпывающим или типичным для всех медицинских изделий, в него можно добавлять вопросы, относящиеся к конкретному медицинскому изделию, и избегать рассмотрения вопросов, не относящихся к данному изделию. Предлагается также рассматривать каждый вопрос не только по отдельности, но и во взаимосвязи с другими вопросами.
С.2 Вопросы
С.2.1 Каково предусмотренное применение и как следует применять медицинское изделие?
Рассматриваемые факторы:
- роль медицинского изделия:
- в диагностике, профилактике, мониторинге, лечении или облегчении заболевания,
- в компенсации травм или физических недостатков,
- в замещении или модификации частей тела или управлении зачатием;
- показания к применению (например, предполагаемые группы пользователей);
- предназначено ли медицинское изделие для сохранения или поддержания жизни;
- необходимость вмешательства специалистов при отказе медицинского изделия.
С.2.2 Предусмотрена ли имплантация медицинского изделия?
Рассматриваемые факторы: местонахождение имплантата; характеристики предполагаемых пользователей: возраст, вес, физическая активность; влияние старения на рабочие характеристики имплантата; предполагаемый срок действия имплантата; возможность извлечения имплантата.
С.2.3 Предусмотрен ли контакт медицинского изделия с пациентом или другими лицами?
Рассматриваемые факторы: характер предполагаемого контакта (поверхностный контакт, инвазивный контакт или имплантация) и в каждом случае - длительность и частота контакта.
С.2.4 Какие материалы или компоненты входят в состав медицинского изделия, используются совместно либо контактируют с ним?
Рассматриваемые факторы:
- совместимость с рассматриваемыми веществами;
- совместимость с тканями или биологическими жидкостями;
- характеристики, относящиеся к безопасности;
- наличие в составе медицинского изделия материалов животного происхождения.
Примечание - См. приложение I, а также стандарты серии [11].
С.2.5 Может ли энергия быть передана пациенту или выработана пациентом?
Рассматриваемые факторы:
- вид передаваемой энергии;
- управление энергией, качество, количество, интенсивность и длительность воздействия;
- являются ли уровни энергии выше, чем в уже применяемых подобных изделиях.
С.2.6 Вводят ли пациенту или выводят из него какие-либо вещества?
Рассматриваемые факторы:
- сведения о введении или выведении веществ;
- сведения о том, одно это вещество или группа веществ;
- данные о максимальной и минимальной скорости введения/выведения вещества и управлении этим процессом.
С.2.7 Проводят ли в медицинском изделии обработку биологических веществ для их последующего использования, трансфузии или трансплантации?
Рассматриваемые факторы: вид обработки и обрабатываемое(ые) вещество(а) (например, аутотрансфузия, диализ, обработка компонентов крови или клеточная терапия).
C.2.8 Стерильно ли поставляемое медицинское изделие, или оно предназначено для стерилизации пользователем, или необходимы другие виды микробиологической обработки?
Рассматриваемые факторы:
- предназначено ли медицинское изделие для однократного или многократного применения;
- данные об условиях хранения;
- ограничение числа повторных применений;
- способы стерилизации;
- воздействие способов стерилизации, не предусмотренных изготовителем.
С.2.9 Предназначено ли медицинское изделие для рутинной очистки и дезинфекции, выполняемых пользователем?
Рассматриваемые факторы: виды применяемых чистящих и дезинфицирующих средств, а также любые ограничения числа циклов очистки. Конструкция медицинского изделия может влиять на результативность рутинной очистки и дезинфекции. Кроме того, следует учитывать влияние чистящих и дезинфицирующих средств на безопасность или характеристики медицинского изделия.
С.2.10 Влияет ли медицинское изделие на среду, окружающую пациента?
Рассматриваемые факторы:
- температура;
- влажность;
- состав атмосферных газов;
- давление;
- освещенность.
С.2.11 Предназначено ли медицинское изделие для проведения измерений?
Рассматриваемые факторы: измеряемые переменные, правильность и точность результатов измерений.
C.2.12 Является ли медицинское изделие интерпретирующим?
Рассматриваемые факторы: способность медицинского изделия давать заключение на основе входных или полученных в процессе применения данных, использованных алгоритмов и доверительных интервалов. Особое внимание следует уделить непредусмотренному применению данных или алгоритмов.
С.2.13 Предусмотрено ли применение медицинского изделия в сочетании с другими медицинскими изделиями, медикаментами или другими медицинскими средствами?
Рассматриваемые факторы: идентификация любых других применяемых медицинских изделий, медикаментов или медицинских средств и возможные проблемы, связанные с их взаимодействием; реакция пациента на получаемое лечение.
С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное излучение), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в случае, если они остаются на изделии.
Другие рассматриваемые факторы, связанные с веществами: выведение химических веществ, продуктов жизнедеятельности и биологических жидкостей.
С.2.15 Чувствительно ли медицинское изделие к воздействию окружающей среды?
Рассматриваемые факторы: производственная среда, транспортные средства и условия хранения. К ним относят: освещенность, температуру, вибрации, утечки, чувствительность к изменениям в энергоснабжении и охлаждении, электромагнитные помехи.
С.2.16 Воздействует ли медицинское изделие на окружающую среду?
Рассматриваемые факторы:
- воздействие на средства энергоснабжения и охлаждения;
- выделение токсичных веществ;
- генерирование электромагнитных помех.
С.2.17 Имеются ли расходные материалы или принадлежности, связанные с медицинским изделием?
Рассматриваемые факторы: технические требования к расходным материалам или принадлежностям, связанным с медицинским изделием, и любые ограничения для пользователей в выборе данных материалов или принадлежностей.
С.2.18 Необходимы ли техническое обслуживание или калибровка медицинского изделия?
Рассматриваемые факторы включают сведения о том:
- должны ли техническое обслуживание или калибровка выполняться оператором, пользователем или специалистом;
- необходимы ли специальные вещества или оборудование для надлежащего технического обслуживания или калибровки.
С.2.19 Входит ли в состав медицинского изделия программное обеспечение?
Рассматриваемые факторы: сведения о том, должно ли программное обеспечение быть установлено, верифицировано, модифицировано или заменено оператором, пользователем или специалистом.
С.2.20 Имеет ли медицинское изделие ограниченный срок хранения?
Рассматриваемые факторы: соответствующая маркировка или индикаторы, а также сведения об утилизации медицинского изделия по истечении срока годности.
С.2.21 Существуют ли отсроченные или длительные последствия применения медицинского изделия?
Рассматриваемые факторы: эргономические и кумулятивные эффекты. К ним относят: воздействие коррозии на насосы для физиологического раствора, механическую усталость, ослабление узлов или креплений, воздействие вибрации, истирание или отклеивание маркировок, ухудшение свойств материалов при долгосрочном применении медицинского изделия.
С.2.22 Какие механические силы могут воздействовать на медицинское изделие?
Рассматриваемые факторы: сведения о том, управляет ли механическими силами, оказывающими воздействие на медицинское изделие, только пользователь или пользователь совместно с другими лицами.
С.2.23 Чем определяется срок службы медицинского изделия?
Рассматриваемые факторы: старение изделия и истощение элементов питания.
С.2.24 Предназначено ли медицинское изделие для однократного применения?
Рассматриваемые факторы: возможность самоликвидации медицинского изделия после применения; наличие указаний на то, что данное изделие уже было применено.
С.2.25 Необходимы ли безопасный демонтаж или утилизация медицинского изделия?
Рассматриваемые факторы: отходы, возникающие при утилизации медицинского изделия. Например, следует ответить на вопрос, содержит ли данное изделие токсичные или опасные вещества или вещества, пригодные для переработки.
С.2.26 Необходимы ли специальное обучение или специальные навыки для монтажа или применения медицинского изделия?
Рассматриваемые факторы: новизна медицинского изделия; навыки и знания, необходимые для монтажа данного изделия.
С.2.27 Какая информация по безопасному применению будет предоставлена пользователю?
Рассматриваемые факторы:
- сведения о том, будет ли информация предоставлена конечному пользователю непосредственно изготовителем или будут задействованы другие лица, например специалисты по монтажу/установке, поставщики медицинских услуг, медицинские работники или фармацевты, и как это повлияет на обучение;
- информация о пусконаладочных работах (вводе в эксплуатацию) и передаче изделия конечному пользователю, а также о целесообразности/возможности монтажа лицами, не имеющими специальных навыков;
- сведения о необходимости переобучения или переаттестации операторов или обслуживающего персонала исходя из предполагаемого срока службы изделия.
С.2.28 Будут ли необходимы разработка и внедрение новых производственных процессов?
Рассматриваемые факторы: новые технологии или новые масштабы производства.
С.2.29 Зависит ли успешное применение медицинского изделия от человеческого фактора, такого как пользовательский интерфейс?
С.2.29.1 Могут ли особенности конструкции пользовательского интерфейса привести к ошибкам применения?
Рассматриваемые факторы: особенности конструкции пользовательского интерфейса, которые могут привести к ошибкам применения. К ним относят: элементы управления или индикаторы, используемые символы, эргономические свойства, дизайн изделия и схему расположения элементов управления, иерархию действий, меню для изделий с программным управлением, наглядность предупреждений, хорошую слышимость сигналов тревоги, стандартные цветовые коды. Дополнительное руководство по эксплуатационной пригодности см. [12], по системам сигнализации - [13].
С.2.29.2 Используют ли медицинское изделие в среде, в которой отвлекающие факторы могут привести к ошибкам применения?
Рассматриваемые факторы:
- последствия ошибок применения;
- сведения о том, являются ли отвлекающие факторы привычными для пользователя;
- вероятность появления редко случающихся отвлекающих факторов.
С.2.29.3 Имеет ли медицинское изделие соединительные части или принадлежности?
Рассматриваемые факторы: возможность неправильных соединений, сходство с соединениями в других изделиях, прочность соединения, обратная связь при повреждении соединения, а также слишком прочное или недостаточно прочное соединение.
С.2.29.4 Имеет ли медицинское изделие управляющий интерфейс?
Рассматриваемые факторы: пространственное распределение элементов управления, их кодирование, группировка, схема пространственного распределения, режимы обратной связи, грубые ошибки, незначительные промахи, дифференциация управления, качество изображений, направление активирования или изменения, дискретность или непрерывность функционирования элементов управления, обратимость настроек или действий.
С.2.29.5 Имеет ли медицинское изделие функцию отображения информации?
Рассматриваемые факторы: качество изображения в разных условиях; ориентация, зрительные способности пользователя; степень заполнения экрана монитора и перспективное изображение; четкость представленной информации; единицы отображения информации; цветовые коды; доступ к наиболее важной информации.
С.2.29.6 Имеет ли медицинское изделие меню управления?
Рассматриваемые факторы: сложность и число уровней меню, осведомленность пользователя о состоянии меню, расположение настроек, способ перемещения между объектами, число шагов в одном действии, проблемы четкой последовательности действий и запоминания, соотношение важности и доступности функции управления, а также влияние, оказываемое отклонениями от установленных операционных процедур.
С.2.29.7 Будет ли медицинское изделие применяться лицами с особыми потребностями?
Рассматриваемые факторы: предполагаемый пользователь, его умственные способности и физические возможности, навыки и подготовка, эргономические аспекты, среда применения, требования к установке/монтажу, способность пациента управлять применением медицинского изделия или воздействовать на данное применение. Особое внимание следует уделять пользователям со специфическими потребностями, таким как лица с ограниченными возможностями, пожилые люди и дети. К специфическим потребностям можно также отнести потребность в дополнительной помощи другого лица в процессе применения медицинского изделия. Следует рассмотреть возможность применения медицинского изделия лицами с разным уровнем подготовки и культурными особенностями.
С.2.29.8 Можно ли использовать пользовательский интерфейс для инициирования действий пользователя?
Рассматриваемые факторы: возможность инициирования намеренных действий пользователя для входа в управляемый рабочий режим, что увеличивает риски для пациента; осведомленность пользователя в данных обстоятельствах.
С.2.30 Используют ли в медицинском изделии систему сигнализации?
Рассматриваемые факторы: риск подачи ложных сигналов тревоги, несрабатывание системы сигнализации, отсоединение системы сигнализации, ненадежность удаленных систем сигнализации, а также возможность понимания медицинским персоналом принципов работы системы сигнализации. Руководство по системам сигнализации приведено в [13].
С.2.31 Каковы способы неправильного применения медицинского изделия?
Рассматриваемые факторы: неправильное применение переключателей; выход из строя системы сигнализации или элементов, обеспечивающих безопасность применения; пренебрежение рекомендациями изготовителя в отношении технического обслуживания.
С.2.32 Обеспечивает ли медицинское изделие хранение данных, важных для ведения пациента?
Рассматриваемые факторы: последствия изменения или искажения данных.
С.2.33 Является ли медицинское изделие мобильным или портативным?
Рассматриваемые факторы: необходимые зажимы, рукоятки, колеса, тормоза, механическая устойчивость и прочность медицинского изделия.
С.2.34 Зависит ли применение медицинского изделия от его важнейших эксплуатационных характеристик?
Рассматриваемые факторы: выходные характеристики изделий, обеспечивающих жизнеобеспечение организма; срабатывание системы сигнализации.
В [1] рассмотрены важнейшие эксплуатационные характеристики медицинских электрических изделий и медицинских электрических систем.
Приложение D
(справочное)
Концепции риска, применимые к медицинским изделиям
D.1 Общие положения
Данное приложение содержит руководящие указания применительно к следующим концепциям риска, важным для менеджмента риска медицинских изделий:
- опасности и опасные ситуации;
- определение риска;
- допустимость риска;
- управление риском;
- анализ соотношения риск/польза;
- всестороннее оценивание риска.
Термин "риск" определен в 2.16 как "сочетание вероятности причинения вреда и тяжести этого вреда". Но это не значит, что для определения величины риска данные факторы следует просто перемножить. Одним из способов описания риска и визуального воспроизведения смысла, заложенного в его определении, является построение двумерной диаграммы риска.
Диаграмма риска, как показано на рисунке D.1, обеспечивает визуальное представление тяжести вреда по оси и вероятности наступления вреда по оси
. Для каждой опасности или опасной ситуации числовое значение, рассчитанное исходя из тяжести вреда и вероятности причинения вреда, наносят графически на диаграмму риска в виде отдельной точки. В этом примере на диаграмму нанесены риски, величина которых уже определена (
,
,
, ...).
Условные обозначения: - увеличение тяжести вреда;
- увеличение вероятности причинения вреда
Рисунок D.1 - Пример диаграммы риска
D.2 Опасности и опасные ситуации
D.2.1 Общие положения
Медицинское изделие причиняет вред только в том случае, когда возникает последовательность событий, приводящая к опасной ситуации и впоследствии к причинению вреда. Последовательность событий может включать как отдельное событие, так и комбинацию событий. Опасная ситуация складывается, когда люди, имущество или окружающая среда подвергаются опасности.
Приложение С содержит руководящие указания в виде вопросов, на которые необходимо ответить для определения характеристик медицинских изделий, влияющих на безопасность их применения, что будет способствовать идентификации возможных опасностей. Приложение E содержит руководящие указания относительно идентификации опасностей и последовательностей событий, которые могут привести к опасной ситуации. Приложение H содержит руководящие указания по идентификации опасностей и последовательностей событий, которые могут привести к опасным ситуациям и причинению вреда в отношении медицинских изделий для диагностики in vitro.
Необходимо подчеркнуть, что опасные ситуации могут возникать даже в отсутствие отказов, т.е. в условиях нормального применения медицинского изделия.
D.2.2 Опасные ситуации, являющиеся следствием отказов медицинского изделия
D.2.2.1 Общие положения
В случае, когда опасная ситуация возникает вследствие отказа медицинского изделия, следует различать вероятность отказа и вероятность причинения вреда. Отказ медицинского изделия не всегда приводит к возникновению опасной ситуации, так же как и опасная ситуация не всегда приводит к причинению вреда.
Особое внимание следует уделять опасным ситуациям, являющимся следствием отказов медицинского изделия. Важно понимать, что, как правило, бывают два вида отказов, которые могут привести к возникновению опасной ситуации: случайные и систематические отказы.
D.2.2.2 Опасные ситуации, являющиеся следствием случайных отказов
В отношении многих событий вероятности возникновения отказов можно присвоить числовое значение. Некоторые примеры случайных отказов:
- отказ части изделия, например интегральной схемы в электронном блоке;
- получение неверных результатов из-за загрязнения изделия вследствие старения IVD-реагента (реагента для диагностики in vitro);
- отказ из-за наличия возбудителей инфекций или токсичных веществ в/на медицинском изделии. Количественное определение биологических рисков возможно только в том случае, когда имеется достаточная информация об опасности и обстоятельствах, влияющих на вероятность возникновения опасной ситуации, например, при обеспечении стерильности. Эту ситуацию можно трактовать так же, как случайный отказ аппаратного обеспечения. Во многих других случаях наличие возбудителей инфекций или токсичных веществ следует рассматривать как систематический отказ (см. D.2.2.3). Риск, возникающий из-за наличия токсичного вещества в материале изделия, следует определять в соответствии с [14]. Это может гарантировать, что ожидаемая степень причинения вреда от применения медицинского изделия ниже той, которая способна причинить вред здоровью пользователя.
D.2.2.3 Опасные ситуации, являющиеся следствием систематических отказов
Систематический отказ может быть результатом ошибки при осуществлении любого вида деятельности. Определенное сочетание входных данных или условий окружающей среды будет систематически приводить к отказу медицинского изделия, тогда как в других случаях отказ не произойдет.
Ошибки, приводящие к систематическим отказам, могут быть как в аппаратном, так и в программном обеспечении и на любой стадии разработки, изготовления и технического обслуживания медицинского изделия. Некоторые примеры систематических отказов:
- неверно рассчитанный плавкий предохранитель не позволяет предотвратить опасную ситуацию: параметры плавкого предохранителя могли быть неверно определены или неправильно установлены при изготовлении, или неправильно заменены при ремонте;
- в базе данных программного обеспечения не предусмотрено состояние заполненности: если база данных заполнена, то неясно, что делать программному обеспечению; возможным следствием может стать удаление имеющихся записей в целях высвобождения места для новых записей;
- жидкость, использованная при изготовлении медицинского изделия, имеет точку кипения ниже температуры тела человека: остатки жидкости могут при определенных обстоятельствах попасть в кровь и, возможно, привести к эмболии;
- антитела в пробе на гепатит не распознают некоторые (новые) виды вирусов;
- неадекватное управление окружающей средой или сбой в системах управления окружающей средой приводит к загрязнению данной среды токсичными веществами или возбудителями инфекций.
Точное определение частоты систематических отказов затруднительно в первую очередь по следующим причинам:
- измерение частоты систематических отказов требует больших затрат человеческих и денежных ресурсов. Достижение приемлемой достоверности результатов невозможно без наличия подробных данных о частоте отказов или без знания параметров, относящихся к управлению риском;
- не достигнуто согласия по методу количественного определения частоты систематических отказов.
Поскольку количественное определение риска в данных обстоятельствах затруднено, то для предотвращения опасных ситуаций предпочтительно применение робастных систем.
D.3 Определение риска
D.3.1 Общие положения
Для определения риска можно использовать разные методы. Настоящий стандарт не требует применения конкретного метода определения риска, но он требует проведения процесса определения риска (см. 4.4). При наличии необходимых данных предпочтительнее осуществлять количественное определение риска, однако даже в отсутствие этих данных для определения риска достаточно применения качественных методов.
Концепция риска является комбинацией следующих двух составляющих:
- вероятности причинения вреда;
- последствий причиненного вреда, т.е. его тяжести.
При определении риска следует рассматривать:
- инициирующее событие или обстоятельство [см. E.3 (приложение Е)];
- последовательность событий, которая может привести к возникновению опасной ситуации;
- вероятность возникновения такой ситуации;
- вероятность того, что опасная ситуация приведет к причинению вреда.
В зависимости от области применения возможно рассмотрение только некоторых элементов процесса определения риска. Например, в тех случаях, когда вред минимален или вероятность причинения вреда не может быть определена, нет необходимости идти дальше первичного анализа опасности и ее последствий.
Риск следует определять в выражениях, облегчающих принятие решений по управлению риском, например используя шкалы и единицы измерения вреда и вероятности причинения вреда, отражающие фактическое применение процесса определения риска. При анализе риска его составляющие, а именно вероятность и тяжесть, следует анализировать отдельно.
Диаграмма рисков, как показано на рисунке D.1, отображает риски, которые определены, что имеет значение для дальнейшего принятия решений. Риски наносят на диаграмму по мере их определения. Матрицы риска, созданные на основе рисунка D.1, использованы в примерах, приведенных в настоящем приложении. Это не означает, что данный метод применим ко всем медицинским изделиям, однако он может быть полезен во многих случаях. Если диаграмму рисков или матрицу рисков используют для градации рисков, то применение диаграммы или матрицы конкретного риска для конкретного случая (с соответствующими разъяснениями) должно быть обосновано.
D.3.2 Вероятность риска
D.3.2.1 Общие положения
В конкретных обстоятельствах при наличии необходимых данных предпочтительным является разделение уровней вероятности риска по количественным характеристикам. Если это невозможно, то изготовитель должен предоставить описание по качественным признакам. Хорошее описание по качественным признакам лучше неточного описания по количественным характеристикам. При разделении уровней вероятности риска по качественным признакам изготовитель может использовать дескрипторы, предназначенные для конкретного медицинского изделия.
D.3.2.2 Определение вероятности риска
Несмотря на то, что фактически вероятность представляет собой непрерывное множество, на практике можно использовать дискретное число уровней. В этом случае изготовитель на основе предполагаемой достоверности определения вероятности риска решает, сколько уровней вероятности необходимо. Чем больше предполагаемая достоверность, тем больше уровней вероятности необходимо использовать. Для облегчения принятия решений следует использовать, как минимум, три уровня вероятности. Уровни могут быть описательными (риск маловероятен на протяжении жизненного цикла медицинского изделия, вероятность возникновения риска - несколько раз на протяжении жизненного цикла изделия, вероятно частое возникновение риска и т.д.) или могут быть выражены символами (P1, P2 и т.д.). Изготовителю следует четко определить уровни вероятности, для того чтобы не возникало путаницы. Особенно результативным способом является определение числовых значений для дискретных уровней.
Определение вероятности риска включает все обстоятельства и всю последовательность событий, начиная от возникновения первоначальной причины и заканчивая причинением вреда. Рассмотрение вероятности причинения вреда связано с возможностью возникновения опасности. Если нет опасности, то не может быть и вреда. Таким образом, при определении вероятности причинения вреда необходимо учитывать возможность возникновения опасности, что подразумевает ответы на следующие вопросы:
- возникает ли опасная ситуация в отсутствие отказа медицинского изделия;
- возникает ли опасная ситуация в условиях отказа;
- возникает ли опасная ситуация только в условиях множественных отказов;
- насколько вероятно, что опасная ситуация приведет к причинению вреда.
На вероятность того, что опасная ситуация приведет к причинению вреда, влияют жизненный цикл медицинского изделия и число таких изделий на рынке.
Для определения вероятности риска обычно применяют семь подходов:
- использование соответствующих данных, относящихся к истории медицинского изделия;
- прогнозирование вероятности риска с использованием аналитических методов или моделирования;
- использование экспериментальных данных;
- использование показателей надежности;
- использование данных, полученных в процессе изготовления;
- использование постпроизводственной информации;
- использование экспертных заключений.
Все эти подходы можно использовать как по отдельности, так и в комбинации. Первые три подхода являются взаимодополняющими: каждый из них имеет свои сильные и слабые стороны. Там, где возможно, следует использовать несколько подходов. Таким образом можно обеспечить независимую проверку одного подхода с помощью других, что способствует повышению достоверности результатов. В случае, если эти подходы нельзя использовать или их применение является недостаточным, следует положиться на заключение экспертов.
D.3.2.3 Риски, вероятность которых не может быть определена
Достоверность определения вероятности риска повышается, когда на основе точных и надежных данных может быть проведено количественное определение вероятности риска или когда возможно обоснованное определение вероятности риска по качественным признакам. Однако это не всегда осуществимо. Например, вероятность систематических отказов, таких как описанные в D.2.2.3, определить чрезвычайно трудно. Если точность определения вероятности риска ставится под сомнение, то часто бывает необходимо установить широкий диапазон вероятности риска или установить, что точность определения вероятности риска не хуже некоторой конкретной величины. Можно привести примеры опасностей, когда очень трудно определить вероятность риска:
- отказ программного обеспечения;
- саботаж или несанкционированное вмешательство в конструкцию медицинского изделия;
- новые опасности, природа которых еще плохо изучена (например, недостаточное знание инфицирующей способности возбудителя такого заболевания, как губчатая энцефалопатия крупного рогатого скота, мешает количественному определению риска передачи данного заболевания);
- некоторые токсикологические опасности, такие как наличие генотоксичных канцерогенных веществ и сенсибилизаторов, когда бывает невозможно установить пороговую величину воздействия, ниже которой не наступает эффект токсичности.
В отсутствие данных о вероятности причинения вреда невозможно определить величину риска и обычно необходимо оценивать риск только на основании характера вреда. Если можно сделать вывод о том, что опасность имеет незначительные практические последствия, то риск может быть сочтен допустимым и в мерах по управлению риском нет необходимости. Однако для значительных опасностей, таких как приведенные в D.3.2.3, которые могли бы причинить вред высокой степени тяжести, не может быть установлен уровень воздействия, соответствующий настолько низкому риску, которым можно было бы пренебречь. В таких случаях величину риска следует определять исходя из вероятности возникновения наиболее неблагоприятного прогнозируемого варианта. В некоторых случаях удобно по умолчанию установить значение вероятности отказов, равным единице, и меры по управлению риском основывать на исключении опасности, уменьшая вероятность вреда до допустимого уровня или уменьшая тяжесть вреда (см. D.4).
Обычно считается, что существует обратная зависимость между отлаженностью процессов, используемых при проектировании и разработке сложных систем, и вероятностью выявления или невыявления систематических отказов. Часто целесообразно определить необходимую степень отлаженности процесса проектирования и разработки с учетом тяжести последствий систематических отказов и результатов мер по управлению риском, внешних по отношению к медицинскому изделию. Чем тяжелее последствия отказов и незначительнее результативность внешних мер по управлению риском, тем более отлаженным должен быть процесс проектирования и разработки.
D.3.3 Тяжесть вреда
Для разделения тяжести возможного вреда на уровни изготовителю следует использовать дескрипторы, подходящие для медицинского изделия. Понятие тяжести вреда фактически представляет собой непрерывное множество, однако на практике для облегчения анализа можно использовать дискретное число уровней тяжести. В таких случаях изготовитель сам устанавливает необходимое число уровней тяжести и способ их определения. Уровни могут быть описательными (не требуется медицинское вмешательство, требуется медицинское вмешательство, требуется госпитализация, возможен летальный исход и т.д.). Они также могут быть выражены символами (S1, S2 и т.д.), но в таком случае каждый символ должен быть четко определен. В любом случае определение уровней тяжести не должно включать какой-либо элемент вероятности (см. примеры в D.3.4).
Изготовителю необходимо выбрать и обосновать уровни тяжести для конкретного медицинского изделия при четко установленных условиях его применения.
D.3.4 Примеры
D.3.4.1 Анализ риска по качественным признакам
Для анализа риска по качественным признакам можно использовать несколько подходов. Типичным подходом является применение матрицы ![]() для описания вероятности и тяжести риска для каждой опасной ситуации. Определяют
для описания вероятности и тяжести риска для каждой опасной ситуации. Определяют уровней вероятности и
уровней тяжести. Каждый элемент матрицы представляет собой подгруппу полного набора возможных рисков. Элементы матрицы образованы пересечением диапазона возможных вероятностей и диапазона возможной тяжести (возможных последствий). Простым примером является матрица 3х3, основанная на определениях, взятых из таблиц D.1 и D.2. Изготовителю следует давать определения в соответствии со спецификой конкретного изделия; определения должны быть настолько точными, чтобы можно было обеспечить воспроизводимость их применения.
Таблица D.1 - Уровни тяжести по качественным признакам
Определение тяжести | Описание |
Значительная | Смерть, или утрата функций организма, или изменение анатомического строения тела |
Умеренная | Обратимая или незначительная травма (поражение) |
Пренебрежимо малая | Отсутствие травмы (поражения) или очень незначительная травма (поражение) |
Таблица D.2 - Уровни вероятности по качественным признакам
Определение вероятности | Описание |
Высокая | Очень вероятный, часто встречающийся риск |
Средняя | Возможный, но не частый риск |
Низкая | Обычно невозможный, редкий, отдаленный риск |
При использовании в строках матрицы обозначений вероятности, а в столбцах - обозначений тяжести можно создать матрицу риска 3х3. Риски, которые определены (,
,
, ...), необходимо занести в соответствующие элементы матрицы. Полученный результат изображен на рисунке D.2.

Рисунок D.2 - Пример матрицы риска 3х3 по качественным признакам
D.3.4.2 Полуколичественный анализ
В данном пункте приведен пример полуколичественного анализа. Шкала является полуколичественной, т.к. значения вероятности определены неточно, но известно, что они находятся в границах конкретного диапазона (т.е. определен порядок величины). Решения принимают, соотнося значения вероятности с уровнями тяжести, а не с числовой шкалой. На практике уровень тяжести редко определяется количественно, т.к. это невозможно по отношению к таким событиям, как летальный исход, стойкое нарушение состояния здоровья или поражение, угрожающее жизни.
В данном примере используют матрицу риска 5х5. Уровни вероятности и тяжести установлены в таблицах D.3 и D.4 соответственно.
Таблица D.3 - Пять уровней тяжести по качественным признакам
Определение тяжести | Описание |
Катастрофическая | Ведет к смерти пациента |
Критическая | Ведет к стойким нарушениям состояния здоровья или к поражениям, угрожающим жизни |
Значительная | Ведет к поражениям или нарушениям состояния здоровья, требующим профессионального медицинского вмешательства |
Незначительная | Ведет к временным нарушениям функций организма или к нарушениям состояния здоровья, не требующим профессионального медицинского вмешательства |
Пренебрежимо малая | Выражается в неудобстве или временном дискомфорте |
Таблица D.4 - Уровни вероятности при полуколичественном анализе
Определение вероятности | Примеры диапазона значений вероятности |
Частая |
|
Возможная |
|
Эпизодическая |
|
Отдаленная |
|
Невозможная |
|
Определения вероятности могут быть разными для разных видов продукции. Например, изготовитель может выбрать один набор определений для рентгеновских аппаратов и другой - для стерильной одежды одноразового применения. В зависимости от области применения могут быть использованы разные показатели вероятности. Шкала вероятности может включать "вероятность причинения вреда при однократном применении", "вероятность причинения вреда при применении одного изделия", "вероятность причинения вреда за один час применения изделия" и т.д.
Различают несколько статистических факторов, важных для анализа вероятности причинения вреда. Эти статистические факторы включают помимо прочего ответы на следующие вопросы:
- Как часто применяют рассматриваемое медицинское изделие?
- Каков срок жизни медицинского изделия?
- Каков состав популяций пользователей и пациентов?
- Сколько пользователей/пациентов подвергались воздействию медицинского изделия?
- Как долго и при каких обстоятельствах пользователи/пациенты подвергались воздействию медицинского изделия?
Риски, которые определены (,
,
,...), заносят в соответствующие элементы матрицы. Пример заполненной матрицы 5х5 приведен на рисунке D.3.
Рисунок D.3 - Пример матрицы риска при полуколичественном анализе
Помимо матриц 3х3 и 5х5 можно применять другие матрицы, однако для матриц с более чем пятью уровнями может потребоваться значительно больше данных, для того чтобы данные уровни существенно отличались друг от друга. Необходимо документально обосновать выбор матриц и значения показателей на выходе. Следует также учесть, что трехуровневые матрицы не всегда являются достаточно точными для принятия правильного решения. Несмотря на то, что в качестве примеров приведены матрицы 3х3 и 5х5, нет необходимости выбирать исключительно между ними. Например, матрица 4х5 также может быть уместна в данном случае.
D.4 Оценивание риска и допустимость риска
Настоящий стандарт не устанавливает уровни допустимости риска. Это решение остается за изготовителем. Методы определения допустимости риска включают помимо прочих:
- использование применимых стандартов, устанавливающих требования, соответствие которым указывает на достижение допустимого риска в отношении конкретных видов медицинских изделий или отдельных рисков;
- сравнение уровней риска, очевидных для уже применяемых медицинских изделий;
- оценивание данных клинических исследований, особенно при поиске новых технологий или новых предусмотренных применений, с учетом современного уровня научно-технического развития и имеющейся информации по новым технологиям и практическому применению подобных изделий на момент проектирования рассматриваемого медицинского изделия.
Термин "современный уровень научно-технического развития" используют здесь для обозначения того, что в настоящий момент является общепризнанной надлежащей практикой. Для определения соответствия конкретного медицинского изделия современному уровню научно-технического развития можно использовать разные методы:
- стандарты, применяемые для аналогичных или подобных изделий;
- сведения о наилучшем практическом применении аналогичных или подобных изделий;
- результаты общепризнанных научных исследований.
Современный уровень научно-технического развития необязательно означает использование самого прогрессивного технического решения.
Хорошо известно, что восприятие риска часто отличается от количественного определения риска, полученного опытным путем. По этой причине при принятии решения о допустимости риска следует учитывать восприятие риска возможно большим числом заинтересованных сторон. Принимая во внимание общественное мнение, бывает необходимо уделить особое внимание некоторым рискам. При этом иногда единственно возможным вариантом может быть следующий: считать, что интересы идентифицированных заинтересованных сторон являются общественно значимыми и учтены изготовителем при использовании методов, приведенных в D.4.
Одним из способов применения критериев допустимости риска является демонстрация на примере матрицы допустимых и недопустимых комбинаций вероятности вреда и тяжести вреда, как показано на рисунках D.4 и D.5. Такого рода матрицы обычно (но не всегда) создают для конкретного медицинского изделия и его предусмотренного применения.
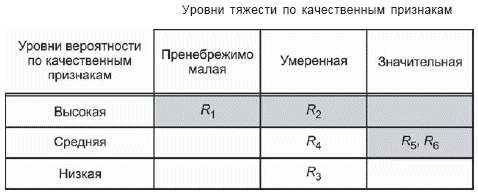
Условные обозначения:
- серый цвет - недопустимый риск;
- белый цвет - допустимый риск
Рисунок D.4 - Пример матрицы оценивания риска 3х3 по качественным признакам
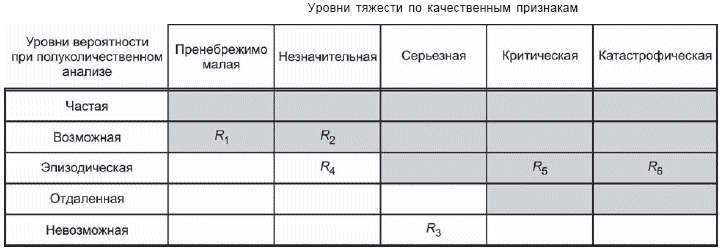
Условные обозначения:
- серый цвет - недопустимый риск;
- белый цвет - допустимый риск
Рисунок D.5 - Пример матрицы оценивания риска при полуколичественном анализе
Следует учесть, что изготовитель может и дальше подразделять область матрицы, обозначающую допустимый риск (например, на "незначительный" и т.д.), исследуя дальнейшее уменьшение риска (см. D.8.5).
D.5 Управление риском
D.5.1 Анализ возможностей управления риском
Различают несколько подходов к уменьшению риска, которые можно применять по отдельности или в комбинации друг с другом. Соответственно разработчик/инженер должен рассмотреть возможности обоснованного практического уменьшения риска(ов) до допустимого уровня. Далее приведен неполный перечень общепризнанных способов управления риском:
a) конструкция медицинского изделия, обеспечивающая его внутреннюю безопасность посредством:
- устранения конкретной опасности,
- уменьшения вероятности причинения вреда или
- уменьшения тяжести вреда;
b) добавление средств защиты посредством:
- применения автоматических предохранителей или клапанов безопасности,
- применения визуальных и звуковых сигналов тревоги для предупреждения оператора об опасных состояниях;
c) обеспечение информацией по безопасности посредством:
- размещения предупреждений на маркировке медицинского изделия,
- ограничения применения или области применения медицинского изделия,
- обмена информацией о неправильном применении, возможных опасностях или другой информацией, способствующей уменьшению риска,
- более широкого применения средств индивидуальной защиты, например перчаток или очков, при работе с токсичными или опасными материалами,
- предоставления информации о мерах по уменьшению вреда,
- обучения операторов в целях улучшения их работы или способности к обнаружению ошибок,
- описания надлежащего технического обслуживания и его периодичности, максимального срока службы медицинского изделия или его правильной утилизации.
Способы, описанные в перечислениях а)-с), приведены в порядке убывания их общепризнанной результативности в уменьшении рисков. Разработчикам/инженерам следует учитывать эти факторы, прежде чем принять решение о применении конкретной комбинации мер по управлению риском.
D.5.2 Компоненты и изделия, спроектированные без учета требований настоящего стандарта
Известно, что изготовитель не всегда имеет возможность следовать всем процессам, установленным в настоящем стандарте для каждого компонента рассматриваемого медицинского изделия, а именно для составных частей данного изделия, немедицинских подсистем, а также для медицинских изделий, которые были спроектированы до опубликования настоящего стандарта. В этом случае изготовителю следует обратить особое внимание на необходимость дополнительных мер по управлению риском.
D.5.3 Примеры управления риском
В таблице D.5 представлены некоторые широко применяемые меры по управлению риском. Решение о применении одной из данных мер зависит от специфики продукта (медицинского изделия) и процесса.
Таблица D.5 - Некоторые меры по управлению риском
Продукт/ процесс | Медицинское изделие | Опасность | Безопасность, заложенная в конструкции | Защитная мера/ средство | Информация по безопасности |
Медицинское изделие однократного применения | Катетер | Биологическое (перекрестное) заражение | Саморазрушение после применения | Четкая индикация после первого применения | Предостережение против негативных последствий повторного применения |
Активный имплантат | Водитель ритма | Электрические поля | Применение неэлектрических приводов и элементов управления | Применение дифференциаль- ных усилителей и дополнительных фильтрующих алгоритмов | Предупреждение о часто возникающих опасных ситуациях |
Медицинское изделие для диагностики in vitro | Анализатор крови | Неправильный результат вследствие методических погрешностей | Применение прослеживаемых калибраторов | Применение прослеживаемых элементов управления | Информирование потребителей о недопустимом отклонении от заданных значений |
Программное обеспечение | Менеджмент данных пациентов | Неверные данные | Программное обеспечение с высоким уровнем интеграции | Применение контрольного суммирования | Появление на экране предупреждений для пользователя |
Стерилизация паром | Инструмент для биопсии, хирургические щипцы | Высокая температура (результат - старение материала) | Применение материалов, устойчивых к высоким температурам | Мониторинг и регистрация давления и температуры | Инструкции по упаковыванию и загрузке |
D.5.4 Процесс изготовления и управление риском
Ненадлежащее управление процессом изготовления может поставить под угрозу безопасность медицинского изделия вследствие:
- наличия отходов производства или нежелательных загрязняющих частиц;
- воздействия на существенные физические и химические свойства материала, такие как покрытие поверхности, прочность на разрыв, сопротивляемость старению, однородность и т.д.;
- превышения критичных допустимых отклонений или
- нарушения целостности паяных, клеевых или других соединений компонентов изделия.
Для управления данными рисками важно идентифицировать элементы процесса изготовления.
Управление некоторыми рисками наиболее результативно при тщательном соблюдении процесса изготовления с помощью таких методов, как, например, анализ опасностей в критических контрольных точках (Hazard Analysis on Critical Control Points - HACCP) [см. G.6 (приложение G)].
D.5.5 Стандарты и управление риском
Применяя соответствующие стандарты, изготовитель может упростить задачу анализа остаточного риска, однако следует подчеркнуть, что области применения используемых стандартов не могут охватить все риски, связанные с рассматриваемым медицинским изделием.
Многие стандарты рассматривают безопасность, заложенную в конструкции изделия, защитные меры/средства и информацию по безопасности для медицинских изделий. В соответствующих стандартах по безопасности могут быть рассмотрены некоторые или все риски, применимые в отношении конкретного медицинского изделия. Считается, что в отсутствие объективных доказательств обратного эффекта выполнение требований соответствующих стандартов приводит к уменьшению конкретных рисков до допустимого уровня, однако ответственность за достоверность такой аргументации для конкретного медицинского изделия несет изготовитель.
D.6 Анализ соотношения риск/польза
D.6.1 Общие положения
Настоящий стандарт не требует анализа соотношения риск/польза для каждого риска. Анализ соотношения риск/польза применяется для обоснования остаточного риска после выполнения всех практически осуществимых мер по уменьшению риска. Если после выполнения данных мер риск все еще считается недопустимым, то необходим анализ соотношения риск/польза, для того чтобы установить, действительно ли польза от применения медицинского изделия превышает причиняемый им вред.
Как правило, если всех практически осуществимых мер по управлению риском недостаточно для удовлетворения установленных в плане менеджмента риска критериев допустимости риска, то разработка изделия должна быть прекращена. Однако в некоторых случаях даже значительные риски могут быть оправданы, если они меньше ожидаемой пользы от применения медицинского изделия. Настоящий стандарт предоставляет изготовителям возможность провести анализ соотношения риск/польза, для того чтобы определить, является ли остаточный риск допустимым с учетом имеющейся пользы от применения медицинского изделия.
Решение относительно соотношения риск/польза принимают главным образом на основании экспертизы, проводимой опытными и компетентными специалистами. Важным соображением в пользу допустимости остаточного риска является возможность достижения ожидаемой клинической эффективности с помощью альтернативных проектных решений или возможных методов лечения, исключающих конкретный остаточный риск или уменьшающих совокупный остаточный риск. При рассмотрении ожидаемой пользы (см. D.8.4) следует учитывать практическую осуществимость дальнейшего уменьшения риска. В настоящем стандарте описано, как можно идентифицировать риски, для того чтобы получить достоверное количественное определение риска. К сожалению, не существует стандартизованного метода количественного определения ожидаемой пользы от применения медицинского изделия.
D.6.2 Определение пользы
Польза от применения медицинского изделия зависит от ожидаемой вероятности и степени улучшения здоровья пациента. Пользу можно определить, зная:
- предполагаемое функционирование медицинского изделия при его клиническом применении;
- предполагаемую клиническую эффективность при вышеуказанном функционировании;
- факторы, относящиеся к рискам и пользе при использовании других способов лечения.
Достоверность определения пользы существенно зависит от точного знания вышеперечисленных факторов и включает признание существования следующих возможных последствий и факторов, которые необходимо учитывать:
- сложности сравнения разных по характеру последствий (например, что хуже: боль или потеря подвижности?). Разные последствия могут быть обусловлены разными побочными эффектами при решении одной и той же проблемы;
- трудности учета нестабильных результатов, которые могут возникать как в период выздоровления пациента, так и при длительном воздействии на него.
Из-за трудностей, возникающих ввиду отсутствия точного подхода, в большинстве случаев необходимо использовать упрощенные допущения. Поэтому часто бывает целесообразно сосредоточиться на наиболее вероятных последствиях каждого варианта - как самых благоприятных, так и самых неблагоприятных.
Определение клинической пользы может заметно отличаться на разных стадиях проектирования медицинского изделия. При наличии надежных клинических данных, демонстрирующих надлежащие функционирование и клиническую эффективность медицинского изделия, можно достоверно определить клиническую пользу от применения данного изделия. Если клинических данных недостаточно или их качество не внушает доверия, то клиническую пользу определяют с меньшей достоверностью на основе любой доступной соответствующей информации. Например, иногда необходимо определить ожидаемую степень улучшения состояния здоровья пациента на ранних стадиях процесса проектирования медицинского изделия. Однако в отсутствие достоверных клинических данных вероятность достижения предполагаемого функционирования и ожидаемой клинической эффективности медицинского изделия следует прогнозировать исходя из мер по обеспечению качества, а также на основе сведений о функционировании медицинского изделия in vivo или in vitro.
При наличии значительных рисков и высокой степени недостоверности в определении клинической пользы необходимо как можно быстрее верифицировать предполагаемое функционирование или клиническую эффективность медицинского изделия с помощью исследований на модели или клинических испытаний. Это важно для подтверждения того, что соотношение риск/польза соответствует ожидаемому результату, а также для предотвращения нежелательного воздействия существенного остаточного риска на пациентов. В [15] и [16] установлены процедуры организации и проведения клинических испытаний медицинских изделий.
D.6.3 Критерии оценивания соотношения риск/польза
Лица, проводящие оценку соотношения риск/польза, несут ответственность за понимание и учет технических, медицинских, регулирующих, экономических, социальных и политических составляющих принимаемых ими решений по менеджменту риска. К таким решениям можно отнести интерпретацию основополагающих требований, установленных в соответствующих нормативных документах или стандартах и применимых к рассматриваемой продукции в предполагаемых условиях применения. Поскольку данный вид анализа сильно зависит от специфики продукции, то дальнейшие руководящие указания общего свойства невозможны. Вместе с тем требования безопасности, установленные в стандартах на конкретные изделия или стандартах, областью применения которых являются конкретные риски, можно считать совместимыми с допустимым уровнем риска, особенно если применение данных стандартов санкционировано действующими регулирующими органами. Необходимо помнить, что для верификации допустимости соотношения между медицинской пользой и остаточным риском может потребоваться проведение клинических испытаний в соответствии с юридически признанной процедурой.
D.6.4 Сопоставление риска и пользы
Прямое сопоставление риска и пользы возможно только при использовании единой шкалы. В этом случае риск и пользу можно сопоставлять посредством их количественного определения. При непрямом сопоставлении риска и пользы единая шкала неприменима, и данные показатели определяют по качественным признакам. В обоих случаях при сравнении риска и пользы следует:
- изучить литературные источники о возможных опасностях для рассматриваемого класса продукции с точки зрения соотношения риск/польза;
- рассмотреть медицинские изделия с высокой степенью потенциального риска применения/высокой клинической эффективностью, обычно основанные на использовании лучшей технологии, обеспечивающей максимальную медицинскую пользу, но не устраняющей полностью риск травмы или поражения. Поэтому для достоверного анализа соотношения риск/польза необходимо понимание используемой технологии и особенностей ее применения в медицинской практике. Для сопоставления риска и пользы могут быть использованы выражения, применяемые в аналогичных случаях для другой выпущенной в обращение продукции;
- для валидации того, что медицинское изделие удовлетворяет критериям допустимости соотношения риск/польза, часто необходимо провести клинические испытания. Клинические испытания позволяют количественно определить ожидаемую пользу и риски от применения медицинского изделия. В ходе клинических испытаний можно установить допустимость соотношения риск/польза для пациентов, пользователей и медицинских работников;
- в отношении медицинских изделий высокого риска применения/высокой клинической эффективности следует использовать маркировку, предоставляющую соответствующим пользователям, т.е. пациентам или медицинским работникам, информацию, необходимую для принятия ими решений в отношении соотношения риск/польза до применения данных изделий;
- как правило, медицинские изделия высокого риска применения/высокой клинической эффективности должны удовлетворять дополнительным регулирующим требованиям до введения их в обращение.
До выпуска новых или усовершенствованных медицинских изделий, требующих проведения анализа соотношения риск/польза, изготовитель должен суммировать доступную информацию, относящуюся к определению соотношения риск/польза, и документировать свои заключения по поводу данного соотношения (с обоснованием при необходимости). Руководящие указания в отношении анализа литературных источников, содержащих необходимые клинические данные, можно найти в приложении А, [15].
D.6.5 Примеры решений с учетом соотношения риск/польза
Примеры
1 Неправильное размещение нейтрального электрода прибора для высокочастотной хирургии на теле пациента может привести к ожогу. Соблюдение требований соответствующего стандарта на изделие сводит к минимуму, но не исключает вероятность ожогов. Тем не менее, польза от применения такого прибора для высокочастотной хирургии в сравнении с другими хирургическими методами превышает остаточный риск от ожогов.
2 Известно, что применение рентгеновского излучения причиняет вред пациентам, но клиническая эффективность стандартной цифровой рентгенографии почти всегда оправдывает ее применение. Однако нежелательное воздействие излучения на пациентов нельзя игнорировать. Существуют стандарты, соблюдение требований которых помогает свести к минимуму нежелательное воздействие излучения на пациентов и принять решение по поводу соотношения риск/польза. Если рассматривается возможность нового применения ионизирующего излучения для получения диагностических изображений, а имеющиеся стандарты неприменимы, то изготовителю следует верифицировать тот факт, что результаты анализа соотношения риск/польза, как минимум, также благоприятны, как для альтернативных изделий и альтернативных методов лечения.
3 Некоторые компоненты кохлеарного имплантата, например имплантированный стимулятор приемника с матрицей электродов, трудно заменить после их имплантации. Они должны оставаться имплантированными на протяжении всей жизни пациента, и от них требуется надежное функционирование в течение многих лет и даже десятилетий, что особенно важно для молодого человека или ребенка. Можно проводить ускоренные испытания надежности данных компонентов для конкретных механизмов отказа. Однако нецелесообразно проводить валидацию надежности компонентов, которые должны функционировать на протяжении десятилетий. Поэтому совокупный остаточный риск, включая риск отказа изделия, сравнивают с приносимой им пользой, т.е. потенциальной возможностью улучшения слуха. Совокупный остаточный риск будет зависеть от количественного определения надежности компонентов и достоверности определения надежности тех компонентов, которые не могут быть валидированы. В некоторых случаях остаточный риск будет больше пользы, в других случаях польза будет больше остаточного риска.
D.7 Оценивание совокупного остаточного риска
D.7.1 Общие положения
Оценивание совокупного остаточного риска означает, что остаточный риск рассматривают во всем его многообразии. Изготовитель должен решить, как оценивать имеющийся остаточный риск с учетом критериев допустимости.
Оценивание совокупного остаточного риска должны проводить лица, обладающие необходимыми знаниями, опытом и полномочиями для выполнения таких задач. Часто желательно привлекать узких специалистов, имеющих знания и опыт работы с конкретным медицинским изделием (см. 3.3).
Не существует предпочтительного метода оценивания совокупного остаточного риска, и изготовитель несет ответственность за выбор соответствующего метода. В настоящем приложении описаны некоторые возможные методы и даны рекомендации по их выбору.
D.7.2 Анализ дерева событий
Определенная последовательность событий может привести к возникновению нескольких отличных друг от друга рисков, каждый из которых будет частью совокупного остаточного риска. Например, повторное применение изделия однократного применения может привести к заражению пациента, выделению токсичных веществ и биологически несовместимых остатков дезинфицирующих веществ, механическому отказу из-за старения материалов. Дерево событий может стать подходящим методом анализа перечисленных рисков. Для определения допустимости совокупного остаточного риска необходимо рассмотреть отдельные остаточные риски в совокупности.
D.7.3 Анализ противоречивых требований
Меры по управлению отдельными рисками могут порождать конфликтующие (противоречивые) требования. Например, предупреждение медицинскому работнику в отношении пациента, находящегося в бессознательном состоянии, во избежание риска падения данного пациента с кушетки "Никогда не оставляйте без присмотра пациента, находящегося в бессознательном состоянии" может противоречить предупреждению "Находитесь на предусмотренном расстоянии от пациента, подвергающегося воздействию рентгеновского излучения", предназначенному для защиты оператора от воздействия рентгеновского излучения.
D.7.4 Анализ дерева неисправностей
Причинение вреда пациенту или пользователю может стать следствием опасных ситуаций (см. приложение E). В таких случаях вероятность причинения вреда, применяемая для определения совокупного остаточного риска, основана на комбинации отдельных вероятностей. Анализ дерева неисправностей может быть подходящим методом установления совокупной вероятности причинения вреда.
D.7.5 Анализ предупреждений
Каждое предупреждение, рассматриваемое по отдельности, может обеспечить требуемое уменьшение риска, однако слишком большое число предупреждений может снизить эффективность воздействия каждого отдельного предупреждения. Может даже возникнуть необходимость в анализе чрезмерного доверия, испытываемого пользователями в отношении предупреждений, а также влияния, оказываемого подобным доверием, на уменьшение риска и совокупный остаточный риск.
D.7.6 Анализ рабочих инструкций
Анализ рабочих инструкций, прилагаемых к изделию, позволяет обнаружить противоречивость и чрезмерную сложность содержащейся в них информации.
D.7.7 Сравнение рисков
Данный метод заключается в сопоставлении отдельных остаточных рисков, возможных при применении рассматриваемого медицинского изделия, с рисками, возникающими при применении подобных изделий (последовательное сопоставление рисков с учетом разных вариантов применения изделия). При проведении таких сравнений следует использовать актуальную информацию о нежелательных событиях, связанных с применяемыми изделиями.
D.7.8 Анализ с привлечением экспертов
Для демонстрации допустимости рисков может потребоваться оценка пользы для пациента от применения медицинского изделия. Один из подходов заключается в свежем взгляде на совокупный остаточный риск практикующих специалистов, которые непосредственно не участвовали в разработке данного изделия. Практикующие специалисты могут провести оценивание допустимости совокупного остаточного риска, рассматривая такой фактор, как эксплуатационная пригодность при применении изделия в надлежащих клинических условиях. В подобном случае оценивание изделия в надлежащих клинических условиях может стать подтверждением допустимости рисков, связанных с его применением.
D.8 Подход "Настолько малый, насколько практически осуществимо"
D.8.1 Общие положения
При разработке политики в отношении допустимости рисков изготовитель может применять подход "Настолько малый, насколько практически осуществимо" (ALARP-подход).
После применения конкретной меры по управлению риском возможны три результата:
a) остаточный риск превышает критерии, установленные изготовителем в отношении допустимости риска;
b) остаточный риск допустим, т.к. он настолько мал, что им можно пренебречь;
c) величина остаточного риска занимает промежуточное положение между величинами, указанными в перечислениях a) и b); в таком случае остаточный риск допустим при условии применения мер, уменьшающих риск до практически осуществимого уровня, с учетом пользы для пациента и затрат на дальнейшее уменьшение данного риска.
Подход "Настолько малый, насколько практически осуществимо" (ALARP-подход) можно использовать как часть анализа возможностей управления риском (см. 6.2). ALARP-подход обычно применяют в отношении рисков, вероятность которых не может быть определена.
D.8.2 Уровни риска
Остаточный риск, находящийся ниже конкретного уровня, можно оценивать как настолько незначительный, что он станет сравнимым с каждодневными рисками, которые мы все испытываем. Такой риск можно назвать пренебрежимо малым.
Имеется существенная разница между остаточным риском, являющимся настолько низким, что нет необходимости его рассматривать, и более высоким остаточным риском, который является допустимым вследствие приносимой изделием пользы и практической невозможности уменьшения данного риска.
Когда риск определен, возникает главный вопрос: "Является ли данный риск настолько малым, что нет необходимости анализировать возможности уменьшения риска?" Такое решение принимают для каждого риска в отдельности.
D.8.3 Анализ возможностей управления риском
Для каждого остаточного риска, которым нельзя пренебречь, должны быть проанализированы возможности уменьшения риска. Уменьшение риска может быть или не быть практически осуществимым, но его следует рассмотреть. При этом можно получить следующие выводы:
- одна или несколько мер по управлению риском уменьшают риск до пренебрежимо малого уровня, и нет необходимости в его дальнейшем рассмотрении;
- уменьшение риска до пренебрежимо малого уровня практически нецелесообразно независимо от имеющихся возможностей его уменьшения.
Каждый остаточный риск, сохранившийся после выполнения мер по управлению риском, следует оценивать по критериям, установленным в плане менеджмента риска. Если остаточный риск отвечает критериям, установленным изготовителем в отношении допустимости риска, и был применен ALARP-подход, то в дальнейшем уменьшении риска нет необходимости.
D.8.4 Практическая осуществимость уменьшения риска
Может сложиться впечатление, что любой риск, связанный с медицинским изделием, является допустимым, если последует улучшение прогноза состояния здоровья пациента. Но такой посыл нельзя использовать как обоснование допустимости ненужного риска. Все риски следует уменьшать до самого низкого практически достижимого уровня, учитывая современный уровень научно-технического развития, пользу от допустимого риска и практическую осуществимость его дальнейшего уменьшения.
Практическая осуществимость отражает способность изготовителя уменьшить риск и имеет две составляющие:
- техническую осуществимость;
- экономическую осуществимость.
Техническая осуществимость отражает способность изготовителя уменьшить риск без учета материальных затрат. Ниже перечислены случаи, когда техническая осуществимость является спорной:
- большое число маркировок с предупреждениями/предостережениями, затрудняющими применение пользователем медицинского изделия;
- большое число сигналов тревоги, приводящих пользователя в замешательство;
- сообщение о таком большом числе остаточных рисков, что оператору сложно понять, какие из них являются действительно значимыми;
- слишком сложные процедуры применения медицинского изделия, противоречащие его предусмотренному применению;
- выполнение мер по управлению риском, противоречащих предусмотренному применению (например, уменьшение мощности электрохирургического устройства до уровня ниже эффективного).
Экономическая осуществимость отражает способность изготовителя уменьшить риск, не делая разработку и изготовление медицинского изделия экономически невыгодным проектом. Принятые решения обязательно должны базироваться на оптимальном соотношении между допустимостью рисков и пригодностью медицинского изделия для лечения и диагностики. Материальные затраты и пригодность медицинского изделия рассматривают при принятии решения о практической осуществимости уменьшения рисков, когда речь идет о сохранении или улучшении состояния здоровья пациента. Однако экономическую осуществимость не следует использовать как обоснование допустимости ненужного риска. Например, экономическая осуществимость является спорной при рассмотрении необходимости дублирования каждого критичного компонента дефибриллятора.
Обычно следует уменьшать риски, незначительно превышающие критерии, установленные изготовителем в отношении допустимости риска, даже при условии существенных затрат. Если уровень риска приближается к "незначительному", то дальнейшее уменьшение риска может не понадобиться, если только оно не может быть легко достигнуто.
В некоторых случаях (например, для защиты от радиации) применяют ALARP-подход. При этом вместо практической осуществимости принимают во внимание достижимость, что в действительности означает принятие во внимание только технической осуществимости и игнорирование экономической осуществимости.
D.8.5 Пример
На рисунке D.6 приведен пример матрицы риска, где область допустимого риска матрицы имеет дальнейшее подразделение. Риски, количественное значение которых определено (,
,
, ...), занесены в соответствующие элементы матрицы.
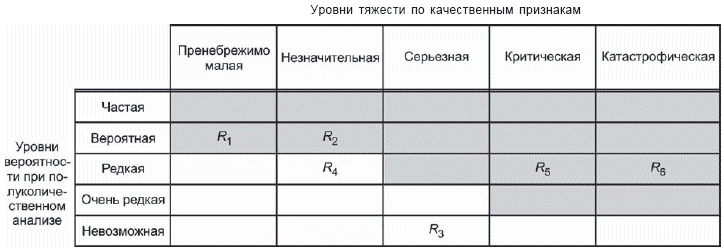
Условные обозначения:
- темно-серый цвет - недопустимый риск;
- светло-серый цвет - необходимо исследовать возможности дальнейшего уменьшения риска;
- белый цвет - незначительный риск
Рисунок D.6 - Пример матрицы оценивания риска по трем критериям
Приложение E
(справочное)
Примеры опасностей, прогнозируемых последовательностей событий и опасных ситуаций
Е.1 Общие положения
Требование к изготовителю составить перечень известных и прогнозируемых опасностей, связанных с медицинским изделием, как для нормальных условий, так и для условий отказа содержится в 4.3. Требование к изготовителю рассмотреть прогнозируемые последовательности событий, следствием которых может стать возникновение опасной ситуации и причинение вреда, содержится в 4.4. Согласно определениям данных терминов опасность не может привести к причинению вреда до тех пор, пока последовательность событий или другие обстоятельства (в том числе условия нормального применения) не приведут к возникновению опасной ситуации. На данном этапе риск может быть оценен путем определения тяжести и вероятности причинения вреда, в который могла бы развиться опасная ситуация (см. рисунок E.1).
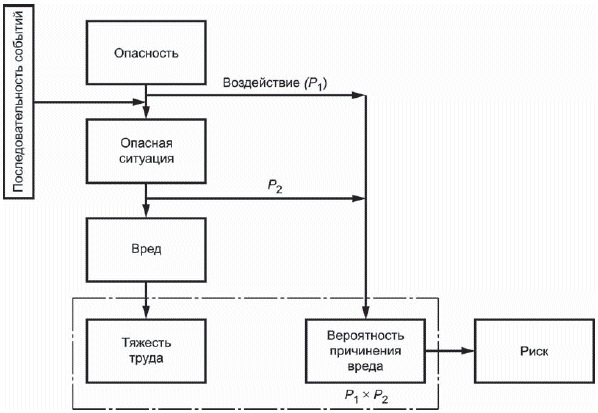
Условные обозначения:
- Р1 - вероятность возникновения опасной ситуации;
- Р2 - вероятность возникновения опасной ситуации, ведущей к причинению вреда
Рисунок Е.1 - Схема взаимосвязи между опасностью, последовательностью событий, опасной ситуацией и вредом
Хорошей отправной точкой для составления перечня опасностей является анализ опыта работы с такими же или подобными видами изделий. При анализе следует учитывать собственный опыт изготовителя, а также опыт других изготовителей, нашедший отражение в базе данных нежелательных событий, публикациях и других доступных источниках. Данный вид анализа очень полезен для идентификации и составления перечня типичных опасных ситуаций и связанных с ними возможных рисков для рассматриваемого изделия. Далее перечень опасных ситуаций и такие вспомогательные средства, как перечень примеров из таблицы E.1, можно использовать для составления первоначального перечня опасностей.
Таблица Е.1 - Примеры опасностей
Примеры энергетических опасностей | Примеры биологических и химических опасностей | Примеры эксплуатационных опасностей | Примеры информационных опасностей |
Опасности, связанные с электромагнитной энергией: - сетевое напряжение; - токи утечки: - ток утечки на корпус; - ток утечки на землю; - ток утечки на пациента; - электрические поля; - магнитные поля - ионизирующее излучение; - неионизирующее излучение - высокие температуры; - низкие температуры - сила тяжести: - при падении; - в подвешенном состоянии; - вибрация; - накопленная энергия; - наличие движущихся частей; - сила скручивания, усилие сдвига и сила растяжения; - перемещение пациента и придание ему требуемого положения; - акустическая энергия: - ультразвуковая энергия; - инфразвуковая энергия; - звук; - вливание жидкости под высоким давлением | Биологические опасности: - бактерии; - вирусы; - прочие возбудители заболеваний (например, прионы); - повторная или перекрестная инфекция - воздействие на дыхательные пути, ткани, окружающую среду или имущество, например, на инородные субстанции: - кислотами или основаниями; - отходами; - загрязняющими веществами; - добавками или вспомогательными веществами; - чистящими и дезинфицирующими средствами, тестирующими веществами; - продуктами распада; - медицинскими газами; - анестетиками - токсичность химических составляющих, например: - аллергенность/ раздражающее действие; - пирогенность | Опасности, связанные с функционированием: - неправильные или ненадлежащие результаты или функционирование; - некорректные измерения; - ошибки при передаче данных; - утрата или ухудшение функции - недостаток внимания; - забывчивость; - незнание правил; - недостаточность знаний; - нарушение установленного порядка | Опасности, связанные с маркировкой: - недостаточно полные инструкции по эксплуатации; - ненадлежащее описание эксплуатационных характеристик; - ненадлежащее описание предусмотренного применения; - ненадлежащая информация об ограничениях - ненадлежащая спецификация на принадлежности, применяемые совместно с медицинским изделием; - ненадлежащие спецификации на проверочные испытания перед применением медицинского изделия; - слишком сложные рабочие инструкции - о побочных эффектах; - об опасностях, связанных с повторным применением медицинских изделий однократного применения |
Следующим шагом является идентификация некоторых последовательностей событий, которые вместе с опасностями могут привести к возникновению опасных ситуаций и причинению вреда. Т.к. многие опасности могут никогда не причинить вред и поэтому их можно далее не рассматривать, то полезно проводить анализ, начиная с вреда, который медицинское изделие действительно может причинить, и далее следовать в обратном направлении. Однако несмотря на полезность такого подхода, следует признать, что подобный анализ не является совершенным. Многие последовательности событий возможно идентифицировать только при систематическом применении методов анализа риска, описанных в приложении G. Анализ и идентификация в дальнейшем усложняются из-за необходимости учета многих событий и обстоятельств, подобных приведенным в таблице E.2. Таким образом, для завершения всестороннего анализа необходимо использовать более чем один метод анализа риска, а иногда и дополнительные методы. Таблица E.3 содержит примеры взаимосвязи между опасностями, последовательностями событий, опасными ситуациями и вредом.
Таблица Е.2 - Примеры инициирующих событий и обстоятельств
Категория | Примеры инициирующих событий и обстоятельств |
Недостаточно подробные требования | Ненадлежащая спецификация на: - параметры конструкции; - эксплуатационные параметры; - эксплуатационные требования; - требования к обслуживанию (например, к текущему ремонту, переработке); - утилизацию |
Производственные процессы | Недостаточное управление изменениями производственных процессов. Недостаточное управление информацией о материалах/совместимости материалов. Недостаточное управление производственными процессами. Недостаточное управление субподрядчиками |
Транспортирование и хранение | Ненадлежащее упаковывание. Загрязнение или изнашивание (порча). Ненадлежащие условия окружающей среды |
Факторы окружающей среды | Физические (тепло, давление, время). Химические (коррозия, старение материалов, загрязнение). Электромагнитные поля (например, чувствительность к электромагнитным помехам). Недостаточное энергоснабжение. Недостаточная подача хладагента |
Очистка, дезинфекция и стерилизация | Отсутствие или недостаточно подробные требования к валидированным процедурам очистки, дезинфекции и стерилизации. Ненадлежащее проведение очистки, дезинфекции и стерилизации |
Вывод из эксплуатации и утилизация | Отсутствие информации или недостаточная информация. Ошибка применения |
Химический состав | Биологический распад. Биологическая совместимость. Отсутствие информации или недостаточно подробные технические требования. Ненадлежащее предупреждение об опасностях, связанных с неправильным химическим составом. Ошибки применения |
Человеческий фактор | Возможные ошибки применения, обусловленные дефектами конструкции, например: - недостаточно четкие инструкции по применению или их отсутствие; - сложная или недостаточно четкая система управления; - неоднозначное или неясное состояние изделия; - неправильная или нечеткая визуализация установочных параметров, результатов измерений или другой информации; - неправильное представление результатов; - недостаточные видимость, слышимость и тактильность; - ненадлежащее расположение элементов управления действиями или ненадлежащее представление информации о фактическом состоянии; - режимы работы или расположение элементов управления, противоречащие имеющемуся оборудованию; - применение медицинского изделия неквалифицированным/необученным персоналом; - недостаточное предупреждение о побочных эффектах; - недостаточное предупреждение об опасностях повторного применения медицинских изделий однократного применения; - неточные измерения и другие метрологические аспекты; - несовместимость с расходными материалами/принадлежностями/другими медицинскими изделиями; - промахи, упущения и заблуждения |
Режим отказа | Внезапная потеря электрической или механической целостности. Ухудшение функциональных свойств (например, постепенная окклюзия магистралей для жидкости и газа, изменение гидравлического сопротивления, электропроводности) как результат старения, износа и многократного применения изделия. Отказ вследствие усталости |
Таблица Е.3 - Взаимосвязь между опасностями, прогнозируемыми последовательностями событий, опасными ситуациями и вредом
Опасность | Прогнозируемая последовательность событий | Опасная ситуация | Вред |
Электромагнитная энергия (сетевое напряжение) | Электродный кабель случайно включен в сетевую розетку | На электродах появилось сетевое напряжение | Серьезное возгорание. Фибрилляция сердца. Летальный исход |
Химическая опасность (летучий растворитель) | Неполная очистка летучего растворителя, использованного при изготовлении изделия. Переход остатка растворителя в газовое состояние при температуре тела | Образование пузырьков газа в крови во время диализа | Газовая эмболия. Повреждение головного мозга. Летальный исход |
Биологическая опасность (бактериальное заражение) | Ненадлежащие инструкции по обеззараживанию наркозных трубок многократного применения. Использование загрязненных наркозных трубок | Высвобождение бактерий и попадание их в дыхательные пути пациента во время анестезии | Бактериальная инфекция. Летальный исход |
Электромагнитная энергия (ESD - электростатический заряд) | Пациент, имеющий электростатический заряд, касается инфузионного насоса. Наличие ESD приводит к отказу насоса и системы сигнализации насоса. Отсутствует подача инсулина пациенту | Незнание пациента с высоким уровнем сахара в крови о прекращении подачи инсулина | Незначительное повреждение органов. Помутнение сознания. Кома, летальный исход |
Ненадлежащее функционирование (нет выхода электроэнергии) | Истечение срока службы элемента питания имплантируемого дефибриллятора. Слишком большой интервал между клиническими обследованиями | При возникновении аритмии может не последовать разряд дефибриллятора | Летальный исход |
Несмотря на то, что составление перечня опасностей, опасных ситуаций и последовательностей событий желательно завершить на ранних стадиях процесса проектирования и разработки, для того чтобы облегчить управление риском, на практике идентификация и составление перечня - это непрерывная деятельность, которая продолжается, включая постпроизводство.
Настоящее приложение содержит неполный перечень возможных опасностей, связанных с различными медицинскими изделиями (таблица E.1), а также перечень инициирующих событий и обстоятельств (таблица E.2), которые могут привести к возникновению опасных ситуаций, а те, в свою очередь, - к причинению вреда. Таблица E.3 содержит логическую последовательность примеров того, как опасность может развиться в опасную ситуацию, а опасная ситуация - в причинение вреда через последовательность событий или обстоятельств.
Понимание возможности развития опасности в опасную ситуацию является важным для определения вероятности причинения и тяжести вреда. Целью процесса является составление как можно более исчерпывающего перечня опасных ситуаций. Идентификация опасностей и последовательностей событий является средством достижения данной цели. Перечни, содержащиеся в таблицах настоящего приложения, могут применяться для содействия в идентификации опасных ситуаций. Для проведения надлежащего анализа изготовитель должен сам определить, что считать опасностью.
Е.2 Примеры опасностей
Перечень, приведенный в таблице Е.1, можно использовать как помощь в идентификации связанных с медицинским изделием опасностей, которые в итоге могут привести к причинению вреда пациенту или другим лицам.
Е.3 Примеры инициирующих событий и обстоятельств
Для идентификации прогнозируемых последовательностей событий часто полезно проанализировать их причины, а именно инициирующие события и обстоятельства. В таблице Е.2 приведены примеры инициирующих событий и обстоятельств, разделенных на категории. Несмотря на то что данный перечень не является исчерпывающим, он содержит много разных видов инициирующих событий и обстоятельств, которые необходимо учитывать при идентификации прогнозируемых последовательностей событий для рассматриваемого медицинского изделия.
Е.4 Примеры взаимосвязей между опасностями, прогнозируемыми последовательностями событий, опасными ситуациями и вредом
Таблица Е.3 демонстрирует взаимосвязь между опасностями, прогнозируемыми последовательностями событий, опасными ситуациями и вредом в упрощенном виде. Другой обобщенный пример последовательности событий, включающей косвенные риски, представлен для медицинских изделий для диагностики in vitro на рисунке H.1 (приложение Н).
Следует помнить, что одна опасность может причинить несколько видов вреда, а более чем одна последовательность событий могут способствовать возникновению опасной ситуации.
Для проведения надлежащего анализа необходимо установить, в чем заключается опасная ситуация. В некоторых обстоятельствах примером опасной ситуации может быть снятие кожуха с устройства, находящегося под высоким напряжением, тогда как при других обстоятельствах примером опасной ситуации можно считать непосредственный контакт человека с устройством, находящимся под высоким напряжением.
Приложение F
(справочное)
План менеджмента риска
F.1 Общие положения
План менеджмента риска может представлять собой самостоятельный документ или быть связан с другими документами, например с документами системы менеджмента качества. Он может также содержать ссылки на другие документы, что соответствует требованиям, установленным в 3.4.
Структура и степень детализации плана должны быть сопоставимы с уровнем риска, связанным с рассматриваемым медицинским изделием. Требования, установленные в 3.4, являются минимальным набором требований к плану менеджмента риска. Изготовители могут включать в план менеджмента риска другие элементы, такие, как календарный план, средства анализа риска или обоснование установленных критериев допустимости риска.
F.2 Область применения плана
В области применения идентифицируют и описывают медицинское изделие и стадии его жизненного цикла, для которых применим каждый элемент плана.
Все элементы процесса менеджмента риска связаны с жизненным циклом медицинского изделия, установленным изготовителем. Некоторые элементы процесса менеджмента риска будут присутствовать на разных стадиях процессов жизненного цикла изделия, установленных изготовителем (см. [3]), например, при управлении проектированием и разработкой. Остальные элементы проявятся на других стадиях жизненного цикла, вплоть до вывода изделия из эксплуатации. В плане менеджмента риска эта связь для рассматриваемого медицинского изделия установлена непосредственно или посредством ссылок на другие документы.
Несмотря на необходимость планирования всех видов деятельности по менеджменту риска, изготовитель может иметь несколько планов для разных стадий жизненного цикла медицинского изделия. Давая пояснения относительно области применения каждого плана, можно подтвердить, что планом менеджмента риска охвачен весь жизненный цикл медицинского изделия.
F.3 Распределение ответственности и полномочий
В плане менеджмента риска необходимо идентифицировать сотрудников, ответственных за выполнение конкретных видов деятельности по менеджменту риска, например: проверяющего(их), эксперта(ов), независимого(ых) специалиста(ов) по верификации, лицо, имеющее право утверждения (см. 3.2). Распределение полномочий может быть включено в матрицу распределения ресурсов, разработанную для стадии проектирования и разработки.
F.4 Требования к анализу деятельности по менеджменту риска
В плане менеджмента риска следует подробно описать, как и когда будет проводиться анализ деятельности по менеджменту риска применительно к конкретному медицинскому изделию. Требования к данному анализу могут быть частью требований к анализу системы менеджмента качества (см. [3], 7.3.4).
F.5 Критерии допустимости риска, в том числе в тех случаях, когда вероятность причинения вреда не может быть определена
Критерии допустимости риска разрабатывают исходя из политики изготовителя в отношении определения допустимого риска [см. D.4 (приложение D)]. Критерии допустимости риска могут быть частью разработанной изготовителем системы менеджмента качества, на которую могут быть ссылки в плане менеджмента риска (см. [3], 7.1).
F.6 Деятельность по верификации
План менеджмента риска устанавливает процедуры проведения двух разных верификаций согласно требованиям настоящего стандарта [см. также А.2.6.3 (приложение А)]. Верификация результативности мер по управлению риском может потребовать сбора клинических данных, данных по эксплуатационной пригодности и т.п. (см. также 2.28). План менеджмента риска может включать подробное описание данного вида деятельности по верификации или ссылку на планы по другим видам деятельности по верификации.
F.7 Метод(ы) получения постпроизводственной информации
Метод(ы) получения постпроизводственной информации может быть частью установленных процедур системы менеджмента качества (см. [3], 8.2). Изготовителям следует установить универсальные процедуры сбора информации из таких источников, как пользователи, обслуживающий персонал, обучающий персонал, сообщения об инцидентах и обратная связь с пользователями. В большинстве случаев достаточно ссылки на процедуры системы менеджмента качества, однако любые специальные требования к медицинскому изделию следует внести непосредственно в план менеджмента риска.
План менеджмента риска должен включать принятые на основе анализа риска документированные решения по способу наблюдения, пригодному для сбора постпроизводственной информации о медицинском изделии, например, необходимо ли наблюдение на основе обратной связи или же требуется активное наблюдение. План менеджмента риска должен также включать подробное описание предусмотренных клинических исследований.
Приложение G
(справочное)
Информация о методах анализа риска
G.1 Общие положения
Настоящее приложение содержит руководящие указания в отношении некоторых доступных методов анализа риска, применяемых согласно 4.3. Данные методы дополняют друг друга, и может возникнуть необходимость в использовании нескольких из них. Основной принцип анализа риска заключается в поэтапном анализе последовательности событий.
Предварительный анализ опасностей (Preliminary Hazard Analysis - PHA) - это метод, применяемый на ранних стадиях процесса разработки для идентификации опасностей, опасных ситуаций и событий, которые могут привести к причинению вреда, когда конструкция медицинского изделия известна лишь в общем виде.
Анализ диагностического дерева неисправностей (Fault Tree Analysis - FTA) - это метод, который может быть особенно полезен для обеспечения безопасности на ранних стадиях разработки медицинского изделия в целях идентификации и установления приоритета опасностей и опасных ситуаций, а также для анализа нежелательных событий.
Анализ характера и последствий отказов (Failure Mode and Effect Analysis - FMEA) и анализ характера, последствий и критичности отказов (Failure Mode, Effects and Criticality Analysis - FMECA) являются методами, пригодными для систематической идентификации влияния и последствий отказов отдельных компонентов; эти методы больше подходят для практически завершенных конструкций.
Исследование опасностей и эксплуатационной пригодности (Hazard and Operability Study - HAZOP) и анализ опасностей в критических контрольных точках (Hazard Analysis on Critical Control Points - HACCP) - это методы, обычно применяемые на более поздних стадиях разработки медицинского изделия для верификации, а затем для оптимизации концепций проекта или внесения изменений.
G.2 Предварительный анализ опасностей (PHA)
Предварительный анализ опасностей (PHA) - это индуктивный метод анализа, целью которого является идентификация опасностей, опасных ситуаций и событий, которые могут привести к причинению вреда определенному виду деятельности, устройству или системе. Чаще всего данный метод применяют на ранней стадии разработки проекта, когда имеется недостаточно информации о деталях конструкции или производственных процессах; зачастую PHA определяет направление дальнейших исследований. Он может быть полезен при анализе действующих систем или установлении приоритета опасностей в условиях, когда обстоятельства препятствуют применению другого метода.
При использовании РНА составляют перечень опасностей и связанных с ними опасных ситуаций с учетом следующих факторов:
a) применяемые или изготавливаемые материалы и их реактивность;
b) применяемое оборудование;
c) производственная среда;
d) проектная схема;
e) взаимодействие компонентов медицинского изделия или системы.
В завершение определяют вероятность несчастного случая, выполняют оценивание возможной травмы или возможного ущерба здоровью по качественным признакам и идентифицируют применимые восстановительные меры. Полученные результаты можно наглядно представить в виде таблиц или древовидных схем.
Более подробная информация о процедурах РНА приведена в [8], А.5.
G.3 Анализ диагностического дерева неисправностей (FTA)
Метод FTA является в первую очередь средством анализа опасностей, идентифицированных с помощью других методов; его начинают с постулата о нежелательном последствии, называемом также "главное событие". Методом дедукции, начиная с главного события, идентифицируют возможные причины или характер неисправностей, вызывающие нежелательные последствия (отказы системы). Каждый раз идентификацию выполняют на все более низком функциональном уровне системы. Следующая за этим поэтапная идентификация нежелательного функционирования системы с последовательным понижением уровней приводит к искомому уровню системы, который обычно содержит источник неисправности или является самым низким уровнем, на котором могут применять меры по управлению риском. Это позволяет установить цепочки событий, с наибольшей вероятностью приводящие к заявленному нежелательному последствию (отказу). Результаты представляются графически - в виде дерева неисправностей. На каждом уровне схемы комбинации видов неисправностей описаны логическими операторами (И, ИЛИ и т.д.). Виды неисправностей, идентифицированные на дереве неисправностей, могут быть связаны с неисправностью аппаратного обеспечения, с человеческим фактором или с любыми другими событиями, ведущими к нежелательным последствиям (отказам). Они не ограничены условиями единичного отказа.
Метод FTA позволяет систематически и в то же время достаточно гибко анализировать разные факторы, в том числе человеческий фактор. Данный метод используют при анализе риска в качестве инструмента определения вероятностей отказов или для идентификации единичных или множественных отказов одного вида, приводящих к развитию опасных ситуаций. Графическое представление в виде дерева неисправностей позволяет легко понять поведение системы и входящих в нее факторов, но если дерево неисправностей становится большим, то его обработка может потребовать применения компьютерных систем, что, впрочем, не представляет сложности.
Более полная информация о процедурах FTA приведена в [17].
G.4 Анализ характера и последствий отказов (FMEA)
Анализ характера и последствий отказов (FMEA) является методом, с помощью которого систематически идентифицируют и оценивают последствия отдельной неисправности (единичного отказа). Это индуктивный метод, при использовании которого отвечают на вопрос: "Что произойдет, если...?" Компоненты анализируют последовательно, один за другим, что в основном соответствует условию единичного отказа. Анализ выполняют в режиме "снизу вверх", т.е. следуя процедуре перехода к очередному, более высокому функциональному уровню медицинского изделия или системы.
Метод FMEA не сводится к анализу отказов, являющихся следствием ошибки проектирования компонентов медицинского изделия, но может также включать анализ отказов в процессе изготовления и монтажа компонентов изделия или при применении или ошибочном применении изделия конечным пользователем (применение метода FMEA). Метод FMEA может быть расширен за счет исследования отказов отдельных компонентов, вероятности их возникновения и возможности выявления (до того уровня, когда выявление позволяет применять предупреждающие действия), а также степени тяжести последствий отказов. Анализ характера и последствий отказов (FMEA) может стать анализом характера, последствий и критичности отказов (FMECA). Для выполнения FMECA конструкция медицинского изделия должна быть известна во всех подробностях.
Метод FMEA может быть также полезен при рассмотрении ошибок применения. Недостатками данного метода являются трудности его применения при наличии избыточной информации, при включении в анализ действий по ремонту и профилактическому техническому обслуживанию медицинского изделия, а также ограничение данного метода условиями единичного отказа.
Более подробная информация о процедурах FMEA приведена в [18].
G.5 Исследование опасностей и эксплуатационной пригодности (HAZOP)
Метод исследования опасностей и эксплуатационной пригодности (HAZOP) аналогичен методу FMEA. Исследование опасностей и эксплуатационной пригодности основано на теоретической предпосылке, что инциденты обусловлены отклонениями от проекта или предусмотренного применения. Это систематический метод идентификации опасностей и проблем эксплуатационной пригодности. Он был первоначально разработан для применения в химической промышленности. Поскольку применение метода в химической промышленности сфокусировано на отклонениях от проекта, то существуют возможности его альтернативного использования в области разработки медицинских изделий. Метод HAZOP можно использовать в отношении применения/функционирования медицинских изделий (например, применительно к существующим способам диагностики, лечения или облегчения заболеваний, выступающим в качестве цели проекта) или в отношении процессов, применяемых при изготовлении или профилактическом уходе/техническом обслуживании медицинских изделий (например, стерилизация), которые могут оказывать значительное влияние на их функционирование. Для данного метода характерно:
a) использование группы специалистов, компетентных в области проектирования и применения рассматриваемого медицинского изделия;
b) применение соответствующих ключевых слов (NONE - ничто, никто, нисколько; PART OF - часть чего-либо и т.д.) для идентификации отклонений от нормального применения.
Целями данного метода являются:
- составление полного описания медицинского изделия и его предусмотренного применения;
- систематический анализ каждого аспекта предусмотренного применения медицинского изделия в целях определения причины отклонения от нормальных рабочих условий и утвержденного проекта;
- определение последствий таких отклонений и принятие решения о том, могут ли данные последствия привести к возникновению опасностей или создать проблемы с функционированием медицинского изделия.
Достижение последней цели особенно важно применительно к процессам, используемым при изготовлении медицинских изделий, когда характеристики медицинского изделия зависят от процесса изготовления.
Более подробная информация о процедурах HAZOP приведена в [19].
G.6 Анализ опасностей в критических контрольных точках (НАССР)
Анализ опасностей в критических контрольных точках (НАССР) - это систематический метод идентификации, оценивания и управления опасностями. Первоначально он был разработан Национальным агентством по аэронавтике и исследованию космического пространства (NASA) для предупреждения пищевого отравления астронавтов. Метод основан на использовании установленного набора принципов и терминов с определениями. Применительно к медицинским изделиям метод HACCP используют для мониторинга и управления причинами опасностей, связанных с медицинским изделием и инициированных сопутствующими процессами, особенно процессами изготовления.
Метод НАССР основан на семи принципах:
1) проведение анализа опасностей согласно 4.3 и идентификация предупреждающих действий согласно 6.2;
2) определение критических контрольных точек (ССРs) согласно 6.2;
3) установление предельно допустимых значений согласно 4.2 и разделу 5;
4) мониторинг каждой критической контрольной точки (ССР) согласно 6.3 и разделу 9;
5) разработка корректирующих действий согласно разделу 9;
6) разработка процедур верификации согласно 6.3 и разделу 9;
7) разработка процедур ведения и хранения записей и других документов согласно 3.5 и разделу 8.
Каждое медицинское изделие подвергается опасностям, связанным с его предусмотренным применением. Опасные ситуации могут быть вызваны событиями (причинами или сопутствующими факторами), происходящими на разных стадиях жизненного цикла изделия, таких, как проектирование, изготовление, обслуживание, применение, утилизация и т.д. Примеры некоторых видов опасностей приведены в приложении Е.
Залогом результативности НАССР служат непрерывный мониторинг и управление идентифицированными опасностями (принципы НАССР 2, 3 и 4). Изготовитель демонстрирует результативность установленных мер по управлению опасностями (принципы НАССР 5 и 6) посредством разработки документированной схемы процесса, анализа опасностей и плана контроля критических точек (принцип НАССР 7).
В системе НАССР в качестве документального подтверждения используют следующие средства ведения записей:
a) блок-схема процесса.
Целью блок-схемы является простое и четкое графическое воспроизведение последовательности стадий процесса. Блок-схема необходима для последующей работы по методу НАССР. Она может стать в будущем руководством для тех, кому необходимо будет разобраться в процессе в целях его верификации. Область распространения блок-схемы должна охватывать все стадии процесса, непосредственно управляемые изготовителем;
b) рабочая карта для анализа опасностей.
Анализ опасностей включает идентификацию опасностей и вызывающих их причин. Записи по анализу опасностей должны содержать:
- идентификацию и перечень тех стадий процесса, на которых возникают значительные опасности;
- перечень всех идентифицированных опасностей с указанием их значимости на каждой стадии процесса;
- перечень всех предупреждающих действий, необходимых для управления каждой опасностью;
- идентификацию всех критических контрольных точек (ССРs), средств их мониторинга и управления ими;
c) план НАССР.
План НАССР представляет собой документ в письменной форме, основанный на семи принципах НАССР и описывающий процедуры, которым необходимо следовать для обеспечения управления проектом, изделием, процессом или процедурой. План включает:
- идентификацию критических контрольных точек и предельно допустимых значений;
- виды деятельности по мониторингу и непрерывному управлению;
- идентификацию и мониторинг корректирующих действий, видов деятельности по верификации и ведению записей.
Приложение H
(справочное)
Руководство по менеджменту риска медицинских изделий для диагностики in vitro
H.1 Общие положения
Настоящее приложение содержит дополнительные руководящие указания по применению менеджмента риска к медицинским изделиям для диагностики in vitro (IVD). Основное внимание уделено менеджменту риска в отношении пациентов на основании применения результатов исследований медицинских изделий для диагностики in vitro. Приведенные примеры не являются исчерпывающими, они предназначены для разъяснения общих принципов и служат отправной точкой при менеджменте риска.
Медицинские изделия для диагностики in vitro предназначены для отбора, подготовки и исследования проб, взятых из тела человека. К данным изделиям относят реагенты, приборы, программное обеспечение, изделия для отбора проб и емкости для их хранения, калибраторы, контрольные материалы и соответствующие принадлежности. Перечисленные изделия можно применять как отдельно, так и комбинируя их в систему.
Результаты, полученные с помощью медицинских изделий для диагностики in vitro, можно использовать для диагностики заболеваний или других состояний, в том числе состояния здоровья пациента, для восстановления здоровья и облегчения состояния пациента, лечения или предупреждения заболевания, а также для мониторинга лекарственной терапии и определения безопасности донорской крови или донорских органов. Данные изделия могут использовать лица с разным уровнем образования, подготовки и разным практическим опытом, а также в разной окружающей среде и при разной степени управления окружающей средой. Например, одни медицинские изделия для диагностики in vitro предназначены для применения специалистами в медицинских лабораториях, другие - медицинскими работниками на местах, третьи - непрофессиональными пользователями в домашних условиях.
С одной стороны, результаты проведенных в лаборатории IVD-исследований передают врачу, который интерпретирует данные и ставит диагноз, проводит лечение или наблюдает пациента; с другой стороны, IVD-исследования может выполнять сам пациент, когда использует результаты исследований для наблюдения за состоянием своего здоровья и последующего лечения.
Ввиду многообразия медицинских изделий для диагностики in vitro и их предусмотренного применения настоящие руководящие указания можно применять не во всех случаях. В отношении медицинских изделий для диагностики in vitro, предназначенных для самотестирования, термины "пациент" и "непрофессиональный пользователь" будут взаимозаменяемыми, хотя могут обозначать и разных пользователей (например, определение уровня сахара в крови у ребенка, страдающего диабетом, может провести его родственник). Если используют термин "врач", то следует понимать, что любой медицинский работник также может запрашивать, получать, интерпретировать результаты in vitro-исследований, проведенных в лаборатории, и действовать на их основании.
Медицинские изделия для диагностики in vitro потенциально могут способствовать причинению вреда пациенту. Ошибочные или запоздалые результаты могут привести к ошибочным или запоздалым медицинским решениям и действиям, которые могут навредить пациенту. Ошибочные результаты, полученные при применении медицинских изделий для диагностики in vitro, предназначенных для скрининга переливания крови и трансплантации органов, потенциально могут причинить вред реципиентам крови или органов. Ошибочные результаты, полученные при применении медицинских изделий для диагностики in vitro, предназначенных для выявления инфекционных заболеваний, могут представлять потенциальную опасность для общественного здоровья.
На рисунке H.1 представлена одна из моделей рисков применения медицинских изделий для диагностики in vitro в лабораторных условиях. В этом примере отказ системы менеджмента качества (СМК) изготовителя (например, на стадии проектирования, разработки, изготовления, упаковывания, маркирования, реализации или обслуживания) инициирует последовательность событий, которая начинается с неисправности или ненадлежащего функционирования медицинского изделия для диагностики in vitro. Если отказ медицинского изделия происходит в медицинской лаборатории, то это приводит к ошибочным результатам исследований. Если результат не идентифицирован в лаборатории как ошибочный, то он попадает к медицинскому работнику. Если медицинский работник также не идентифицирует результат как ошибочный, то это может привести к неправильному диагнозу и создать опасную ситуацию для пациента.
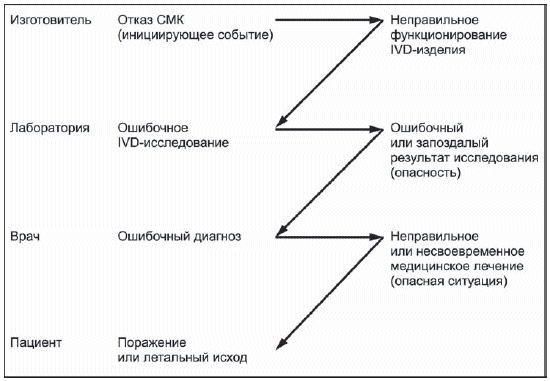
Рисунок H.1 - Модель рисков применения медицинского изделия для диагностики in vitro в лабораторных условиях
Врач применяет результаты IVD-исследований совместно с другой доступной медицинской информацией, для того чтобы оценить состояние здоровья пациента, поставить диагноз или назначить лечение. В некоторых случаях результаты in vitro-исследований могут быть главным или даже единственным основанием для принятия клинического решения. Вероятность причинения вреда пациенту складывается из вероятностей наступления каждого события, показанного на рисунке H.1. Каждая отдельная вероятность наступления события частично компенсируется вероятностью обнаружения опасности или опасной ситуации изготовителем, лабораторией или врачом, что позволяет вмешаться и предотвратить причинение вреда пациенту. Фактическая последовательность событий будет зависеть от конкретного медицинского изделия для диагностики in vitro и его применения.
На рисунке H.1 видно, что ошибочные или запоздалые результаты могут быть получены в лаборатории, например, вследствие несоблюдения процедур, графиков технического обслуживания или калибровки, игнорирования предупреждений и предостережений. События, которые могут привести к причинению вреда пациенту, также могут быть инициированы в лаборатории. Поэтому признана необходимость уменьшения числа ошибок с помощью применения процесса менеджмента риска к медицинской лаборатории, а информация по безопасности, получаемая на выходе процесса менеджмента риска изготовителя, может стать входом процесса менеджмента риска применительно к лаборатории.
H.2 Анализ риска
H.2.1 Идентификация предусмотренного применения
Н.2.1.1 Общие положения
Медицинские изделия для диагностики in vitro, предназначенные для лабораторных исследований или исследований на месте проведения лечения, имеют двух пользователей: медицинского работника, проводящего исследование, и медицинского работника, получающего результаты, интерпретирующего их и действующего на их основании. В случае IVD-изделия для самотестирования пациент может быть единственным пользователем.
При идентификации предусмотренного применения следует рассматривать заявленные цели изготовителя в отношении обоих вариантов применения:
1) применение IVD-изделия для получения результатов исследования; 2) применение результатов исследования для диагностики, лечения или наблюдения за пациентом.
В настоящем приложении применены следующие термины с соответствующими определениями:
- оператор - лицо, проводящее in vitro-исследования; данное лицо может быть как сотрудником лаборатории, так и медицинским работником или непрофессиональным пользователем с минимальной подготовкой или ее отсутствием;
- медицинский работник - лицо, запрашивающее, получающее результаты исследований или действующее на основе данных результатов от имени пациента; медицинским работником является врач, медсестра, фельдшер "скорой помощи" или любое другое лицо, принимающее клиническое решение на основе результатов IVD-исследований.
Н.2.1.2 Предусмотренное применение
Предусмотренное применение медицинского изделия для диагностики in vitro может включать требования к измерительной системе, анализируемым пробам (образцам), лабораторному оборудованию, матрице проб, процедуре исследования (анализ по качественным, полуколичественным или количественным признакам), оператору и месту применения.
Например, может возникнуть необходимость в исследовании содержания хорионического бета-гонадотропина человека (-hCG) в образцах сыворотки, плазмы крови или мочи. Не каждая процедура исследования
-hCG имеет те характеристики, которые соответствуют всем трем видам матрицы проб.
Н.2.1.3 Показания к применению
Показания к применению включают медицинское применение и определение популяций пользователей, для которых предназначено медицинское изделие для диагностики in vitro.
Например, результаты исследования содержания -hCG можно использовать для подтверждения беременности, скрининга беременных женщин на наличие у плода синдрома Дауна, а также для мониторинга некоторых форм рака. Каждое медицинское применение может выдвигать разные требования к чувствительности, специфичности, точности и погрешности измерений.
H.2.2 Идентификация возможных ошибок применения
Н.2.2.1 Ошибки применения
Ошибки применения включают действия, не предусмотренные изготовителем, такие, как ускорение процедур, попытки оптимизации и импровизации, а также невыполнение действий, предусмотренных изготовителем и описанных в инструкциях по применению.
Н.2.2.2 Примеры возможных ошибок применения со стороны сотрудников лаборатории
Ниже приведены примеры возможных ошибок применения в лабораторных условиях. Данные примеры являются показательными, но неисчерпывающими:
- применение медицинского изделия для диагностики in vitro с ненадлежащим калибратором, реагентом, прибором или анализируемым образцом;
- попытка оптимизации процедуры исследования для улучшения эксплуатационных характеристик;
- сокращение процедуры исследования (ускорение процедуры);
- невыполнение технического обслуживания прибора;
- неспособность или невозможность осуществления мер по обеспечению безопасности;
- функционирование в неблагоприятных условиях окружающей среды.
Н.2.2.3 Примеры возможных ошибок применения со стороны медицинского персонала
Ниже приведены примеры возможных ошибок применения со стороны медицинского персонала. Данные примеры являются показательными, но неисчерпывающими:
- применение результатов in vitro-исследования для скрининга населения на наличие заболевания, в то время как процедура исследования предназначена для диагностики данного заболевания (эксплуатационные характеристики IVD-изделия могут не соответствовать задаче скрининга населения);
- применение результатов IVD-исследования для диагностики заболевания, в то время как процедура исследования предназначена для наблюдения за состоянием здоровья (эксплуатационные характеристики IVD-изделия могут не соответствовать задачам диагностики);
- применение результатов IVD-исследования для нового способа медицинского применения, не заявленного изготовителем (эксплуатационные характеристики IVD-изделия могут оказаться непригодными для нового способа клинического применения).
Н.2.2.4 Примеры возможных ошибок применения пациентами при самотестировании
Ниже приведены примеры возможных ошибок применения пациентами при самотестировании. Эти примеры являются показательными, но неисчерпывающими:
- использование недостаточного объема образца;
- неспособность правильно вставить модуль с реагентом;
- разделение реагентных стрипов (для уменьшения материальных затрат);
- неспособность или невозможность осуществления мер по обеспечению безопасности;
- хранение реагента в ненадлежащих условиях.
H.2.3 Идентификация характеристик, относящихся к безопасности
Н.2.3.1 Общие положения
Помимо химических, механических, электрических и биологических характеристик и наряду с другими медицинскими изделиями медицинские изделия для диагностики in vitro имеют эксплуатационные характеристики, определяющие точность результатов исследования. Неспособность данных медицинских изделий соответствовать эксплуатационным характеристикам, необходимым для конкретного медицинского применения, может привести к созданию опасной ситуации и соответственно риска для пациентов.
Н.2.3.2 Характеристики процедур исследования по количественным признакам
Процедуры исследования по количественным признакам предназначены для определения количества или концентрации анализируемого вещества (аналита). Результаты отмечают на интервальной шкале. Основными аналитическими характеристиками процедур исследования по количественным признакам являются точность (неточность), погрешность (систематическая ошибка оценки, обусловленная субъективным фактором или неточностью прибора), специфичность анализа и диапазон количественного определения. Эксплуатационные требования зависят от вида медицинского применения. Ошибочно высокий или ошибочно низкий результат может привести к неправильному диагнозу или запоздалому лечению, а последующий вред, причиняемый пациенту, может зависеть от концентрации анализируемого вещества и значения систематической погрешности.
Н.2.3.3 Характеристики процедур исследования по качественным признакам
Процедуры исследования по качественным признакам предназначены только для выявления присутствия или отсутствия аналита. Результаты процедур классифицируют как положительные, отрицательные или неопределенные. К характеристикам процедур исследования по качественным признакам обычно относят диагностическую чувствительность и специфичность. Положительный результат при отсутствии аналита или отрицательный результат при наличии аналита может привести к постановке неправильного диагноза или запоздалому лечению и причинению вреда пациенту.
Н.2.3.4 Характеристики функциональной надежности
Если лечащему врачу необходимы результаты in vitro-исследования для принятия срочного клинического решения, например, о направлении пациента в отделение интенсивной терапии, то своевременность результатов может быть так же важна, как и их точность. Невозможность получения результата, когда он необходим, может привести к созданию опасной ситуации.
Н.2.3.5 Дополнительная информация о пациенте
В некоторых случаях для правильной интерпретации результатов исследования могут потребоваться демографические данные пациента, а также данные о пробе или ее исследовании. Примерами таких данных, вводимых вручную лаборантом-аналитиком или автоматически с помощью лабораторной компьютерной системы, являются идентификация пациента, идентификация пробы, вид пробы, описание пробы, единицы измерения, образцовый интервал, возраст, пол и генетические факторы. Если медицинское изделие для диагностики in vitro спроектировано так, чтобы вместе с результатами исследования сообщать дополнительную информацию (т.е. является интерпретирующим), то неспособность изделия выдать правильную информацию совместно с результатами исследования может нарушить правильность интерпретации данных результатов и привести к созданию опасной ситуации.
H.2.4 Идентификация известных и прогнозируемых опасностей
Н.2.4.1 Опасности для пациентов
С точки зрения пациента, результат IVD-исследования представляет собой опасность, если он может привести к несоответствующему медицинскому вмешательству, следствием которого может стать причинение вреда здоровью или смерть, или к невозможности оказать медицинскую помощь, которая могла бы предотвратить причинение вреда здоровью или смерть. Ошибочный или запоздалый результат IVD-исследования может быть обусловлен ненадлежащим функционированием медицинского изделия для диагностики in vitro, что является инициирующей опасностью в прогнозируемой последовательности событий, приводящей к созданию опасной ситуации. Идентификация опасностей и последовательностей событий помогает изготовителю в составлении подробного перечня опасных ситуаций. В процессе анализа риска изготовитель устанавливает, что именно необходимо рассматривать как опасность.
Как показано на рисунке Н.1, опасная ситуация может возникнуть, если медицинский работник получает ошибочный результат исследования и принимает решения на его основании. Опасная ситуация может также возникнуть в отсутствие результата, когда он необходим. В случае с изделиями для самотестирования опасная ситуация может возникнуть при получении ошибочного результата пациентом или отсутствии результата, когда он необходим.
При проведении исследования по количественным признакам результат можно рассматривать как ошибочный, если разница по сравнению с правильным значением превышает предельное значение, основанное на клинической пользе от применения медицинского изделия. Клиническая значимость неправильного результата может зависеть от величины разницы между измеренным и правильным значениями, а также от физиологического состояния пациента (например, наличия у него гипогликемии или гипергликемии).
При проведении исследования по качественным признакам, когда может быть получен только положительный или отрицательный результат (например, исследования на ВИЧ или на беременность), данный результат является также либо верным, либо ошибочным.
Следующие опасности могут обусловить неправильную диагностику и возможность причинения вреда или запоздалое медицинское вмешательство:
- ошибочные результаты (см. Н.2.3.2 и Н.2.3.3);
- запоздалые результаты (см. Н.2.3.4);
- ошибочная информация, сообщаемая прибором в дополнение к результату исследований (см. Н.2.3.5).
Н.2.4.2 Взаимосвязь с эксплуатационными характеристиками
Необходимо оценивать несоответствие любой из эксплуатационных характеристик, относящихся к безопасности (см. Н.2.3), предъявляемым изготовителем требованиям, для того чтобы определить возможность создания опасной ситуации.
Методы анализа опасностей, такие как предварительный анализ опасностей (Preliminary Hazard Analysis - PHA), анализ диагностического дерева неисправностей (Fault Tree Analysis - FTA), анализ характера и последствий отказов (Failure Mode and Effect Analysis - FMEA) и анализ опасностей в критических контрольных точках (Hazard Analysis on Critical Control Points - HACCP), описаны в приложении G.
Н.2.4.3 Идентификация опасностей в условиях отказа
При идентификации IVD-опасностей в условиях отказа следует рассматривать режимы отказов, которые могут привести к несоответствию эксплуатационных характеристик, требуемых для применения медицинского изделия (например, погрешность, точность, специфичность и т.д.), а именно:
- неоднородность внутри одной серии медицинских изделий;
- несовместимость разных серий медицинских изделий;
- непрослеживаемость значений калибратора;
- невозможность замены калибратора;
- неспецифичность (например, факторы, создающие помехи);
- использование остатков проб или реагентов для нового исследования;
- неточность измерений (связанная с прибором);
- нарушение стабильности (при хранении, транспортировании, применении).
При идентификации IVD-опасностей в условиях отказа медицинского изделия следует рассматривать те режимы отказов, которые могут привести к запоздалым результатам при необходимости оказания срочной медицинской помощи, например:
- нестабильность реагентов;
- отказ аппаратного или программного обеспечения;
- ненадлежащее упаковывание.
При идентификации IVD-опасностей в условиях отказа медицинского изделия следует рассматривать те режимы отказов, которые могут привести к выдаче ошибочной информации о пациенте, например:
- ошибочные Ф.И.О. или идентификационный номер пациента;
- ошибочная дата рождения или возраст;
- неправильно указанный пол.
Н.2.4.4 Идентификация опасностей в условиях нормального применения
Ошибочные результаты могут появиться также в условиях нормального применения, даже если эксплуатационные характеристики медицинского изделия для диагностики in vitro отвечают требованиям изготовителя. Это может быть следствием недостоверности результатов исследования, биологической вариабельности проб пациентов, выбора границы отсекания или других факторов. Ошибочный результат в условиях нормального применения может привести к созданию опасной ситуации в отношении отдельного пациента, например:
- недостаточно четкое различие между положительными и отрицательными результатами: процедуры исследования по качественным признакам демонстрируют типичные ложно положительные и ложно отрицательные оценки, частично обусловленные погрешностями измерения, связанными с определением подходящей границы отсекания;
- погрешность измерения: современный уровень научно-технического развития может ограничивать точность определения значения параметра медицинскими изделиями для диагностики in vitro (глюкометры, описанные в [20]); если критерии функционирования требуют, чтобы не менее 95% результатов находились в границах установленного диапазона значений, основанного на понятии клинической пользы, то до 5% результатов могут выходить за рамки данного диапазона;
- непредвиденное влияние других элементов (факторов, создающих помехи) в основном анализируемом образце: новые лекарственные средства, биохимические метаболиты, гетерофильные антитела и вещества для подготовки проб могут воздействовать на функциональные характеристики процедуры IVD-исследования;
- естественная гетерогенность аналита: антитела и другие белки в образцах крови являются смесями разных изоформ; известные функциональные характеристики процедуры IVD-исследования могут быть неприменимы ко всем составным частям такой смеси.
Н.2.4.5 Идентификация опасных ситуаций
К опасным ситуациям, создаваемым медицинскими изделиями для диагностики in vitro, относят:
- ложно отрицательный результат анализа на ВИЧ (HIV) или на поверхностный антиген гепатита В (HbsAg) при скрининге переливаемой крови, получаемый банком крови;
- диагностирование лечащим врачом заболевания печени на основе результатов исследования функции печени, которые были искажены под влиянием билирубина;
- ошибочное измерение прибором для самотестирования концентрации сахара в крови (завышенное значение) у больного гипогликемией.
H.2.5 Определение риска для пациента
Н.2.5.1 Общие положения
Определение риска основано на определении тяжести и вероятности причинения вреда каждой идентифицированной опасной ситуацией, связанной с медицинским изделием для диагностики in vitro, как в нормальных условиях, так и в условиях отказа.
Для получения ошибочного результата IVD-исследования ключевыми определяющими факторами являются: а) вероятность того, что результат будет признан ошибочным, и b) вероятность того, что результат приведет к нежелательному медицинскому воздействию.
При получении результатов, ошибочно показывающих, что в медицинском вмешательстве нет необходимости (например, ложно отрицательные результаты или ложно "нормальные" результаты), в процессе оценивания риска следует учитывать: 1) прогноз для невылеченного заболевания; 2) возможность диагностирования данного заболевания другими средствами; 3) последствия для лиц, не являющихся пациентами (перенос возбудителя инфекции или наследственного заболевания, воздействие на плод опасных веществ).
При получении результатов, ошибочно показывающих, что требуется медицинское вмешательство (например, ложно положительные результаты или ложно "анормальные" результаты), в процессе оценивания риска следует учитывать: 1) возможный вред от ненадлежащего лечения; 2) возможность лечения другими средствами; 3) последствия для других лиц (исследование или лечение в связи с опасностью инфицирования, диагностирование или лечение наследственного заболевания).
Н.2.5.2 Определение тяжести вреда
Медицинское применение результатов IVD-исследования позволяет определить возможный вред, который может быть причинен пациенту вследствие получения ошибочного результата. Следует рассматривать предусмотренное применение и возможное неправильное применение, описанные в Н.2.1 и Н.2.2.
Для определения тяжести вреда необходимо понимание медицинских последствий применения результатов IVD-исследования, аналитических эксплуатационных требований для каждого случая применения и того, в какой степени клинические решения основаны на результатах IVD-исследования. По этой причине решающее значение имеют надлежащие клинические входные данные для процесса определения риска.
Н.2.5.3 Определение вероятности причинения вреда
Как показано в приложении Е, вероятность причинения вреда при применении медицинского изделия для диагностики in vitro зависит от суммы вероятностей, связанных с последовательностью событий.
При применении медицинского изделия для диагностики in vitro в лабораторных условиях, как показано на рисунке Н.1, данные вероятности включают:
- вероятность того, что медицинское изделие для диагностики in vitro выдаст ошибочный результат;
- вероятность того, что лаборатория не идентифицирует полученный результат как ошибочный и передаст данный ошибочный результат врачу;
- вероятность того, что лечащий врач не идентифицирует полученный результат как ошибочный и предпримет (или не предпримет) надлежащие действия;
- вероятность того, что пациенту будет причинен вред в результате действия или бездействия лечащего врача.
Лаборатория может идентифицировать результат как ошибочный в следующих случаях:
- система управления качеством идентифицировала изменение в процедуре исследования;
- значение измеренного параметра несовместимо с жизнью;
- результат превысил критическое значение, что требует проведения верификации;
- разница по сравнению с предыдущим результатом для данного пациента превысила ожидаемую или возможную величину.
При определении вероятности причинения вреда следует учитывать, что не все лаборатории имеют результативные системы обнаружения ошибок, которые в состоянии предотвратить сообщение неправильных результатов.
Врач может идентифицировать результат как ошибочный в следующих случаях:
- данный результат физиологически невозможен;
- данный результат не согласуется с состоянием здоровья пациента;
- данный результат вступает в противоречие с другой информацией.
Если медицинское изделие для диагностики in vitro применяют за пределами лаборатории, то надлежащие или результативные системы обнаружения ошибок при этом зачастую отсутствуют. Непрофессиональные пользователи могут не знать, какие результаты являются неправдоподобными. Для подобных медицинских изделий для диагностики in vitro примеры, приведенные в данном пункте, следует видоизменить, исключив неприменимые события и вероятности.
Для того чтобы определить количественные значения вероятностей причинения вреда, редко имеются достаточные данные. Вопросы, приведенные в Н.2.5.4, могут быть полезными для получения качественных и полуколичественных оценок вероятностей. В первую очередь данные вопросы относятся к медицинским изделиям для диагностики in vitro, применяемым в лабораторных условиях, но подобные вопросы могут быть составлены и для других видов медицинских изделий для диагностики in vitro.
Н.2.5.4 Проблемы, которые необходимо рассмотреть при определении риска для пациента
Н.2.5.4.1 Вероятность получения ошибочного результата при применении медицинского изделия для диагностики in vitro:
- в предполагаемых условиях отказа;
- в нормальных условиях;
- при обоснованно прогнозируемом неправильном применении.
Н.2.5.4.2 Вероятность идентификации ошибочного результата IVD-исследования пользователем/лабораторией:
- возможность поставки контрольных материалов вместе с медицинским изделием для диагностики in vitro;
- наличие в составе медицинского изделия элементов управления для выявления неисправности;
- результативность элементов управления при выявлении неисправности;
- наличие других средств обеспечения качества, способных выявить ошибочный результат (например, предельно допустимых значений, проверки достоверности результата);
- возможность для пользователя откорректировать проблему на основании сообщения об ошибке и получить достоверный результат исследования при повторном исследовании. Например, сообщение "Недостаточное количество крови" на дисплее прибора для самотестирования дает пользователю подсказку о необходимости повторного исследования;
- наличие в лаборатории результативной системы обнаружения ошибочных результатов, если медицинское изделие предназначено для применения в лабораторных условиях.
Н.2.5.4.3 Вероятность того, что ошибочный результат IVD-исследования будет выявлен врачом:
- требуют ли действующие стандарты медицинской практики подтверждающего исследования аналита;
- проводит ли лаборатория подтверждающее исследование положительного результата скринингового исследования автоматически;
- можно ли распознать ошибочный результат исходя из других результатов, признаков, симптомов и медицинского анамнеза пациента;
- подтверждает ли обычно врач результаты, полученные для аналита, с помощью других средств и ставит ли под вопрос результаты, не соответствующие клинической картине заболевания;
- существуют ли другие способы проверки надежности результатов, полученных для аналита, помогающие предотвратить врачебную ошибку;
- является ли исследование единственным основанием для принятия важных клинических решений и в какой мере диагноз основан на результате исследования (т.е. как исследование влияет на принятие клинического решения);
- требует ли ситуация принятия срочного решения в отсутствие возможности получения подтверждающих данных или информации; приводит ли результат исследования непосредственно к принятию клинического решения или к назначению лечения;
- доступны ли альтернативные исследования, например, в центральной лаборатории в случае отказа медицинского изделия на месте оказания медицинской помощи.
Н.2.5.4.4 Вероятность того, что врач будет действовать (или не сможет действовать) в соответствии с результатом исследования:
- является ли медицинское изделие для диагностики in vitro определяющим фактором в терапии тяжелых состояний, например злокачественных опухолей или опасных для жизни инфекций;
- предназначено ли медицинское изделие для диагностики in vitro для трансфузии, трансплантации или другого медицинского применения, при котором возможно заражение реципиента;
- предназначено ли медицинское изделие для диагностики in vitro для мониторинга жизненно важных функций организма, когда ошибка или промедление могут привести к смерти или тяжелому поражению здоровья пациента.
Н.2.5.4.5 Вероятность того, что действие или бездействие врача приведет или будет способствовать причинению вреда пациенту:
- является ли действие необратимым, как, например, хирургическая резекция или прерывание беременности;
- в какой мере действие является обратимым;
- в какой мере действие может причинить вред пациенту;
- в какой мере бездействие ведет к летальному исходу или ухудшению состояния здоровья;
- какие физиологические условия способствуют причинению вреда.
Н.2.5.4.6 Тяжесть причиненного вреда:
- летальный исход;
- поражение, опасное для жизни;
- сокращение ожидаемой продолжительности жизни;
- необратимое ухудшение состояния здоровья;
- стойкое нарушение состояния здоровья;
- постоянное нарушение функции организма или анатомического строения тела;
- поражение, требующее медицинского вмешательства для предупреждения причинения серьезного вреда;
- обратимое ухудшение состояния здоровья;
- незначительная травма (поражение);
- временное ограничение физических возможностей, не требующее медицинского вмешательства;
- временный дискомфорт.
Н.2.5.5 Информация о риске для медицинских изделий для диагностики in vitro
Н.2.5.5.1 Базы данных о неблагоприятных событиях
Программы контроля медицинских изделий собирают от изготовителей и конечных пользователей данные, включающие примеры нежелательных последствий ошибочных или запоздалых результатов IVD-исследования. Изготовитель может анализировать сообщения о подобных медицинских изделиях для диагностики in vitro, для того чтобы определить их возможное соответствие своим изделиям и идентифицировать ранее не обнаруженные опасности или тенденции. Однако, делая выводы на основании отдельных сообщений, необходимо соблюдать осторожность. Информация в базах данных о неблагоприятных событиях не верифицирована, а отдельные сообщения могут содержать неполные, ошибочные или вводящие в заблуждение данные.
Н.2.5.5.2 Изучение согласованного заключения экспертов
Согласованное заключение экспертов было впервые изучено при классификации воздействия ошибочного определения уровня сахара в крови у пациентов, прибегающих к самоконтролю сахарного диабета. В [21] описан метод систематического исследования для получения клинических входных данных о риске для пациентов. Авторы метода разработали классификацию погрешностей с использованием категорий разной клинической достоверности, смоделированную на основании графического подхода, примененного в [22]. Метод изучения согласованного заключения экспертов, описанный в [21], можно применять и для других измеряемых величин.
Н.2.5.5.3 Беседы с врачами
Традиционным способом получения клинических входных данных о риске для пациента являются беседы с практикующими врачами для определения того: 1) как они применяют результаты IVD-исследования; 2) могут ли они выявить ошибочные результаты; 3) какие действия они могут предпринять при получении ненадлежащих результатов; 4) какие последствия может иметь ненадлежащее медицинское действие. Хотя данный способ является более субъективным по сравнению с методом изучения согласованного заключения экспертов, но можно разработать стратегию проведения беседы, помогающую определить величину систематической погрешности или неточности, представляющую риск для пациентов.
Н.3 Оценивание риска
Глубина оценивания риска должна быть пропорциональна тяжести возможного вреда. Оценивание риска при получении каждого ошибочного результата, идентифицированного как опасный, следует проводить в соответствии с требованиями D.3 и D.4 (приложение D).
Н.4 Управление риском
Н.4.1 Общие положения
Тяжесть вреда, причиняемого пациенту, определяется медицинским вмешательством (или его отсутствием), обусловленным результатом IVD-исследования. Способность изготовителя оказывать влияние на тяжесть вреда зависит от конкретного IVD-исследования.
Если медицинское вмешательство зависит от значения конкретного физического параметра, например от содержания в крови сахара, электролитов, терапевтических лекарственных средств или конкретных ферментов, то тяжесть вреда может быть уменьшена с помощью мер по управлению риском, предназначенных для уменьшения погрешностей, неточностей и помех. Однако если результат является положительным или отрицательным, то тяжесть вреда, причиняемого пациенту, не может быть уменьшена изготовителем.
Риски для пациентов вследствие получения ошибочных результатов IVD-исследования обычно можно уменьшить посредством снижения вероятности их возникновения. Виды деятельности по уменьшению рисков, являющихся следствием ошибочных результатов, должны быть распределены в порядке приоритета согласно иерархии, приведенной в 6.2. В отношении медицинских изделий для диагностики in vitro это означает:
a) попытку уменьшить вероятность получения ошибочного результата благодаря внутренней безопасности изделия, обусловленной его конструкцией, улучшению соответствующих эксплуатационных характеристик (например, аналитической и диагностической специфичности, подлинности или точности), что может быть необходимо для обеспечения соответствия результатов медицинским требованиям;
b) если внутренняя безопасность изделия, обеспечиваемая проектом и конструкцией, практически неосуществима, то следует предпринять защитные меры для снижения вероятности передачи ошибочного результата врачу или пациенту преимущественно путем выявления таких результатов самим изделием или с помощью процедур управления качеством, предоставляемых вместе с изделием;
c) если защитные меры практически неосуществимы, то следует предоставить пользователям информацию по безопасности, например инструкции, предупреждения и другую информацию, необходимую, чтобы избежать опасных ситуаций.
Примечания
1 Предусмотренные для применения отдельно от прибора методы обнаружения возможных рисков, например рекомендованное управление качеством, осуществляемое лабораторией, или рекомендованные подтверждающие исследования, затребованные врачом, следует рассматривать как информацию по безопасности, а не как защитные меры.
2 Минимальная информация, которую должен предоставлять изготовитель с медицинским изделием для диагностики in vitro, определена в нормативных документах и международных стандартах (см. Н.4.2.4).
Н.4.2 Анализ возможностей управления риском
Н.4.2.1 Внутренняя безопасность, обеспечиваемая проектом и конструкцией
Если медицинское изделие для диагностики in vitro не постоянно соответствует предъявляемым медицинским требованиям, то, возможно, его конструкция может быть изменена во избежание получения ошибочных результатов, например, посредством улучшения при необходимости одной или нескольких нижеследующих характеристик:
- точности измерительной системы;
- соответствия значений калибратора нормативным требованиям;
- аналитической специфичности IVD-реагентов (например, использования более качественных антител);
- диапазона обнаружения или диапазона предельно допустимых количественных значений;
- надежности прибора (например, возможности предупреждения ошибочных результатов);
- разграничения между положительными и отрицательными пробами;
- автоматизации процедурных этапов, имеющих высокую предрасположенность к ошибкам;
- идентификации положительных проб (например, с помощью штрих-кодов);
- простоты применения (например, посредством изучения человеческого фактора).
Подобным образом процесс изготовления можно усовершенствовать, для того чтобы избежать выпуска медицинских изделий для диагностики in vitro, обеспечивающих получение ошибочных результатов (т.е. не соответствующих медицинским требованиям). Анализ опасностей в критических контрольных точках [Hazard Analysis on Critical Control Points - HACCP, см. G.6 (приложение G)] может помочь идентифицировать элементы процесса изготовления, влияющие на предотвращение изготовления несоответствующей продукции, а именно:
- реагенты с очень большой разницей результатов для разных серий изделий;
- компоненты приборов, влияющие на получение неверных результатов;
- калибраторы со значениями, превышающими технические требования в отношении систематической погрешности;
- контрольные материалы, калибраторы или реагенты, не удовлетворяющие заявленным требованиям к сроку хранения.
Н.4.2.2 Защитные меры
Если улучшение конструкции медицинского изделия для диагностики in vitro является практически неосуществимым, то для выявления условий, приводящих к ошибочным результатам, можно встроить в изделие дополнительные элементы, например:
- средства контроля сохранности для выявления неприемлемых проб (например, гемолизированных);
- устройства для удаления с пробы пены (если устройство для отбора проб снабжено датчиком уровня жидкости) или сгустков фибрина;
- встроенные датчики и средства контроля программного обеспечения для выявления неблагоприятных состояний системы (например, ненадлежащей температуры, отклонения спектрофотометра, встроенного дозирующего устройства);
- встроенные элементы управления для выявления отказа калибратора, реагента или прибора;
- системы сигнализации, сообщения об ошибках или алгоритмы для подавления ненадлежащих результатов;
- алгоритмы достоверности для идентификации недостоверных результатов.
Если совершенствование процесса изготовления практически неосуществимо, то для предотвращения выпуска несоответствующей продукции могут понадобиться дополнительные средства управления процессом или более жесткие технические требования, например:
- проверка входных материалов на соответствие техническим требованиям к качеству;
- эксплуатационные испытания в ходе процесса изготовления для выявления несоответствующих компонентов;
- применение контрольных материалов для обеспечения метрологической прослеживаемости калибраторов (см. [23] и [24]);
- эксплуатационные характеристики, удовлетворяющие требованиям пользователей;
- окончательные испытания перед выпуском продукции.
Н.4.2.3 Информация по безопасности
Н.4.2.3.1 Эксплуатационные характеристики
Руководителям лабораторий и медицинским работникам необходимо знать эксплуатационные характеристики медицинского изделия для диагностики in vitro, для того чтобы определить их соответствие рассматриваемому медицинскому изделию. Данную информацию предоставляет изготовитель. Надежные числовые значения эксплуатационных характеристик позволяют выявить остаточные риски в основные моменты принятия клинического решения и надлежащим образом интерпретировать результаты исследования. Это следующие характеристики:
- аналитическая специфичность (например, интерферирующих или перекрестно реагирующих веществ);
- погрешность (т.е. допустимая систематическая погрешность);
- точность;
- диапазон обнаружения или предельно допустимые количественные значения;
- правильность (сочетание точности и погрешности);
- диагностическая чувствительность (доля правильных положительных результатов исследования у пациентов с заболеваниями);
- диагностическая специфичность (доля правильных отрицательных результатов исследования у пациентов без заболеваний).
Н.4.2.3.2 Информация, необходимая для предотвращения получения ошибочных результатов
Инструкции по применению, процедурные ограничения и требования к окружающей среде помогают пользователю предотвратить получение ошибочных (опасных) результатов, а именно:
- требования к отбору, хранению и подготовке проб;
- вещества, известные как обеспечивающие помехи;
- валидированный измерительный интервал;
- предупреждения в отношении неправильного применения, способствующего получению ошибочных результатов;
- ограничения в отношении некоторых категорий пациентов;
- предупреждения о ненадлежащих клинических условиях или ненадлежащих видах проб;
- надлежащие методы очистки;
- процедуры профилактического технического обслуживания и их периодичность;
- требования к хранению реагентов и дата истечения срока их годности.
Н.4.2.3.3 Информация, способствующая выявлению ошибочных результатов
Следующие дополнительные инструкции и рекомендации помогают уменьшить вероятность сообщения врачу ошибочных (опасных) результатов:
- по процедурам контроля для выявления условий, способствующих получению ошибочных результатов (см. [25]);
- по процедуре монтажа, необходимого для верификации надлежащего функционирования IVD-изделия;
- руководящие указания по пригодности системы для идентификации отказа НРLC- или GC-колонок (колоночная жидкостная хроматография высокого давления и колоночная газовая хроматография);
- по подтверждающей процедуре исследования, основанной на принципе измерения, отличном от использованного.
H.4.2.3.4 Подготовка и квалификация пользователя
Во избежание ошибок применения изготовитель может предложить пользователю пройти необходимую подготовку.
Пользователю медицинских изделий для диагностики in vitro могут быть предоставлены учебные материалы для программ непрерывного обучения. В отношении некоторых критичных медицинских изделий для диагностики in vitro (например, систем мониторинга перорального приема антикоагулянтов, пригодных для домашнего применения) можно предложить официальные программы повышения квалификации пользователей, финансируемые изготовителем (см. [26]).
H.4.2.4 Обязательная информация по безопасности
Во многих странах установлены регулирующие требования к информации, предоставляемой изготовителем. К ним можно отнести требования к управлению рисками, связанными с возможными ошибками применения и другими опасностями, типичными для медицинских изделий для диагностики in vitro. Соответствие применимым регулирующим документам и стандартам может служить свидетельством управления рисками, возникающими при конкретных ошибках применения, при условии верификации результативности такого управления (см. Н.4.3).
Таблица Н.1 содержит примеры возможных ошибок применения и соответствующую информацию, обычно поставляемую изготовителем, для того чтобы помочь пользователям избежать данных ошибок.
Таблица Н.1 - Примеры возможных ошибок применения и идентифицированных мер по управлению риском
Ошибка применения | Мера по управлению риском |
Некалиброванный прибор | Установление межкалибровочного интервала |
Реагенты, потерявшие реакционную способность | Указание срока годности на упаковке реагента |
Ненадлежащее техническое обслуживание прибора | Надлежащие инструкции по техническому обслуживанию |
Смешивание несовместимых серий реагентов | Маркирование серий и надлежащие инструкции по применению |
Исследование невзаимозаменяемых биологических жидкостей | Технические требования к соответствующим видам проб |
Неправильное приготовление проб | Инструкции по приготовлению проб |
Неправильное хранение реагентов | Требования к хранению, включающие критические факторы (температуру, освещение, влажность и т.д.) |
Путаница в единицах измерения (например, ммоль/л или мг/дл) | Визуализация или распечатка единиц измерения по каждому результату |
Ненадлежащая установка прибора | Инструкции по установке; квалификационные испытания |
Ненадлежащее функционирование прибора | Инструкции по применению с идентификацией основных этапов |
Ненадлежащее разведение проб | Требования к разведению, включающие приемлемые разбавители |
H.4.2.5 Предупреждения, предостережения и ограничения
Четкие предупреждения, инструкции и противопоказания могут служить результативными мерами по управлению риском при профессиональном применении медицинских изделий для диагностики in vitro, поскольку последствия невыполнения данных мер опубликованы и являются очевидными. Информация, не отражающая опасных последствий несоблюдения инструкций по применению, не может быть результативной мерой по управлению риском.
Например, медицинское изделие для диагностики in vitro может быть предназначено для исследования образцов плазмы или сыворотки крови, но не мочи. Если в инструкциях по применению не содержится информация относительно применения медицинского изделия для исследования образцов мочи, то некоторые лаборатории могут применять данное изделие для подобных исследований, особенно если медицинские изделия для диагностики in vitro, отвечающие современному уровню технического развития, способны проводить исследование образцов мочи. При отсутствии указаний на невозможность удовлетворительного исследования образцов мочи исследование подобных образцов будет являться прогнозируемой ошибкой применения.
Подобным же образом результаты исследования могут быть использованы для не предусмотренного изготовителем медицинского применения, не пригодного для данного медицинского изделия для диагностики in vitro. Изготовитель должен оценить риски от такого применения с учетом опыта работы с подобными изделиями, подобных условий применения других изделий и возможности данных условий применения. Изготовитель должен предоставить пользователям предупреждения, предостережения и ограничения в целях уменьшения возможных рисков.
H.4.2.6 Стандарты по медицинским изделиям для диагностики in vitro
По некоторым видам медицинских изделий для диагностики in vitro имеются международные стандарты, национальные стандарты, нормативные и регулирующие документы. Соблюдение общепризнанных стандартов на изделия, регулирующих требований и руководящих указаний, рассматривающих внутреннюю безопасность, обеспечиваемую проектом и конструкцией, защитные меры и информацию по безопасности, способствует установлению требований к проектированию и проведению испытаний, соответствие которым служит свидетельством управления риском (например, [20], [26], [27]).
H.4.3 Верификация результативности мер по управлению риском
Осуществление и результативность мер по управлению риском, включая информацию по безопасности, необходимо верифицировать. Степень верификации зависит от степени управления риском.
Для рисков с низкой тяжестью или вероятностью причинения вреда достаточной верификацией может служить анализ жалоб потребителей. Если целесообразно, то данная верификация может включать анализ имеющейся информации по медицинским изделиям для диагностики in vitro с подобными элементами управления риском. Для рисков с высокой тяжестью или вероятностью причинения вреда может потребоваться перспективное исследование в целях верификации результативности управления риском. Например, посредством изучения человеческого фактора можно оценить степень осознания пользователем и соблюдения им предупреждений и инструкций, а также верифицировать результативность информации по безопасности. Человеческий фактор включает формат печати, уровень читаемости предупреждений и предостережений, выделение предупреждающей информации надлежащим образом и т.д.
Допущения в отношении результативности информации по безопасности необходимо делать с осторожностью. При определении возможности уменьшения риска в результате предоставленной изготовителем информации следует принимать во внимание следующее:
- требования к аккредитации лабораторий, нормативные документы и меры по обеспечению их выполнения не являются универсальными; практика управления качеством и обеспечения качества сильно отличается в разных странах;
- инструкции по применению, прилагаемые к медицинским изделиям для диагностики in vitro для профессионального применения, предназначены для медицинских лабораторий; информация о противопоказаниях, несовместимых медицинских средствах, а также другая информация, относящаяся к применению результатов IVD-исследования, может быть неизвестна лечащим врачам, назначающим исследования.
Н.5 Мониторинг производственной и постпроизводственной информации
Н.5.1 Мониторинг функционирования медицинского изделия для диагностики in vitro на основании данных из внешних источников
Изготовители медицинских изделий для диагностики in vitro обычно имеют доступ к данным из внешних источников, которые можно использовать для мониторинга некоторых аспектов функционирования медицинского изделия. К подобным источникам можно отнести:
- отчеты о неблагоприятных событиях;
- жалобы в отношении ошибочных результатов, неправильно идентифицированных проб, функциональной надежности приборов и т.д.;
- данные по управлению качеством в пределах лаборатории;
- схемы оценки качества на основании данных из внешних источников (External Quality Assessment Schemes - EQAS), называемые также обзорами профессиональной пригодности;
- оценивание функционирования медицинского изделия независимыми лабораториями (результаты часто публикуют в научной литературе).
Н.5.2 Внутренний мониторинг эксплуатационных характеристик
Данные, регулярно получаемые от изготовителей, можно применять для мониторинга некоторых эксплуатационных характеристик медицинского изделия для диагностики in vitro в регулируемых условиях, а именно:
- мониторинга процесса;
- мониторинга стабильности;
- приписывания значений калибраторам;
- испытания надежности оборудования;
- видов деятельности по валидированию.
Приложение I
(справочное)
Руководящие указания по процессу анализа риска в отношении биологических опасностей
I.1 Общие положения
Настоящее приложение содержит руководящие указания по применению анализа риска к биологическим опасностям. Диапазон воздействия возможных биологических опасностей велик и может включать как кратковременные воздействия (острая токсичность, раздражение кожи, глаз и слизистых оболочек, гемолиз и тромбогенность), так и долговременные или специфические токсические воздействия (подострые и хронические токсические воздействия, сенсибилизация, генотоксичность, канцерогенность или онкогенность и воздействие на репродуктивную способность, в том числе тератогенность).
Стандарт [28] устанавливает общие принципы оценки биологического действия материалов/медицинских изделий.
I.2 Определение биологических рисков
I.2.1 Факторы, которые необходимо рассматривать
При проведении анализа биологических рисков следует учитывать:
- физические и химические свойства рассматриваемых материалов;
- историю клинического применения или данные о воздействии на человека;
- имеющиеся данные по биологической и токсикологической безопасности в отношении материалов, используемых в медицинском изделии и его компонентах;
- методику испытаний.
Количество необходимых данных и глубина исследования будут отличаться в зависимости от предусмотренного применения, а также от характера и продолжительности контакта с пациентом. Требования к данным обычно являются менее строгими для упаковочных материалов; медицинских изделий, находящихся в контакте с неповрежденной кожей; любых компонентов медицинских изделий, не вступающих в непосредственный контакт с тканями тела, растворами для внутривенного вливания, слизистыми оболочками или поврежденной кожей.
Для определения потребности в дополнительных данных следует проанализировать имеющуюся в научной литературе информацию о применяемых материалах/медицинских изделиях, предшествующий клинический опыт их применения и другие соответствующие данные. В некоторых случаях необходимо получить данные о химическом составе, остаточных продуктах (отходах) (например, мономерах, отходах при стерилизации), данных биологических испытаний и т.д.
I.2.2 Химическая природа материалов
Информация, характеризующая химическую идентичность и биологическое воздействие материалов, полезна при оценивании предусмотренного применения медицинского изделия. К факторам, способным повлиять на биологическую совместимость материалов, относят:
- идентичность, концентрацию, доступность и токсичность всех ингредиентов (добавок, средств обработки, мономеров, катализаторов, продуктов реакции);
- влияние биодеградации и коррозии на материалы.
Если при производстве, обработке, хранении или деградации материалов были применены или могли быть получены реагирующие или опасные ингредиенты, то следует рассмотреть возможное воздействие остаточных продуктов. Может понадобиться информация о концентрации или выщелачивании остаточных продуктов. Это могут быть экспериментальные данные или информация о химических свойствах применяемых материалов.
Если необходимые данные (например, о полном химическом составе) недоступны изготовителю по причине конфиденциальности, то следует верифицировать оценивание соответствия материала на предполагаемое применение.
I.2.3 Предыдущий опыт применения
Необходимо анализировать доступную информацию о предыдущем опыте применения каждого материала, предусмотренных добавках и о любых нежелательных реакциях. Однако предыдущий опыт применения ингредиента или материала необязательно гарантирует его пригодность для аналогичных применений. Следует учитывать предусмотренное применение, концентрацию ингредиентов и текущую токсикологическую информацию.
I.2.4 Данные испытаний на биологическую безопасность
Серия международных стандартов [28] содержит руководящие указания о том, какие испытания необходимо провести в каждом конкретном случае применения. Необходимость испытаний следует рассматривать в свете имеющихся данных для каждого случая отдельно, для того чтобы избежать проведения необязательных испытаний.
Приложение J
(справочное)
Информация по безопасности и остаточному риску
J.1 Общие положения
В настоящем приложении содержатся руководящие указания в отношении того, как:
- информация по безопасности [см. 6.2, перечисление с), и D.5.1, перечисление c) (приложение D)] может быть мерой по управлению риском;
- информировать об остаточном(ых) риске(ах) (см. 6.4 и раздел 7) в целях управления и осознания риска(ов).
Информация по безопасности является последним по степени предпочтения способом управления риском(ами); ее следует использовать только в том случае, когда исчерпаны другие меры по управлению риском(ами). Информация по безопасности служит руководством к выполнению (невыполнению) действия (действий) в целях избежания риска(ов).
Информирование о каждом отдельном и совокупном остаточном риске служит основой и дает необходимые сведения для объяснения остаточного(ых) риска(ов) пользователям, чтобы они могли заранее предпринять надлежащие действия в целях минимизации воздействия остаточного(ых) риска(ов).
По возможности необходимо учитывать как структуру и содержание информации по безопасности, так и способы ее применения.
Информацию по безопасности можно сообщать разными способами в зависимости от того, на какой стадии жизненного цикла медицинского изделия данная информация должна быть доведена до сведения пользователя, например в виде предупреждений в сопроводительной документации или в виде примечания или через пользовательский интерфейс изделия, управляемого с помощью меню.
J.2 Информация по безопасности
При подготовке информации по безопасности важно установить, кому предназначена данная информация и как ее следует сообщать. Изготовителю необходимо предоставить описание риска, его последствий и действий, необходимых для предотвращения причинения вреда.
При подготовке информации изготовителю следует рассмотреть:
- степень приоритета, необходимую для классификации действий (опасность, предупреждение, предостережение, примечание и т.д.);
- степень детализации необходимой информации;
- местоположение информации по безопасности (например, предупреждающая маркировка);
- словесные формулировки или изображения, применяемые для обеспечения ясности и доходчивости информации;
- непосредственных получателей информации (пользователи, обслуживающий персонал, монтажники, пациенты);
- соответствующие средства обеспечения информации (инструкции по эксплуатации, маркировки, сигналы тревоги, предупреждения в пользовательском интерфейсе);
- регулирующие требования и т.д.
J.3 Информирование об остаточном(ых) риске(ах)
При разработке способа информирования о каждом отдельном или совокупном остаточном риске важно установить, что именно необходимо сообщить и кому предназначена данная информация, для того чтобы довести ее до сведения пользователя, объяснить и помочь пользователю безопасно и результативно применять медицинское изделие. Изготовителю следует рассмотреть остаточный(ые) риск(и), идентифицированный(ые) согласно 6.4 и разделу 7, и определить, о чем следует информировать пользователя.
Изготовитель также должен рассмотреть:
- необходимую степень детализации информации;
- формулировки, применяемые для обеспечения ясности и доходчивости информации;
- непосредственных получателей информации (пользователей, обслуживающий персонал, монтажников, пациентов);
- используемые средства сообщения/источники информации.
Библиография
[1] | МЭК 60601-1:2005 | Электроаппаратура медицинская. Часть 1. Общие требования к общей безопасности и существенные рабочие характеристики | |
(IEC 60601-1:2005) | (Medical electrical equipment - Part 1: General requirements for basic safety and essential performance) | ||
[2] | Руководство ИСО/МЭК 51:1999 | Аспекты безопасности. Руководящие указания по включению их в стандарты | |
(ISO/IEC Guide 51:1999) | (Safety aspects - Guidelines for their inclusion in standards) | ||
[3] | ИСО 13485:2003 | Изделия медицинские. Системы менеджмента качества. Требования к регулированию | |
(ISO 13485:2003) | (Medical devices - Quality management systems - Requirements for regulatory purposes) | ||
[4] | Документ N N029R11 от 2 февраля 2002 г. | Целевая группа по глобальной организации, исследовательская группа 1 | |
(Document No. N029R11, dated 2 Feb. 2002) | (Global Harmonization Task Force (GHTF) - Study Group 1 (SG1) | ||
[5] | ИСО 9000:2005 | Системы менеджмента качества. Основные положения и словарь | |
(ISO 9000:2005) | (Quality management systems - Fundamentals and vocabulary) | ||
[6] | МЭК 62366:2007 | Аппаратура медицинская. Использование технологий по применимости к медицинской аппаратуре | |
(IEC 62366:2007) | (Medical devices - Application of usability engineering to medical devices) | ||
[7] | МЭК 60601-1-4:1996 | Электроаппаратура медицинская. Часть 1-4. Общие требования к безопасности: Дополняющий стандарт. Программируемые медицинские электрические системы | |
(IEC 60601-1-4:1996) | (Medical electrical equipment - Part 1-4: General requirements for safety - Collateral standard: Programmable electrical medical systems) | ||
[8] | МЭК 60300-3-9:1995 | Управление общей надежностью. Часть 3. Руководство по применению. Раздел 9. Анализ риска для технических систем | |
(IEC 60300-3-9:1995) | (Dependability management - Part 3: Application guide - Section 9: Risk analysis of technological systems) | ||
[9] | МЭК/ТО 60513:1994 | Электрооборудование медицинское. Основные аспекты безопасности | |
(IEC/TR 60513:1994) | (Fundamental aspects of safety standards for medical electrical equipment) | ||
[10] | Директива 93/42/ЕEС от 14 июня 1993 г. | О медицинских изделиях (с изменениями, внесенными Директивой 98/79/ЕС о медицинских изделиях для диагностики in vitro) | |
(Council Directive 93/42/ЕEС of 14 June 1993) | (Directive concerning medical devices as amended by Directive 98/79/ЕС of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices) | ||
[11] | ИСО 22442:2007 (все части) | Медицинские изделия, использующие ткани и их производные животного происхождения | |
(ISO 22442:2007) (all parts) | (Medical devices utilizing animal tissues and their derivatives) | ||
[12] | МЭК 60601-1-6:2010 | Электрооборудование медицинское. Часть 1-6. Общие требования безопасности и основные рабочие характеристики. Дополняющий стандарт. Возможность использования | |
(IEC 60601-1-6:2010) | (Medical electrical equipment - Part 1-6: General requirements for basic safety and essential performance - Collateral standard: Usability) | ||
[13] | МЭК 60601-1-8:2006 | Электроаппаратура медицинская. Часть 1-8. Общие требования к базовой безопасности и существенные рабочие характеристики. Дополняющий стандарт. Общие требования, испытания и руководство системами сигнализации в электрической медицинской аппаратуре и системах | |
(IEC 60601-1-8:2006) | (Medical electrical equipment - Part 1-8: General requirements for basic safety and essential performance - Collateral standard: General requirements, tests and guidance for alarm systems in medical electrical equipment and medical electrical systems) | ||
[14] | ИСО 10993-17:2002 | Оценка биологическая медицинских изделий. Часть 17. Установление допустимых пределов выщелачиваемых веществ | |
(ISО 10993-17:2002) | (Biological evaluation of medical devices - Part 17: Establishment of allowable limits for leachable substances) | ||
[15] | ИСО 14155-1:2003 | Испытания клинические медицинских изделий для людей. Часть 1. Общие требования | |
(ISO 14155-1:2003) | (Clinical investigation of medical devices for human subjects - Part 1: General requirements) | ||
[16] | ИСО 14155-2:2003 | Испытания клинические медицинских изделий для людей. Часть 2. Схемы клинических испытаний | |
(ISO 14155-2:2003) | (Clinical investigation of medical devices for human subjects - Part 2: Clinical investigation plans) | ||
[17] | МЭК 61025:2006 | Анализ диагностического дерева неисправностей | |
(IEC 61025:2006) | (Fault tree analysis (FTA) | ||
[18] | МЭК 60812:2006 | Техника анализа надежности систем. Метод анализа вида и последствий отказа | |
(IEC 60812:2006) | (Analysis techniques for system reliability - Procedures for failure mode and effects analysis (FMEA) | ||
[19] | МЭК 61882:2001 | Исследования опасности и работоспособности (HAZOP). Руководство по применению | |
(IEC 61882:2001) | (Hazard and operability studies (HAZOP studies). Application guide) | ||
[20] | ИСО 15197:2003 | Системы диагностические in vitro. Требования к системам мониторного наблюдения за концентрацией глюкозы в крови для самоконтроля при лечении сахарного диабета | |
(ISO 15197:2003) | (In vitro diagnostic test systems - Requirements for blood-glucose monitoring systems for self-testing in managing diabetes mellitus) | ||
[21] | Паркс Дж. Л. и др. (Лечение диабета. 2000. Том 23) | Новая согласованная таблица ошибок для оценивания клинической значимости неточностей при измерении концентрации глюкозы в крови | |
(Parkes J.L. et al., Diabetes Care, 23, 2000) | (A new consensus error grid to evaluate the clinical significance of inaccuracies in the measurement of blood glucose | ||
[22] | Кларк и др. (Лечение диабета. 1987. 10(5) | Оценивание клинической точности систем для самоконтроля концентрации глюкозы в крови | |
(Clarke W.L. et al., Diabetes Care, 10(5), 1987) | (Evaluating Clinical Accuracy of Systems for Self-Monitoring of Blood Glucose) | ||
[23] | ИСО 17511:2003 | Оборудование медицинское для диагностики in vitro. Количественные измерения в биологических образцах. Метрологическая прослеживаемость величин, заданных для калибраторов и контрольных материалов | |
(ISO 17511:2003) | (In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values assigned to calibrators and control materials) | ||
[24] | ИСО 18153:2003 | Оборудование медицинское для диагностики in vitro. Количественные измерения в биологических образцах. Метрологическая прослеживаемость величин каталитической концентрации ферментов, заданных для калибраторов и контрольных материалов | |
(ISO 18153:2003) | (In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values for catalytic concentration of enzymes assigned to calibrators and control materials) | ||
[25] | ИСО 15198:2004 | Клиническая лабораторная медицина. Медицинские приборы для диагностики in vitro. Валидация пользовательских процедур контроля качества изготовителем | |
(ISO 15198:2004) | (Clinical laboratory medicine - In vitro diagnostic medical devices - Validation of user quality control procedures by the manufacturer) | ||
[26] | ИСО 17593:2007 | Клинические лабораторные исследования и медицинские устройства in vitro. Требования к системам контроля in vitro для самоконтроля пероральной антикоагуляционной терапии | |
(ISO 17593:2007) | (Clinical laboratory testing and in vitro diagnostic test systems - Requirements for in vitro monitoring systems for self-testing of oral-anticoagulant therapy) | ||
[27] | ИСО 19001:2002 | Приборы медицинские для диагностики in vitro. Информация, предоставляемая изготовителем вместе с диагностическими реактивами in vitro для окрашивания в биологии | |
(ISO 19001:2002) | (In vitro diagnostic medical devices. Information supplied by the manufacturer with in vitro diagnostic reagents for staining in biology) | ||
[28] | ИСО 10993-1:2009 | Оценка биологическая медицинских изделий. Часть 1. Оценка и испытания процесса менеджмента риска | |
(ISO 10993-1:2009) | (Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management system) |
Электронный текст документа
и сверен по:
, 2011

{kind=link}