ГОСТ Р ИСО 14708-4-2016
Группа Р29
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИМПЛАНТАТЫ ДЛЯ ХИРУРГИИ
Активные имплантируемые медицинские изделия
Часть 4
Имплантируемые инфузионные насосы
Implants for surgery. Active implantable medical devices. Part 4. Implantable infusion pumps
ОКС 11.040.40
ОКП 944480
Дата введения 2017-10-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "Научно-технический центр "МЕДИТЭКС" (ООО "НТЦ "МЕДИТЭКС") на основе собственного перевода англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 011 "Медицинские приборы, аппараты и оборудование"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 21 октября 2016 г. N 1488-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14708-4:2008* "Имплантаты для хирургии. Активные имплантируемые медицинские изделия. Часть 4. Имплантируемые инфузионные насосы" (ISO 14708-4:2008 "Implants for surgery - Active implantable medical devices - Part 4: Implantable infusion pumps", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется вместо ссылочных международных стандартов использовать соответствующие им национальные стандарты Российской Федерации и действующие в этом качестве межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта в ближайшем выпуске информационного указателя "Национальные стандарты" будет опубликовано соответствующее уведомление. Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт определяет частные требования к активным имплантируемым медицинским изделиям, предназначенным для введения в организм человека лекарственного вещества в целях обеспечения безопасности использования как для пациентов, так и для специалистов. Настоящий стандарт вносит изменения и дополнения в ИСО 14708-1:2000, именуемый в дальнейшем ИСО 14708-1. Требования настоящего стандарта являются приоритетными по отношению к требованиям ИСО 14708-1.
Имплантируемый инфузионный насос - изделие, которое подает постоянный или периодический поток лекарственного вещества в организм через введенный катетер. Для настройки изделия может использоваться внешнее программное средство.
Действие настоящего стандарта распространяется на все части и принадлежности имплантируемых инфузионных насосов, включая катетеры, наборы для заправки, наборы прямого доступа к порту, программаторы и соответствующее программное обеспечение. Не все детали и принадлежности предназначены для полной или частичной имплантации, но поскольку некоторые неимплантируемые части и принадлежности могут повлиять на безопасность или стабильность работы изделия, заявленные изготовителем, то к ним также необходимо определить ряд требований.
В настоящую редакцию стандарта не включены требования к функциям имплантируемых инфузионных насосов в части измерения физиологических параметров. Они могут быть рассмотрены в последующих редакциях.
В настоящем стандарте для внесения изменений и дополнений в ИСО 14708-1 используются следующие термины:
- "Замена" означает, что соответствующие пункты ИСО 14708-1 полностью заменены текстом настоящего стандарта;
- "Дополнение" означает, что текст настоящего стандарта является дополнительным к требованиям ИСО 14708-1;
- "Изменение" означает, что соответствующие пункты ИСО 14708-1 изменены в соответствии с текстом настоящего стандарта;
- "Не применяют" означает, что соответствующий раздел ИСО 14708-1 в настоящем стандарте не применяется.
Дополнительные подпункты, рисунки или таблицы к ИСО 14708-1 пронумерованы начиная со 101; дополнительные приложения обозначены буквами АА, ВВ и т.д.
1 Область применения
Настоящий стандарт распространяется на активные имплантируемые медицинские изделия, предназначенные для введения лекарственных веществ в организм человека.
Настоящий стандарт также распространяется на некоторые неимплантируемые части и принадлежности имплантируемых инфузионных насосов, как определено в разделе 3.
Методы испытаний, приведенные в настоящем стандарте, являются типовыми для оценки соответствия образцов изделий и не предназначены для регулярных испытаний выпускаемой продукции.
Примечание - Настоящий стандарт не распространяется на неимплантируемые системы инфузии. Тем не менее, он распространяется на изделия, предназначенные для пробных измерений, вследствие их тесного взаимодействия с имплантируемыми инфузионными насосами.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на стандарты и другие нормативные документы, которые необходимо учитывать при его использовании*. Для датированных ссылок применяют только указанные издания. Для недатированных ссылок применяют самые последние издания (включая любые изменения и поправки).
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ISO 14708-1, Implants for surgery - Active implantable medical devices - Part 1: General requirements for safety, marking and for information to be provided by the manufacturer (Имплантаты хирургические. Активные имплантируемые медицинские изделия. Часть 1. Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем)
IEC 60601-1:2005, Medical electrical equipment - Part 1: General requirements for basic safety and essential performance (Электроаппаратура медицинская. Часть 1. Общие требования к общей безопасности и существенные рабочие характеристики)
IEC 60601-1-2:2007, Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral standard: Electromagnetic compatibility - Requirements and tests (Электроаппаратура медицинская. Часть 1-2. Общие требования к базовой безопасности и основной эксплуатационной характеристике. Дополняющий стандарт. Электромагнитная совместимость. Требования и испытания)
IEC 61000-4-3(2002), Electromagnetic compatibility (EMC) - Part 4-3: Testing and measurement techniques - Radiated, radio-frequency electromagnetic field immunity test (Электромагнитная совместимость. Часть 4-3. Методики испытаний и измерений. Испытание на невосприимчивость к воздействию электромагнитного поля с излучением на радиочастотах)
ANSI/AAMI РС69:2000, Active implantable medical devices - Electromagnetic compatibility - EMC test protocols for implantable cardiac pacemakers and implantable cardioverter defibrillators (Активные имплантируемые медицинские устройства. Электромагнитная совместимость. Программа испытаний электромагнитной совместимости для имплантируемых кардиостимуляторов и имплантируемых кардиовертер-дефибрилляторов)
3 Термины и определения
В настоящем стандарте применены термины по ИСО 14708-1, а также следующие термины с соответствующими определениями:
3.101 имплантируемый инфузионный насос (implantable infusion pump): Активное вживляемое медицинское изделие, предназначенное для доставки лекарственного вещества в определенную часть организма человека.
Примечание - Согласно настоящему стандарту имплантируемый инфузионный насос может являться как единым изделием, так и системой, состоящей из частей и принадлежностей, которые взаимодействуют друг с другом для обеспечения производительности изделия, заявленной изготовителем. Не все из этих компонентов или принадлежностей могут быть частично или полностью имплантированы, например программатор, устройство пробных измерений.
3.102 механизм насоса (pump gear): Имплантируемая часть имплантируемого инфузионного насоса, состоящая из резервуара, источника энергии и в некоторых случаях из электронных контроллеров.
3.103 система циркуляции жидкости (fluid path): Внутренние поверхности имплантируемого инфузионного насоса, которые находятся в непосредственном контакте с лекарственным веществом.
3.104 выпускной порт (outlet port): Порт, через который жидкость попадает в катетер доставки.
3.105 порт заправки (refill access port): Порт доступа к резервуару с жидкостью.
3.106 прямой порт доступа (direct access port): Порт доступа к катетеру доставки.
3.107 внутренний объем (internal volume): Объем жидкости, заполняющей область от резервуара до выпускного порта.
3.108 объем резервуара (reservoir volume): Объем жидкости в резервуаре, который может быть израсходован при условии сохранения точности инфузии в пределах технических характеристик.
3.109 остаточный объем (residual volume): Объем жидкости, удаление которого из механизма насоса невозможно.
3.110 расчетный срок службы (projected service life): Период после имплантации, в течение которого имплантируемый инфузионный насос соответствует заявленным техническим характеристикам.
3.111 интервал стабильности (stability interval): Рассчитанный максимальный интервал пополнения резервуара для обеспечения стабильности поступления медицинского вещества.
3.112 точность инфузии (infusion accuracy): Степень близости фактического уровня инфузии к заданному уровню.
3.113 повторяемость (repeatability): Максимальное значение абсолютного расхождения между результатами двух последовательных испытаний одного и того же имплантируемого инфузионного насоса с одним и тем же инфузатом в одинаковых условиях при доверительной вероятности 95%.
Примечание 1 - Наиболее качественным критерием оценки повторяемости является способность постоянно воспроизводить одинаковые результаты в течение длительного времени (например, уровень инфузии). Повторяемость - показатель, независимый от точности.
Примечание 2 - Метод расчета повторяемости приведен в приложении В ИСО 11631-1998 [9].
3.114 минимальная скорость (minimum rate): Наименьшая скорость, выбираемая пользователем.
3.115 средняя скорость (intermediate rate): Скорость, указанная изготовителем в качестве стандартной для имплантируемого инфузионного насоса.
Примечание - Скорость может зависеть от применения.
3.116 максимальная скорость (maximum rate): Предельная скорость, выбираемая пользователем.
3.117 болюс (bolus): Количество жидкости, поступающее порционно через короткие промежутки времени.
3.118 требуемая производительность (essential performance): Производительность, необходимая для отсутствия недопустимого риска.
Примечание - Подробнее об основных требованиях производительности см. МЭК 60601-1.
4 Обозначения и сокращения
Применены обозначения и сокращения по ИСО 14708-1, а также нижеследующее:
ИУ (DUT) - испытуемое устройство (изделие).
5 Общие требования к неимплантируемым частям
Применяют соответствующий раздел ИСО 14708-1.
6 Требования к активным имплантируемым медицинским изделиям определенных видов
Дополнительные подпункты:
6.101 Характеристики имплантируемого инфузионного насоса
Технические требования и характеристики изделия (например, точность инфузии и повторяемость), заявленные изготовителем в сопроводительной документации (см. 28.8), должны поддерживаться в течение гарантийного срока службы и в условиях окружающей среды, указанных изготовителем.
Примечание 1 - Минимально допустимый уровень атмосферного давления определен в разделе 25.
Для всех выбираемых скоростей инфузии (включая скорость болюса) должна быть обеспечена точность инфузии в диапазоне объема резервуара. Для определенной скорости инфузии насоса в диапазоне объема резервуара должна быть обеспечена точность постоянного потока инфузии имплантируемого насоса.
Изготовитель должен предоставить график зависимости точности инфузии от объема резервуара. График для насосов с несколькими скоростями инфузии должен содержать кривые для минимальной и максимальной скоростей, а также для одной или более промежуточных скоростей.
Сопроводительная документация должна содержать детальный метод вычисления и определения точности инфузии. Также должны быть указаны условия окружающей среды, которые должны соблюдаться во время определения точности инфузии.
Примечание 2 - Точность является общепринятым термином и может включать в себя влияние систематических и случайных ошибок. Несмотря на то, что удобно использовать термин "точность" для объединения всех этих ошибок, данный термин по-прежнему остается качественным.
Для всех возможных скоростей инфузии должна быть определена повторяемость фактической скорости. В сопроводительной документации должен быть детально описан метод вычисления и определения повторяемости скорости.
Соответствие должно быть подтверждено контролем методик испытаний и результатов, предоставленных изготовителем и подкрепленных вычислениями изготовителя в соответствующей форме.
6.102 Испытание мембраны методом прокола
Мембрана, обеспечивающая вход в порт доступа (порт заправки или прямой порт доступа), должна выдерживать неоднократное прокалывание иглой шприца для подкожных инъекций с обеспечением сохранности резервуара для жидкости в течение гарантийного срока службы.
Метод тестирования. Для достижения теплового равновесия ИУ должно находиться при температуре (37±1)°C не менее 12 ч. Каждая мембрана имплантируемого насоса должна быть проколота случайным образом с помощью иглы, указанной изготовителем в качестве иглы для пункции мембраны, и в соответствии с инструкциями изготовителя. Игла должна полностью проткнуть мембрану. Необходимо соблюдать осторожность для предотвращения повреждения наконечника иглы во время теста. Прокол должен быть осуществлен прямолинейным движением, параллельным оси симметрии мембраны, как показано на рисунке 101. Если оператор заметит повреждение иглы или наконечника, ее необходимо заменить.
Для выявления утечки мембраны необходимо погрузить испытуемый объект в ванну с водой температурой (37±1)°C как минимум на 30 мин для стабилизации. Затем, постепенно нагнетая давление воздуха до значения, вдвое превышающего максимальное рабочее давление насоса или составляющего как минимум 276 кПа, необходимо в течение 1 мин проверить поверхности мембраны на пропускание пузырьков воздуха.
Соответствие подтверждается в том случае, если срок службы порта доступа соответствует пределам, указанным изготовителем. Кроме этого, должно быть указано максимальное число проколов, рекомендованное изготовителем (см. 28.8).
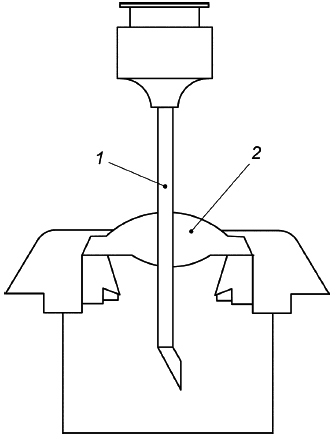
1 - игла;
2 - мембрана
Рисунок 101 - Испытание мембраны методом прокола
7 Общие требования к упаковке
Применяют соответствующий раздел ИСО 14708-1.
8 Общая маркировка активных имплантируемых медицинских изделий
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
Дополнительные подпункты:
8.101 В случае особых условий транспортирования изделия на упаковку должна быть нанесена соответствующая маркировка (см., например, ИСО 780 [1] или ИСО 15223 [2]).
Соответствие проверяют в ходе осмотра.
8.102 Допустимые условия окружающей среды транспортирования должны быть обозначены маркировкой на внешней стороне транспортной упаковки.
Соответствие проверяют в ходе осмотра.
9 Маркировка на товарной упаковке
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
9.4 Дополнение:
Для следующих компонентов должна быть указана дополнительная информация, приведенная ниже:
а) Механизм насоса:
- объем резервуара;
- скорость инфузии для насоса с постоянным потоком;
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
b) Катетер:
- длина (в сантиметрах);
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
c) Набор для заправки:
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
d) Набор прямого порта доступа:
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
10 Конструкция товарной упаковки
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
10.3 Изменение:
Испытание заменено пунктом 7.1.3, перечисление b) МЭК 60601-1:2005.
Примечание - Предметом испытания не являются съемные наклейки (например, временные наклейки, используемые в процессе производства), которые содержат дополнительные данные, помимо информации, определенной в разделе 9.
11 Маркировка на стерильном пакете
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
Дополнительный подпункт:
11.101 Для приведенных ниже компонентов маркировка на стерильном пакете должна содержать следующую дополнительную информацию:
a) Механизм насоса:
- объем резервуара;
- скорость инфузии для насосов с постоянным потоком;
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
b) Катетер:
- длина (в сантиметрах);
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
c) Набор для заправки:
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
d) Набор порта прямого доступа:
- любая дополнительная информация и соответствующие характеристики, необходимые для идентификации изделия.
Соответствие проверяют в ходе осмотра.
12 Конструкция одноразовой упаковки
Применяют соответствующий раздел ИСО 14708-1.
13 Маркировка активных имплантируемых медицинских изделий
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
13.1 Изменение:
Тест на влажное протирание, после которого маркировка должна оставаться читаемой, заменен пунктом 7.1.3, перечисление b) МЭК 60601-1:2005.
14 Защита от непреднамеренных биологических воздействий, вызванных активным имплантируемым медицинским изделием
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
14.2 Замена:
Любая часть активного имплантируемого медицинского изделия, которая при нормальном использовании контактирует с жидкими средами организма человека, должна быть оценена на отсутствие выделения опасного числа твердых частиц.
Методика тестирования. Активное имплантируемое медицинское изделие асептически извлекают из одноразовой упаковки. Имплантируемую часть погружают в пригодный для инъекций раствор соли 9 г/л, находящийся в нейтральном стеклянном контейнере. Объем соляного раствора должен примерно в (5±0,5) раз превышать объем испытуемой части. Контейнер накрывают стеклянной пластиной и выдерживают в течение временного промежутка от 8 до 18 ч при температуре (37±1)°C, периодически перемешивая. Готовят контрольный раствор аналогичного объема с той же концентрацией соли, поддерживая те же условия, что и в контейнере с образцом. Образец жидкости из контейнера сравнивают с образцом контрольного раствора с помощью аппаратуры для автоматического измерения размеров частиц, работающей по принципу оптического затенения (например, метод 2.9.19 Европейской Фармакопеи, 3 издание, 1977 (Совет Европы) [3]).
Отношение среднего числа частиц, выделяющихся из образца, к среднему числу частиц, выделяющихся из контрольного раствора, не должно превышать число, определенное изготовителем как небезопасное. Если изготовитель не определил это число, среднее число частиц не должно превышать 100 частиц/мл для частиц размерами более 5,0 мкм и 5 частиц/мл для частиц размерами более 25 мкм.
Соответствие должно быть подтверждено проверкой анализа конструкции, предоставленного изготовителем, при необходимости - дополнительно расчетами изготовителя и данными тестовых исследований.
14.3 Дополнение:
Это требование также относится к материалам системы циркуляции жидкости имплантируемого насоса (косвенный контакт с жидкостью).
Биологическая совместимость может быть оценена в соответствии с одной или несколькими частями ИСО 10993, например, ИСО 10993-1 [4].
Дополнительный подпункт:
14.101
Для имплантируемых инфузионных насосов, требующих периодического пополнения лекарственного вещества, изготовитель должен установить максимальный интервал, в течение которого лекарственное вещество будет поддерживать активность на уровне по крайней мере 90% от исходного, имеющегося после заполнения резервуара насоса. Для каждого заявленного лекарственного вещества изготовителем должен быть определен интервал стабильности при температуре тела человека (см. 28.8). Кроме того, должно быть оценено наличие потенциально опасных продуктов распада лекарственного вещества в течение установленного интервала стабильности.
Соответствие считается подтвержденным, если записи, предоставленные изготовителем, подтверждают, что безопасность и качество лекарственного препарата были проверены с использованием надлежащих методов.
15 Защита пациента или пользователя от вреда, вызванного внешними физическими особенностями активного имплантируемого медицинского изделия
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
15.1 Изменение:
Раздел 23 МЭК 60601-1:1988 заменен на подраздел 9.3 МЭК 60601-1:2005 (см. раздел 5).
Соответствие должно быть проверено согласно МЭК 60601-1.
16 Защита пациента от вреда, вызванного электрическим током
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
16.1 Изменение:
Раздел 19 МЭК 60601-1:1988 заменен на подраздел 8.7 МЭК 60601-1:2005 (см. раздел 5).
16.2 Дополнение:
Если результаты оценки степени риска или другие данные (например, опубликованные данные исследований, тестовые исследования, расчеты) укажут, что для отдельных видов применения предел не должен превышать 1 мкА, для снижения риска допустимый предел должен быть изменен.
17 Защита пациента от вреда, вызванного тепловыделениями
Замена:
Ни одна из наружных поверхностей имплантируемой части насоса не должна нагреваться более чем на 2°C выше нормальной температуры тела пациента как при стабильной работе, так и в условиях единичного отказа, если изготовитель не укажет, что более значительное повышение температуры целесообразно для применения в особых случаях.
Для подтверждения соответствия необходимо проанализировать документацию изготовителя, включая результаты моделирования, анализ конструкции или оценку степени риска, тестовые исследования или другие соответствующие документы.
Примечание - Современные исследования показывают, что в зависимости от местоположения определенной ткани в теле человека температурный предел 2°C накладывает излишние ограничения. Учитывая это обстоятельство, изготовитель может привести свое обоснование.
18 Защита от ионизирующего излучения, выделяемого или испускаемого активным имплантируемым медицинским изделием
Применяют соответствующий раздел ИСО 14708-1.
19 Защита от непреднамеренных воздействий, вызванных изделием
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
19.2 Замена:
Если срок службы (см. 3.110) имплантируемого инфузионного насоса зависит от имплантированного источника электроэнергии, такого как батарея, изготовитель должен предусмотреть в изделии индикатор, предупреждающий о низком заряде источника электроэнергии. Изготовитель также должен определить продолжительность срока службы источника электроэнергии после предупреждения о низком заряде.
Для подтверждения соответствия используется анализ конструкции, представленный изготовителем и подтвержденный результатами испытаний в соответствующей форме.
Примечание - Настоящий подпункт также применим к перезаряжающимся источникам энергии.
19.3 Замена:
Конструкция имплантируемого инфузионного насоса должна исключать возможность возникновения недопустимого риска в случае отказа любого из компонентов, части изделия или программного обеспечения (если изделие имеет встроенную электронную систему программирования).
Метод оценки. Оценка рисков и менеджмент рисков осуществляются в соответствии с опубликованными стандартами, такими как ИСО 14971 [5].
Соответствие должно быть подтверждено отчетами менеджмента рисков или эквивалентными документами изготовителя.
19.4 Изменение:
Система оценки изменена для возможности проведения клинических исследований в соответствии с опубликованными стандартами, такими как ИСО 14155-1 [6] или ИСО 14155-2 [7].
Дополнительный подраздел:
19.101 Для имплантируемого инфузионного насоса с различными скоростями инфузии, содержащего имплантированный источник электроэнергии, срок службы должен быть определен для диапазона скоростей инфузии, для различных объемов при ежедневной непрерывной инфузии и т.д. Скорости краткосрочных инфузий, превышающие типичные терапевтические скорости, но не влияющие на долговечность изделия, исключаются из расчета срока службы изделия (см. 28.19).
Соответствие должно быть подтверждено оценкой анализа конструкции, проведенного изготовителем, с необходимыми расчетами и данными испытаний в соответствующей форме.
20 Защита изделия от повреждений, вызванных применением наружных дефибрилляторов
Применяют соответствующий раздел ИСО 14708-1.
21 Защита изделия от изменений, вызванных электрическими полями высокой энергии, приложенными непосредственно к пациенту
Применяют соответствующий раздел ИСО 14708-1.
22 Защита активного имплантируемого медицинского изделия от изменений, вызванных применением разнородных видов лечения
Дополнение:
Должно быть рассмотрено влияние других методов диагностики и лечения, таких как магнитно-резонансная томография (МРТ), позитронно-эмиссионная томография (ПЭТ), литотрипсия и т.д.
23 Защита активного имплантируемого медицинского изделия от воздействия механических сил
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
23.1 Замена:
Неимплантируемые части имплантируемого инфузионного насоса должны соответствовать подразделу 15.3 МЭК 60601-1:2005 (см. раздел 5). Неимплантируемые части, переносимые пациентом, должны выдерживать по три удара в трех различных направлениях, которые могут случиться при нормальном использовании (см. подпункт 15.3.4.1 МЭК 60601-1:2005).
Соответствие должно быть проверено согласно МЭК 60601-1.
23.2 Изменение:
Конструкция механизма насоса должна выдерживать механическое воздействие, которое может возникнуть при нормальных условиях использования:
а) рабочая полоса частот: от 5 до 500 Гц;
b) спектральная плотность мощности: 0,7 (м/с)
/Гц;
c) форма спектральной плотности мощности: плоская горизонтальная от 5 до 500 Гц;
d) продолжительность тестирования: 30 мин по каждой из трех взаимно перпендикулярных осей.
Если после выполнения процедуры проверки механизм насоса будет соответствовать заявленным техническим характеристикам, соответствие подтверждается.
24 Защита активного имплантируемого медицинского изделия от повреждений, вызванных электростатическим разрядом
Замена:
Неимплантируемые части имплантируемого инфузионного насоса должны соответствовать пункту 6.2.2 МЭК 60601-1-2:2007 (см. раздел 5).
Соответствие проверяется по МЭК 60601-1-2.
25 Защита активного имплантируемого медицинского изделия от повреждений, вызванных изменениями атмосферного давления
Применяют соответствующий раздел ИСО 14708-1.
26 Защита активного имплантируемого медицинского изделия от повреждений, вызванных изменениями температуры
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
26.1 Изменение:
Раздел 42 МЭК 60601-1:1998 заменен на подраздел 11.1 МЭК 60601-1:2005 (см. раздел 5).
27 Защита активного имплантируемого медицинского изделия от электромагнитного неионизирующего излучения
Замена:
27.101 Помехоустойчивость
Имплантируемые части инфузионного насоса не должны наносить вред или вызывать локальное увеличение плотности электрического тока в теле пациента вследствие восприимчивости к электрическим воздействиям внешних электромагнитных полей, неисправности, повреждения или нагрева изделия.
Соответствие должно быть подтверждено анализом результатов испытаний и документацией, подготовленной изготовителем, для испытаний согласно 27.103-27.106.
27.102 Общие условия испытаний
a) Режим работы
Во время испытания на помехоустойчивость все функции имплантируемого инфузионного насоса, связанные с основными функциональными характеристиками, должны быть испытаны в режимах, наиболее важных с точки зрения состояния пациента, определенных на основании анализа рисков. В испытательной документации должны быть перечислены функции и используемые режимы.
Примечание 1 - Например, основные функциональные характеристики могут быть связаны с точностью и повторяемостью, а насос с различными скоростями инфузии может иметь несколько режимов работы (например, инфузия с постоянной скоростью). Поэтому целесообразно протестировать несколько режимов.
Примечание 2 - Скорости инфузии, выбранные для испытаний, могут быть связаны со стандартным использованием, например, промежуточные скорости. Это целесообразно в том случае, когда результат испытаний насоса на помехоустойчивость не зависит от скорости инфузии. Более высокая скорость инфузии облегчит обнаружение изменений в доставке инфузата. Выбранная скорость должна соответствовать времени, отведенному на испытание. Длина катетера, определяющая скорость при постоянном потоке, не имеет никакого влияния на помехоустойчивость при работе, но должна быть достаточной для проведения испытания.
b) Критерии эффективности
В условиях проведения испытаний, указанных в разделе 27, каждая функция имплантируемого инфузионного насоса [см. 27.102, перечисление а)] должна быть оценена с точки зрения общей производительности с применением соответствующих критериев, указанных в таблице 101. Если производительность испытуемого изделия удовлетворяет требуемым критериям, регламент теста (тестов) выполнен. По критерию В ухудшение характеристик, потеря функции или непредусмотренная реакция являются допустимыми, если это не приводит к недопустимому риску.
Примечание - Оценка степени риска может показать, что опасность, созданная в результате ухудшения характеристик, потери функции или непредвиденной реакции, не приводит к недопустимому риску.
Следующие отклонения недопустимы:
- отказ элементов;
- изменения программируемых параметров настроек;
- сброс к заводским настройкам;
- изменение режима работы;
- ложные тревоги;
- начало любой непреднамеренной работы.
Таблица 101 - Общие критерии качества испытуемого изделия для испытаний помехоустойчивости согласно разделу 27
Критерий | Во время теста | После теста | Результаты теста |
A | Работа в соответствии с назначением | Работа в соответствии с назначением | 27.103 - 1 мТл |
Отсутствие потерь функции | Отсутствие потерь функции | 27.104 - А-линия | |
Отсутствие непреднамеренной реакции | Отсутствие ухудшений характеристик. | 27.105 - 16 В/м | |
Соответствует спецификации устройства | 27.106 - 40 мВт | ||
B | Допускается, если отсутствует неприемлемый риск | Работа в соответствии с назначением | 27.103 - 50 мТл |
Снижение производительности | Отсутствие потерь функции | 27.104 - В-линия | |
Потеря функции | Отсутствие ухудшений характеристик | 27.105 - 140 В/м | |
Непредусмотренная реакция | Соответствует спецификации устройства. | ||
C | Определяется изготовителем | Определяется изготовителем | 27.106 - дополнительные значения |
Тестовая документация должна включать в себя подробную информацию об используемых критериях качества, описание методов, используемых для проверки производительности, обоснование любых используемых допущений настоящего подпункта и отчет о результатах испытаний, в котором указаны характеристики ИУ относительно критериев A, B или C.
В сопроводительной документации должны быть описаны выявленные в результате проверки помехоустойчивости ИУ электромагнитные помехи, о которых пациенту необходимо знать или избегать их (см. 28.22).
c) Конструкция ИУ
Перед каждым испытанием необходимо заполнить резервуар насоса соответствующей жидкостью (окрашенную жидкость легче контролировать), которая в течение испытаний перекачивается в контейнер. Оценку производительности ИУ проводят контролем поступления жидкости из катетера.
Объем жидкости и, при необходимости, размер и длина катетера должны быть достаточными для поддержания рабочего режима [см. 27.102, перечисление а)] во время проведения испытания.
ИУ должно состоять из механизма насоса и любой другой имплантируемой части, необходимой для выполнения целевого назначения.
Документация по тестированию должна содержать описание конфигурации ИУ и условий окружающей среды, в которых проводится испытание (например, температура и давление).
d) Проверка не наблюдаемых в норме функций
Необходимо предусмотреть методы проверки функций, которые не наблюдаются при испытаниях [см. 27.102, перечисление а)]. Для этого может потребоваться использование специальных аппаратных или программных средств.
Работу функции необходимо проверить во время испытаний. Если это невозможно, проверку следует провести позднее.
e) Имплантируемые инфузионные насосы, использующие беспроводную телеметрию
Критерий B для проверки функции телеметрии применяется в виде исключения для удовлетворения требований 27.102, перечисление а). Все остальные функции должны соответствовать указанным требованиям.
Группа исключений не должна быть больше, чем это необходимо для работы в соответствии с назначением.
f) Имплантируемые инфузионные насосы без электрической функции
На имплантируемые инфузионные насосы без электрической функции (например, насосы под давлением с постоянной скоростью инфузии, не работающие от электричества) испытания по 27.104, 27.105 и 27.106 не распространяются.
27.103 Защита от постоянного магнитного поля
Оценку работы имплантируемого инфузионного насоса при постоянных магнитных полях проводят путем воздействия на ИУ двух уровней статических (не изменяющихся во времени) полей.
Методика тестирования. Общие условия испытаний приведены в 27.102.
Уровни испытаний. Используют два уровня напряженности электромагнитного поля, к каждому применяют различные критерии качества. ИУ должно быть подвергнуто воздействию магнитного поля нижнего уровня индукции 1 мТл с применением критерия качества A, как указано в 27.102, перечисление b). Второй уровень магнитной индукции, равный 50 мТл, должен быть использован с применением критерия качества В.
Схема измерения. Устройство генерирования магнитного поля должно воспроизводить поле с однородностью от 0 до плюс 3 дБ на площади радиусом минимум 7,5 см, лежащей в параллельной аппарату плоскости. Эта плоскость будет называться центральной. Однородность магнитного поля задана только в центральной плоскости, содержащей мнимые оси Y и Z. Однородность магнитного поля не сохраняется вдоль положительного и отрицательного направлений мнимой оси X, которая проходит через центр изделия перпендикулярно к центральной плоскости.
Для большинства схем испытаний (см. 27.102) площадь равномерного поля радиусом 7,5 см достаточна для покрытия ИУ. Иначе площадь равномерного поля следует увеличивать до удовлетворения требованиям настоящего подпункта. ИУ размещается в центре центральной плоскости, в котором магнитное поле является наиболее равномерным. Плоскость поверхности ИУ должна быть расположена параллельно центральной плоскости (это обеспечивает размещение поверхности инфузионного насоса в области наибольшей концентрации первичных линий магнитного потока, которые перпендикулярны к центральной плоскости). Такое расположение является обязательным для ИУ. Положение катетера должно облегчать контроль доставки жидкости во время испытаний (см. 27.102). Расположение любого дополнительного оборудования, необходимого для работы насоса или контроля его работы во время испытаний, должно быть выбрано таким образом, чтобы вносить минимальные изменения в однородность поля.
Методика испытаний. Контроль производительности ИУ следует проводить как минимум в течение 10 мин на каждом уровне испытаний.
Оценка результатов испытаний. Необходимо использовать критерий качества А (при 1 мТл) и критерий качества В (при 50 мТл) согласно 27.102, перечисление b).
27.104 Защита от магнитных полей в диапазоне от 10 Гц до 30 МГц
Оценку имплантируемого инфузионного насоса в диапазоне частот от 10 Гц до 30 МГц проводят путем воздействия постоянных и переменных магнитных полей на ИУ.
Методика испытаний. Общие условия испытаний приведены в 27.102.
Уровни испытания. Тестовые уровни магнитного поля показаны графически на рисунке 102 (среднеквадратичное значение в А/м). Уровни испытаний варьируются в зависимости от частоты и критериев качества [см. 27.102, перечисление b)]. Сплошной линией на рисунке 102 обозначены уровни воздействия на ИУ с использованием критерия качества А, как указано в 27.102, перечисление b). Пунктирной - уровни воздействия с использованием критерия качества В. В рамках этого подпункта линии именуются как А-линия и В-линия соответственно. К ИУ применяют оба набора тестовых уровней.
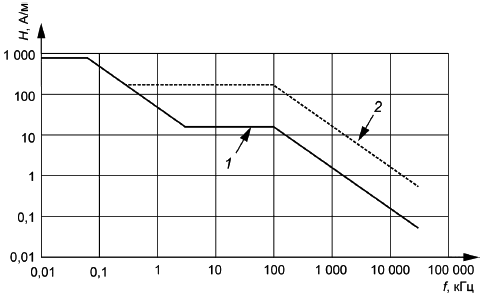
1 - А-линия (тестовые уровни по критерию качества А); 2 - В-линия (тестовые уровни по критерию качества В)
Рисунок 102 - Тестовые уровни магнитного поля (среднеквадратичное значение)
Численные значения тестовых уровней магнитного поля в зависимости от частоты приведены в таблице 102.
Таблица 102 - Тестовые уровни магнитного поля
Диапазон частот, кГц | Уровень магнитного поля для линии А | Уровень магнитного поля для линии В | ||
Напряженность магнитного поля Н, А/м | Магнитная индукция В, мкТл | Напряженность магнитного поля Н, А/м | Магнитная индукция В, мкТл | |
0,01-0,06 | 795 | 1000 | - | - |
0,06-0,3 | 47,7/ | 60/ | - | - |
0,3-3,0 | 47,7/ | 60/ | 159 | 200 |
3,0-100 | 15,9 | 20 | 159 | 200 |
100-30000 | 1590/ | 2000/ | 15900/ | 20000/ |
Примечание - | ||||
Схема измерения. Однородность магнитного поля, генерируемого испытательной катушкой (катушками), должна соответствовать данным таблицы 103. Однородное поле должно существовать на площади радиусом минимум 7,5 см параллельно катушке. Эта плоскость будет называться центральной. Однородность магнитного поля задана только в центральной плоскости, содержащей мнимые оси Y и Z. Она не сохраняется вдоль положительного и отрицательного направлений мнимой оси X, которая проходит через центр катушки перпендикулярно к центральной плоскости.
Для большинства схем испытания площадь радиусом 7,5 см равномерного поля достаточна для покрытия ИУ. Иначе площадь следует увеличить до удовлетворения требованиям настоящего подпункта.
Таблица 103 - Требование к однородности магнитного поля
А-линия | В-линия |
| 300 Гц |
Примечание - | |
ИУ помещают в солевую ванну проводимостью 0,27 См/м (эквивалентной объемному сопротивлению 370 Ом·см) в центре центральной плоскости, где магнитное поле наиболее равномерно. Плоскость большей поверхности ИУ располагается параллельно центральной (это обеспечивает размещение поверхности насоса в области наибольшей концентрации первичных линий магнитного потока, которые перпендикулярны к центральной плоскости). Такое расположение является обязательным для ИУ. Положение катетера должно облегчать контроль доставки жидкости во время теста (см. 27.102). Дополнительное оборудование, необходимое для работы и контроля выходных характеристик насоса, следует выбирать и располагать таким образом, чтобы оно вносило минимальные изменения в однородность поля.
Методика испытания. Диапазон частот тестовых сигналов от 10 Гц до 30 МГц может меняться непрерывно или пошагово. Если используется непрерывная развертка по частоте, то скорость развертки должна быть не более чем 0,0003 декада/с. Если пошаговая - размер шага в декаде не должен превышать F, где F
- начальная частота каждой декады. Начальная частота каждой декады равна: 10 Гц, 100 Гц, 1 кГц, 10 кГц, 100 кГц, 1 МГц, 10 МГц. Время задержки на каждом шаге должно быть достаточно велико, чтобы ИУ адекватно реагировало на тест-сигнал, но не менее чем 15 с.
Испытательные частоты, полученные с использованием метода пошаговой частотной развертки и удовлетворяющие минимальным требованиям, приведены в таблице 104.
Таблица 104 - Тестовые частоты, с учетом требования минимального шага развертки, кГц
0,01 | 0,02 | 0,03 | 0,04 | 0,05 | 0,06 | 0,07 | 0,08 | 0,09 |
0,1 | 0,2 | 0,3 | 0,4 | 0,5 | 0,6 | 0,7 | 0,8 | 0,9 |
1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 |
100 | 200 | 300 | 400 | 500 | 600 | 700 | 800 | 900 |
1000 | 2000 | 3000 | 4000 | 5000 | 6000 | 7000 | 8000 | 9000 |
10000 | 20000 | 30000 | ||||||
Примечание - Частоты выражены в кГц. Эта таблица основана на методе использования минимального шага развертки по частоте. Использование непрерывной развертки или малого шага развертки приведет к большему числу тестовых частот. Перечисленные частоты находятся в следующей зависимости: n·10 | ||||||||
Тестовые сигналы, соответствующие А-линии (см. тестовые уровни), должны применяться в виде непрерывной синусоидальной волны (НСВ) сигнала во всем диапазоне частот. Тестовые сигналы, соответствующие В-линии, должны применяться в виде НСВ на частотах <3 кГц и как сигнал с импульсной модуляцией на частотах
3 кГц. Частота модуляции должна соответствовать 125 Гц, в течение 20% рабочего цикла (время нарастания синусной составляющей - 1,6 мс, время затухания - 6,4 мс в каждом цикле модуляции).
Если во время тестирования частот 3 кГц на уровнях В-линии (на которых используется импульсная модуляция) ухудшаются характеристики или происходит утрата функционирования, чего не было при начальном тестировании сигнала на уровнях А-линии (использующих тестовые сигналы НСВ), необходимо повторить тестирование импульсной модуляции сигнала (125 Гц, в течение 20% рабочего цикла) на частотах, при которых проявлялись ухудшение или потеря функций.
Испытания проводят при одной ориентации ИУ согласно описанию схемы измерения.
Оценка результатов испытаний. Критерий качества А (А-линия) и критерий качества В (В-линия) применяют согласно 27.102, перечисление b).
27.105 Защита от электромагнитных полей в диапазоне от 30 до 450 МГц
Оценку имплантируемого инфузионного насоса в диапазоне частот от 30 до 450 МГц проводят посредством воздействия электромагнитных полей с использованием методов испытаний и оборудования, указанных в МЭК 61000-4-3. Для целей настоящего стандарта некоторые части МЭК 61000-4-3 были изменены.
Методика испытаний. Общие условия испытаний описаны в 27.102. Применяются требования МЭК 61000-4-3 за исключением изменений, перечисленных ниже.
Уровни испытания (МЭК 61000-4-3, раздел 5). Используются два тестовых уровня поля с учетом соответствующего критерия качества. Низкий уровень поля со среднеквадратичным значением напряженности 16 В/м должен воздействовать на ИУ в диапазоне тестовых частот с учетом критерия качества А, согласно 27.102, перечисление b). Второй уровень поля со среднеквадратичным значением напряженности 140 В/м должен быть использован на определенных частотах, таких как 30 МГц, 50 МГц, 75 МГц, 150 МГц и 450 МГц, с учетом критерия качества В. Оба уровня поля представляют собой немодулированный тестовый сигнал. Ниже описаны требования к модуляции в тестовых процедурах (МЭК 61000-4-3, раздел 8).
Схема измерения (МЭК 61000-4-3, раздел 7). ИУ помещают в солевую ванну проводимостью 0,27 См/м (эквивалентной объемному сопротивлению 370 Ом·см). Положение катетера должно облегчать контроль инфузии во время испытаний (см. 27.102). Дополнительное оборудование, необходимое для работы и контроля работы насоса, необходимо выбирать и располагать таким образом, чтобы изделия вносили минимальные изменения в однородность поля.
Методика испытания (МЭК 61000-4-3, раздел 8). Испытание должно быть проведено с генерирующей антенной, обращенной к передней грани насоса и одному краю насоса. Кроме того, должна быть учтена поляризация генерируемого поля. Например, каждая сторона и край ИУ, расположенные лицом к антенне, должны подвергаться воздействию поля с горизонтальной и вертикальной поляризацией. В типичной безэховой камере с поляризованной антенной понадобится четыре сканирования ИУ. Если используется гигагерцовая поперечная электромагнитная испытательная камера, то достаточно использовать три ортогональные ориентации ИУ.
Для тестового уровня, равного среднеквадратичному значению 16 В/м в диапазоне необходимых тестовых частот от 30 до 450 МГц, можно применять непрерывную или пошаговую развертку. Если используется непрерывная частота развертки, то скорость развертки не должна превышать 0,0003 декады/с. Если развертка пошаговая, то размер шага должен быть не больше, чем 5% предыдущей частоты, и время пребывания на каждом этапе должно быть достаточно большим для адекватной реакции ИУ на тест-сигнал, но не менее 15 с. Амплитудная модуляция тестовых сигналов должна составлять 80% с частотой модуляции 2 Гц. На рисунке 1 МЭК 61000-4-3:2008 определены уровень сигнала тестирования и форма волны.
Тестовый уровень со среднеквадратичным значением 140 В/м, применяется на определенных частотных участках, таких как 30 МГц, 50 МГц, 75 МГц, 150 МГц и 450 МГц. Чтобы ИУ адекватно реагировало на тест-сигнал, время задержки на каждом этапе должно быть достаточно большим (не менее 15 с). Форма немодулированного сигнала НСВ синусоидальна и показана на рисунке 1 а) МЭК 61000-4-3:2002. Тестовые сигналы должны сопровождаться импульсной модуляцией частотой 2 Гц в течение 20% времени рабочего цикла. Длительность переднего фронта синусоидальной несущей при этом составляет 100 мс, а заднего - 400 мс каждый цикл модуляции.
Оценка результатов испытаний (МЭК 61000-4-3, раздел 9). Критерий качества А (16 В/м) и критерий качества B (140 В/м) (применяются согласно 27.102, перечисление b).
27.106 Защита от электромагнитных полей в диапазоне от 450 МГц до 3 ГГц
Проверку имплантируемого инфузионного насоса для диапазона частот от 450 МГц до 3 ГГц проводят путем воздействия излучаемых электромагнитных полей с использованием методов испытаний и оборудования, указанных в ANSI/AAMI РС69. РС69 предназначен для имплантируемых кардиологических изделий; часть установок и процедур испытаний не применяются и были изменены.
Метод тестирования. Общие условия испытаний описаны в 27.102. Применяются требования раздела 6 ANSI/AAMI РС69:2000, кроме изменений, перечисленных ниже (цифры в фигурных скобках относятся к номерам пунктов ANSI/AAMI РС69). Требования, связанные с подачей сигнала и программированием параметров, используемые в ANSI/AAMI РС69, не применяются.
Объем выборки (МЭК 61000-4-3, подраздел 6.2). Объем выборки не применим.
Схема испытания (МЭК 61000-4-3, подраздел 6.3). Устанавливают насос приблизительно в центре пересечения осей Х и Y, как показано на рисунке В.1 ANSI/AAMI РС69:2000. Такое положение насоса необходимо соблюдать для всех тестов. Положение катетера должно обеспечить наилучший контроль инфузии во время испытаний (см. 27.102).
Методика испытания (МЭК 61000-4-3, раздел 8). Выполняют испытания, требуемые для тестирования по оси X (МЭК 61000-4-3, подпункт 6.4.1.1). Продолжительность воздействия должна быть не менее 15 с для того, чтобы ИУ адекватно реагировало на тест-сигнал. Повторяют с антенной, параллельной оси Y. Повторяют испытания по осям Х и Y для всех частот, перечисленных в МЭК 61000-4-3 (подпункт 6.3.4.2), используя соответствующую дипольную антенну.
Дополнительная характеристика испытания (МЭК 61000-4-3, пункт 6.4.2). Дополнительно, по усмотрению изготовителя, могут быть проверены более высокие уровни мощности.
Критерии качества (МЭК 61000-4-3, подраздел 6.5). Должен применяться критерий качества А, как указано в 27.102, перечисление b). Критерии качества для дополнительной характеристики испытаний (МЭК 61000-4-3, пункт 6.4.2) определяются изготовителем.
28 Сопроводительная документация
Применяют соответствующий раздел ИСО 14708-1, за исключением следующего.
28.1 Дополнение:
В документации должна быть предоставлена дополнительная контактная информация, например номер телефона или адрес электронной почты, на случай если изделие требует немедленного обслуживания или пользователю понадобятся дополнительные инструкции для правильного использования.
28.8 Дополнение:
Определенная дополнительная информация должна быть предоставлена для следующих компонентов:
а) Механизм насоса:
- масса (в граммах);
- основные размеры (в миллиметрах);
- объем резервуара (в миллилитрах);
- остаточный объем (в миллилитрах);
- внутренний объем (если применяется);
- срок службы мембраны;
- доступные скорости инфузии;
- максимальный и минимальный выход жидкости (в миллилитрах на применимую единицу времени);
b) Катетер:
- длина (в сантиметрах);
- объем на единицу длины.
28.10 Дополнение:
Руководство по эксплуатации (инструкция по эксплуатации) должно включать следующие данные:
- инструкции по пополнению резервуара;
- список сигналов тревог и их обозначение;
- комментарий о том, что при некоторых обстоятельствах заявленные точность и повторяемость не будут соблюдаться.
28.12 Дополнение:
- предупреждение о возможной опасности, связанной с магнитно-резонансной томографией, если такое применение допускается.
28.22 Дополнение:
- предупреждение о возможной опасности, связанной с барокамерами, если такое применение допускается;
- предупреждение о возможной опасности, связанной с электронным наблюдением, металлодетектором и другими системами безопасности, если такое применение допускается;
- информация о возможной опасности от электромагнитных помех (см. раздел 27).
Дополнительные подпункты:
28.101 Сопроводительная документация должна включать рекомендуемые методы для определения нормальной работы имплантируемого инфузионного насоса.
Соответствие проверяют в ходе осмотра.
28.102 На каждой отдельной части сопроводительной документации должен быть указан год выпуска.
Соответствие проверяют в ходе осмотра.
28.103 Сопроводительная документация для имплантируемых частей имплантируемого инфузионного насоса должна включать в себя карту пациента со следующим:
a) инструкция, разрешающая сохранение карты пациентом;
b) место для следующей информации:
- модель и наименование изделия;
- серийный номер или номер партии изделия;
- идентификационные данные пациента;
- дата имплантации;
- наименование и адрес центра имплантации;
- запись о наличии у пациента имплантированного медицинского изделия.
Примечание - Карту можно предъявить представителю службы безопасности или медперсонала. Если сотрудники будут проинформированы о том, что пациент имеет имплантированное медицинское изделие, то они смогут предотвратить воздействие электромагнитных помех и полей высокой мощности.
Соответствие проверяют осмотром.
Приложение АА
(справочное)
Соответствие между фундаментальными принципами ИСО/ТО 14283 [8] и разделами настоящего стандарта
Таблица АА.1
Фундаментальные принципы ИСО/ТО 14283 | Разделы | Разделы настоящего стандарта и охватываемые аспекты |
3 Общие принципы | ||
3.1 Имплантаты должны быть разработаны и сконструированы таким образом, чтобы при использовании в условиях и с целью, для которых они созданы, не возникало угрозы безопасности пациентов или безопасности и жизни пользователей, а также других лиц, гарантируя тем самым, что любой риск, который может быть связан с их использованием, является приемлемым наряду с пользой для пациента или сопоставим с высоким уровнем защиты его здоровья и безопасности | 8.1 | Действует |
3.2 Решения, принятые изготовителем для разработки и конструирования имплантатов, должны соответствовать принципам безопасности с учетом общего уровня знаний и технического развития. При выборе наиболее подходящего решения изготовитель должен руководствоваться перечисленными ниже принципами в следующем порядке: | Примечание 1 | - |
3.3 Имплантаты должны достигать рабочих характеристик, предусмотренных изготовителем, и должны быть разработаны, изготовлены и упакованы таким образом, чтобы они были пригодны для выполнения одной или более функций, приведенных в 3.1 (из ИСО/ТО 14283:2004), как указано изготовителем | 10.4 | Действует. |
3.4 На характеристики и изделия, приведенные в 3.1, 3.2 и 3.3 ИСО/ТО 14283:2004), не должно быть негативного влияния в такой степени, чтобы нарушались клинические условия и безопасность пациентов и, где применимо, других лиц, в течение срока служба имплантата, указанного изготовителем, при воздействии на имплантат нагрузок, возникающих при нормальном использовании имплантата | 19.2 | Заменен. |
19.3 | Заменен. | |
23.1 | Заменен. | |
23.2 | Заменен. | |
23.3 | Действует. | |
23.4 | Действует. | |
23.5 | Действует. | |
23.6 | Действует. | |
26.1 | Изменение. | |
28.4 | Действует. | |
28.23 | Действует. | |
6.102 Испытание мембраны методом прокола. | ||
19.101 Срок службы должен быть определен для диапазона скоростей инфузии. | ||
3.5 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы на их характеристиках и работе в течение периода предполагаемого использования отрицательно не сказывались транспортирование и хранение, принимая во внимание инструкции и информацию, указанную изготовителем | 7.2 | Действует. |
10.1 | Действует. | |
10.2 | Действует. | |
10.3 | Изменение. | |
12.3 | Действует. | |
26.2 | Действует. | |
8.101 Маркировка упаковки для специального обращения во время транспортирования. | ||
8.102 Маркировка упаковки для допустимых условий окружающей среды во время транспортирования | ||
3.6 В отношении любого нежелательного побочного эффекта необходимо оценить приемлемость риска относительно предполагаемых рабочих характеристик | 19.3 | Заменен. |
19.4 | Изменение | |
4 Специальные принципы, касающиеся дизайна конструкции | ||
4.1 Химические, физические и биологические свойства | ||
4.1.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы гарантировать характеристики и производительность, требуемые разделом 3 "Основные принципы". Особое внимание необходимо уделить: | ||
а) выбору используемых материалов, в частности оценке токсичности и, где применимо, свойствах возгораемости; | 14.3 | Дополнение. |
b) совместимости используемых материалов и биологических тканей, клеток и тканевых жидкостей, принимая во внимание предполагаемое назначение имплантата | 14.3 | Дополнение |
4.1.2 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы минимизировать риск, вызванный загрязнением и наличием остаточных веществ, для лиц, занятых транспортированием, хранением и использованием имплантатов, и пациентов, принимая во внимание предполагаемое назначение изделия. Особое внимание необходимо уделить незащищенным тканям, продолжительности и частоте воздействия | 14.2 | Заменен. |
14.3 | Дополнение | |
4.1.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы они могли быть использованы безопасно с материалами, веществами и газами, с которыми они контактируют при нормальном использовании или при рутинных процедурах; если предполагается использование имплантатов для доставки лекарственных веществ, то они должны быть разработаны и изготовлены таким образом, чтобы быть совместимыми с лекарственным веществом в соответствии с положениями и ограничениями для этих веществ и чтобы их рабочие характеристики поддерживались в соответствии с предполагаемым использованием | 19.5 | Действует |
4.1.4 В случае если в имплантаты включено как неотъемлемая часть вещество, которое, если применяется отдельно, может быть рассмотрено как лекарственное вещество согласно определению 2.7 (ИСО/ТО 14283:2004) и которое может воздействовать на организм дополнительно к влиянию, оказываемому изделием, его безопасность должна быть подтверждена, принимая во внимание предполагаемое использование имплантированного изделия | 14.4 | Действует. |
4.1.5 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы свести к минимуму риск, вызванный утечкой вещества из имплантата | 25 | Действует. |
4.1.6 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы уменьшить, насколько возможно, риски, вызванные случайным проникновением вещества в имплантат, принимая во внимание имплантат и характер окружающей среды, в которой предполагается его использовать | 25 | Действует |
4.1.7 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски пациента или пользователя системы, включая программное обеспечение | 19.3 | Заменен |
4.2 Инфекция и микробное заражение | ||
4.2.1 Имплантаты и процесс их изготовления должны быть разработаны таким образом, чтобы уменьшить или устранить, насколько возможно, риск инфекции для пациента, пользователя или третьего лица. Конструкция должна позволять легкость манипулирования и, если необходимо, минимизировать инфицирование имплантата пациентом или, наоборот, во время использования | 14.1 | Действует |
4.2.2 Ткани животного происхождения должны быть поставлены от животных, подвергнутых ветеринарным контролю и надзору, адаптированным к предполагаемому применению ткани. Информация о географическом происхождении животного должна быть сохранена изготовителем. Обработка, сохранение, испытания и манипуляции с тканями, клетками и веществами животного происхождения должны осуществляться в соответствии с оптимальным уровнем безопасности. В частности, безопасность в отношении вирусов и других передающихся патогенов должна быть достигнута применением проверенных методов удаления или вирусной инактивации в рамках производственного процесса | Примечание 2 | - |
4.2.3 Имплантаты, поставляемые стерильными, должны быть разработаны, изготовлены и упакованы в защитную упаковку, которая создает антимикробный барьер, для обеспечения их стерильности при поступлении на рынок и при условиях хранения и транспортирования, оговоренных изготовителем, до тех пор, пока упаковка не будет вскрыта или повреждена | 7.1 | Действует. |
7.2 | Действует. | |
10.1 | Действует. | |
10.2 | Действует. | |
11.7 | Действует. | |
11.9 | Действует. | |
12.1 | Действует. | |
12.2 | Действует. | |
14.1 | Действует | |
4.2.4 Имплантаты, поставляемые стерильными, должны быть изготовлены и стерилизованы, если применимо, валидированным методом | 14.1 | Действует |
4.2.5 Имплантаты, для которых предполагается стерилизация, должны быть изготовлены в соответствующих контролируемых условиях (например, окружающей среды) | 14.1 | Действует. |
14.2 | Заменен | |
4.2.6 Система упаковки нестерильных имплантатов должна поддерживать на оговоренном уровне чистоты изделие без ухудшения и, если имплантаты стерилизуют перед использованием, минимизировать риск микробного заражения; система упаковки должна соответствовать методу стерилизации, указанному изготовителем | Примечание 3 | - |
4.2.7 Упаковка и/или этикетка имплантата должны устанавливать различие между идентичными или похожими изделиями в стерильном и нестерильном состояниях | Примечание 3 | - |
4.3 Свойства конструкции и окружающей среды | ||
4.3.1 Если имплантат предназначен для использования совместно с другим изделием или оборудованием, вся комбинация, включая систему соединений, должна быть безопасной и не должна ухудшать специфических свойств изделия. Любые ограничения по использованию должны быть указаны на этикетке или в инструкции по применению | 9.9 | Действует. |
11.8 | Действует. | |
23.6 | Действует. | |
28.4 | Действует. | |
28.5 | Действует | |
4.3.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы устранить или минимизировать, насколько это возможно: | ||
а) риск травмы с учетом их физических особенностей, таких как соотношение объем/давление, размеры и, если применимо, эргономические особенности; | 15.1 | Изменение. |
15.2 | Действует. | |
b) риски, связанные с разумными, предсказуемыми условиями окружающей среды, такими как магнитные поля, воздействиями внешнего электричества, электростатического разряда, давления, температуры или изменений давления и ускорения; | 23.1 | Заменен. |
23.2 | Заменен. | |
24 | Заменен. | |
25 | Действует. | |
26.2 | Действует. | |
27 | Заменен. | |
27.101 Требование к помехоустойчивости от электромагнитных полей. | ||
27.102 Общие условия испытаний. | ||
27.103 Защита от постоянного магнитного поля. | ||
27.104 Защита от магнитных полей в диапазоне от 10 Гц до 30 МГц. | ||
27.105 Защита от электромагнитных полей в диапазоне от 30 до 450 МГц. | ||
27.106 Защита от электромагнитных полей в диапазоне от 450 МГц до 3 ГГц. | ||
28.103 Требование к идентификационной карте пациента, имеющего имплантат. | ||
с) риски обоюдных помех при использовании с другими изделиями (такими как дефибрилляторы или высокочастотные хирургические инструменты), обычно используемыми для исследований или при данном виде лечения; | 20.1 | Действует. |
20.2 | Действует. | |
21 | Действует. | |
22 | Дополнение. | |
28.12 | Дополнение. | |
28.13 | Действует. | |
28.14 | Действует. | |
28.15 | Действует. | |
d) риски, которые могут возникнуть при условии, что обслуживание и калибровка невозможны, включая (если применимо): чрезмерное увеличение тока утечки, старение материалов, усиление нагрева, генерируемого имплантатом, снижение точности любого измерительного или контрольного механизма | 17 | Заменен. |
19.1 | Действует. | |
19.2 | Заменен. | |
19.101 Срок службы должен быть определен для диапазона скоростей инфузии | ||
4.3.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски возгорания или взрыва при нормальных условиях эксплуатации или в случае единичного отказа. Риски "при нормальных условиях или в случае единичного отказа" означают риски, которые можно оценить путем анализа рисков. Особое внимание следует обратить на имплантаты, предполагаемое использование которых включает воздействие огнеопасных веществ или веществ, которые могут вызвать возгорание | 5 | Действует |
4.4 Имплантаты с функцией измерения | ||
4.4.1 Имплантаты с функцией измерения должны быть разработаны и изготовлены таким образом, чтобы обеспечить установленную точность и стабильность в заданных пределах точности с учетом предполагаемого использования имплантата. Пределы точности должны быть установлены изготовителем | 5 | Действует |
4.4.1.1 Методы выполнения измерений с помощью шкалы на дисплее или в режиме наблюдения должны быть разработаны в соответствии с принципами эргономики, с учетом предполагаемого использования имплантата | 5 | Действует |
4.4.1.2 Если на имплантате или его принадлежностях приведены инструкции по работе с имплантатом или работе индикации, или параметрам настройки с помощью визуальной системы, эта информация должна быть понятна пользователю и, если необходимо, пациенту | 13.4 | Действует. |
5 | Действует | |
4.4.2 Измерения, осуществляемые с помощью имплантатов, снабженных функцией измерения, должны быть выражены в единицах, соответствующих положениям серии ИСО 31 [30] | 5 | Действует |
4.5 Защита от излучения | ||
4.5.1 Общие положения | (См. более подробную информацию ниже) | - |
4.5.2 Предусмотренное излучение | Примечание 2 | - |
4.5.3 Непредусмотренное излучение | 9.1 | Действует. |
18.1 | Действует. | |
18.2 | Действует. | |
18.3 | Действует. | |
28.2 | Действует | |
4.5.4 Инструкции | Примечание 2 | - |
4.6 Ионизирующее излучение | Примечание 2 | - |
4.7 Принципы, касающиеся имплантатов, подсоединенных или оборудованных источником энергии | ||
4.7.1 Общие положения | ||
4.7.1.1 Имплантаты со встроенными электронными программируемыми системами должны быть разработаны таким образом, чтобы обеспечивать воспроизводимость, надежность и эффективность этих систем в соответствии с предполагаемым использованием. При возникновении рисков (рисков системы), установленных при анализе рисков для данного изделия/системы, должны быть приняты соответствующие меры по уменьшению или устранению риска, насколько возможно | 19.3 | Заменен |
4.7.1.2 Имплантаты, в которых безопасность пациента зависит от внутреннего источника энергии, должны быть оснащены средствами индикации состояния источника питания | 19.2 | Заменен. |
4.7.1.3 На имплантатах, насколько возможно, должен быть нанесен код, с помощью которого тип имплантата или изготовители могут быть идентифицированы (в частности, в соответствии с типом имплантата); при необходимости код должен быть читаем без хирургического вмешательства | 13.3 | Изменение. |
28.6 | Действует | |
4.7.1.4 Для имплантатов, в которых безопасность пациента зависит от внешнего источника энергии, внешний источник энергии должен включать в себя систему сигнализации для подачи сигнала в случае отказа питания | 5 | Действует |
4.7.1.5 Внешние изделия, предназначенные для мониторинга одного или более клинических параметров имплантата, должны быть оснащены системой сигнализации для подачи сигнала пользователю в ситуации, которая может привести к смерти или серьезному ухудшению состояния пациента | 5 | Действует |
4.7.2 Защита от электрических рисков | ||
4.7.2.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы избежать, насколько возможно, риска случайного поражения электрическим током при нормальных условиях и в случаях отказа, которые могут возникнуть, несмотря на правильную установку имплантата. В понятие "риски при нормальных условиях и в случаях отказа" входят риски, которые определяются анализом рисков для данного вида изделия | 5 | Действует. |
16.1 | Изменение | |
4.7.2.2 Активные имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, связанные с использованием источников энергии, в первую очередь при использовании электроэнергии, уделяя особое внимание изоляции, токам утечки и перегреву изделия | 16.2 | Дополнение. |
16.3 | Действует. | |
17 | Заменен. | |
26.1 | Изменение | |
4.7.3 Защита от механических рисков | ||
4.7.3.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы защитить пользователя и пациента от механических рисков, связанных, например, с сопротивлением, устойчивостью, движущимися частями изделий | 5 | Действует |
4.7.3.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие от вибрации, генерируемой изделием, принимая во внимание технический прогресс и средства, доступные для снижения вибрации, особенно источника энергии, если только вибрация не является частью специальных рабочих характеристик изделий | 5 | Действует |
4.7.3.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие из-за шумов, принимая во внимание технический прогресс и средства, доступные для снижения шумов, особенно от источника энергии, если только шумы не являются частью специальных рабочих характеристик изделий | 5 | Действует |
4.7.3.4 Терминалы и коннекторы для источников электрической, газовой или гидравлической и пневматической энергии, с которыми приходится работать пользователю, должны быть разработаны и сконструированы таким образом, чтобы минимизировать все возможные риски | 5 | Действует |
4.7.4 Защита пациента от рисков, вызванных источником энергии или веществами | ||
4.7.4.1 Имплантаты должны быть разработаны и сконструированы таким образом, чтобы собственно функционирование программируемых и контрольных систем, включая программное обеспечение, не ухудшало безопасность пациента и пользователя, принимая во внимание предусмотренные назначением изделия условия применения | 19.3 | Заменен |
4.7.4.2 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы таким образом, чтобы параметры потока могли устанавливаться и поддерживаться достаточно точно для минимизации риска для пациента | 5 | Действует. |
4.7.4.3 Имплантаты, разработанные для введения лекарственных веществ, должны включать в себя соответствующие средства предотвращения и/или индикации любой некорректной скорости потока, которая может привести к опасности | 5 | Действует. |
4.7.4.4 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы со встроенными средствами минимизации рисков случайного превышения опасного уровня энергии или лекарственного вещества | 5 | Действует. |
4.8 Информация, предоставляемая изготовителем | ||
4.8.1 Каждый имплантат должен поставляться с сопровождающей информацией, необходимой для его безопасного использования и идентификации изготовителя, с учетом уровня подготовки и знаний потенциального пользователя. | 10.4 | Действует. |
12.3 | Действует | |
10.4 | Действует. | |
12.3 | Действует | |
4.8.2 Если применимо, информацию следует представлять в виде символов. Любой символ или цветовая индикация должны соответствовать национальным стандартам. Если стандарта не существует, символ должен быть описан в документации, поставляемой с имплантатом | 4 | Действует |
4.8.3 Этикетка должна содержать следующие сведения: | 5 | Действует. |
а) наименование или фирменный знак и адрес изготовителя; | 9.2 | Действует. |
11.1 | Действует. | |
b) подробные сведения, необходимые пользователю для идентификации имплантата и содержимого упаковки; | 9.3 | Действует. |
9.4 | Дополнение. | |
9.8 | Действует. | |
9.10 | Действует. | |
11.6 | Действует. | |
11.7 | Действует. | |
11.101 Нанесение дополнительной информации на стерильном пакете | ||
28.103 Требования к карте пациента с обозначением модели имплантата и информацией о центре имплантации. | ||
с) если применимо, индикацию, что содержимое упаковки стерильно (например, "СТЕРИЛЬНО"); | 9.5 | Действует. |
11.2 | Действует. | |
11.3 | Действует. | |
d) если применимо, номер партии или серийный номер, которому предшествует соответствующая идентификация (например, "LOT" или "SN"); | 9.3 | Действует. |
11.6 | Действует. | |
28.103 Требования к карте пациента с указанием серии и номера имплантата. | ||
е) если применимо, дата, до которой имплантат может быть использован; | 9.7 | Действует. |
11.5 | Действует. | |
f) индикацию, что имплантаты предназначены для одноразового использования; | 28.18 | Действует. |
д) если применимо, указание на специальное назначение (например, "изделие, сделанное на заказ" или "только для клинических исследований"); | 9.12 | Действует. |
11.10 | Действует. | |
h) любые специальные условия хранения и/или транспортирования; | 9.11 | Действует. |
i) любые специальные рабочие инструкции; | Примечание 4 | - |
j) любые предупреждения или меры предосторожности, которые следует предпринять; | Примечание 5 | - |
k) для активных имплантатов - месяц и год производства; | 9.6 | Действует. |
11.4 | Действует. | |
28.102 Требование к указанию на каждой части документации года выпуска. | ||
I) если применимо, метод стерилизации. | 11.2 | Действует |
4.8.4 Если предполагаемое использование имплантата не очевидно для пользователя, изготовитель должен четко указать назначение изделия на этикетке и в инструкции по применению | 9.10 | Действует |
4.8.5 Если это разумно и применимо, имплантаты и съемные компоненты должны быть идентифицированы по возможности в терминах серийного номера или номера партии для обеспечения возможности проведения всех надлежащих действий после установления любых потенциальных рисков, обусловленных применением как имплантата, так и съемных компонентов | 8.2 | Действует. |
13.1 | Изменение. | |
13.2 | Действует | |
4.8.6 Если применимо, инструкция по применению должна содержать следующие сведения: | ||
а) детали, упомянутые в 4.8.3, за исключением перечислений d), е) и k); | 28.1 | Дополнение. |
28.3 | Действует. | |
28.16 | Действует. | |
28.18 | Действует. | |
28.21 | Действует. | |
b) рабочие характеристики, приведенные в ИСО/ТО 14283:2004, подраздел 3.3, и любые нежелательные побочные эффекты; | 28.8 | Действует. |
с) если имплантат используется в сочетании с другими медицинскими изделиями или оборудованием, как требует его предполагаемое использование, необходимые подробности о его характеристиках для определения корректных используемых имплантатов или оборудования для получения безопасной комбинации; | 28.4 | Действует. |
28.5 | Действует. | |
28.9 | Действует. | |
d) всю информацию, необходимую для подтверждения того, что имплантат используется надлежащим образом и может работать правильно и безопасно, а также, где применимо, сведения, позволяющие оценить срок службы источника энергии; | 28.10 | Дополнение. |
е) если применимо, информацию, необходимую для предотвращения специальных рисков, связанных с имплантацией имплантата; | 28.11 | Действует. |
f) информацию, относящуюся к рискам перекрестного влияния, вызванного присутствием имплантата во время проведения специфического лечения или обследований; | 28.12 | Дополнение. |
g) необходимые инструкции на случай повреждения стерильной упаковки и, если применимо, подробные сведения о допустимом методе повторной стерилизации; | 28.17 | Действует. |
h) если имплантаты поставляются с условием, что они будут стерилизованы перед использованием, инструкция по очистке и стерилизации должна быть такой, что при ее соблюдении имплантат будет соответствовать требованиям ИСО/ТО 14283:2004, раздел 3; | 28.17 | Действует. |
i) детали последующей обработки или обслуживания, требуемые до использования имплантата (например, стерилизация, окончательная сборка и т.д.); | Примечание 3 | - |
j) в случае если имплантат испускает радиоактивное излучение с лечебными целями, сведения о характере, типе, интенсивности и распространении этого излучения. | Примечание 2 | - |
В инструкцию по применению также должны быть включены подробные сведения, позволяющие медицинскому персоналу оповестить пациента о любых противопоказаниях и любых необходимых мерах предосторожности. Эти сведения должны, в частности, охватывать: | ||
k) меры предосторожности, соблюдаемые в случае изменения рабочих характеристик имплантата; | 28.19 | Действует |
28.20 | Действует | |
I) меры предосторожности в ответ на воздействие в разумно предсказуемых условиях окружающей среды, например, магнитного поля, внешних источников электроэнергии, электростатических разрядов, давления или перепада давлений, ускорения, источников тепла и т.д.; | 28.22 | Дополнение |
m) достаточный объем информации, касающейся лекарственных веществ или веществ, которые должны доставляться с помощью данного имплантата, включая любые ограничения при выборе веществ, которые необходимо доставлять; | 28.7 | Действует |
n) меры предосторожности, предпринимаемые против особых, необычных рисков, связанных с удалением имплантата; | 28.24 | Действует |
о) лекарственные вещества, включенные в имплантат как неотъемлемая часть в соответствии с ИСО/ТО 14283:2004, пункт 4.1.4; | 28.8 | Дополнение |
р) уровень точности измерений, заявленный для имплантатов, снабженных функцией измерения | 5 | Действует |
4.9 Клиническая оценка | ||
а) сбора данных из соответствующей научной литературы, доступной в настоящее время, по назначению изделия, предусмотренного изготовителем; | 19.4 | Изменение |
b) результатов всех клинических исследований, проводимых таким образом, чтобы защищать пациентов и обеспечивать при этом научный характер исследования | 19.4 | Изменение |
Примечание 1 - Этот принцип является основополагающим для всех аспектов активного имплантируемого медицинского изделия, адресованного ИСО 14708. Примечание 2 - Не применимо к активным имплантируемым медицинским изделиям. Примечание 3 - Не применимо, поскольку 14.1 требует, чтобы имплантируемые части активного имплантируемого медицинского изделия предоставлялись стерильными. Примечание 4 - Для имплантируемых частей активного имплантируемого медицинского изделия все инструкции по эксплуатации приведены в сопроводительной документации. Примечание 5 - В общем случае предупреждения и меры предосторожности для тех, кто занимается с особыми условиями обработки, кроме (см. 4.8.3.h)), должны быть описаны в сопроводительной документации, а не на этикетке. | ||
Приложение ВВ
(справочное)
Связь между разделами настоящего стандарта и фундаментальными принципами, перечисленными в приложении АА
Таблица ВВ.1
Подраздел ИСО 14708-4 | Соответствующий фундаментальный принцип ИСО/ТО 14283 |
4 | 4.8.2 |
5 | 4.4.1, 4.4.1.1, 4.4.1.2, 4.4.2, 4.7.1.4, 4.7.1.5, 4.7.3.1, 4.7.3.2, 4.7.3.3, 4.7.3.4, 4.7.4.2, 4.7.4.3, 4.7.4.4, 4.8.3, 4.8.6 р) |
6.101 | 3.3, 4.7.4.2 |
6.102 | 3.3, 3.4, 4.1.5 |
7.1 | 4.2.3 |
7.2 | 3.5, 4.2.3 |
8.1 | 3.1 |
8.2 | 4.8.5 |
8.101 | 3.5 |
8.102 | 3.5 |
9.1 | 4.5.3 |
9.2 | 4.8.3 а) |
9.3 | 4.8.3 b), 4.8.3 d) |
9.4 | 4.8.3 b) |
9.5 | 4.8.3 с) |
9.6 | 4.8.3 k) |
9.7 | 4.8.3 е) |
9.8 | 4.8.3 b) |
9.9 | 4.3.1 |
9.10 | 4.8.3 b), 4.8.4 |
9.11 | 4.8.3 h) |
9.12 | 4.8.3 g) |
10.1 | 3.5, 4.2.3 |
10.2 | 3.5, 4.2.3 |
10.3 | 3.5 |
10.4 | 3.3, 4.8.1 |
11.1 | 4.8.3 а) |
11.2 | 4.8.3 с), 4.8.3 I) |
11.3 | 4.8.3 с) |
11.4 | 4.8.3 k) |
11.5 | 4.8.3 е) |
11.6 | 4.8.3 b), 4.8.3 d) |
11.7 | 4.8.3 b), 4.2.3 |
11.8 | 4.3.1 |
11.9 | 4.2.3 |
11.10 | 4.8.3 g) |
11.101 | 4.8.3 b) |
12.1 | 4.2.3 |
12.2 | 4.2.3 |
12.3 | 3.5 |
13.1 | 4.8.5 |
13.2 | 4.8.5 |
13.3 | 4.7.1.3 |
13.4 | 4.4.1.2 |
14.1 | 4.2.1, 4.2.3, 4.2.4, 4.2.5 |
14.2 | 4.1.2, 4.2.5 |
14.3 | 4.1.1 a), 4.1.1 b), 4.1.2 |
14.4 | 4.1.4 |
14.101 | 4.1.4 |
15.1 | 4.3.2 a) |
15.2 | 4.3.2 a) |
16.1 | 4.7.2.1 |
16.2 | 4.7.2.2 |
16.3 | 4.7.2.2 |
17 | 4.7.2.2, 4.3.2 d) |
18.1 | 4.5.3 |
18.2 | 4.5.3 |
18.3 | 4.5.3 |
19.1 | 4.3.2 d) |
19.2 | 3.4, 4.3.2 d), 4.7.1.2 |
19.3 | 3.4, 3.6, 4.1.7, 4.7.1.1, 4.7.4.1, 4.7.4.3, 4.7.4.4 |
19.4 | 3.6, 4.9 а), 4.9 b) |
19.5 | 4.1.3 |
19.101 | 3.4, 4.3.2 d), 4.7.1.2 |
20.1 | 4.3.2 с) |
20.2 | 4.3.2 с) |
21 | 4.3.2 с) |
22 | 4.3.2 с) |
23.1 | 3.4, 4.3.2 b) |
23.2 | 3.4, 4.3.2 b) |
23.3 | 3.4 |
23.4 | 3.4 |
23.5 | 3.4 |
23.6 | 3.4, 4.3.1 |
24 | 4.3.2 b) |
25 | 4.3.2 b) |
26.1 | 3.4, 4.7.2.2 |
26.2 | 3.5, 4.3.2 b) |
27.101 | 4.3.2 b) |
27.102 | 4.3.2 b) |
27.103 | 4.3.2 b) |
27.104 | 4.3.2 b) |
27.105 | 4.3.2 b) |
27.106 | 4.3.2 b) |
28.1 | 4.8.6 a) [4.8.3 a)] |
28.2 | 4.5.3 |
28.3 | 4.8.6 a) [4.8.3 b)] |
28.4 | 3.4, 4.3.1, 4.8.6 c) |
28.5 | 4.3.1, 4.8.6 с) |
28.6 | 4.7.1.3 |
28.7 | 4.8.6 m) |
28.8 | 4.8.6 b), 4.8.6 о) |
28.9 | 4.8.6 с) |
28.10 | 4.8.6 d) |
28.11 | 4.8.6 е) |
28.12 | 4.3.2 с), 4.8.6 f) |
28.13 | 4.3.2 с) |
28.14 | 4.3.2 с) |
28.15 | 4.3.2 с) |
28.16 | 4.8.6 а) [4.8.3 с)] |
28.17 | 4.8.6 g), 4.8.6 h) |
28.18 | 4.8.6 а) [4.8.3 f)] |
28.19 | 4.8.6 k) |
28.20 | 4.8.6 k) |
28.21 | 4.8.6 а) [4.8.3 h)] |
28.22 | 4.8.6 I) |
28.23 | 3.4 |
28.24 | 4.8.6 n) |
28.101 | 4.8.6 d) |
28.102 | 4.8.3 k) |
28.103 | 4.3.2 b), 4.8.3 b), 4.8.3 d) |
Примечание - Цифры в скобках указывают косвенное отношение к цитируемому подпункту. | |
Приложение СС
(справочное)
Обоснование
СС.1 Общие положения
Изложенные ниже замечания к некоторым положениям настоящего стандарта призваны облегчить его понимание. Содержание настоящего приложения относится не ко всем разделам настоящего стандарта; именно поэтому нумерация параграфов в приложении не является последовательной.
СС.2 Замечания по специальным разделам и подразделам
1 Система для пробных измерений включена в область применения из-за обычной практики проводить инфузию в течение испытательного срока до принятия решения об имплантации изделия. Система пробных измерений должна отвечать тем же требованиям безопасности, что и фактически имплантируемое изделие. Внешние инфузионные насосы, применяемые для других целей и не относящиеся к принадлежностям инфузионного насоса, в настоящем стандарте не рассматриваются.
6.101 Настоящий подпункт определяет соотношение между заявленными изготовителем техническими требованиями, характеристиками и сроком службы.
Точность инфузии является основным фактором безопасности, хорошо известно, что в некоторых имплантируемых насосах точность связана с объемом резервуара. Запрашиваемая информация важна для врача для определения скорости инфузии и настроек изделия.
В стандартах ИСО (например, ИСО 11631 [9]) приведены определения понятиям "точность" и "повторяемость", в соответствии с которыми они применяются в настоящем стандарте. В то время как повторяемость может быть количественно измерена и выражена численно (относится к самому набору данных, но не к сравнению полученного и истинного значений), фактическая точность, в сравнении с "истинным" значением, не может быть известна. Может быть определен термин "погрешность измерения", но этого не требуется для настоящего стандарта, т.к. термин не является широко известным за пределами статистической и инженерной практики и может привести к путанице. Термин "точность", напротив, является легко узнаваемым (хотя, возможно, обычно неправильно истолковывается) и подходит для целей настоящего стандарта. Тем не менее важно, чтобы изготовитель указал, как именно вычислялась точность.
Процедура определения точности инфузии проводится на установке, утвержденной для определения измеряемых требований. Из-за небольших объемов инфузии, требуемых для измерения, ошибки в испытательной установке и подготовке насоса могут быть значительными, если процедуры испытания не полностью утверждены. Поэтому необходимо контролировать процедуру проверки испытательной установки, предоставленную изготовителем, и методы испытаний для установления точности инфузии имплантируемого инфузионного насоса. Для испытания может использоваться вещество, такое как дистиллированная вода, до тех пор, пока сохраняется эквивалентность фактическому предполагаемому употреблению лекарственного вещества.
Изготовителю необходимо разработать подходящие испытания на долговечность для гарантирования безотказной работы изделия весь гарантийный срок службы. Возможно использовать ускоренное испытание для запуска насоса и заполнения резервуара для установления ожидаемой производительности в течение долгого времени. Эти испытания могут быть проведены на образцах, аналогичных конечному изделию, пока есть надлежащее подтверждение того, что оцениваемые параметры не искажаются при использовании этих образцов.
6.102 Порт доступа к мембране, такой как те, которые используются для пополнения резервуара или предоставления прямого доступа к катетеру доставки, является важнейшим компонентом, поскольку повреждение мембраны может привести к тому, что резервуар опорожнится или жидкость будет просачиваться во внутренние ткани человека, окружающие имплантируемый насос. Испытание мембраны методом прокола предназначено для обеспечения стандартизированного метода получения данных для оценки надежности мембраны.
8.101 Настоящее требование распространяется на маркировку во время транспортировки ко всем частям, включая имплантируемые. К неимплантируемым частям, имеющим электрическое питание, применяются требования раздела 5.
8.102 Настоящее требование распространяется на маркировку с указанием требований к окружающей среде во время транспортирования ко всем частям, включая имплантируемые части. К неимплантируемым частям с электрическим питанием применяют требования раздела 5.
10.3 Испытания в настоящем стандарте согласованы с испытаниями по МЭК 60601-1. В примечании объясняется, что сменные наклейки не должны подвергаться испытаниям, если они содержат информацию вне требований настоящего стандарта.
13.1 Испытание влажным протиранием приведено в соответствии с 10.3.
14.2 Требования ИСО 14708-1 были первоначально сформулированы в отношении передающихся через кровь частиц. Настоящий стандарт позволяет изготовителю определять требования для твердых частиц, которые могут отличаться в зависимости от предполагаемого применения инфузионного насоса. При этом изготовитель должен провести анализ рисков, анализ конструкции, испытания или использовать другие подходящие средства.
Так как система циркуляции жидкости находится не в прямом контакте с жидкостями организма, а посредством доставки лекарственного вещества, она тоже включена в настоящий подпункт.
14.3 Особое внимание должно быть уделено выбору материалов, используемых в производстве медицинского изделия. Для проведения биологической оценки безопасности имплантируемых инфузионных насосов конструкционные материалы должны быть оценены на остаточные мономеры, добавки, примеси, вымываемые вещества и продукты распада.
Материалы системы циркуляции жидкости могут косвенно соприкасаться с жидкостями в организме человека, поэтому в качестве меры предосторожности эти материалы также должны быть оценены на биологическую совместимость.
14.4 Подразумевается, что лекарственные вещества, обычно используемые с имплантируемыми инфузионными насосами, прошли многочисленные исследования и проверки до того, как были допущены к применению.
14.101 Должны быть использованы общепринятые испытания для установления стабильности лекарственного вещества в имплантируемом инфузионном насосе. В том случае, если тестирование проводилось третьей стороной, определение показателей стабильности или обеспечение необходимой документации для поддержки интервала стабильности является ответственностью изготовителя.
15.1 Применяется действующая редакции ссылочного стандарта.
16.1 Применяется действующая редакции ссылочного стандарта.
16.2 Требует, чтобы изготовитель определил, являются ли пределы утечки приемлемыми для конкретного применения изделия.
17 Основываясь в целом на распределении температуры тела человека, ограничение повышения температуры на 2°C можно считать чрезмерно строгим. При перфузии может возникнуть большее локальное увеличения температуры. См. [10].
19.2 Пункт изменен в соответствии с используемыми терминами в настоящем стандарте (т.е. срок службы) и уточнено, что имеется в виду под источником энергии. Первоначальное требование ИСО 14708-1 относится к резервуару под давлением, как к источнику энергии; не стоит путать с резервуаром для жидкости в случае имплантируемого инфузионного насоса.
19.3 Пункт изменен, чтобы отразить текущую методику оценки степени риска согласно ИСО 14971. Заменяет старый и менее общий метод, такой как FMEA (Failure Mode Effects Analysis - анализ видов и последствий отказов).
19.4 Изменяет прежнее требование для обеспечения клинических исследований, проводимых в соответствии с опубликованными стандартами, помимо ИСО 14155-1 или ИСО 14155-2.
19.5 Имплантируемый резервуар и катетер содержат материалы, которые могут оказать влияние на стабильность и эффективность лекарственного вещества. Все материалы устройства системы циркуляции жидкости имплантируемого инфузионного насоса, которые подвергаются воздействию лекарственного вещества, должны быть оценены с точки зрения их пригодности в конструкции. Оценка должна предусматривать возможность изменений физических характеристик материалов и существенного ухудшения материалов от выделения лекарственного вещества в течение долгого времени. Любое вымываемое вещество или известный продукт распада должны быть оценены с точки зрения их воздействия на лекарственное вещество. В случае наличия вымываемого вещества или продукта распада необходимо вычислить их объем, и общая суточная доза, которая может быть введена, должна быть оценена токсикологическим образом, либо посредством испытания или литературного обоснования. Необходимо отметить, уменьшается ли количество вымываемых веществ или продуктов распада в течение долгого времени.
19.101 Для пользователя важно понимать, какой будет срок службы на основе не только обычной скорости инфузии, потому что скорость инфузии может оказать существенное влияние на срок службы.
21 Имплантируемые инфузионные насосы могут оказаться под воздействием мощных электрических полей, приложенных непосредственно к пациенту. При этом изготовитель должен доказать, что требование настоящего пункта выполняется. Частные требования могут быть определены в будущих редакциях настоящего стандарта.
22 Утверждается, что должны быть оценены новые методы лечения и процедуры. Частные требования могут быть определены в будущих редакциях настоящего стандарта.
23.1 Согласовано с МЭК 60601-1:2005, но увеличено число падений для ручных приспособлений для учета механического воздействия.
23.2 Изменен ИСО 14708-1, чтобы отразить обычные условия использования только для имплантируемых частей. Неимплантируемые части подвергаются механическому воздействию согласно 23.1 и разделу 10, в которых требуется учесть возможность вибрации во время хранения и использования.
24 Испытательные уровни электростатического разряда, указанные в ИСО 14708-1, являются устаревшими. Новое требование, указанное в МЭК 60601-1-2, будет гарантировать, что изделия проверены на уровнях, которые в настоящее время считаются подходящими. Раздел 24 также согласован с разделом 5.
26.1 Применяется действующая редакции ссылочного стандарта.
27.102 Наиболее важные функции имплантируемого инфузионного насоса с точки зрения безопасности должны быть подвергнуты испытаниям на помехоустойчивость. Функции, не связанные с основными функциональными характеристиками, не должны проверяться испытаниями. Как правило, функции можно рассматривать как клинически существенные признаки, для обеспечения которых предназначено изделие. Раздел 3 настоящего стандарта не определяет понятие "функция", потому что функция, которая не является клинически значимой, может быть каким-то образом связана с основными функциональными характеристиками и, следовательно, должна подвергаться испытаниям на помехоустойчивость.
Большинство испытаний на помехоустойчивость включает два набора испытательных уровней. Следовательно, существует также два определенных набора критериев эффективности (третий уровень критерия определяется изготовителем).
Испытательные уровни, которые были выбраны для всех тестов, основаны на общих условиях проживания населения и, в ряде случаев, связаны с нормами биологического воздействия. Испытательные уровни не наносят необратимое повреждение и не вызывают долгосрочный эффект. В связи с этим разработаны соответствующие критерии производительности (одинаковые критерии используются для обоих тестовых уровней).
Критерии эффективности во время испытания устанавливаются в соответствии с испытательным уровнем. Нижние уровни испытаний основаны на общих, повседневных условиях проживания населения. Эти уровни характерны для бытовых условий, в том числе линий электропередачи, транспорта, мест общего пользования (школы, магазины, офисы и больницы) и офисного оборудования, и мест, где изделие находится под влиянием других устройств длительное время. Таким образом, в этой среде следует ожидать, что имплантируемый инфузионный насос будет работать полностью согласно предназначению без потери функции и без каких-либо непредвиденных ответных реакций.
Верхние уровни испытаний представляют собой среды, в которых пациент оказывается редко, обычно в течение короткого времени, и которых, как правило, возможно избежать. Источниками этой категории являются системы досмотра и мобильное оборудование с высоким уровнем излучения. Для магнитных полей более высокие уровни испытаний помехоустойчивости равны десяти нижним уровням. Общий уровень и для электрических полей составляет пять нижних уровней. Существуют три уровня систем аварийной сигнализации и пять уровней мобильных приемопередатчиков. В этой среде изделие, как ожидается, не будет повреждено или подвергнуто неприемлемому риску. Так как невозможно предусмотреть все ситуации, установлены нормы временного ухудшения характеристик и непреднамеренной работы до того момента, пока пациент находится в безопасности. Таким образом, использование насоса может быть разрешено, чтобы пациент мог использовать клинические преимущества изделия. Это разрешение должно зависеть от мнения изготовителя и врачей. Как правило, не предполагается непредвиденная работа изделия, но необходимо это проверить экспериментально при десятикратном увеличении тестового уровня.
Некоторые ухудшения не допустимы из соображений безопасности и потому, что они не оправданы тяжестью испытательных уровней.
27.103 Нижний уровень, равный 1 мТл (10 Гс), был выбран на основании обычно встречающихся напряженностей поля. На практике многие имплантируемые изделия имели внутренние переключатели, которые активировались магнитным полем. Эти переключатели обычно активировались магнитным полем около 1 мТл. Таким образом, можно ожидать, что все изделия будут функционировать нормально в присутствии поля 1 мТл.
Высокий уровень 50 мТл (500 Гс) редко, но может встречаться. Индукция постоянного магнитного поля, образованного на поверхности обычных бытовых магнитов (например, магниты на холодильнике) и от магнитов в медицинских изделиях, необходимых для активации переключателя, обычно составляет 50 мТл (магнитные поля резко сжимаются, поэтому сила поля в нескольких сантиметрах, как правило, незначительна). Таким образом, изделия не будут подвержены необратимым повреждениям или изменениям состояния после воздействия этих уровней. Поскольку некоторые имплантируемые медицинские изделия также зависят от активации переключателей для некоторых функций управления, изменение режима работы может произойти от постоянного магнитного поля индукцией более чем 1 мТл.
Требование к однородности поля 3 дБ оправдано известными техническими возможностями.
Существует одна обязательная ориентация ИУ, при которой наибольшая поверхность подвергается воздействию вектора магнитного поля. Другой ориентации не требуется.
Десяти минут должно быть достаточно для получения реакции, если был выбран нормальный уровень инфузии для проведения испытания.
27.104 Общее облучение населения электромагнитной энергией в диапазоне частот от 10 Гц до 30 МГц состоит из электрического и магнитного полей. Считается, что только магнитные поля способны нарушить работу имплантируемых инфузионных насосов.
Электрические поля в данном диапазоне частот оказывают незначительное влияние на изделия размером с нейростимулятор, имея очень короткую электрическую длину величиной 1/20 длины волны. Учитывая следующее соотношение:
![]() (СС.101)
(СС.101)
ткани с диэлектрической проницаемостью 80 и средним максимальным размером 6 см (типичный для имплантируемого инфузионного насоса), при частоте менее чем 27 МГц должны незначительно влиять на насос. При частоте свыше 27 МГц электрическое поле в корпусе ослабляется более чем на 35 дБ, за исключением области разъема блока. Помехи от излучателей в этом диапазоне частот, такие как любительское радио, АМ-радио, время и частота вещания, ISM (Industrial, Scientific, Medical - индустриальный, научный и медицинский диапазон), портативные рации и системы морской радионавигации, как известно, не влияют на имплантируемые инфузионные насосы.
Воздействие магнитных полей, способных вызвать нарушения, в первую очередь происходят от источника частоты оборудования и приборов. Большинство воздействий происходит в доме, но также вблизи линий электропередачи, транспортных средств, оргтехники и в местах общего пользования, таких как школы, магазины, офисы и больницы.
На рисунке СС.101 проиллюстрирован вывод А-линии и B-линии из исходных данных окружающей среды (см. [10], [19]), общего уровня Международной комиссией по защите от неионизирующих излучений (ICNIRP) [20], неконтролируемых уровней IEEE С95.1 [21] и IEEE С95.6 [22], MIL 461Е [23] уровней RS101, данных МЭК 61000-2-5:1995 [24] (таблица 6, степень нарушения 4) и испытательных А-уровней настоящего стандарта.
А-линия показывает ежедневное общее воздействие магнитных полей. A-линия совпадает с MIL 461 в полосе частот от 30 Гц до 3 кГц. Начиная с 3 кГц она практически повторяет траекторию ICNIRP базового уровня за исключением того, что проходит на 3,2 выше в полосе до 100 кГц и приблизительно на 2,2 выше в полосе до 30 МГц. Это может быть использовано для расчета пульсации. Данные, представленные на рисунке, уже рассчитаны для локализованных эффектов, основанных на однородности источника поля (включая размер и близость) в соответствии с IEC 62226-2-1 [25]. Общее воздействие магнитных полей, представленное А-линией, считается вероятным, обычным и неизбежным. Функционирование имплантируемого изделия в этих условиях воздействия, как ожидается, будет нормальным.
В-линия используется для дополнительной гарантии защиты от воздействия, превышающего А-линию. Это не представляет собой частный случай влияния окружающей среды как таковой, но соответствует рекомендациям IEEE С95.1 для максимально допустимого уровня воздействия на человека. Запас прочности, обеспечиваемый В-линией, в десять раз превышает А-линию в полосе 3 кГц. Потенциальные источники такого уровня воздействия достаточно редки и для достижения таких уровней воздействия необходима близость к подобному источнику. Таким образом, общее воздействие магнитных полей, представленных B-линией, считается возможным, относительно редким и, в случае возникновения, краткосрочным, и в целом предотвратимым, если источники известны. Функционирование имплантируемого изделия в этих условиях воздействия, как ожидается, будет проходить без повреждений и недопустимого риска.

Рисунок СС.101 - Данные источников магнитного излучения ICNIR, неконтролируемых уровней IEEE С95.1 [21] и IEEE С95.6 [22], MIL 461Е [23] уровней RS101, данные МЭК 61000-2-5 и испытательного А-уровня
Поле должно быть однородным до 100 кГц, чтобы охватить нормальные условия окружающей среды. При таких низких частотах проще генерировать однородное поле в заданном диапазоне. Однородность на более высоких частотах и уровнях испытаний связана с ограничениями испытательного стенда.
Солевая ванна используется для имитации естественных условий окружающей среды нейростимулятора при нормальном использовании. Значение проводимости 0,27 См/м является компромиссным для различных тканей тела и консистенции во всем диапазоне тестовых частот всех испытаний.
Размеры шага настоящего стандарта позволяют охватить весь диапазон частот, не создавая избыточное число испытательных измерений.
А-линия использует тестовую синусоидальную непрерывную волну для моделирования окружающей среды. B-линия - импульсная модуляция частотой не менее 3 кГц имитирует виды сигналов, присутствующих в окружающей среде и создает дополнительную нагрузку на ИУ. Ниже уровня 3 кГц B-линия - синусоидальная, т.к. импульсные поля с частотой модуляции 200 Гц не распространены в практике, находится слишком близко к начальной тестовой частоте 300 Гц. Выбор частоты модуляции для имитации известных источников магнитного поля основан на том, чтобы не создавать помех на частоте питания или стандартной частоте стимуляции, которая, как правило, меньше, чем 200 Гц.
Повторное тестирование А-линии на частотах, обладающих эффектом импульсной модуляции, обеспечивает дополнительную уверенность в безопасности.
Для проверки воздействия вектора магнитного поля на наибольшую поверхность тестируемого изделия достаточно лишь одной определенной ориентации в испытательной установке. Для других расположений профиль изделия слишком мал, чтобы наблюдать взаимодействия, не замеченные при обязательной ориентации. Максимальное вызванное напряжение будет наблюдаться при перпендикулярном расположении петли провода к магнитному полю.
27.105 Удаленные источники электромагнитной энергии воздействуют на население в диапазоне частот от 30 до 450 МГц. К первичным преобразователям в этом частотном диапазоне относят излучающие колебательные источники, которые, как правило, являются телерадиовещательными передатчиками, портативными и мобильными передатчиками и ISM оборудованием.
__________________
ISM диапазон (англ. Industrial, Scientific, Medical: индустриальный, научный и медицинский диапазон) занимает полосу частот от 5725 до 5875 МГц. ISM диапазон используют многие популярные системы, такие как беспроводные телефоны и другие системы.
Как правило, в населенных или посещаемых широкой общественностью местах напряженности поля от этих источников менее 10 В/м. Сравнительный стандартный тест помехоустойчивости МЭК 60601-1-2 используется для неимплантируемых медицинских изделий, определяя напряженности тестового поля 3 В/м для вспомогательного нежизненно необходимого оборудования 10 В/м для питания оборудования. В зависимости от близости источника напряженность поля может быть выше.
Примечание - Для напряженности электрического поля приведены среднеквадратичные значения, если не указано иное.
Биологические стандарты безопасности могут быть использованы для целей сравнения, чтобы оценить потенциальную угрозу для безопасности человека. Пределы, представленные в этих стандартах, могут быть использованы в качестве ориентира для установки уровней испытаний помехоустойчивости, основанью на предположении, что воздействие электромагнитных полей должно быть ограничено. Недавно некоторые страны предприняли шаги, чтобы принять законодательство по контролю выбросов для защиты общественности.
IEEE С95.1 [21] в диапазоне частот от 30 до 450 МГц устанавливает пределы риска электрических полей от 27,5 до 33,6 В/м (неконтролируемых) и от 61,4 до 75 В/м (управляемых). ICNIRP [20] в том же диапазоне частот ограничивает воздействие электрического поля от 28 до 29 В/м (широкой общественности) и от 61 до 63,6 В/м (профессиональной).
Пределы излучений от ISM оборудования, установленные CISPR 11 [26], различаются в зависимости от группы, класса и расстояния, но могут быть сведены к 60 дБ мкВ/м (1000 мкВ/м), в худшем случае до 30 м, от 30 до 450 МГц. Пределы для бытовых приборов, электрических инструментов и аналогичных устройств, установленные CISPR 14 [27], приведены ниже. Пределы ITE (информационные технологии) оборудования, установленные CISPR 22 [28], не более 37 дБ мкВ/м (71 мкВ/м). Пределы класса A цифровых устройств, установленные FCC [29], не более 46,5 дБ мкВ/м (210 мкВ/м).
Примеры напряженности поля авторизованных передатчиков приведены в таблице СС.101 и таблице 1 МЭК 61000-2-3:1992 [16].
Таблица СС.101 - Примеры напряженности полей для авторизованных передатчиков
Служба | Частотный диапазон, МГц | Типовой диапазон пространственного разнесения, м | Расчетный диапазон значений напряженности поля на соответствующем расстоянии, В/м |
Низкочастотное и морское вещание | 0,014-0,5 | 1000-20000 | 5,5-0,55 |
Радиовещание с амплитудной модуляцией | 0,2-1,6 | 500-2000 | 12,5-0,78 |
Любительские высокие частоты | 1,8-30 | 10-100 | 22,1-2,21 |
Высокочастотные системы связи, включая коротковолновое | 1,6-30 | 1000-20000 | 0,7-0,04 |
Гражданский диапазон | 27-28 | 10-100 | 2,4-0,24 |
Любительские высокие частоты и сверхвысокие частоты | 50-52 | 10-500 | 63-0,44 |
144-146 | 39 | 16 | |
434-438 | |||
1290-1300 | |||
Стационарная и мобильная связь | 29-40 | 2-200 | 40-0,25 |
68-87 | 5 | 16 | |
146-174 | |||
422-432 | |||
438-470 | |||
860-990 | |||
Портативные телефоны, в том числе беспроводные | 1880-1990 | 1-100 | 15,6-1,56 |
ТВ-вещание очень высокой частоты | 48-68 | 500-2000 | 8-1,11 |
Радиовещание с частотной модуляцией | 88-108 | 250-1000 | 8,9-2,2 |
ТВ-вещание сверхвысокой частоты | 470-853 | 500-3000 | 10-1,6 |
МЭК 61000-2-5:1995 [24] (таблица В.1) содержит значения вероятности того, что напряженности электрического поля не будут превышать установленные значения на определенных расстояниях от общих передатчиков. В соответствии с этой таблицей, вероятность того, что напряженность поля 10 В/м будет превышать установленное значение для данного излучателя и расстояния, указанных в таблице СС.102, незначительна (0,0001%).
Таблица СС.102 - Электрические расстояния напряженности поля для 10 В/м
AM вещание | Переносной приемопередатчик | Гражданский диапазон | ТВ-вещание очень высокой частоты |
495 м | 1,6 м | 2,4 м | 313 м |
Принципы установки уровней для испытания помехоустойчивости в настоящем стандарте принимают во внимание то, что имплантат неотделим от своего носителя без хирургического вмешательства. Требование, чтобы тестовый уровень помехоустойчивости был выше уровня, необходимого для неимплантируемых медицинских изделий, но, в то же время, соответствовал реальным встречающимся источникам, является обоснованным.
Уровень среднеквадратичного значения 16 В/м при амплитудной модуляции 80% в соответствии с приведенной выше информацией (28 В/м максимального среднеквадратичного значения) обеспечивает повышение на пять уровней над не жизненно необходимыми требованиями МЭК 60601-1-2 (на 1,6 уровня - над требованиями жизнеобеспечения).
Амплитудная модуляция используется в соответствии с требованиями МЭК 60601-1-2 для неимплантируемых частей. Согласно обоснованиям, в МЭК 61000-4-3 амплитудная модуляции имеет преимущества по сравнению с другими методами. Частота модуляции равна 2 Гц, что близко к физиологическим полосам пропускания.
Размер шага 5% обеспечивает рациональное покрытие диапазона частот, оптимизируя количество тестовых измерений.
Пульсация и локализация воздействия электромагнитных полей учитываются в биологических стандартах безопасности, где нет фактора локализации для электрического поля. В теории коэффициент пульсации для электрического поля может быть таким же высоким, как у людей без имплантатов: 32. На практике нет никаких известных источников в открытом доступе, которые используют такие уровни. Коэффициент пульсации 5 был выбран в качестве разумного предела, в результате чего верхний испытательный уровень равен среднеквадратичному значению 140 В/м (28 В/м5).
Верхний уровень импульсной модуляции (вместо амплитудной), прежде всего, ограничивается возможностями оборудования. Шаги частоты были выбраны, чтобы получить показательную выборку источников, занимающих частотный диапазон.
Солевая ванна используется для моделирования естественных условий среды имплантируемого инфузионного насоса при нормальной эксплуатации. Необходимо применять показатель проводимости 0,27 См/м по всему диапазону частот во время испытания.
Использование МЭК 61000-4-3 для обеспечения соответствия процедуры испытания требованиям для неимплантируемых частей является признанной нормой. Целесообразно использовать стандартную методологию МЭК из-за сходства электромагнитного воздействия окружающей среды, с которыми сталкиваются внешние и имплантируемые медицинские изделия.
27.106 Население подвергается воздействию электромагнитной энергии в диапазоне частот от 450 МГц до 3 ГГц, прежде всего, электрического поля. Источники с достаточно высокой напряженностью, чтобы вызвать возможные помехи у имплантированных медицинских изделий, состоят в основном из ручных беспроводных передатчиков (например, сотовые телефоны) на близком расстоянии.
Стандарт по электромагнитной совместимости американского института национальных стандартов/ассоциации по развитию медицинского приборостроения (ANSI/AAMI - The American National Standards Institute/The Association for the Advancement of Medical Instrumentation) ANSI/AAMI PC69, написанный рабочей группой Комитета по кардиостимулятору, затрагивает прежде всего этот тип оборудования. Настоящий стандарт включает требования и методы испытаний ANSI/AAMI РС69 из-за сходства между имплантируемыми кардиостимуляторами и имплантируемыми инфузионными насосами в части среды пациента, адекватных механических и электрических элементов конструкции и использования изделий в естественных условиях.
Несмотря на то, что ANSI/AAMI РС69 предназначен, прежде всего, для оценки воздействия беспроводных устройств на близком расстоянии, требования этого стандарта применимы для оценки воздействия и на отдаленном расстоянии в пределах 3 или 10 В/м. Угроза от карманных беспроводных устройств выше, чем от других устройств, например от телевидения или радио.
ANSI/AAMI РС69 дает обоснование для выбора тестовых частот в этом диапазоне и для испытательных уровней. Стандартный уровень 40 мВт предназначен для имитации ручного беспроводного передатчика на расстоянии 15 см от имплантата, которое является общепринятым расстоянием. Дополнительные, более высокие уровни 8 Вт менее 1000 МГц и 2 Вт менее 1000 МГц имитаций на более близких расстояниях позволяют изготовителю делать выводы о повышенной работоспособности или помехоустойчивости. Так как это тестирование является дополнительным, изготовитель имеет право по своему усмотрению установить критерии эффективности, на основе которых их произвести.
28.1 В дополнительной информации изготовитель обязан предоставить пользователю альтернативные пути установления контакта, особенно в экстренных ситуациях.
28.12 МРТ становится очень важной и широко применяемой процедурой диагностики. Очень важно, чтобы пациент и врач понимали риски, связанные с имплантируемым изделием. Если изготовитель не может обеспечить убедительного доказательства совместимости, необходимо предупреждение о возможности риска до того момента, пока не будут разработаны стандартизированные процедуры и требования для оценки работы изделия и безопасности пациентов.
28.22 Электронные системы защиты от кражи и подобные им системы видеонаблюдения находятся повсюду в общественной среде, и их не всегда можно избежать или обнаружить. Также существуют другие потенциальные источники электромагнитных помех, которые могут быть невидимыми для пациентов в повседневной жизни. По результатам испытаний в разделе 27 может быть необходимо включить предупреждения в документацию пользователя.
28.103 Запись в карте пациента о том, что он имеет имплантированное медицинское изделие, будет служить напоминанием для пациента и персонала об обеспечении безопасности, принятии мер предосторожности и возможной необходимости избегать потенциально опасных электромагнитных помех или электромагнитного поля высокой мощности.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ISO 10993-1:2003 | IDT | ГОСТ ISO 10993-1-2011 "Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования" |
ISO 15223-1:2007 | IDT | ГОСТ Р ИСО 15223-1-2010 "Изделия медицинские. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации" |
ISO 14971:2007 | IDT | ГОСТ ISO 14971-2011 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
ISO 14155-1:2003 | IDT | ГОСТ Р ИСО 14155-1-2008 "Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования" |
ISO 14155-2:2003 | IDT | ГОСТ Р ИСО 14155-2-2008 "Руководство по проведению клинических испытаний медицинских изделий. Часть 2. Планирование клинических испытаний" |
ISO 14708-1:2000 | IDT | ГОСТ Р ИСО 14708-1-2012 "Имплантаты хирургические. Активные имплантируемые медицинские изделия. Часть 1. Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем" |
ISO 780:2015 | - | * |
ISO/TR 14283:2004 | - | * |
ISO 11631:1998 | - | * |
IEC 60601-1:2005 | - | * |
IEC 60601-1-2:2007 | IDT | ГОСТ Р МЭК 60601-1-2-2014 "Изделия медицинские электрические. Часть 1-2. Общие требования безопасности с учетом основных функциональных характеристик. Параллельный стандарт. Электромагнитная совместимость. Требования и испытания" |
IEC 61000-4-3:2002 | - | * |
IEC 61000-2-3:1992 | - | * |
IEC 61000-2-5:1995 | IDT | ГОСТ Р 51317.2.5-2000 "Совместимость технических средств электромагнитная. Электромагнитная обстановка. Классификация электромагнитных помех в местах размещения технических средств" |
ANSI/AAMI PC69:2000 | - | * |
IEC/TR 61000-2-7:1998 | - | * |
IEC/TR 61000-2-3:1992 | - | * |
IEEE C95.1:1999 | - | * |
IEEE C95.6:2002 | - | * |
MIL-STD-461E:1999 | - | * |
IEC 62226-2-1 (2004) | - | * |
IEC/TS 61000-2-5:1995 | - | * |
CISPR 11:2004 | MOD | ГОСТ Р 51318.11-2006 "Совместимость технических средств электромагнитная. Промышленные, научные, медицинские и бытовые (ПНМБ) высокочастотные устройства. радиопомехи индустриальные. Нормы и методы измерений" |
CISPR 14-1:2005 | MOD | ГОСТ 30805.14.1-2013 "Совместимость технических средств электромагнитная. Бытовые приборы, электрические инструменты и аналогичные устройства. Радиопомехи индустриальные. Нормы и методы измерений" |
CISPR 22:2006 | MOD | ГОСТ 30805.22-2013 "Совместимость технических средств электромагнитная. Оборудование информационных технологий. Радиопомехи индустриальные. Нормы и методы измерений" |
* "Соответствующий национальный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного международного стандарта. Примечание - В настоящей таблице использованы следующие условные обозначения степени соответствия стандартов: - IDT - идентичные стандарты; - MOD - модифицированные стандарты. | ||
Библиография
[1] | ISO 780, Packaging - Pictorial marking for handling of goods |
[2] | ISO 15223-1, Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements |
[3] | European Pharmacopoeia, 3rd edition, Council of Europe, 1977 |
[4] | ISO 10993-1, Biological evaluation of medical devices - Part 1: Evaluation and testing |
[5] | ISO 14971, Medical devices - Application of risk management to medical devices |
[6] | ISO 14155-1:2003, Clinical investigation of medical devices for human subjects - Part 1: General requirements |
[7] | ISO 14155-2:2003, Clinical investigation of medical devices for human subjects - Part 2: Clinical investigation plans |
[8] | ISO/TR 14283:2004, Implants for surgery - Fundamental principles |
[9] | ISO 11631:1998, Measurement of fluid flow - Methods of specifying flowmeter performance |
[10] | WUST et al 1995, Int. J. Hyperthermia 11, pp.151-167 |
[11] | Electric and magnetic fields associated with the use of electric power, NIEHS, June 2002 |
[12] | World Health Organization (WHO) web site (http://www.who/int/peh-emf/en/) |
[13] | National Grid Company (Warwick, England) EMF web site (http://www.emfs.info/default.asp) |
[14] | Children's exposure to magnetic fields produced by U.S. television sets used for viewing programs and playing video games, Bioelectromagnetics 21, pp.214-227 (2000), Tables 3 and 4 combined |
[15] | IEC/TR 61000-2-7, Electromagnetic compatibility (EMC) - Part 2: Environment - Section 7: Low frequency magnetic fields in various environments |
[16] | IEC/TR 61000-2-3:1992, Electromagnetic compatibility (EMC) - Part 2: Environment - Section 3: Description of the environment - Radiated and non-network-frequency-related conducted phenomena |
[17] | RICKLI H, et al., Induction ovens and electromagnetic interference: What is the risk for patients with implanted pacemakers? PACE 2002; 26:1494-1497 |
[18] | BINGGELI, et al., Induction ovens and electromagnetic interference: What is the risk for patients with implanted cardioverter defibrillators? J of Cardio Electrophysiol 2005; 16, pp.399-401 |
[19] | HIROSE, et al., Electromagnetic interference of implantable unipolar cardiac pacemakers by an induction oven. PACE 2005; 28, pp.540-548 |
[20] | ICNIRP Guidelines:1998, Guidelines for limiting exposure to time-varying electric, magnetic, and electromagnetic fields (up to 300 GHz), Health Physics Society |
[21] | IEEE C95.1:1999, IEEE Standard for Safety Levels with Respect to Human Exposure to Radio Frequency Electromagnetic Fields, 3 kHz to 300 GHz |
[22] | IEEE C95.6:2002, IEEE Standard for Safety Levels with Respect to Human Exposure to Electromagnetic Fields, 0-3 kHz |
[23] | MIL-STD-461E:1999, Test Specifications - Requirements for the control of electromagnetic interference characteristics of subsystems and equipment |
[24] | IEC/TS 61000-2-5:1995, Electromagnetic compatibility (EMC) - Part 2: Environment - Section 5: Classification of electromagnetic environments |
[25] | IEC 62226-2-1, Exposure to electric or magnetic fields in the low and intermediate frequency range - Methods for calculating the current density and internal electric field induced in the human body - Part 2-1: Exposure to magnetic fields - 2D models |
[26] | CISPR 11:2004, Industrial, scientific and medical (ISM) radio-frequency equipment - Electromagnetic disturbance characteristics - Limits and methods of measurement |
[27] | CISPR 14-1:2005, Electromagnetic compatibility - Requirements for household appliances, electric tools and similar apparatus - Part 1: Emission |
[28] | CISPR 22:2006, Information technology equipment - Radio disturbance characteristics - Limits and methods of measurement |
[29] | FCC 47 CFR, Part 15, Telecommunication - Radio frequency devices |
[30] | ISO 31 (all parts), Quantities and units |
УДК 621.65.03:006.354 | ОКС 11.040.40 | Р29 | ОКП 944480 |
Ключевые слова: имплантируемое медицинское изделие, имплантаты хирургические, имплантируемые инфузионные насосы | |||
Электронный текст документа
и сверен по:
, 2016

{kind=link}