ГОСТ Р 53918-2010
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИЗДЕЛИЯ МЕДИЦИНСКИЕ
Руководство по интеграции принципов менеджмента риска в систему менеджмента качества
Medical devices. Implementation of risk management principles and activities within a quality management system
ОКС 11.040.01
ОКП 94 4000
Дата введения 2011-11-01
Предисловие
Цели и принципы стандартизации Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "Медитест" на основе аутентичного перевода на русский язык стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством и соответствующие аспекты для медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 12 ноября 2010 г. N 383-ст
4 Настоящий стандарт идентичен международному документу Целевой группы по глобальной гармонизации (Global Harmonization Task Force - GHTF) "Внедрение принципов и видов деятельности по менеджменту риска в систему менеджмента качества" (GHTF/SG3/N15R8:2005* "Implementation of risk management principles and activities within a quality management system")
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
Введение
К изготовителям медицинских изделий часто предъявляется требование наличия систем менеджмента качества и менеджмента риска. Могут быть выбраны две отдельные системы, но рациональнее объединить их в одну, что позволит уменьшить расходы и создать более результативную систему менеджмента качества организации.
Настоящее руководство предназначено для оказания помощи изготовителям медицинских изделий на основе практических разъяснений и примеров в интеграции системы менеджмента риска (или принципов и видов деятельности в этой области) в систему менеджмента качества организации.
Настоящее руководство базируется на основных принципах систем менеджмента качества и риска, а не на частных требованиях отдельных стандартов:
- применимо к системам менеджмента качества изготовителей медицинских изделий;
- рассматривает менеджмент риска в основном с точки зрения обеспечения безопасности медицинских изделий, не учитывая финансового и других рисков;
- не предполагает наличия специального метода внедрения принципов и видов деятельности;
- не содержит требований, которые должны быть положены в основу контролирующей деятельности регулирующих органов или деятельности по сертификации.
Результативная система менеджмента качества важна для надлежащего функционирования медицинских изделий и обеспечения их безопасности, включающей разные аспекты. При приоритетном внимании к аспектам безопасности полезно идентифицировать некоторые основные виды деятельности, влияющие на применение медицинских изделий и обеспечивающие надлежащие входные данные, а также надлежащую обратную связь для системы менеджмента качества. Степень внимания к аспектам безопасности следует соотносить со степенью риска. Существуют изделия с относительно низкой степенью риска или изделия, риски применения которых хорошо изучены, в то время как другие требуют мер по управлению риском, основанных на современных достижениях науки и техники.
В некоторых регулирующих документах установлена иерархия управления риском, которую следует рассматривать в следующем порядке:
- меры, обеспечиваемые внутренней безопасностью, заложенной при проектировании;
- средства защиты, имеющиеся в изделии или использованные при его изготовлении;
- информация по безопасности, например предупреждения и т.д.
На протяжении всего жизненного цикла медицинского изделия изготовитель должен отслеживать допустимость идентифицированных рисков и отсутствие новых опасностей. Частью мониторинга может быть информация, получаемая с помощью системы менеджмента качества, например производственная, жалобы и обратная связь с потребителем. Если риск становится недопустимым, то его анализ следует повторить и предпринять действия, позволяющие достичь соответствия установленным критериям допустимости риска. При идентификации новой опасности следует выполнить заново все этапы управления риском.
Все указанные виды деятельности можно осуществлять в рамках системы менеджмента качества организации.
В рамках настоящего руководства определения:
- "должен" означает, что соответствие требованиям или тексту является обязательным;
- "следует", "рекомендуется" показывают, что соответствие требованиям или тексту не является обязательным;
- "может" используется для описания способов достижения соответствия.
1 Область применения
Настоящее руководство рассматривает принципы и возможности интеграции системы менеджмента риска в систему менеджмента качества изготовителей медицинских изделий на основе разъяснений и примеров практического применения.
Настоящее руководство предназначено для использования в сфере обращения медицинских изделий с целью обучения, но не в контролирующей деятельности регулирующих органов, при проведении сертификации или аудита на соответствие регулирующим требованиям (т.е. требованиям, установленным в регулирующих документах).
2 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
2.1 вред (harm): Физическая травма или ущерб здоровью людей или имуществу и окружающей среде (заимствовано из [1], 3.1).
2.2 опасность (hazard): Потенциальный источник вреда (заимствовано из [1], 3.5).
2.3 остаточный риск (residual risk): Риск, остающийся после выполнения защитных мер (заимствовано из [1], 3.9).
2.4 риск (risk): Сочетание вероятности причинения вреда и тяжести этого вреда (заимствовано из [1], 3.2).
2.5 анализ риска (risk analysis): Систематическое использование имеющейся информации для выявления опасностей и определения риска (заимствовано из [1], 3.10).
2.6 оценка риска (risk assessment): Полный процесс анализа и оценивания риска (заимствовано из [1], 3.12).
2.7 управление риском (risk control): Процесс принятия решений и выполнения мер по уменьшению рисков до установленных уровней или поддержания рисков на установленных уровнях (заимствовано из [2], 2.19).
2.8 оценивание риска (risk evaluation): Принятие решения на основе анализа риска и существующих общественных ценностей о возможности достижения допустимого риска в рассматриваемом случае.
Примечание - Основано на [1], 3.11 и 3.7.
2.9 менеджмент риска (risk management): Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания, управления и мониторинга риска (заимствовано из [2], 2.22).
3 Общие положения
3.1 Документация
Документы или записи, являющиеся следствием таких видов деятельности по менеджменту риска, как процедуры и отчеты, можно хранить (или ссылаться на них) в файле менеджмента риска или других соответствующих файлах (например, в файле истории изделия, техническом файле/файле технической документации, в досье на изделие, записях разработчика изделия, записях истории изделия или в файлах процесса валидации).
Изготовителю следует рассмотреть пользу от внедрения процедур, документов и записей менеджмента риска непосредственно в процедуры, документы и записи системы менеджмента качества. Главным преимуществом может стать создание единой системы управления документацией, простой в применении, легкоанализируемой, легкодоступной и удобной для хранения документов, записей и т.д. Если изготовитель принимает решение о внедрении системы менеджмента риска в систему менеджмента качества, то в файле менеджмента риска следует иметь ссылки или указатель позиций, удовлетворяющих требованиям менеджмента риска.
Рекомендуются те же средства управления документацией системы менеджмента риска, включая средства управления изменениями в документации, что и при управлении документацией системы менеджмента качества.
3.2 Внешний и внутренний обмен информацией
В рамках системы менеджмента качества необходимо на протяжении всего жизненного цикла медицинского изделия рассматривать внутренний и внешний обмен информацией, виды и глубина которого зависят от предусмотренного пользователя.
Внутренний обмен информацией требуется для того, чтобы персонал организации знал о рисках, оставшихся после выполнения мер по управлению ими (остаточных рисках). В приложении А приведена диаграмма риска для обмена информацией о внутренних видах деятельности по менеджменту риска.
Для предоставления необходимой информации о риске следует также использовать внешние методы обмена ею: предупреждающие надписи, руководства по эксплуатации, пояснительные уведомления и т.д.
4 Ответственность высшего руководства
Высшее руководство несет ответственность за внедрение менеджмента риска внутри организации. Его действия включают в себя разработку политики, обеспечивающей результативное внедрение принципов и видов деятельности по менеджменту риска.
Рекомендуется, чтобы цели, направленные на обеспечение безопасного применения медицинского изделия, стали для изготовителя главной составляющей в области качества.
Чтобы достичь указанных целей, высшему руководству следует также обеспечить выполнение плана деятельности по менеджменту риска как части плана в области качества, куда необходимо включить:
- разработку критериев допустимости риска;
- анализ риска;
- оценивание риска;
- управление риском и его мониторинг.
Высшее руководство несет ответственность за обеспечение ресурсами, достаточными для осуществления деятельности по менеджменту риска.
Высшему руководству следует также распределить ответственность и полномочия среди квалифицированного персонала организации за виды деятельности по менеджменту риска, в том числе за мониторинг производственных и постпроизводственных данных.
Изготовителю медицинских изделий необходимо планировать и проводить внутренние аудиты качества с целью верификации соответствия видов деятельности по менеджменту риска и полученных результатов запланированным и установленным процедурам. Внутренние аудиты качества гарантируют постоянную результативность применяемой системы менеджмента риска.
При анализе высшим руководством системы менеджмента качества организации следует рассматривать информацию, полученную при проведении внутренних аудитов качества, в том числе (если целесообразно) о деятельности по менеджменту риска и полученным результатам.
5 Внешние источники
Изготовитель может использовать внешние процессы (стерилизация, обработка металла, окрашивание, испытание, проектирование, изготовление) и изделия (компоненты, отдельные узлы или изделия в целом) и должен управлять ими. Изготовитель несет ответственность за осуществление соответствующих видов деятельности по менеджменту риска, относящихся к данным процессам и изделиям, посредством планирования и надлежащего выполнения мер по управлению риском. Прежде чем одобрить внешний процесс или изделие и внести какое-либо изменение в него, изготовитель должен:
- проанализировать изменение;
- оценить возможность появления новых рисков;
- определить допустимость имеющихся и/или новых отдельных остаточных рисков и/или совокупного остаточного риска в соответствии с установленными критериями допустимости риска.
При наличии мер по управлению риском, относящихся к внешним процессам или изделиям, их выполнение и степень важности следует документировать и доводить до сведения поставщика в контексте информации по закупкам.
6 Планирование
Планирование менеджмента риска должно охватывать весь жизненный цикл медицинского изделия. При этом не обязательно разрабатывать отдельный план менеджмента риска, если изготовителем надлежащим образом внедрены виды деятельности по менеджменту риска при их планировании в рамках системы менеджмента качества.
7 Проектирование и разработка
Проектирование и разработка медицинских изделий являются эволюционными процессами, включающими в себя многие технические и управленческие виды деятельности. Идентификация рисков и управление ими могут быть интегрированы в процессы проектирования и разработки и стать частью составляющих их видов деятельности. На управление рисками могут оказывать влияние технические и коммерческие факторы, а также функциональные свойства и связанная с ними клиническая эффективность медицинских изделий. Целью менеджмента риска редко является его снижение во всем объеме, скорее речь идет об уменьшении отдельных рисков до допустимого уровня при сохранении клинической эффективности и функциональных свойств медицинских изделий.
Деятельность по менеджменту риска следует начинать на самой ранней стадии этапа проектирования и разработки, когда легче предупредить возникновение проблем, чем корректировать их. Для каждой идентифицированной опасности необходимо оценивать риски как в нормальных условиях применения, так и в условиях отказа (неисправности), чтобы решить вопрос о возможности их уменьшения. Результаты оценивания риска, например необходимость выполнения мер по управлению риском, становятся затем частью входных данных для этапа проектирования.
Меры по управлению риском являются частью выходных данных этапа проектирования и оцениваются в процессе верификации проекта. Описанный цикл (получение входных/выходных данных/их верификация) повторяется на протяжении всего процесса управления проектированием медицинского изделия до тех пор, пока остаточные риски не будут уменьшены и поддержаны на допустимом уровне. Общая результативность мер по управлению рисками подтверждается при валидации проекта.
При управлении рисками недостаточно полагаться исключительно на проектирование и разработку. Даже при наличии оптимальных процессов трудно обеспечить свободные от ошибок выходные данные проектирования.
После выпуска медицинского изделия в продажу деятельность по менеджменту риска следует связать с менеджментом качества, например с управлением производственными процессами, корректирующими и предупреждающими действиями, процессами обслуживания и обратной связи с потребителями.
Управление рисками при проектировании и разработке рекомендуется поддерживать соответствующей документацией, которая должна обеспечить объективное свидетельство о соответствии характера и степени управления проектированием степени идентифицированных рисков.
Блок-схема, приведенная в приложении В, иллюстрирует внедрение видов деятельности по менеджменту риска в процессы проектирования и разработки как составляющие системы менеджмента качества организации.
7.1 Планирование проектирования и разработки
При планировании проектирования и разработки следует координировать виды деятельности по менеджменту риска при осуществлении этих процессов, а также идентифицировать:
- взаимосвязь(и) между видами деятельности по менеджменту риска, проектированию и разработке;
- необходимые ресурсы (в том числе экспертизы), требующиеся для достаточного освещения возможных проблем безопасности.
7.2 Входные данные проектирования и разработки
Входные данные проектирования и разработки медицинского изделия должны быть представлены в документах, являющихся основой дальнейших видов деятельности, и включать рассмотрение предусмотренного применения, требования к функционированию, эксплуатационным свойствам и безопасности, а также регулирующие требования.
Меры по управлению риском являются одним из видов выходных данных процесса менеджмента риска и одновременно входными данными проектирования и разработки.
Анализ риска заключается в идентификации возможных опасностей и причиняемого ими вреда, а также в оценивании рисков от причинения. Идентификация опасностей начинается с рассмотрения предусмотренного применения медицинского изделия, его характеристик и факторов окружающей обстановки. При наличии риска применения данного изделия рекомендуется рассматривать постпроизводственную информацию и информацию, относящуюся к технологиям его изготовления. Эти действия обычно заканчиваются составлением предварительного перечня известных и прогнозируемых опасностей, описанных в соответствующих стандартах или других источниках, например в базах данных системы наблюдения и оповещения об инцидентах, независимых отчетах об испытаниях изделий и т.д.
Идентифицированные опасности могут приводить к причинению вреда, являющегося следствием нескольких факторов. Необходимо определить вероятность причинения вреда и его тяжесть (см. приложение А). Риски, возникающие при наличии опасностей, оцениваются по предварительно установленным критериям допустимости для определения необходимости мер по управлению риском.
Все предлагаемые изменения идентифицированных характеристик проекта, спецификаций, а также меры по управлению риском и связанные с ними опасности, выявленные при текущем анализе риска, должны быть оценены с точки зрения постоянной безопасности и функционирования медицинского изделия до внесения изменений.
Если изделие предназначено для применения в комплекте с другим изделием или оборудованием либо монтируется или подсоединяется к нему, то опасности и меры по управлению риском следует оценивать для каждого изделия отдельно и для системы или комбинации изделий в целом.
При рассмотрении входных данных проектирования и разработки следует также учитывать необходимость мер по управлению риском. Если они признаны необходимыми и определены на начальном этапе, то становятся выходными данными как часть итеративного цикла.
7.3 Выходные данные проектирования и разработки
Меры по управлению риском, идентифицированные на начальном этапе, должны быть включены в выходные данные проектирования и разработки и оцениваться с точки зрения их осуществимости.
Эти данные обычно бывают трех типов: первого - включают в себя подробные сведения о характеристиках медицинского изделия, особенно относящихся к его безопасности и предусмотренному применению; второго - связаны с требованиями к закупкам, изготовлению, обращению, распространению и обслуживанию; третьего - определяют критерии приемки изделия. Данные всех типов могут содержать информацию, существенную для безопасности и предусмотренного применения. Меры по управлению риском могут относиться к любой из этих категорий. В таблице 1 приведены примеры мер по управлению риском для выходных данных каждого типа.
Таблица 1
Тип выходных данных проектирования и разработки | Примеры мер по управлению риском |
Характеристики медицинского изделия, включая относящиеся к его безопасному и предусмотренному применению | Соответствие требованиям [3]. Сигнал тревоги при превышении температуры. Информация, относящаяся к остаточным рискам (предупреждающая маркировка на изделии, руководство для операторов или по обслуживанию). Дополнительный источник питания на медицинском изделии, поддерживающем жизненно важные функции организма. Блокирующий переключатель на двери в рентгеновский кабинет. Контрольный таймер (в микропроцессорном изделии). Обучение пользователя |
Требования к закупкам, изготовлению, обращению, распространению и обслуживанию | Специальные требования к качеству, указанные в договоре. Обеспечение строгих средств контроля процессов. Обязательная замена отдельных частей медицинского изделия или оборудования, используемого в ходе процессов, через запланированные периоды во время профилактического обслуживания (ремонта). Ограничение количества изделий в партии. Требования к окружающей среде, например к температуре, давлению, влажности и т.д. |
Критерии приемки медицинского изделия | Спецификация момента затяжки резьбового соединения. Допустимые отклонения размеров при монтаже вакуумной магистрали (трубопровода). Уровни загрязнения или требования к стерильности для медицинского изделия или принадлежностей. Предельные технические характеристики при обеспечении электрической безопасности (например, ток утечки, прочность изоляции) |
Выходные данные проектирования должны включать в себя специальные меры по управлению риском и указания по их применению.
При проектировании и разработке, если невозможно или практически неосуществимо проектирование внутренней безопасности изделия и/или средств защиты, выходными данными могут стать дополнительные меры по управлению риском, такие как маркирование (этикетирование), обучение и информирование об остаточном риске(ах), которые следует применять в течение всего жизненного цикла медицинского изделия.
7.4 Анализ проекта и разработки
При анализе проекта и разработки необходимо определить надлежащее информирование заинтересованных лиц (включая пользователей) об отдельных остаточных рисках и о совокупном остаточном риске, а также установить обоснованность решений о соотношении риск/польза с точки зрения допустимости совокупного остаточного риска. Лица, проводящие анализ, должны быть компетентными в оценивании решений, принятых в процессе проектирования, о допустимости риска.
В процессе анализа проекта на соответствующих этапах проектирования и разработки рекомендуется оценить:
- идентификацию всех опасностей, определение рисков и идентификацию мер по управлению потенциальными рисками;
- результативность мер по управлению отдельными рисками;
- результативность определения совокупного остаточного риска, связанного с применением медицинского изделия предусмотренным пользователем, при валидации проекта;
- наличие управляемости и верификации в процессе передачи проекта новых идентифицированных выходных данных, связанных с риском.
7.5 Верификация проекта и разработки
В результате верификации проекта и разработки необходимо получить объективное доказательство того, что идентифицированные риски определены, меры по управлению ими реализованы надлежащим образом и верифицированы на результативность соответствия конечных результатов установленным критериям допустимости риска.
Следует разработать процедуры, определяющие аналитические методы и методы испытаний, обусловленные требованиями безопасности. Рекомендуется, чтобы данные процедуры обеспечивали прослеживаемость идентифицированных опасностей, мер по управлению риском, требований к проектированию и разработке медицинского изделия, планов испытаний и их результатов.
В приложении С приведена сводная таблица по менеджменту риска, демонстрирующая соответствие требованию прослеживаемости.
7.6 Валидация проекта и разработки
Валидация должна подтвердить, что медицинское изделие удовлетворяет потребностям пользователей и предусмотренному применению, а совокупный остаточный риск - всем критериям допустимости риска. Для выполнения мер по управлению риском в соответствии с планом валидации данный план должен включать в себя достаточное количество пользователей из предусмотренных групп населения и все возможные случаи предусмотренного применения, подтверждающие соответствие совокупного остаточного риска ожиданиям. Необходимо оценивать непредвиденные опасности, возникающие в процессе валидации, и управлять ими.
Примечание - Следует разработать и адресовать меры по управлению риском до проведения клинических испытаний/исследований.
7.7 Управление изменениями проекта и разработки
Кажущиеся незначительными изменения медицинского изделия и/или процессов его изготовления могут иметь непредвиденные и иногда катастрофические последствия, которые нужно оценивать с точки зрения их воздействия на безопасность на основе критериев допустимости риска, установленных в записях и документах по менеджменту риска, проектированию и разработке.
Необходимость изменений может возникнуть на любой стадии жизненного цикла медицинского изделия, некоторые из них могут инициировать новые опасности, уменьшать существующие или изменять связанный с ними уровень риска. Изменение может быть результатом многих факторов, включая меры по управлению риском или его повторное оценивание. При внедрении изменения следует проанализировать и при необходимости пересмотреть имеющуюся оценку риска.
Указанные изменения предусматривают:
- замену материала (даже идентичным по наименованию, но от другого поставщика);
- замену используемого в процессе механизма другим;
- незначительные изменения процесса, что может иметь кумулятивный эффект;
- замену поставщиков;
- изменения, внесенные поставщиками;
- изменение предусмотренного применения или предусмотренного пользователя.
Если изделие является частью системы, то при изменении любых характеристик рекомендуется заново оценивать ее как единое целое.
До внедрения предложенного изменения важно гарантировать определение и сохранение допустимости каждого остаточного риска отдельно и совокупного остаточного риска.
7.8 Передача проекта и разработки
В процессе передачи проекта изготовитель должен обеспечить выполнение и результативность установленных мер по управлению риском и решить связанные с ним имеющиеся или вновь идентифицированные проблемы до передачи проекта в производство.
8 Прослеживаемость
Данные по менеджменту риска следует использовать для определения того, какие изделия, компоненты, материалы и условия производственной среды нуждаются в прослеживаемости.
Для установления ее критериев виды деятельности по менеджменту риска рекомендуется применять во взаимодействии с регулирующими требованиями. При этом необходимо рассмотреть:
- происхождение компонентов и материалов;
- историю технологических процессов;
- распределение и местонахождение медицинского изделия после поставки (первому грузополучателю);
- предусмотренное применение изделия, например для поддержания жизни, жизнеобеспечения или имплантируемое;
- возможность отказа;
- необходимость коррекций, связанных с безопасностью (отзыв медицинского изделия, рассылка пояснительного уведомления, уточнение области применения и т.д.);
- последствия отказа изделия для пациентов, пользователей или других лиц.
Чтобы определить, какие записи необходимы для осуществления прослеживаемости, изготовителю следует рассмотреть все изделия, компоненты, материалы и условия производственной среды, которые могут стать причиной несоответствия медицинского изделия предъявляемым к нему требованиям, включая требования безопасности.
9 Управление деятельностью по закупкам и приемке продукции
9.1 Управление закупками
При выполнении видов деятельности по менеджменту риска рекомендуется идентифицировать опасности и оценить риски, в том числе потенциально возможные по вине поставщиков, на ранних этапах жизненного цикла продукции.
Частью требований к закупкам является определение ролей и ответственности изготовителя и поставщика в процессе менеджмента риска, включая предписанные меры по управлению риском в ходе процессов жизненного цикла продукции как часть информации по закупкам.
Установление критериев выбора, оценивания и повторного оценивания поставщиков закупленной продукции и услуг следует также основывать на возможности возникновения идентифицированных опасностей, связанных с этой продукцией и услугами и определенных в процессе менеджмента риска.
9.2 Виды деятельности по приемке продукции
Изготовителю необходимо осуществлять обмен информацией в области политики его организации по менеджменту риска, а также разрабатывать и выполнять процедуры для обеспечения соответствия закупленной продукции и услуг установленным требованиям. При разработке критериев для приемки закупленной продукции и услуг следует учитывать результаты деятельности по менеджменту риска. Особое внимание при установлении критериев для верификации и деятельности по приемке продукции необходимо уделять идентифицированным опасностям и связанным с ними мерам по управлению риском.
10 Управление процессами жизненного цикла продукции
Производственный процесс может быть источником идентифицированных опасностей, которые возникают из-за оборудования, производственной среды, персонала, технологических и прочих факторов или из-за внесенных в них изменений. Опасности рекомендуется идентифицировать уже при проектировании и разработке или обнаруживать в процессе производства/постпроизводства. Меры по управлению риском, необходимые для данных опасностей, должны быть включены в документированные процедуры управления жизненным циклом продукции.
Выходные данные видов деятельности по менеджменту риска могут стать входными данными для разработки соответствующих методов измерения и мониторинга процессов изготовления, что важно для понимания всем привлеченным персоналом организации значимости мер по управлению риском и их внедрения в производственные процессы с целью обеспечения результативности.
Оценка риска с использованием таких методов, как анализ опасностей в критических контрольных точках (Hazard Analysis on Critical Control Points - HACCP), исследование опасностей и эксплуатационной пригодности (Hazard and Operability Study - HAZOP), анализ "древа неисправностей" (Fault Tree Analysis - FTA), анализ характера и последствий отказов (Failure Mode and Effect Analysis - FMEA), процессно-аналитическая технология (Process Analytical Technology - PAT) и др., может помочь разработать или улучшить средства управления производственными процессами с помощью определения:
- наиболее уязвимых мест в каждой стадии процесса;
- влияния отказов на изделие;
- вероятности отказов;
- средств управления обнаружением и предупреждением отказов и их причин.
Такую производственную информацию, как процент несоответствий, процент перерабатываемых изделий и лома (утиля), производительность труда и другие характеристики качества, следует оценивать и сравнивать с имеющимися выходными данными по менеджменту риска для подтверждения соответствия и полноты управления риском.
10.1 Производственное, измерительное и контрольное оборудование
Пригодность оборудования и периодичность его очистки, профилактического ухода и калибровки необходимо рассматривать с учетом рисков, связанных с процессами.
Рабочие инструкции также следует анализировать и актуализировать с учетом соответствующих мер по управлению риском.
10.2 Производственная среда и персонал
Если воздействие производственной среды или персонала на медицинское изделие или процесс приводит к возникновению риска, то рекомендуется определять, документировать и выполнять меры по управлению им и периодически оценивать их результативность.
10.3 Валидация процессов
На валидацию процессов и определение необходимости повторной валидации могут влиять результаты видов деятельности по менеджменту риска. Валидацию процессов рекомендуется осуществлять с помощью таких средств, как FTA, FMEA, HAZOP, HACCP, PAT и др. Результаты валидации или повторной валидации могут выявить необходимость дополнительных мер по управлению риском. В качестве примера можно назвать подтверждение или усовершенствование параметров и средств управления конкретным процессом, если источником идентифицированной опасности является его изменчивость.
После внесения изменений в процесс имеющиеся меры по управлению риском следует проанализировать на пригодность, что должно гарантировать отсутствие возникновения новых опасностей.
11 Обслуживание
В контексте настоящего раздела "обслуживание" означает профилактический ремонт и уход за медицинскими изделиями. Если оно является установленным требованием, то следует учитывать информацию по менеджменту риска. Периодические обслуживание и уход как средства обеспечения безопасного функционирования изделия могут быть способом управления риском.
Если для какого-либо процесса жизненного цикла продукции требуются меры по управлению риском, то может возникнуть необходимость в таких же (или подобных) мерах в процессе обслуживания.
При наличии опасности необходимо включить четкие инструкции в руководство по эксплуатации или документацию и провести соответствующее обучение обслуживающего персонала.
12 Анализ данных
Производственную и постпроизводственную информацию о медицинских изделиях изготовителя необходимо постоянно отслеживать и анализировать при повторном оценивании риска и пересмотре имеющихся оценок для поддержания результативного процесса менеджмента риска в рабочем состоянии.
Дополнительные источники включают в себя информацию:
- об изделиях конкурентов;
- о подобных медицинских изделиях на рынке;
- опубликованную (отзывы, отчеты о медицинских изделиях, об инцидентах и т.д.);
- о научной литературе.
При анализе данных решения и меры по управлению риском, идентифицированные в процессе менеджмента риска, должны соответствовать регулирующим требованиям.
13 Корректирующие и предупреждающие действия
Рисунок 1 иллюстрирует возможности интеграции менеджмента риска в корректирующие и предупреждающие действия.
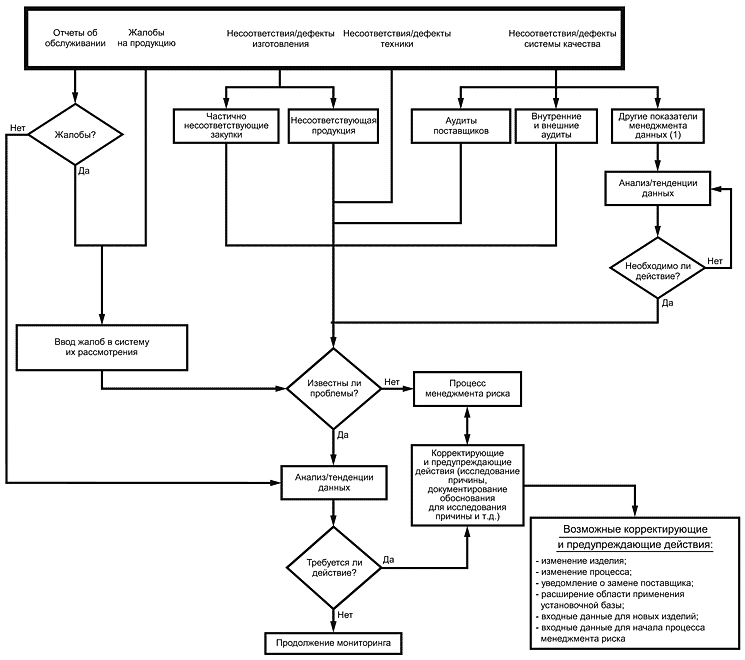
1) Такие, как возврат готовых изделий, возобновление кредита.
2) Взаимосвязь будет зависеть от выходных данных исследований (данный процесс может быть итеративным).
Рисунок 1
Анализ корректирующих и предупреждающих действий должен обнаружить все ранее не идентифицированные риски и подтвердить результативность мер по управлению ими. Данную информацию рекомендуется использовать для определения результативности видов деятельности по менеджменту риска и необходимых действий для коррекции выявленных проблем и предупреждения их повторения.
Например, отчет об обслуживании, содержащий указания на проблему, связанную с безопасностью применения медицинского изделия, должен быть доведен до сведения изготовителя и идентифицирован как жалоба, которую следует проанализировать и провести исследование. В его ходе может быть обнаружено, что в процесс изготовления были внесены изменения, возможные причины которых:
- непредвиденное воздействие на медицинское изделие;
- неадекватная оценка изменений процесса;
- ненадлежащая повторная валидация;
- отсутствие повторной валидации;
- ненадлежащие меры по управлению риском;
- отсутствие оценки мер по управлению риском после внесения изменений.
При любой комбинации указанных причин в рамках системы менеджмента риска для его повторного оценивания будет использована послепродажная информация. Глубина повторного оценивания риска будет зависеть от результатов исследования жалобы, которые следует документировать. Все новые или пересмотренные меры по управлению риском будут частью корректирующих и предупреждающих действий.
Приложение А
(справочное)
Пример использования диаграммы риска для обмена информацией по менеджменту риска внутри организации
Для проведения результативного и эффективного процесса менеджмента риска изготовитель должен постоянно идентифицировать конкретные риски при выполнении различных видов деятельности. Некоторые стандарты содержат простые и ясные описания рисков.
Например, риск можно представить в виде простой двухмерной диаграммы. При этом определяют и обосновывают идентифицированные категории тяжести и вероятности риска. Изготовитель может разработать диаграмму, представляющую три уровня риска (см. рисунок А.1).
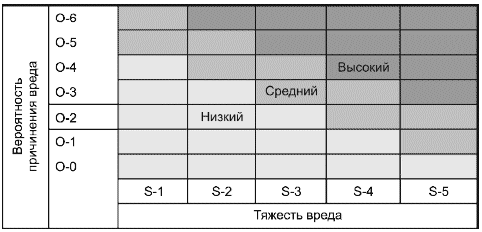
Рисунок А.1 - Диаграмма риска для обмена информацией по его менеджменту внутри организации
Комментарий к диаграмме
Тяжесть вреда | Вероятность причинения вреда | ||
S-5 | Катастрофическая | O-6 | Постоянная |
S-4 | Критическая | O-5 | Частая |
S-3 | Серьезная | O-4 | Вероятная |
S-2 | Незначительная | O-3 | Возможная |
S-1 | Пренебрежимо малая | O-2 | Отдаленная |
O-1 | Невероятная | ||
O-0 | Не наблюдалась | ||
Такая диаграмма риска может быть использована как средство обмена информацией среди персонала организации.
Приложение В
(справочное)
Блок-схема "Внедрение видов деятельности по менеджменту риска в проектирование и разработку"
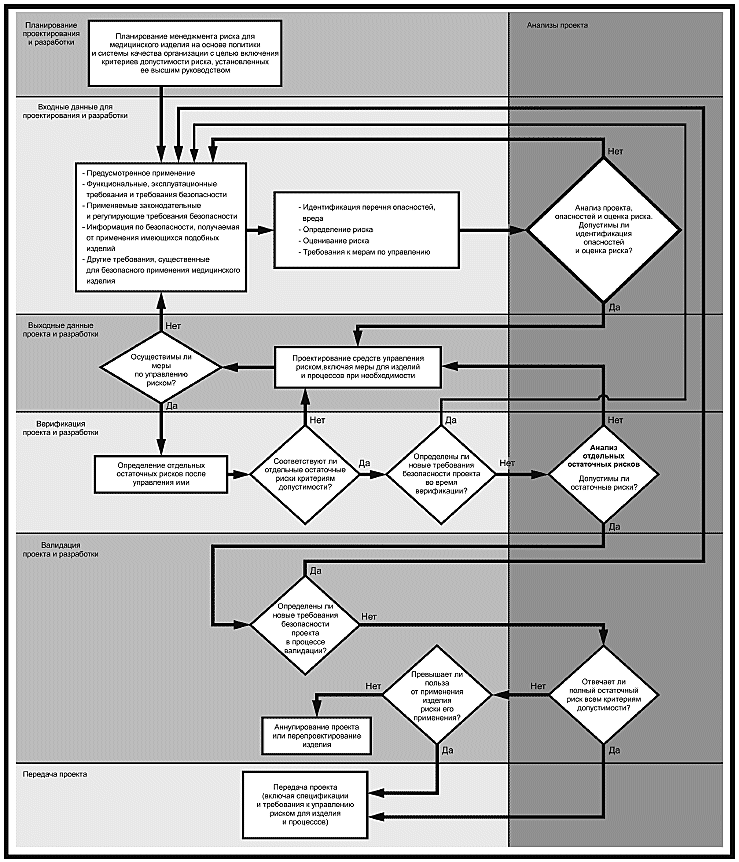
Примечание - Несмотря на невозможность продемонстрировать итеративную природу процессов с помощью приведенной блок-схемы, изготовителям следует предусмотреть обратную связь между происходящими на более ранних стадиях и последующими процессами.
Приложение С
(справочное)
Пример сводной таблицы по менеджменту риска
Ниже приведен пример документирования видов деятельности по менеджменту риска. Однако в распоряжении изготовителей имеется много разных способов их документирования или суммирования с целью осуществления прослеживаемости. Сводная таблица позволяет обоснованно и быстро идентифицировать документацию, отображающую виды деятельности по менеджменту риска.
В данном приложении представлена сводная таблица по менеджменту риска для гипотетического инфузионного насоса. В тексте после таблицы описана ее структура и расшифрованы сокращения.
Таблица С.1 - Сводная таблица по менеджменту риска
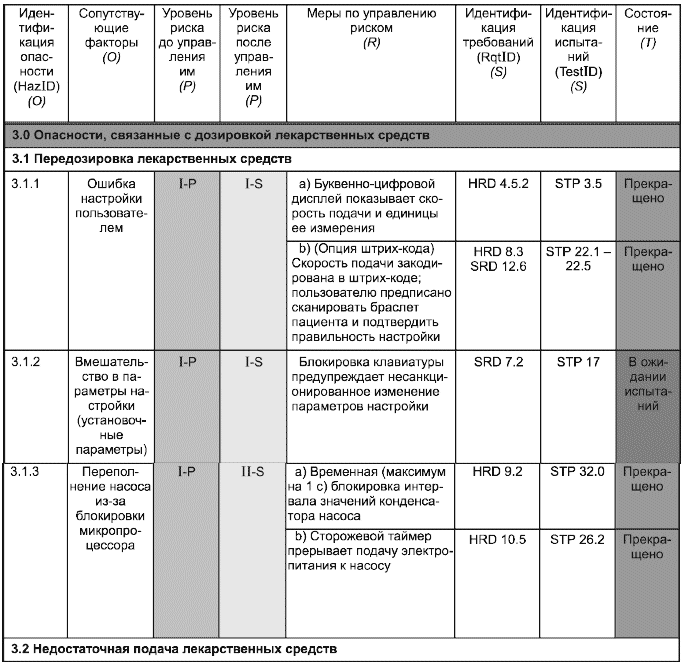
А. Идентификация опасности (HazID)
В настоящем примере (отличается от метода, приведенного в приложении А) риски идентифицируются с помощью трехуровневой иерархии (столбцы (O), верхний уровень которой отражает основные классы опасностей. Например, для инфузионного насоса - это опасности:
1.0 энергетические;
2.0 механические;
3.0 связанные с дозировкой лекарственных средств.
Второй уровень классификации идентифицирует конкретные опасности. Например, для инфузионного насоса это две опасности, связанные с дозировкой лекарственных средств:
3.1 передозировка;
3.2 недостаточная подача.
Третий уровень классификации опасностей является частным случаем сопутствующего фактора. Обычно у данной опасности может быть много причин или сопутствующих факторов, комбинации которых приводят к результатам, проиллюстрированным данным примером (с инфузионным насосом).
B. Оценивание риска
В двух столбцах (P) приведены результаты оценивания риска до и после управления им. В данном примере уровень риска характеризуется алгоритмом кодирования с использованием римских цифр и букв, обозначающих оценки тяжести и вероятности риска соответственно. Цвет или затенение ячейки отражает градацию изготовителем допустимости риска (недопустимый, нежелательный, допустимый, пренебрежимо малый). Детали схемы не важны для данного примера. Другие изготовители могут применять разные методы оценивания риска, но в любом случае полученные результаты рекомендуется суммировать в двух столбцах (P).
C. Меры по управлению риском
В столбце (R) описаны меры по управлению риском, являющиеся основой для его уменьшения. Разработанные программа действий и меры по управлению риском могут быть гораздо более сложными, чем указанные в суммирующей ячейке таблицы. В этом случае данные заносят в столбец (R) в другом документе, более подробно описывающем меры по управлению риском.
D. Данные прослеживаемости (RqtID и TestID)
В двух столбцах (S) отражена прослеживаемость мер по управлению риском, требований к проектированию медицинского изделия и видов деятельности по верификации/валидации. В столбце "Идентификация требований" (RqtID) указаны соответствующие пункты документации на проект медицинского изделия, в которых установлены требования к мерам по управлению риском, в столбце "Идентификация испытаний" (TestID) - пункты, соответствующие процедурам испытаний либо другим документам по верификации или валидации, подтверждающим надлежащее выполнение мер по управлению риском.
В данном примере "HRD" означает: "документ, содержащий требования к аппаратному обеспечению", "SRD" - "документ, содержащий требования к программному обеспечению", "STP" - "процедура испытания системы" для конкретного медицинского изделия (системы).
E. Информация о состоянии
Последний столбец (T) применяют при разработке медицинского изделия для прослеживания прогресса при выполнении видов деятельности по менеджменту риска. В данном примере цвет или затенение ячейки отражает незавершенность вида деятельности.
Пример использования таблицы - В качестве примера использования сводной таблицы по менеджменту риска рассмотрим ввод данных для идентификации опасности HazID 3.1.2, которые описывают применение блокировки клавиатуры для предупреждения внесения несанкционированных изменений в параметры настройки (установочные параметры). Механизм блокировки как мера по управлению риском позволяет уменьшить его с уровня I-P до I-S, демонстрируя снижение вероятности возникновения опасного события. Требование к блокировке клавиатуры имеется в документе по программному обеспечению (подраздел 7.2), а действие функции блокировки описано в процедуре испытания системы (раздел 17). Информация, содержащаяся в последнем столбце, показывает, что результаты этого испытания еще не получены.
Библиография
[1] | Руководство ИСО/МЭК 51:1999 | Руководящие указания по включению аспектов безопасности в стандарты |
(ISO/IEC Guide 51:1999) | (Safety aspects - Guidelines for the inclusion in standards) | |
[2] | ИСО 14971:2007 | Изделия медицинские. Применение менеджмента риска к медицинским изделиям |
(ISO 14971:2007) | (Medical devices - Application of risk management to medical devices) | |
[3] | МЭК 60601-1:2005 | Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик |
(IEC 60601-1:2005) | (Medical electrical equipment - Part 1: General requirements for basic safety and essential performance) | |
ИСО 9000:2005 | Системы менеджмента качества. Основные положения и словарь | |
(ISO 9000:2005) | (Quality management systems - Fundamentals and vocabulary) | |
ИСО 13485:2003 | Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования | |
(ISO 13485:2003) | (Medical devices - Quality management systems - Requirements for regulatory purposes) | |
МЭК 62366:2007 | Изделия медицинские. Обеспечение эксплуатационной пригодности медицинских изделий | |
(IEC 62366:2007) | (Medical devices - Application of usability engineering to medical devices) | |
МЭК 60601-1-6:2004 | Изделия медицинские электрические. Часть 1-6. Общие требования безопасности. Дополнительный стандарт Эксплуатационная пригодность | |
(IEC 60601-1-6:2004) | (Medical electrical equipment - Part 1-6: General requirements for safety - Collateral standard: Usability) | |
FDA/CDRH, опубликовано 11.01.2002 | Общие принципы валидации программного обеспечения | |
(FDA/CDRH, issued January 11, 2002) | (General Principles of Software Validation) | |
FDA/CDRH, опубликовано 09.09.1999 | Программное обеспечение внешнего происхождения в медицинских изделиях | |
(FDA/CDRH, issued September 9, 1999) | (Off-The-Shelf Software Use in Medical Devices) | |
FDA/CDRH, опубликовано в декабре 1996 | Учет человеческого фактора при проектировании медицинских изделий. Введение | |
(FDA/CDRH, issued Deсеmber 1996) | (Do It by Design - An Introduction to Human Factors in Medical Devices) | |
МЭК 62304:2006 | Программное обеспечение медицинских изделий - Процессы жизненного цикла программного обеспечения | |
(IEC 62304:2006) | (Medical device software - Software life cycle processes) | |
ИСО/ТС 19218:2005 | Изделия медицинские. Структура кодов видов и причин неблагоприятных событий | |
(ISO/TC 19218:2005) | (Medical devices - Coding structure for adverse event type and cause) |
Электронный текст документа
и сверен по:
, 2011

{kind=link}