ГОСТ Р 50326-2020/IEC/TR 60513:1994
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ОСНОВНЫЕ ПРИНЦИПЫ БЕЗОПАСНОСТИ ЭЛЕКТРИЧЕСКОГО ОБОРУДОВАНИЯ, ПРИМЕНЯЕМОГО В МЕДИЦИНСКОЙ ПРАКТИКЕ
Basic aspects of the safety philosophy of electrical equipment, used in medical practice
ОКС 11.040.01
Дата введения 2021-03-01
Предисловие
1 ПОДГОТОВЛЕН Федеральным государственным унитарным предприятием "Российский научно-технический центр информации по стандартизации, метрологии и оценке соответствия" (ФГУП "") и Обществом с ограниченной ответственностью "Медтехстандарт" (ООО "Медтехстандарт") на основе официального перевода на русский язык англоязычной версии документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 011 "Медицинские приборы, аппараты и оборудование"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 9 октября 2020 г. N 784-ст
4 Настоящий стандарт идентичен международному документу IEC/TR 60513:1994* "Фундаментальные аспекты стандартов безопасности для медицинских электрических изделий" (IEC/TR 60513:1994 "Fundamental aspects of safety standards for medical electrical equipment", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Наименование настоящего стандарта изменено относительно наименования указанного международного документа для увязки с наименованиями, принятыми в существующем комплексе национальных стандартов Российской Федерации
5 ВЗАМЕН ГОСТ Р 50326-92 (МЭК 513-76)
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
IEC/TR 60513:1994 был подготовлен подкомитетом 62А МЭК "Общие аспекты электрического оборудования, используемого в медицинской практике", Технического комитета 62 "Электрооборудование в медицинской практике".
Текст IEC/TR 60513:1994 основан на следующих документах:
Проект комитета | Отчет о голосовании |
62A(SEC)136 | 62A(SEC)148 |
Полная информация о голосовании для утверждения IEC/TR 60513:1994 приведена в отчете о голосовании, указанном в приведенной выше таблице.
Первое издание МЭК 513 "Основные аспекты философии безопасности для электрического оборудования, используемого в медицинской практике" было опубликовано в 1976 году и послужило основой для разработки:
- первого и второго изданий МЭК 601-1 (исходный стандарт безопасности для медицинских электрических изделий);
- серии дополнительных стандартов МЭК 601-1-хх для медицинских электрических изделий, и
- серии частных стандартов МЭК 601-2-хх для конкретных видов медицинских электрических изделий.
Первоначальный подход МЭК заключался в том, чтобы подготовить отдельные стандарты "безопасности" и "функциональных характеристик" для медицинских электрических изделий. Это было естественным продолжением исторического подхода, принятого на национальном и международном уровне, с другими стандартами для электрического оборудования (например, для бытового оборудования), где основная физическая безопасность регулируется обязательными стандартами, а другие технические характеристики регулируются рыночным давлением. В этом контексте было сказано: "Способность электрочайника кипятить воду не имеет решающего значения для его безопасного использования!"
В настоящее время признано, что это утверждение не относится ко многим элементам медицинских электрических изделий, и пользователи должны опираться на стандарты, чтобы обеспечить его основные функциональные характеристики, а также основную безопасность. Медицинские организации в равной степени обеспокоены способностью оборудования работать правильно и предотвращением других опасностей.
Хотя структура и содержание первого и второго изданий МЭК 601-1 были ограничены вопросами основной безопасности, растущее клиническое участие в подготовке частных стандартов подтвердило, что для многих типов медицинских электрических изделий пользователь должен зависеть от формальных стандартов для обеспечения надлежащих функциональных характеристик, а также основной безопасности. К таким областям относится точность, с которой оборудование контролирует доставку энергии или терапевтических веществ пациенту, или обрабатывает и отображает физиологические данные, которые влияют на ведение пациента.
Исходя из признания того, что разделение "безопасности" и "функциональных характеристик" несколько неуместно при устранении опасностей, возникающих в результате несовершенной конструкции медицинского электрического изделия, многие частные стандарты в серии МЭК 601-2-xx охватывают ряд основных требований к основным функциональным характеристикам, которые не могут быть непосредственно оценены пользователем без применения таких стандартов. Тем не менее текущая серия МЭК 601 включает гораздо меньше требований к функциональной безопасности, чем к основной безопасности.
Также существует и получает все большую поддержку мнение о том, что все требования к безопасности и основным функциональным характеристикам для медицинских электрических изделий должны быть сгруппированы в рамках одного набора международных стандартов. Предложенная европейская директива о медицинских изделиях также подчеркивает необходимость единой серии стандартов, охватывающих основные требования для всех таких продуктов.
История первого издания МЭК 513 и серии МЭК 601 приведена в приложении Е.
Предполагается, что второе издание МЭК 513 послужит руководством для разработки третьего издания МЭК 601-1 и последующей разработки стандартов МЭК 601-1-xx и МЭК 601-2-xx.
Для достижения согласованности с международными стандартами, учета текущих ожиданий в сообществе здравоохранения и согласования с разработками в МЭК 601-2-xx второе издание МЭК 513 включает в себя два основных изменения:
- первое изменение состоит в том, что понятие "безопасность" было расширено по сравнению с простыми, основными положениями безопасности в первом и втором изданиях МЭК 601-1 путем включения в него основных функциональных характеристик (например, точности оборудования для физиологического мониторинга);
- второе изменение заключается в том, что при определении минимальных требований безопасности предусматривается оценка соответствия процесса проектирования, которая позволит обеспечить надлежащую альтернативу лабораторным испытаниям с конкретными критериями "годен/не годен" (например, при оценке безопасности таких новых технологий, как программируемые электронные системы).
Второе издание отменяет и заменяет первое издание, опубликованное в 1976 году.
1 Область применения
В настоящем стандарте приведены основные положения, которые необходимо учитывать при разработке стандартов для обеспечения безопасности медицинских электрических изделий.
Примечание - Настоящий стандарт полностью следует рекомендациям Руководства ИСО/МЭК 51 и расширяет вопросы, которые являются уникальными или имеют решающее значение для применения медицинских электрических изделий.
________________
Руководство ИСО/МЭК 51:1990 "Руководство по включению вопросов безопасности в стандарты".
Настоящий стандарт в первую очередь предназначен для использования:
- теми, кто разрабатывает стандарты на проектирование, установку и использование медицинских электрических изделий;
- регулирующими органами здравоохранения, испытательными центрами и другими организациями, ответственными за внедрение стандартов для медицинских электрических изделий;
- изготовителями медицинских электрических изделий;
- теми, кто собирает системы, включающие медицинские электрические изделия.
Настоящий стандарт также может помочь:
- пользователям или операторам медицинских электрических изделий;
- администраторам больниц;
- нетехническому персоналу, занимающемуся закупкой медицинских электрических изделий;
- преподавателям и студентам;
- больничным и клиническим инженерам, отвечающим за строительство медицинских организаций или за закупку или обслуживание медицинских электрических изделий.
2 Понятия
Следующий материал не предназначен для введения новых определений. Это общее обсуждение терминологии и понятий, используемых в настоящем стандарте. Если конкретный термин определен в международной публикации, определение воспроизводится после соответствующего обсуждения в настоящем стандарте.
2.1 Основная безопасность
Основная безопасность обеспечивает защиту от прямых физических опасностей, когда медицинское электрическое изделие используется в нормальных или других достаточно предсказуемых условиях (например, механическая прочность, ток утечки и пожарная безопасность).
2.2 Эффективность
Эффективность элемента медицинского электрического изделия заключается в его способности выполнять предусмотренные функции (например, точность доставленной энергии от кардиодефибриллятора).
Этот термин определен в IEV следующим образом:
________________
МЭК 50(191):1990 "Международный электротехнический словарь. Глава 191. Надежность и качество обслуживания" (191-02-01).
Эффективность (результативность): способность элемента удовлетворять потребность в заданных количественных характеристиках.
2.3 Действенность
Действенность элемента медицинского электрического изделия заключается в его способности достигать ожидаемый результат в ходе диагностики или лечения (например, способность кардиодефибриллятора обнаруживать фибрилляцию).
2.4 Основные функциональные характеристики
Эта концепция менее понятна, чем основная безопасность. Многие элементы медицинского электрического изделия жизненно необходимы или имеют решающее значение для правильной диагностики. Медицинское электрическое изделие, не работающее надлежащим образом, может представлять неприемлемый риск, и, следовательно, оно может быть небезопасным. Например, большинство людей считают небезопасным дефибриллятор, который не разряжается. Кроме того, неправильный диагноз в результате неисправности оборудования может привести к неправильному лечению.
Для медицинских электрических изделий различие между стандартами безопасности и стандартами на функциональные характеристики часто неясно, и возникает концепция основных функциональных характеристик.
Как правило, основные функциональные характеристики ограничивают или устанавливают критические параметры, такие как частотная характеристика диагностического электрокардиографа и выходная энергия кардиодефибриллятора. Тем не менее в некоторых случаях основные функциональные характеристики лучше всего рассматривать с помощью требований к раскрытию информации.
2.5 Основные требования
Невозможно провести полное различие между безопасностью, эффективностью и действенностью медицинского электрического изделия. Успешная диагностика и лечение, и даже жизнь пациента могут быть поставлены под угрозу устройством, которое:
a) физически опасно;
b) не работает в соответствии с указаниями изготовителя; или же
c) неправильно используется.
Соответственно, в настоящем стандарте понятие "основные требования" определяются как сочетание понятий "основная безопасность" (см. 2.1) и "основные функциональные характеристики" (см. 2.4).
2.6 Пригодность для использования
Некоторые аспекты производительности, не влияющие на основные функциональные характеристики, могут определять пригодность для использования. Пригодность для использования может включать вопросы, касающиеся эффективности или надежности, и требовать раскрытия достаточного количества информации, чтобы позволить потребителю оценить пригодность конструкции для предполагаемого применения.
2.7 Опасность
Обычно слово "опасность" относится к источнику риска или угрозы. В настоящем стандарте слово "опасность", как правило, используется для описания ситуации потенциального вреда людям или имуществу (то есть состояния, ситуации, процедуры, объекта или материала, способного причинить вред).
Опасность определяется в Руководстве ИСО/МЭК 51 как: "Потенциальный источник вреда".
2.8 Обеспечение качества
Обеспечение качества (QA) включает в себя надлежащую производственную практику (GMP) и контроль качества (QC).
Обеспечение качества определено в ИСО 8402 как: "Все запланированные и систематические действия, необходимые для обеспечения достаточной уверенности в том, что продукт или услуга будут соответствовать заданным требованиям к качеству".
________________
ИСО 8402:1986 "Качество. Словарь".
Контроль качества определен в ИСО 8402 как: "Операционные методы и действия, которые используются для выполнения требований к качеству".
________________
ИСО 8402:1986 "Качество. Словарь".
2.9 Риск
Применение медицинского электрического изделия может привести к "прямым" рискам, когда несовершенная конструкция, отказ или неправильное использование оборудования приведут к физическому ущербу для пациента, оператора, оборудования или окружающей среды. Другие "косвенные" риски, возникающие из-за некорректной работы оборудования, могут отрицательно повлиять на пациента (например, выход из строя систем жизнеобеспечения и неправильная диагностика).
Риск определяется в Руководстве ИСО/МЭК 51 как: "Вероятная частота возникновения опасности, вызывающей вред, и степень серьезности ущерба".
2.10 Уровень риска
Уровень риска является качественной мерой риска (см. таблицы 1 и 2).
2.11 Безопасность
Обычно слово "безопасность" означает свободу от опасности или риска. Абсолютная безопасность - это полная свобода от условий и обстоятельств, которые могут причинить вред.
Нецелесообразно ожидать абсолютной безопасности при использовании медицинского электрического изделия. В настоящем стандарте слово "безопасность" обычно относится к свободе от недопустимого риска.
"Безопасность" использовалась в первом и втором изданиях МЭК 601-1 для обозначения "основной" безопасности, связанной с "прямыми" рисками, когда несовершенная конструкция или неправильное использование, или отказ оборудования приводят к физическому ущербу для пациента, оператора, оборудования или окружающей среды.
Тем не менее "безопасность" также используется в настоящем стандарте в связи с более широким спектром "косвенных" рисков, при которых пациент может пострадать из-за неисправности оборудования или оборудования, которое не подходит для предусмотренного назначения. Предполагается, что "безопасность" будет использоваться в этом более широком смысле в будущих изданиях МЭК 601-1.
Безопасность определяется в Руководстве ИСО/МЭК 51 следующим образом: "Свобода от недопустимого риска причинения вреда" (см. также пояснительные примечания в Руководстве ИСО/МЭК 51).
2.12 Уровень целостности безопасности
Уровень целостности безопасности - это мера уровня уверенности в том, что компонент или система будут функционировать должным образом или, в случае неисправности, будут работать безопасным образом. Целостность связана с опасными неисправностями и учитывает нестохастические аспекты, такие как ошибки проектирования, и стохастические аспекты, такие как отказы компонентов.
2.13 Уровень серьезности (опасности)
Уровень серьезности является качественным показателем возможных последствий опасного события. В случае медицинского электрического изделия, как правило, нет необходимости различать более четырех уровней:
- катастрофический, если он может привести к гибели множества людей с серьезными травмами;
- критический, если он может привести к смерти или серьезным травмам;
- предельный, если он может привести к травме; а также
- незначительный, когда потенциальная возможность получения травмы мала или отсутствует.
2.14 Пользователь/оператор
Эти термины определены в МЭК 601-1:1988 следующим образом, чтобы различать лицо, которое (или организацию, которая) несет ответственность за оборудование, и лицо, которое фактически использует его во время нормальной работы:
- ПОЛЬЗОВАТЕЛЬ - авторитетный специалист, ответственный за использование и техническое обслуживание ОБОРУДОВАНИЯ (пункт 2.12.13);
- ОПЕРАТОР - лицо, управляющее ОБОРУДОВАНИЕМ (пункт 2.12.17).
3 Опасность
3.1 Причины опасностей
Применение медицинского электрического изделия может представлять опасность из-за ряда причин, включая следующие:
a) неспособность устройства выполнять предназначенную ему функцию (например, неспособность аппарата искусственной вентиляции легких вентилировать пациента, неспособность монитора апноэ выдать сигнал тревоги);
b) неправильная функция (например, чрезмерная доставка лекарств инфузионным насосом, чрезмерная температура в детском инкубаторе, неточное измерение физиологического параметра);
c) энергии, доставляемые при нормальном функционировании, например:
- ток утечки или функциональный ток, протекающий от кардиодефибриллятора или от блока высокочастотного хирургического оборудования по непредусмотренным путям у пациента или оператора;
- непреднамеренное облучение пациента или оператора;
- воздействие на пациента или оператора ультразвуковой энергией или ускоренными элементарными частицами;
- чрезмерный нагрев или охлаждение пациента;
d) неисправности оборудования, например:
- пожар, поражение электрическим током, взрыв, отказ деталей;
- чрезмерное ионизирующее или неионизирующее излучение в результате неисправности оборудования, утечки или чрезмерного воздействия;
- чрезмерные температуры контактных поверхностей, приводящие к ожогам;
e) пожар или взрыв в результате воспламенения горючего материала в непосредственной близости от медицинского электрического изделия;
f) механические неисправности в нормальных условиях и условиях нарушения;
g) неправильная установка медицинского электрического изделия, например:
- ненадлежащее заземление элемента медицинского электрического изделия класса I;
- наличие опасных поверхностей, углов или ребер;
- физическая нестабильность;
h) неправильный выбор медицинского электрического изделия (например, использование медицинского электрического изделия, имеющего рабочую часть типа BF или B для проведения внутрисердечной процедуры);
i) неправильное использование медицинского электрического изделия (например, выбор неправильной шкалы энергии при использовании имплантируемого кардиодефибриллятора);
j) электромагнитные помехи (например, помехи дисплея ЭКГ из-за высокочастотного хирургического оборудования, генерация помех соседнему медицинскому электрическому изделию под действием сильных магнитных полей, излучаемых дисплеем);
k) выделение едких, ядовитых или горячих жидкостей или газов или контакт с биологически небезопасными материалами;
l) удаление материалов и побочных продуктов, возникающих в результате использования медицинского электрического изделия (например, удаление радиоактивных веществ, используемых в ядерной медицине).
3.2 Опасности, связанные с пациентом
Некоторые опасности характерны для медицинского электрического изделия и возникают или усугубляются следующими обстоятельствами:
a) неспособностью пациента или оператора обнаружить наличие потенциально опасных факторов, таких как ионизирующее или высокочастотное излучение;
b) отсутствием у пациента, который может быть без сознания, под наркозом, под действием мышечно-расслабляющих препаратов, иммобилизованных и т.д. нормальной реакции;
c) отсутствием защиты от электрического тока, обычно обеспечиваемой сопротивлением кожи пациента, когда кожа специально обрабатывается для получения низкого сопротивления проникновению;
d) применением электрических цепей через соединения с низким импедансом к пациенту. Эта опасность существует при достаточно низких уровнях тока, когда оборудование подключено непосредственно к сердцу пациента;
e) осаждением ионов металлов в кожу пациента при прохождении постоянного тока или тока, имеющего постоянный компонент, который может привести к некрозу ткани даже при достаточно низких уровнях тока, если ток протекает в течение более чем ограниченного времени;
f) ненадежностью или отказом оборудования, используемого для поддержки или замены жизненно важных функций организма;
g) одновременным подключением более одного элемента медицинского электрического изделия к пациенту;
h) комбинацией высокочастотного оборудования и оборудования с низким уровнем сигнала, часто в специальных комбинациях;
i) условиями окружающей среды в зонах лечения пациентов, которые могут представлять собой комбинацию влажности, увлажнения и/или опасности пожара или взрыва, вызванной воздухом, кислородом или закисью азота, в сочетании с анестезирующими средами и чистящими средствами.
3.3 Опасности оператора
Операторы медицинских электрических изделий могут подвергаться опасности от воздействия веществ, энергий и ионизирующего или неионизирующего излучения, которые превышают уровни, разрешенные для основной части населения, например:
a) работники в таких областях, как ядерная медицина, радиология и радиотерапия, могут подвергаться воздействию уровней радиации, превышающих уровни, разрешенные для основной части населения;
b) многие работники здравоохранения подвергаются риску поражения электрическим током или ожога от выхода высокого напряжения оборудования, такого как кардиодефибрилляторы и высокочастотное хирургическое оборудование, в то время как пользователи большинства электрооборудования общего назначения защищены от воздействия напряжений выше сверхнизкого напряжения.
Однако эти уровни все же должны быть ограничены приемлемыми пределами, которые должны быть настолько низкими, насколько это разумно достижимо, и иногда должны быть ограничены более низкими пределами, чем допустимые для пациента.
4 Факторы, влияющие на безопасность
Безопасное использование медицинского электрического изделия зависит от ряда факторов, в том числе:
a) конструкции оборудования, которая должна учитывать и включать средства предотвращения опасностей;
b) соответствующей проверки дизайна, как аппаратного, так и программного обеспечения, до начала производства оборудования;
c) применения надлежащих производственных практик при производстве оборудования;
d) выбора соответствующего оборудования для конкретной процедуры;
e) понимания оператором оборудования и его применения, которое может зависеть от обучения или от маркировки оборудования и инструкций изготовителя;
f) использования принадлежностей, совместимых с оборудованием;
g) подключения оборудования к соответствующим источникам (например, к электросети, линиям сжатого газа);
h) профилактического технического обслуживания оборудования;
i) использования предусмотренных запасных частей при ремонте оборудования.
5 Меры по обеспечению безопасности
Безопасность медицинского электрического изделия часто требует комплексного подхода, при котором изготовители и пользователи применяют комплекс мер, включающий:
a) требования, включенные в конструкцию оборудования;
b) дополнительные меры, такие как требования к установке, официальному вводу в эксплуатацию, повседневному обслуживанию и испытаниям на безопасность; а также
c) меры, требующие, чтобы пользователи знали о необходимости принятия особых мер предосторожности при использовании некоторых видов медицинских электрических изделий или при выполнении определенных процедур.
Для достижения общей безопасности следует рассмотреть вопрос о необходимости стандартов в каждой из вышеуказанных областей.
6 Цель стандартов
Целью стандартов безопасности и основных функциональных характеристик для медицинских электрических изделий является повышение безопасности при использовании такого оборудования. Эта цель должна быть достигнута путем разработки стандартов, которые помогают:
a) изготовителям при проектировании и сборке безопасных и эффективных продуктов;
b) изготовителям, испытательным лабораториям и регулирующим органам при оценке соответствия требованиям; а также
c) медицинским работникам в управлении рисками, связанными с использованием этих продуктов.
7 Типы стандартов
Стандарты безопасности и основных функциональных характеристик для медицинских электрических изделий могут относиться к одной или нескольким категориям в соответствии с 19.4.
7.1 Стандарты на продукцию
Эти стандарты относятся к конкретному продукту или группе продуктов и включают в себя:
a) стандарты безопасности, включающие требования безопасности, которые необходимы и применимы к предполагаемому использованию продукта (например, МЭК 601-1 и связанные с ним дополнительные и частные стандарты для медицинских электрических изделий);
b) стандарты на основные функциональные характеристики, которые включают требования, которые:
- необходимы для обеспечения эффективной работы продукта;
- необходимы для многих пользователей, которые не смогут идентифицировать или указать их без ссылки на соответствующий стандарт или оценить их без специализированных лабораторных средств; а также
- могут быть определены в полностью проверяемых технических условиях посредством воспроизводимых испытаний на соответствие или оценки процесса проектирования.
Примечание - Для медицинских электрических изделий требования, описанные в а) и b), часто публикуются как часть одного и того же стандарта;
c) стандарты на неосновные функциональные характеристики, включающие вопросы, которые не попадают в область, описанную в а) или b). Эти стандарты включают требования к производительности или методы испытаний, которые помогают изготовителю или пользователю определить пригодность для использования.
Могут быть разработаны стандарты раскрытия информации, в том числе связанные методы испытаний, где должно быть стандартизировано соблюдение промаркированных или заявленных характеристик.
7.2 Стандарты на процессы
В эту категорию входит ряд типов стандартов, в том числе:
a) стандарты обеспечения качества (QA) [например, стандарты надлежащей производственной практики (GMP) и контроля качества (QC), которые являются частью системы качества и применяются там, где критически важно поддерживать утвержденные типовые требования или уникальные рабочие характеристики процесса];
b) другие стандарты, которые требуют оценки "доказательств процесса" (например, стандарты для программируемых электронных систем, стерильности и эргономики).
7.3 Инсталляционные и экологические стандарты
Эти стандарты включают в себя:
a) стандарты конструкции и монтажа (например, экранирование от рентгеновских лучей, правила электропроводки);
b) системные стандарты (например, критические требования для правильного сопряжения и взаимодействия), такие как:
- МЭК 601-1-1, дополнительный стандарт для медицинских электрических систем;
- стандарты для компьютерных систем архивирования медицинских изображений;
- стандарты для медицинских информационных систем.
c) стандарты ввода в эксплуатацию, где безопасность может быть улучшена путем проведения оценки конструкции или проверки правильности установки и настройки оборудования непосредственно перед первым использованием;
d) стандарты, ограничивающие вероятное влияние оборудования на окружающую среду или влияние внешних воздействий на оборудование (например, стандарты, касающиеся электромагнитной совместимости).
7.4 Стандарты применения
Эти стандарты включают:
a) стандарты на регулярные испытания в процессе эксплуатации, где безопасность может быть поставлена под угрозу из-за износа оборудования или условий инсталляции (например, предлагаемые стандарты МЭК для испытаний в процессе эксплуатации для кардиодефибрилляторов и высокочастотного хирургического оборудования);
b) стандарты постоянства и калибровки, где безопасность зависит от подтвержденного функционирования и точности оборудования;
c) пояснительные руководящие принципы, предоставляющие информацию об опасностях, связанных с конкретными типами оборудования и процедур, вместе с соответствующими рекомендациями по безопасности;
d) руководства пользователя, содержащие информацию о системах классификации безопасности, используемых в соответствующих стандартах на продукцию.
8 Приемлемые уровни риска
Руководство ИСО/МЭК 51 устанавливает, что безопасность - это баланс между свободой от риска и другими требованиями к продукту, процессу или услуге, включая полезность, пригодность и стоимость. Безопасность, определяемая как свобода от какого-либо риска, не может быть абсолютной при использовании медицинского электрического изделия; его применение может быть только относительно безопасным или относительно небезопасным. Требования общества и, следовательно, приемлемая степень безопасности или риска подвержены социальным изменениям.
Очень немногие вещи абсолютно безопасны и предполагают полное отсутствие риска. Реалистичное ожидание должно состоять в том, чтобы риски были бы как можно более низкими, принимая во внимание соотношения затрат, которые будут понесены при последовательном снижении риска, и выгод, получаемых от использования продукта, процесса или услуги.
Общество (включая пользователей медицинских электрических изделий, пациентов, изготовителей, регулирующие органы и общественность) может желать или даже ожидать, что медицинское электрическое изделие будет физически безопасным, работать в соответствии с указаниями и давать соответствующие результаты. Однако при разработке соответствующих стандартов на продукты баланс между риском и затратами должен учитывать неблагоприятное влияние отказа от использования продукта. Медицинское изделие, которое спасает тысячи жизней и ранит одного человека, может быть предпочтительнее устройства, которое спасает сотни жизней и никого не ранит. Связанное с этим соображение - это разница (которую нелегко оценить) между, с одной стороны, травмирующим воздействием на пациента и, с другой стороны, неспособностью спасти его жизнь.
Одной из функций стандартов на медицинские электрические изделия является определение приемлемого уровня риска при надлежащем использовании или даже при неправильном использовании оборудования. В этом отношении конструкция и оценка безопасности продукта зависят от четырех дополнительных действий:
a) определения опасностей, которые необходимо учитывать;
b) оценки рисков, связанных с этими опасностями;
c) установления требуемого уровня безопасности и получения согласия по приемлемым уровням риска;
d) определения требований безопасности для устранения или минимизации этих опасностей и достижения приемлемого уровня риска.
В зависимости от обстоятельств, эти действия могут выполняться одним и тем же человеком или разными людьми, в одно и то же время.
Все эти действия осуществляются при разработке стандартов на продукцию. Тем не менее разработчик или оценщик продукта также должен будет выполнить дополнительную итерацию, чтобы гарантировать, что продукт не представляет дополнительных опасностей, помимо тех, которые описаны в стандарте. Действие с) также является политической и социальной деятельностью, затрагивающей широкий круг интересов.
Поэтому в стандартах на продукцию должны быть указаны требования и испытания, которые соответствующие организации могут использовать для оценки готового продукта или процессов, с помощью которых продукт разработан и изготовлен. Однако зачастую изготовителям разрешается принимать решения, отличные от тех, которые указаны в стандарте, при условии, что они могут продемонстрировать, что достигается эквивалентный уровень безопасности. Рисунок 1 представляет собой блок-схему для определения риска, установления приемлемых уровней риска и разработки требований к безопасности и основным функциональным характеристикам.
|
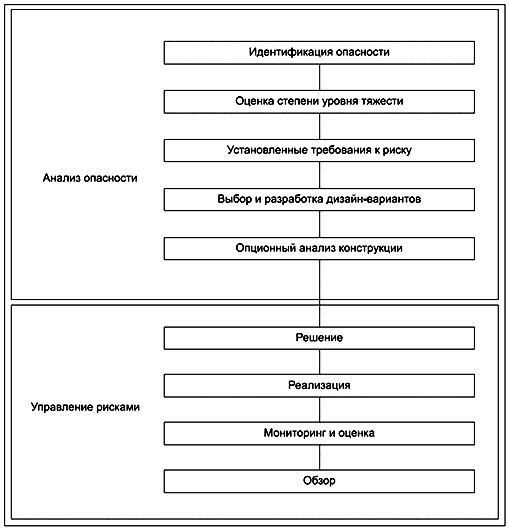
Рисунок 1 - Блок-схема управления рисками
9 Разработка требований к безопасности и основным функциональным характеристикам
9.1 Идентификация опасности
Прежде чем разрабатывать требования к безопасности и основным функциональным характеристикам, необходимо определить опасности, связанные с конкретным видом оборудования. Конкретный элемент медицинского электрического изделия может вызывать или способствовать возникновению различных опасностей (см. 3.1). В МЭК 601-1 также перечислены опасности в соответствии с рядом категорий, которые следует использовать в качестве основы для анализа рисков для каждого конкретного вида оборудования.
Важной функцией частного стандарта является перечисление опасностей, характерных для конкретного вида оборудования, которые необходимо избегать или минимизировать, что позволяет более точно определить общие опасности, которые рассматриваются в МЭК 601-1.
Например, пункт 51 второго издания МЭК 601-1 определяет необходимость защиты от опасных выходов. Однако общий стандарт определяет только общие требования, и конкретное изделие может иметь несколько важных выходных характеристик. Например, производительность инфузионного насоса - это скорость доставки раствора, и любая из следующих конкретных характеристик выхода может вызвать опасность:
a) ошибки в средней скорости относительно установленной скорости при нормальной работе;
b) изменения мгновенной скорости во времени при нормальной работе;
c) ошибки в общем доставленном объеме, с или без включения сигнала тревоги, в случае ошибки.
Как правило, использование новой технологии (например, используемой в программируемых электронных системах) не влияет на диапазон опасностей, связанных с конкретным видом оборудования, хотя может привести к появлению дополнительных механизмов отказа, которые приводят к тем же опасностям. Соответствующие требования к новым технологиям обычно связаны с необходимыми характеристиками, а не с защитой от основных угроз безопасности, таких как пожар и поражение электрическим током.
9.2 Уровень серьезности
Другой функцией частного стандарта является оценка каждой выявленной опасности для определения уровня ее серьезности.
Даже если приемлемый риск не может быть точно определен, процесс систематического перечисления опасностей и определения уровня их серьезности гарантирует, что каждая опасность получает соответствующую степень рассмотрения при разработке продукта.
Может быть несколько способов возникновения одной и той же опасности с разными уровнями тревог. Например, потенциальной опасностью в инфузионном насосе может быть невозможность прекратить инфузию, когда будет доставлен желаемый объем. Это может произойти из-за неисправности насоса или из-за ошибки оператора при настройке желаемого объема. Уровень серьезности одинаков в обоих случаях, но меры по борьбе с риском и эффективность этих мер могут различаться. Вероятность доставки чрезмерного объема из-за неисправности оборудования должна быть низкой, поскольку эту проблему можно решить при проектировании. Однако лучшее, что можно сделать, чтобы избежать ошибки оператора, может потребовать, чтобы оборудование четко указывало установленный объем.
Серьезность опасности может зависеть не только от конструкции оборудования. Например, влияние на пациента чрезмерной скорости потока из инфузионного насоса зависит от того, какая жидкость подается, как долго сохраняется избыточная скорость и каково состояние здоровья пациента. Таким образом, одна и та же опасность может иметь разные уровни серьезности при разных обстоятельствах. Если обстоятельства непредсказуемы, уровень серьезности должен быть определен из худшего вероятного случая.
9.3 Выбор приемлемых уровней риска
Риск - это сочетание частоты и вероятности (в течение определенного периода или при определенных обстоятельствах) и, как следствие, конкретной опасности. Эта концепция всегда имеет два элемента: частоту или вероятность возникновения опасности и последствия (серьезность) опасности.
Хотя безопасность широко упоминается при обсуждении медицинского электрического изделия, важно помнить, что абсолютная безопасность - это полное отсутствие риска. На практике нулевой риск недостижим. Каждый уровень риска должен быть сделан достаточно низким, чтобы быть приемлемым с учетом всех обстоятельств.
Почти любую конструкцию оборудования можно изменить, чтобы сделать ее более безопасной, например, за счет более жесткой конструкции, дополнительных систем безопасности или контроля, или дополнительных процедур производства/испытаний. Однако такие улучшения могут повлечь за собой дополнительные расходы или потерю полезности. Поэтому для каждой опасности необходимо решить, является ли риск приемлемым; то есть приемлема ли комбинация серьезности опасности и вероятности ее возникновения.
Технический комитет МЭК 62 и его подкомитеты не привыкли определять приемлемые уровни риска как таковые, и концепция приемлемого риска может быть незнакомой и даже нежелательной. Тем не менее эта концепция подразумевается в согласованных стандартах, таких как МЭК 601-1. (Одной из областей, где были определены некоторые риски, связанные с медицинским оборудованием, является радиационная защита).
Испытания, которые обычно применяются при принятии решения о приемлемости риска, включают определение:
a) настолько велик риск или настолько неприемлем результат, что от него следует отказаться вообще; или же
b) является ли или был ли сделан риск настолько малым, что является незначительным; или же
c) риск попадает между а) и b), и он был снижен до минимально возможного уровня с учетом выгод, вытекающих из его принятия, и с учетом затрат на любое дальнейшее снижение.
Риски должны быть снижены до уровня, который является настолько низким, насколько это практически возможно (принцип "ALARP"). Если риск попадает между двумя крайностями, то есть "недопустимым" и "незначительным", и применяется принцип "ALARP", результирующий риск приемлем для этого конкретного применения.
Хотя основными соображениями при определении приемлемых уровней риска являются серьезность и вероятность, необходимо учитывать и другие факторы, такие как:
a) частоту ожидаемых предпосылок для возникновения опасности (например, частоту использования оборудования или количество пациентов, которых им лечат);
b) целесообразность дальнейшего улучшения;
c) стоимость дальнейшего улучшения;
d) клинические ограничения;
e) преимущества, связанные с использованием устройства;
f) общественное восприятие.
В отличие от некоторых других отраслей промышленности, не существует общепринятой системы классификации различных опасностей, которые может представлять медицинское электрическое изделие. Например, мнения могут отличаться относительно серьезности следующего:
a) неспособности оборудования для мониторинга пациента подавать звуковой сигнал, когда это необходимо;
b) неспособности оборудования для лучевой терапии доставить правильную дозу облучения;
c) неспособности инфузионного устройства доставить правильное количество лекарства.
Следовательно, еще одной функцией частного стандарта является определение максимально приемлемой вероятности для каждой опасности. При разработке стандарта необходимо принять отдельное решение об уровне серьезности для каждой из общих опасностей для конкретного вида оборудования.
Если вероятность отказов, приводящих к опасности, известна или может быть оценена приблизительно, в качестве отправной точки в оценке риска могут использоваться таблицы 1 и 2.
Эти таблицы были адаптированы для медицинских электрических изделий из документа, который в настоящее время рассматривается SC 65A. Однако несмотря на включение их в первый столбец таблицы 1, катастрофические последствия редко бывают приемлемы для медицинских электрических изделий.
________________
Документ 65A (Секретариат) 123 находится на рассмотрении.
Количество внимания, которое следует уделять оценке рисков, зависит от количества продуктов определенного вида в обслуживании. Однако это число не должно влиять на соотношение рисков и выгод. Если количество единиц больше, риски больше, но выгоды также.
Таблица 1 - Классификация рисков несчастных случаев
Событие | Последствия | |||
Катастрофические | Критические | Предельные | Незначительные | |
Частое | I | I | I | II |
Вероятное | I | I | II | III |
Случайное | I | II | III | III |
Маловероятное | II | III | III | IV |
Невероятное | III | III | IV | IV |
Невозможное | IV | IV | IV | IV |
Таблица 2 - Интерпретация уровней риска
Уровень риска | Интерпретация |
I | Недопустимый риск |
II | Нежелательный риск, допустимый только в том случае, если его снижение нецелесообразно или если затраты на это чрезвычайно несоразмерны получаемому улучшению |
III | Допустимый риск, если стоимость снижения риска превышает получаемое улучшение |
IV | Незначительный риск |
Соотношение затрат и выгод различается для медицинских электрических изделий и большинства других видов оборудования. Может быть лучше успешно лечить большое количество пациентов, чем делать оборудование более дорогим и лечить меньше пациентов. Такие вопросы должны обсуждаться и решаться на основе консенсуса среди всех заинтересованных сторон (изготовителей, пользователей, регулирующих органов и т.д.). Комитет по разработке стандартов, в котором представлены все интересы, является подходящим форумом для определения такого консенсуса.
Комитет, готовящий новый стандарт на продукцию, должен пересмотреть все возможные опасности и указать приемлемый риск для каждой из них. Отправной точкой для этого процесса может стать оценка известных рисков в существующих конструкциях аналогичного оборудования. Комитет, возможно, пожелает указать более высокий или более низкий приемлемый риск, чем в существующих конструкциях. Однако, как правило, в худшем случае целесообразно ожидать того же приемлемого риска, что и раньше, т.е. вероятность каждого вида опасного события одинакова.
Внедрение различных технологий, таких как программируемые электронные системы, само по себе не оправдывает каких-либо изменений по сравнению с принятыми в настоящее время уровнями риска. Однако внедрение новых технологий вместе с изменениями в ожиданиях пользователей может оправдать переоценку баланса между безопасностью и другими соображениями. Однако эта основа для определения приемлемых рисков может оказаться неприемлемой, если оборудование выполняет новые функции, которые были бы невозможны без новой технологии.
9.4 Устранение неисправностей и контроль
Разница между медицинскими электрическими изделиями и другими сопоставимыми электронными устройствами заключается в возможной опасности. Как отмечено выше, одной из функций частного стандарта является выявление конкретных опасностей для соответствующего вида оборудования.
Однако средства, которые можно использовать для предотвращения опасностей, не относятся к медицинским электрическим изделиям. Компоненты и узлы, используемые в медицинских электрических изделиях, в основном являются стандартными компонентами, соответствующими их собственным стандартам. Как правило, также похожи и используемые конфигурации. То же самое относится и к программным компонентам и операционным системам.
В случае случайных или систематических неисправностей можно применять предотвращение неисправностей или контроль неисправностей. Предотвращение неисправности означает предотвращение возникновения ошибки. Контроль неисправности означает принятие дополнительных мер, чтобы в случае возникновения неисправности все еще предотвращались опасные воздействия.
Поскольку сложные системы не могут быть полностью оценены путем испытаний, их правильность и надежность должны оцениваться другими способами. Уверенность достигается применением соответствующих процедур в процессе проектирования, и это оценивается путем анализа доказательств этого процесса. (Оценка "доказательств процесса" также необходима для некоторых других аспектов медицинских изделий, таких как стерильность и эргономика).
Растущее признание того, что абсолютная безопасность не может быть обеспечена, привело к разработке концепций управления рисками. Эти концепции могут быть использованы в рамках более широкого системного подхода к проектированию и производству более качественных медицинских электрических изделий.
ИСО 9004:1987 "Управление качеством и элементы системы качества. Руководящие положения" содержит надлежащее описание этих концепций. Основой этих концепций является то, что качество, включая безопасность и результативность, легче всего внедрить в продукт на самых ранних этапах его проектирования.
Риск-менеджеры должны рассмотреть вопрос об использовании стандартов безопасности в системах качества. Например, раздел 19 ИСО 9004 отмечает, что: "Должны быть предприняты шаги как для ограничения риска ответственности за качество продукции, так и для минимизации количества случаев путем определения соответствующих стандартов безопасности, чтобы повысить эффективность формулировки спецификаций продукта или услуги".
Тщательный и упорядоченный подход ко всем этапам проектирования, включая анализ возможных опасностей, желателен для любого медицинского электрического изделия. Внедрение новых технологий, таких как программное обеспечение, увеличивает потребность в таком подходе по сравнению с традиционными проектами, когда изготовители и разработчики стандартов могут рассчитывать на накопленный опыт основных проблемных областей.
Существует множество методов, с помощью которых вероятность возникновения неисправностей может быть сведена к минимуму, но невозможно применить их все к одному и тому же элементу аппаратного или программного обеспечения. Поэтому изготовитель должен решить, какие методы использовать и в какой степени; и орган по сертификации или регулирующий орган должен решить, принял ли изготовитель разумные решения. Поэтому необходимо установить целевой показатель, который может быть выражен в терминах приемлемого риска в готовом проекте или в отношении целостности, связанной с безопасностью систем(ы).
Кроме того, часто существуют альтернативные подходы к проектированию для достижения эквивалентных, общих уровней безопасности. Например, можно использовать одну систему управления с очень высокой целостностью или несколько независимых систем управления с более низкой индивидуальной целостностью. То, как далеко изготовитель должен зайти с каждой системой, зависит не только от общего уровня требуемой безопасности, но и от конфигурации систем, связанных с безопасностью.
9.5 Уровни целостности
Общие стандарты, разрабатываемые подкомитетом 65A МЭК, вводят концепцию уровней целостности безопасности, которые соответствуют различным уровням гарантии или вероятности того, что программное или аппаратное обеспечение будет функционировать, как предполагалось.
________________
На момент подготовки настоящего стандарта - 65А (Секретариат) 122 и 123.
Эта концепция служит инструментом для связи надежности аппаратных или программных компонентов (которая зависит от методов, используемых при их разработке и изготовлении) с уровнями риска от всего оборудования. Для каждого уровня целостности безопасности необходимо соблюдать набор процедур, чтобы обеспечить необходимую уверенность. При таком подходе конкретный комитет по продукту должен будет только определить соответствующий уровень целостности для каждой системы защиты.
Такие общие стандарты будут необходимы для руководства проектировщиками и оценщиками при определении того, насколько эффективно были предотвращены общие режимы отказов в процессе проектирования.
Однако они не могут указать, как далеко должен зайти конструктор, чтобы избежать конкретной опасности.
Это зависит от серьезности опасности и таких факторов, как общественные ожидания и соображения затрат/выгод, которые варьируются от случая к случаю. Для каждой конкретной опасности комитет по разработке стандартов должен будет установить целевой приемлемый риск, либо в явной форме, либо путем установления требований к допустимым конструктивным особенностям и/или методам проектирования и производства.
Понятие уровней целостности применимо к:
a) общей конфигурации систем, связанных с безопасностью;
b) индивидуальным системам безопасности; а также
c) компонентам (включая программное обеспечение) в рамках отдельной системы, связанной с безопасностью.
Примеры того, как могут быть назначены уровни целостности безопасности, приведены в некоторых дополнительных и частных стандартах.
9.6 Достижение приемлемых уровней риска
Оценка конкретного продукта включает определение того, были ли определенные виды неисправностей сделаны достаточно маловероятными. В целом этот процесс более характерен для технологии, от которой зависит продукт, чем от его применения. Например, медицинское, офисное и бытовое электрооборудование требует одинаковых методов защиты от поражения электрическим током и схожих критериев для проверки диэлектрической прочности и т.д. Аналогичным образом уровень целостности программируемых электронных систем будет оцениваться практически одинаково независимо от вида оборудования, которым они управляют. Различия будут в основном отражать различные уровни целостности, которые подходят для конкретного применения. Поэтому критерии могут быть определены в общих стандартах, охватывающих многие виды оборудования.
Одно из различий между медицинскими электрическими изделиями и большинством других видов оборудования, связанное с безопасностью, заключается в вовлечении пациента в состав системы. Для оборудования, предназначенного для мониторинга, измерения или контроля физиологических параметров, идеальная характеристика результативности может быть определена с уверенностью. В таких случаях все еще необходимо будет установить, что риск того, что оборудование не соответствует его спецификации, является приемлемо низким, но также необходимо будет оценить адекватность и уместность самой спецификации.
Медицинские электрические изделия все чаще используют для выполнения функций, которые ранее выполнялись людьми (например, анализ физиологических данных, принятие решения о доставке терапевтических агентов). Особенно трудно определить, следует ли принимать систему, в которой программное обеспечение провоцирует потенциально опасные действия без вмешательства медицинского персонала.
Предыдущие стандарты безопасности оборудования не должны были решать эту проблему, но теперь они должны будут это сделать. Не имеет смысла указывать некоторые аспекты безопасности, если пренебрегают другими.
9.7 Превышение установленных требований
Изготовитель всегда может произвести продукт, который соответствует требованиям, указанным в стандарте. Если частный стандарт определяет приемлемый риск (в отношении конкретной опасности), изготовитель может обнаружить возможность снизить риск до более низкого уровня по стоимости, которую он считает коммерчески приемлемой. Внедрение такого продукта не обязательно означает, что стандарт не содержит жестких требований.
Однако стандарты должны учитывать то, что является разумно достижимым.
9.8 Оценка риска в серии МЭК 601
МЭК 601-1 и МЭК 601-2-хх идентифицируют большинство общих опасностей, применимых к различным видам медицинских электрических изделий. Для многих видов опасности эти стандарты уже:
a) указывают приемлемую конфигурацию систем, связанных с безопасностью (то есть систем, которые способствуют обеспечению безопасности, таких как базовая изоляция и защитное заземление, в качестве одной допустимой конфигурации для предотвращения поражения электрическим током); или же
b) заявляют, что конкретные события не должны происходить в нормальном состоянии или в состоянии единичного отказа.
Требование, сформулированное любым из этих способов, неявно определяет приемлемый риск.
В некоторых других отраслях вероятность опасных явлений определяется количественно. Стандарт или спецификация проекта требуют, чтобы каждый риск был уменьшен до или ниже указанной вероятности. Фактические вероятности рассчитываются (для случайных отказов оборудования).
9.9 Неисправности
Неисправности, которые необходимо учитывать при разработке стандартов на медицинские электрические изделия, подразделяют на несколько категорий:
a) некоторые неисправности, очевидные для оператора (например, осторожный пользователь может наблюдать внешнее физическое повреждение; обрыв заземляющего провода вызовет явную неисправность в некоторых видах медицинских электрических изделий);
b) некоторые неисправности, которые не могут быть обнаружены даже осторожным оператором, но могут быть обнаружены при регулярном обслуживании (например, частичное повреждение изоляции между питающей сетью и защитным заземлением медицинского электрического изделия);
c) некоторые неисправные состояния, которые не могут быть замечены оператором или обнаружены во время регулярного обслуживания (например, выход из строя одной части двойной изоляции).
9.10 Философия единичного нарушения/единичного отказа
Для медицинских изделий фактические вероятности различных опасностей редко определяются количественно. Вместо этого предлагается "философия единичного нарушения", которая может быть сформулирована следующим образом:
a) опасности не должны возникать ни в одном из перечисленных "условий единичного нарушения";
b) все части оборудования, имеющие отношение к безопасности, должны быть достаточно надежными, чтобы вероятность "единичного нарушения" была низкой;
c) вероятность двух "единичных нарушений" будет тогда очень низкой, поэтому допустимо, чтобы состояние с множественными неисправностями создавало опасность;
d) если одно "единичное нарушение" непосредственно вызывает другие, вероятность такая же, как и у первой неисправности, и оборудование должно оставаться безопасным;
e) если при некоторых обстоятельствах две неисправности могут возникать по общей причине (например, короткое замыкание обоих слоев двойной изоляции проводящими жидкостями или металлическими предметами), вероятность двух неисправностей такая же, как и у общей причины;
f) если неисправность не может быть обнаружена с помощью практических процедур технического обслуживания и вряд ли будет замечена оператором, поскольку она не влияет на работу оборудования, высокая вероятность того, что неисправность останется необнаруженной в течение длительного периода, должна учитываться при разработке требований безопасности.
Существующие требования МЭК 601-1, касающиеся электрической безопасности, иллюстрируют философию единственного отказа. Обеспечением основной безопасности является базовая изоляция. Однако, поскольку она может быть неисправна (может выйти из строя или может быть обойдена), требуется дополнительная защита.
МЭК 601-1 признает три возможности:
a) защитное заземление (доступной части или промежуточной части);
b) дополнительная изоляция; или же
c) усиленная изоляция (которая считается эквивалентной двойной изоляции).
Признано, что соединение защитного заземления может выйти из строя, и, если оно выходит из строя, опасность не допускается. Однако предполагается, что существует незначительная вероятность того, что как базовая изоляция, так и соединение защитного заземления, прервутся в одно и то же время (или, точнее, если один из этих элементов выйдет из строя после того, как другой уже вышел из строя, но не был отремонтирован).
Вероятность двух условий "единичного нарушения" не равна нулю. В некоторых отраслях, в которых потенциальные опасности очень серьезны (например, расплавление ядерного реактора или авиационные катастрофы), вероятность должна быть чрезвычайно низкой. Поэтому необходимы многочисленные защитные системы, а вероятность двойных и даже множественных отказов оценивается и сравнивается с заявленной приемлемой вероятностью.
Для медицинского электрического изделия было сочтено достаточным, чтобы гарантировать, что опасности не могут возникнуть при единичном нарушении. В случае двойной неисправности может возникнуть опасность, но риск считается незначительным.
Комитеты по разработке стандартов должны идентифицировать те угрозы безопасности, связанные с медицинскими электрическими изделиями, которые требуют более высокого уровня защиты, чем тот, который предусмотрен философией единичного нарушения.
Философия единственного отказа подразумевает, что, как правило, медицинское электрическое изделие должно иметь два средства защиты от каждой опасности. Считается, что это дает незначительный риск при условии низкой вероятности отказа отдельных систем. (Иногда может быть предоставлено более двух, но это не требуется существующими стандартами.)
Философия "единичного нарушения" оказалась успешной для большинства традиционных технологий и должна быть сохранена, насколько это возможно, поскольку она в целом подходит для простых аппаратных систем. Например, необходимо только удвоить расстояние утечки, чтобы получить эквивалент двойной или усиленной изоляции. Преодоление такого расстояния исключено по определению. Однако требуется только монета, чтобы соединить такие "две" изоляции одновременно и вызвать электрическую опасность. Этот простой пример показывает, что риск отказа по "общей причине" существует даже при использовании традиционных технологий безопасности.
Медицинское электрическое изделие должно оставаться в допустимых пределах риска при наличии отказов, которые:
a) очевидны для оператора (например, по сигналу или отсутствию функции);
b) могут быть обнаружены путем регулярного осмотра или технического обслуживания, проводимого в соответствии с эксплуатационной документацией; или же
c) не могут быть замечены оператором или обнаружены во время регулярного обслуживания, но могут быть обнаружены или контролироваться с помощью встроенных мер безопасности.
Тем не менее они также должны оставаться в допустимых пределах риска при наличии необнаруженных сбоев.
Ограничения на то, что считается приемлемым риском, должны разрабатываться при подготовке требований к безопасности и основным функциональным характеристикам. Например, решив, что ток утечки, протекающий от медицинского электрического изделия в нормальных условиях, "не должен быть вредным или ощутимым", может быть разумно ослабить это требование в некоторых условиях неисправности, чтобы "быть ощутимым, но не небезопасным".
Безопасность в таких условиях отказа может быть обеспечена резервными системами (резервирование) или использованием защитных устройств, пассивных или активных.
9.11 Уровни целостности, указанные в МЭК 601-1
МЭК 601-1 требует оценки целостности отдельных защитных устройств для подтверждения того, что отказы будут редкими. Например, изоляция должна выдерживать испытание на диэлектрическую прочность; защитные заземления должны иметь низкий импеданс; и тепловые выключатели должны работать несколько раз без сбоев.
Усиленная изоляция считается эквивалентной комбинации основной и дополнительной изоляции. Это означает, что его целостность считается настолько высокой, что вероятностью отказа можно пренебречь.
Токи утечки должны находиться в определенных пределах в нормальных условиях и в более высоких заданных пределах в различных условиях единичного нарушения. Одним из этих условий единичного нарушения является нарушение защитного заземления. Однако выход из строя постоянно установленного защитного заземления считается "очень маловероятным". Другими словами, такое соединение имеет более высокую целостность, чем обычное защитное заземление.
С другой стороны, токи утечки должны соответствовать ограничениям для нормального состояния при любом функциональном заземлении в исправном или разомкнутом состоянии; функциональные заземления имеют низкую целостность, и их отключение считается нормальным состоянием.
Таким образом, МЭК 601-1 уже неявно распознает три уровня целостности для заземления и для изоляции:
a) функциональные заземления/функциональная изоляция - низкая целостность - возможен отказ;
b) защитное заземление/базовая изоляция - средняя целостность - отказ возможен, но маловероятен;
c) постоянно установленные заземляющие соединения/усиленная изоляция - высокая целостность - выход из строя очень маловероятен.
Примечание - Защитные заземляющие провода, которые изгибаются при нормальном использовании, могут выйти из строя независимо от того, установлены они постоянно или нет. Подходящим условием единичного нарушения, при котором следует исследовать токи утечки, является: "Обрыв защитных заземляющих проводов, которые изгибаются при нормальном использовании".
9.12 Приемлемые уровни риска в соответствии с МЭК 601
Философия единичного нарушения обычно применяется к "основной" безопасности (то есть к защите от таких опасностей, как пожар и поражение электрическим током). Однако она не применялась столь последовательно к функциональным опасностям (то есть к тем, в которых оборудование не выполняет надлежащим образом свою предусмотренную функцию). МЭК 601-1 и некоторые частные стандарты требуют учета некоторых ситуаций, которые могут считаться опасными и которых следует избегать в нормальных условиях, но, возможно, не в условиях единичного нарушения.
Например, МЭК 601-2-4:1983 "Изделия медицинские электрические. Часть 2. Частные требования к безопасности кардиодефибрилляторов и дефибрилляторов-мониторов" требует, чтобы энергия, передаваемая в тестовую нагрузку, была в пределах ±15% от установленной энергии. Однако любой дефибриллятор может вообще не дать никакой энергии, если он неисправен. (Чтобы сохранить работоспособность, несмотря на отказ одного компонента, дефибриллятор должен включать в себя практически два комплектных блока в одном корпусе.) Следовательно, требование к точности поставляемой энергии не может применяться в условиях единичного нарушения. Следовательно, согласно стандарту, поставляемая энергия в условии единичного нарушения может быть от нуля до нормального максимума или, возможно, даже выше.
Таким образом, МЭК 601-2-4 подразумевает, что случайная подача неправильного количества энергии представляет собой приемлемый риск; энергия должна быть правильной (в пределах заявленного допуска) при нормальной работе, но при условии единичного нарушения допустим неправильный выходной сигнал (если условие единичного нарушения достаточно редко).
Если требование применяется только в нормальных условиях, это означает, что единой системы управления достаточно.
10 Принадлежности для оборудования
Основные проблемы для испытательных лабораторий и регулирующих органов возникают из-за следующего:
a) всесторонняя оценка некоторых видов медицинских электрических изделий может проводиться только тогда, когда оборудование включает все необходимые принадлежности;
b) некоторые виды медицинских электрических изделий удовлетворяют соответствующим основным требованиям только при использовании с некоторыми доступными принадлежностями;
c) многие принадлежности, используемые совместно с медицинскими электрическими изделиями, производятся не изготовителем оригинального оборудования.
Стандарты на продукцию не могут полностью решить эти проблемы. Однако, где это уместно, в стандартах следует обратить внимание на следующее:
a) изготовитель не может нести ответственность за безопасность или результативность своего продукта, когда он используется с принадлежностями другого изготовителя без его согласия;
b) можно ожидать, что изготовитель оригинального оборудования:
- убедится, что ни одна из его принадлежностей не ставит под угрозу безопасность или сертификацию его продуктов, или идентифицирует те из его принадлежностей, которые это делают;
- предупредит пользователя (с помощью эксплуатационной документации, предоставляемой с оборудованием), если принадлежности других изготовителей могут поставить под угрозу безопасность, производительность или сертификацию его продукта.
Изготовители и испытательные центры должны определить фактические принадлежности, которые включаются при сертификации медицинского электрического изделия.
11 Стандартизация
Одной из важных целей при разработке требований к безопасности и основным функциональным характеристикам является стандартизация определенных физических и эргономических аспектов оборудования, чтобы его можно было использовать и поддерживать в соответствии со стандартными процедурами.
Если в качестве требования включена стандартизация конкретного физического аспекта оборудования, обоснование должно быть доступно пользователям, которые обычно не знакомы с требованиями стандарта на продукцию. Это может быть достигнуто путем:
a) публикации отдельного руководства пользователя; или же
b) включения соответствующего обсуждения в обоснование стандарта на продукцию, чтобы другие комитеты МЭК или ИСО, национальные комитеты или органы здравоохранения могли подготовить соответствующие руководства пользователя.
Стандартизация медицинских электрических изделий в таких областях, как следующие, должна быть сохранена в серии МЭК 601:
а) Классификация в МЭК 601-1 рабочих частей на типы CF/BF/B является примером стандартизации требований к конструкции, испытаниям и маркировке в первичном стандарте, гарантирующая, что одни и те же символы используются для информирования пользователя о степени безопасности пациента относительно всех видов медицинских электрических изделий.
Примечание - Обсуждение этой важной области стандартизации приведено в приложении А;
b) стандартизация медицинской газовой арматуры в медицинских электрических изделиях является еще одним примером требования, которое призвано обеспечить безопасное использование оборудования различных изготовителей.
12 Координация стандартов
12.1 Общий подход к безопасности
При рассмотрении вопросов безопасности медицинского электрического оборудования МЭК, как правило, уделяет приоритетное внимание разработке стандартов на продукцию. Во многих странах больше внимания уделяется внедрению стандартов на продукты, чем руководствам по инсталляции и применению. Этот приоритет является правильным с учетом вопросов, обсуждаемых в разделах 5 и 18 настоящего стандарта. Тем не менее потребность в дополнительных стандартах МЭК, касающихся инсталляции и применения медицинских электрических изделий, хорошо известна.
Несогласованная разработка и внедрение стандартов применения и новых стандартов на продукты могут привести к возникновению новых проблем безопасности. Обсуждение данных проблем приведено в приложении С.
12.2 Общее планирование при разработке стандартов
Создание последовательного подхода к разработке стандартов для медицинских электрических изделий требует соблюдения иерархии стандартов для обеспечения тесной координации. Отдельные стандарты должны быть ограничены конкретными аспектами и должны ссылаться на другие стандарты с другой соответствующей информацией.
13 Стандарты и закон
Безопасность имеет особое значение для регулирующих органов. Во многих областях законы и нормативные акты ссылаются на типы стандартов, упомянутые в настоящем стандарте.
Стандарты могут быть указаны в законодательстве или нормативных актах, и в этом случае стандарты становятся обязательными, а содержание стандартов становится законодательным требованием. Если стандарты ссылаются на законы или правила, ссылка должна быть полной, чтобы заинтересованные стороны могли получить соответствующие правила. В качестве альтернативы законы все больше предоставляют стандартам статус "считается соответствующим", и в этом случае продукт, процедура, действие и т.д., которые соответствуют стандарту, должны соответствовать закону. Соответствие стандарту может быть необязательным в соответствии с такими законами, но тогда может быть сложнее убедить власти в том, что продукт, процедура, действие и т.д. все еще соответствуют закону.
Стандарты также могут быть указаны в судебном процессе в качестве доказательства того, что общество разумно ожидает, и используются для установления соответствия этим ожиданиям.
Из-за тесной взаимосвязи между безопасностью и правилами, а также из-за авторитета ИСО и МЭК и международного признания их стандартов важно, чтобы эти стандарты соответствовали установленным руководящим принципам.
Признавая ограничения, присущие стандартам на продукцию, не следует указывать, что совместимое оборудование является "абсолютно безопасным". Следует просто указать, что "продукт соответствует соответствующим стандартам безопасности МЭК" или "продукт соответствует следующим стандартам безопасности МЭК..."
14 Роль стандартов
Пользователи, покупатели и регулирующие органы могут обоснованно требовать доказательства того, что медицинское электрическое изделие было разработано и изготовлено в соответствии с соответствующими стандартами на продукцию. Изготовители также могут использовать такой стандарт для подтверждения того, что их продукция была разработана и изготовлена безопасно.
15 Несчастные случаи
Изготовители и регулирующие органы должны быть проинформированы об инцидентах и несчастных случаях, происходящих при использовании медицинского электрического изделия, для того, чтобы можно было принять соответствующие меры для предотвращения их повторения. Такие действия могут включать в себя изъятие аналогичного оборудования из обслуживания, внесение изменений в процесс обслуживания или внесение изменений в конструкцию или производственные процессы в последующем производстве.
На момент публикации применяемого в настоящем стандарте международного документа процедуры, принятые для анализа инцидентов, оповещений об опасностях и отзыва оборудования, не стали предметом международных стандартов. Однако они все чаще становятся предметом официальных национальных и международных соглашений.
16 Поддержание стандартов
Важным фактором в поддержании и улучшении стандартов для медицинских электрических изделий является получение обратной связи от органов, разрабатывающих стандарты. Такая связь должна присутствовать в случаях:
a) расследования пользователями и органами здравоохранения несчастных случаев и инцидентов с оборудованием, которое подпадает под действие существующих стандартов; а также
b) определения характеристик опасных устройств в областях, не предусмотренных стандартами.
Когда предоставляется такая обратная связь, важно, чтобы орган, отвечающий за разработку стандартов, оперативно реагировал, чтобы исследовать необходимость улучшения существующего стандарта или разработки нового стандарта.
17 Выявление проблем
Стандарты безопасности и основных функциональных характеристик на медицинские электрические изделия следует разрабатывать только тогда, когда была четко определена проблема, и было установлено, что разработка стандарта поможет в решении этой проблемы.
18 Приоритет
Везде, где это возможно, приоритет следует отдавать обеспечению безопасности путем осуществления мер в следующем порядке, соответствующем уменьшению эффективности таких мер:
a) стандарты на продукцию, устанавливающие требования, которые должны быть включены в конструкцию оборудования;
b) стандарты или руководящие принципы, которые определяют дополнительные меры, касающиеся установки оборудования, ввода в эксплуатацию, технического обслуживания и испытаний на безопасность;
c) стандарты или руководящие принципы, в которых разъясняются меры предосторожности, необходимые для безопасного использования медицинского электрического изделия.
19 Формат и структура
19.1 Наименования
Наименования третьего издания МЭК 601-1 и каждого связанного с ним дополнительного и частного стандарта должны указывать на то, что они являются стандартами безопасности продукции, устанавливающими требования к безопасности и основным функциональным характеристикам медицинского электрического изделия.
19.2 Область применения и обоснование
Каждый стандарт на медицинское электрическое изделие должен четко указывать область применения стандарта (например, "требования к безопасности и основным функциональным характеристикам конкретного продукта или ассортимента продукции").
Каждый стандарт должен также включать общее обоснование его разработки и отдельные обоснования для каждого конкретного требования.
19.3 Форма спецификации
Всякий раз, когда это целесообразно, требования безопасности и основные функциональные характеристики должны выражаться в терминах конкретных требований к проверке и испытаниям, а не в соответствии с требованиями к конструкции.
Требования к испытаниям должны быть четко определены, чтобы гарантировать, что их соответствие может быть адекватно проверено (см. раздел 21). В качестве примеров можно привести хорошо зарекомендовавшие себя и проверенные решения для достижения приемлемых уровней риска. В тех случаях, когда нецелесообразно включать конкретные требования к испытаниям с критериями прохождения/отказа, в будущих стандартах на продукцию должны быть определены опасности и включены требования для оценки соответствия процесса проектирования оборудования (см. раздел 22).
Постоянной целью является разработка требований, которые приведут к последовательной интерпретации стандарта.
Требования должны быть включены в МЭК 601-1 или дополнительный стандарт, если опасность является общей для всех медицинских электрических изделий, или в соответствующий частный стандарт, если опасность является уникальной для одного типа оборудования.
19.4 Структура
Связи между третьим изданием МЭК 601-1, а также его дополнительными и частными стандартами должны соответствовать текущему подходу. Серия должна состоять из:
a) общего стандарта МЭК 601-1, содержащего требования к безопасности и основным функциональным характеристикам, которые являются общими для всех медицинских электрических изделий;
b) ряда дополнительных стандартов, МЭК 601-1-xx, содержащих дополнительные требования к безопасности и основным функциональным характеристикам, которые являются общими для ряда медицинских электрических изделий;
c) ряда частных стандартов, МЭК 601-2-xx, содержащих дополнительные требования к безопасности и основным функциональным характеристикам для определенных типов медицинских электрических изделий.
Опыт показывает, что из-за обширного характера стандарта требуется до десяти лет для полного пересмотра МЭК 601-1 как единого стандарта. Поэтому может быть целесообразно опубликовать третье издание в виде серии отдельных разделов, все из которых упоминаются в сокращенной общей (или первичной) публикации, как показано в таблице 3 и на рисунке 2.
20 Определение требований к безопасности и основным функциональным характеристикам
20.1 Общие положения
Задачи для комитета, отвечающего за стандарт на продукцию:
a) определить/перечислить опасности, которые свойственны конкретному виду оборудования;
b) где это уместно, указать вид действия, которое изготовитель должен предпринять для устранения или уменьшения рисков (например, путем предоставления предупреждений оператору или уменьшения вероятности конкретных режимов отказа);
c) определить, как далеко должен зайти изготовитель, чтобы избежать опасности (например, указав приемлемый риск).
Таблица 3 - Третье издание МЭК 601-1 и его дополнительные стандарты
Наименование стандартов | Сфера охвата материала |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел A - Введение в серию МЭК 601 | Введение в серию МЭК 601 и ее структуру, включая текущий статус первичного, дополнительных и частных стандартов |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел B - Общие требования к испытаниям | Раздел первый (Общее) и раздел второй (Условия окружающей среды) из второго издания МЭК 601-1 |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел C - Защита от поражения электрическим током | Раздел третий (Защита от поражения электрическим током) и электрические требования из десятого раздела (Требования к конструкции) из второго издания МЭК 601-1 |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел D - Защита от механических опасностей | Раздел четвертый (Защита от механических опасностей) и механические требования из десятого раздела (Требования к конструкции) из второго издания МЭК 601-1 |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел E - Защита от возгорания | Раздел шестой (Защита от опасности воспламенения горючих анестезирующих смесей) и соответствующие требования из десятого раздела (Требования к конструкции) из второго издания МЭК 601-1 |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел F - Термические опасности | Соответствующий материал из седьмого раздела (Защита от чрезмерных температур и других угроз безопасности) и десятого раздела (Требования к конструкции) из второго издания МЭК 601-1 |
МЭК 601-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. Раздел G - Дополнительные опасности | Соответствующий материал из седьмого раздела (Защита от чрезмерных температур и других угроз безопасности) и десятого раздела (Требования к конструкции) из второго издания МЭК 601-1 |
МЭК 601-1-1: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. 1. Дополнительный стандарт: Требования к медицинским электрическим системам | Из существующего дополнительного стандарта - на стадии пересмотра |
МЭК 601-1-2: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. 2. Дополнительный стандарт: Электромагнитная совместимость - требования и испытания | Из существующего дополнительного стандарта - на стадии пересмотра |
МЭК 601-1-3: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. 3. Дополнительный стандарт: Радиационная безопасность в медицинских электрических изделиях | Материал, изначально предназначенный для пятого раздела (Защита от чрезмерного излучения) в первом и втором изданиях МЭК 601-1. На рассмотрении |
МЭК 601-1-4: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. 4. Дополнительный стандарт: Требования к программируемым электронным системам | На рассмотрении |
МЭК 601-1-xx: Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик. хх. Дополнительный стандарт: Требования к ... | Будущие дополнительные стандарты |
|
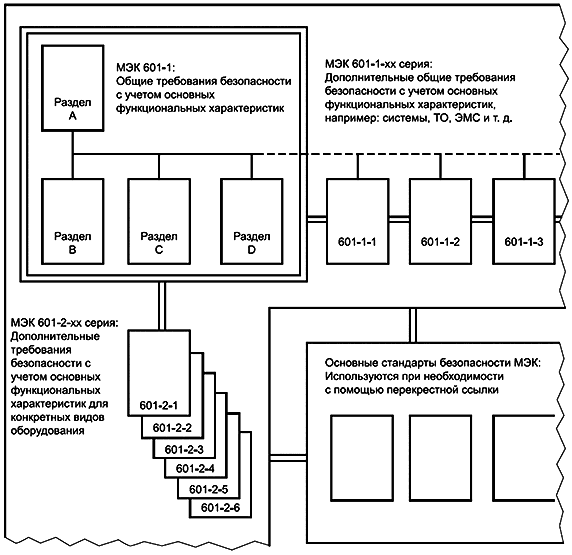
Рисунок 2 - Структура серии МЭК 601 согласно третьему изданию МЭК 601-1
________________
Исключая эксплуатационные стандарты серии МЭК 601-3-xx.
На практике комитеты по стандартизации обычно выполняют эти функции, но в большинстве случаев они не документируются формально и систематически. Основная философия системного подхода ничем не отличается от той, которая используется в настоящее время. Разница заключается в формализации идентификации опасности и оценки риска.
Этапы идентификации опасности могут включать:
a) систематический "мозговой штурм" для перечисления возможных неблагоприятных воздействий соответствующего типа оборудования и их возможных причин в общих чертах;
b) установление для каждой опасности, охватывается ли она уже МЭК 601-1, и если да, то адекватно ли она охвачена;
c) присвоение категории риска для опасностей, которые не соответствуют требованиям МЭК 601-1.
В готовом к публикации стандарте должны быть четко перечислены опасности, которые рассматриваются, и раздел(ы) стандарта, относящиеся к каждой опасности.
Для каждой опасности в стандарте следует либо указать приемлемый(ые) способ(ы) избежать опасности, либо, предпочтительно, указать приемлемый риск, позволяющий определить приемлемые методы избежать опасности из общих стандартов или обосновать другими способами. В последнем случае, если это возможно, следует также включить информативный список приемлемых способов, которыми это требование может быть выполнено.
20.2 Конкретные требования к испытаниям для оценки процесса проектирования
Подход, принятый при разработке стандартов безопасности и основных функциональных характеристик для медицинских электрических изделий, должен быть следующим:
a) анализ рисков должен применяться для выявления опасностей и возможных решений, а также для установления приемлемых уровней риска для каждой опасности;
b) стандарты должны включать требования по устранению каждой выявленной опасности;
c) где это возможно, эти требования должны быть изложены с точки зрения воспроизводимых испытаний с конкретными критериями прохождения/отказа;
d) если критерии прохождения/отказа не могут быть разработаны для устранения конкретной опасности (например, в предлагаемом дополнительном стандарте безопасности программируемых электронных систем), стандарты на продукцию должны:
- определить опасности, которые необходимо учитывать при проектировании оборудования; а также
- установить (или предоставить критерии для установления) приемлемых уровней риска;
e) стандарты безопасности продукции для медицинских электрических изделий также должны требовать от изготовителя:
- проведение соответствующего анализа опасностей во время процесса проектирования, чтобы выявить опасности, которые являются уникальными для конкретного оборудования и которые не были учтены в соответствующем(их) стандарте(ах) на продукцию;
- установления приемлемых уровней риска, если они не были указаны в соответствующем(их) стандарте(ах) на продукцию;
- включения защитных мер в конструкцию для обеспечения соблюдения соответствующих стандартов на продукцию;
- включения дополнительных защитных мер для снижения до приемлемого уровня рисков, возникающих в результате других опасностей, выявленных в ходе анализа рисков изготовителя;
f) стандарты безопасности продукции для медицинских электрических изделий должны также требовать от изготовителя осуществление процесса проектирования в рамках соответствующей системы качества (например, ИСО 9001), чтобы продемонстрировать соответствие этим требованиям.
________________
ИСО 9001:1987 "Системы качества. Модель обеспечения качества при проектировании/разработке, производстве, монтаже и обслуживании".
21 Требования к испытаниям
21.1 Общие положения
Невозможно определить каждую возможную опасность для каждого типа медицинских электрических изделий в общем стандарте. Это приводит к необходимости в частных стандартах, но в равной степени невозможно разработать достаточно частных стандартов, чтобы охватить каждый новый тип оборудования по мере его появления.
Если в частном стандарте учтены все возможные опасности для конкретного оборудования, это может привести к простой оценке оборудования. Тем не менее в отсутствие соответствующих частных требований, открытые критерии в текущей редакции МЭК 601-1, такие как "...где может возникнуть опасность...", способствуют исследованию всех возможных опасностей. Это дает лицам, имеющим соответствующие знания, возможность исследовать все возможные опасности и применять общие требования стандарта, в зависимости от обстоятельств, для устранения этих опасностей. Тем не менее это также приводит к различным интерпретациям разными конструкторами оборудования и испытательными лабораториями.
21.2 "Условие единичного нарушения" и "Угроза безопасности"
В третьем издании МЭК 601-1 каждое требование к испытаниям должно четко указывать:
a) какие конкретные неисправности подлежат испытаниям, где это применимо, без необходимости анализа конкретного риска (например, сломанное защитное заземление); а также
b) в каких других областях изготовитель должен применять анализ рисков (например, безопасность программируемых электронных систем).
Реализация философии, описанной в следующих разделах, может снизить ценность включения списка условий единичного нарушения в третье издание МЭК 601-1. Тем не менее, если "условие единичного нарушения" сохраняется как определенный термин с целью определения конкретных неисправностей, которые нужно исследовать только по одному без необходимости конкретного анализа риска:
i) в стандарте должен быть только один такой список;
ii) термин "условие единичного нарушения" следует использовать только для обозначения неисправностей, которые исследуются по одному;
iii) идентификация этих "условий единичного нарушения" не должна основываться на оценочных суждениях относительно возможности последующей "угрозы безопасности". В связи с этим определение термина "угроза безопасности" не требуется.
21.3 Защитные устройства
Защитные устройства предотвращают опасную работу устройств (например, путем контроля предельных значений). Отказ такого защитного устройства не наносит немедленного вреда. Обнаружение такой неисправности допустимо в течение периода времени, так что вероятность отказа работающего устройства в течение этого периода пренебрежимо мала.
Примером пассивной защиты может быть последовательное включение импедансов с подключениями пациента к рабочей части, чтобы ограничить ток утечки в случае единичного нарушения. Активное защитное устройство может контролировать температуру детали и выключать оборудование в случае перегрева. Чтобы обеспечить пренебрежимо низкую вероятность возникновения второго отдельного и не связанного с этим сбоя до того, как будет распознан первый сбой, вероятность каждого отдельного сбоя должна быть достаточно низкой.
21.4 Компоненты высокой надежности
Не все компоненты могут давать сбой в "безопасном" режиме. Отказ ножки стола (компонента стола) может привести к потенциально опасному разрушению. Такие компоненты должны быть настолько надежными, что возможностью отказа можно пренебречь. Примерами компонентов, к которым обычно применяется эта философия, являются цепи и кабели, поддерживающие подвешенные массы и усиленная изоляция.
21.5 Факторы безопасности
Предельные значения для некоторых характеристик устройства могут быть установлены путем применения факторов безопасности. Однако этот подход следует применять только в том случае, если существует установленный порог, ниже которого не возникает вредного воздействия. Если такой порог может быть установлен, применение коэффициента безопасности определяет предельное значение.
Если пределы устанавливаются с использованием факторов безопасности, в обосновании должно быть указано:
a) как факторы безопасности влияют на выбранные пределы;
b) определенный(ые) используемый(ые) коэффициент(ы) безопасности;
c) данные, использованные при установлении порога, ниже которого наблюдается приемлемо низкий эффект; а также
d) любые другие сделанные предположения.
Стохастические эффекты, для которых порог не существует, требуют других методов оценки риска.
В приложении D обсуждается использование факторов безопасности более подробно.
21.6 Методы испытаний
Технические требования и методы испытаний являются взаимосвязанными элементами стандартов на продукцию и должны всегда рассматриваться совместно.
Стандарты на продукцию должны определять, где необходимы медицинские обоснованные суждения при принятии решения о применении определенного требования.
Везде, где возможно, стандарты должны содержать спецификации испытаний для полной и четкой проверки соответствия техническим требованиям. В некоторых случаях заявление о соответствии, такое как "визуальный осмотр", "ручное тестирование" или подобное, подходит для этой цели, если такой метод дает точную оценку.
Должно быть легко определяемо, какие методы испытаний применимы к каждому техническому требованию. Соответствующие заголовки должны обозначать соответствующее испытание (например, "испытание на термическую выносливость"), и следует сделать ссылку на пункт, содержащий требование. Это также относится к ссылкам на другие соответствующие стандарты испытаний.
При разработке требований к испытаниям:
a) результаты испытаний должны воспроизводиться в определенных пределах. При необходимости, каждый метод испытаний должен включать в себя утверждение предела его относительной точности;
b) где последовательность испытаний может влиять на результаты, должна быть указана правильная последовательность;
c) следует особо отметить испытания, которые должны проводить изготовитель, импортер или другие стороны;
d) может потребоваться дополнительное испытание, например, обычное или случайное тестирование отдельных свойств, особенно важных для безопасности и основных функциональных характеристик.
Дополнительные рекомендации по разработке воспроизводимых испытаний изложены в 6.4.3 Руководства ИСО/МЭК 51.
21.7 Основные стандарты безопасности
В Руководстве МЭК 104 указано, что технические комитеты, разрабатывающие стандарты на продукцию, должны для каждой части, раздела или пункта стандарта, не относящегося к сфере применения только этого стандарта:
a) ссылаться на другие стандарты с четким указанием издания и пункта; или же
b) в случае включения небольших фрагментов текста воспроизводить материал по таким стандартам (с указанием источника).
Эта политика должна соблюдаться при определении требований соответствия для устранения опасностей, при которых риск для пациента, пользователя или оборудования не выше, чем при использовании обычного немедицинского электрического оборудования. В большинстве таких случаев следует ссылаться на основные стандарты безопасности МЭК.
21.8 Маркировка и лейбелинг
Пользователи должны знать о рисках, присущих нормальному использованию определенных типов медицинских электрических изделий, и о тех рисках, которые могут возникнуть в результате неправильного использования, которое обычно происходит.
Меры предосторожности могут принимать форму специальной маркировки, чтобы ограничить условия, при которых продукт может быть установлен или использован, или предупреждений относительно рисков, связанных с обычным использованием или разумно ожидаемым неправильным использованием устройства.
Однако на маркировку не следует полагаться, чтобы предупредить пользователя об опасностях, которые должны быть устранены изготовителем в соответствии с общепринятыми уровнями безопасности конструкции или путем замены опасных ингредиентов на более безопасные, где они одинаково эффективны.
В общем, следует избегать лишних или ненужных маркировок, поскольку они имеют тенденцию уменьшать ценность маркировок, которые необходимы.
21.9 Предупреждения
Требования в предупреждающих уведомлениях должны быть тщательно сформулированы, чтобы стимулировать реалистичные ожидания со стороны пользователей в отношении безопасности, производительности и надежности продукта.
Чтобы обозначить риски и информировать пользователей или установщиков об их характере и способах, которыми их можно избежать, следует использовать следующие слова.
"ОПАСНОСТЬ" следует использовать для функций продукта, представляющих наиболее серьезные риски.
"ПРЕДУПРЕЖДЕНИЕ" и "ВНИМАНИЕ" следует использовать в порядке убывания серьезности предполагаемых рисков.
Утверждение должно следовать за использованием этих слов или должно быть предоставлено в инструкциях по применению с указанием основного(ых) риска(ов), предвиденного в связи с обычным или разумно ожидаемым использованием, или неправильным использованием продукта, таким как "Вызывает ожоги" или "Опасность поражения электрическим током" (или взрыва, пожара, травм людей и т.д.).
Эта формулировка должна также указывать соответствующие меры предосторожности или включать инструкции о том, как снизить риск, например, "Не используйте для ...", "Хранить вдали от ..." и т.д.
21.10 Другая информация, которая должна быть предоставлена
Может потребоваться другая информация, относящаяся к отдельным элементам медицинского электрического изделия. Например, информация может потребоваться:
a) для скрытых или непредвиденных рисков;
b) специальных классов пользователей;
c) инструкции по инсталляции и использованию; или
d) требований безопасной практики пользователя и предостерегающих заявлений.
22 Проверка конструкции
Нынешний подход к разработке требований к проверке или испытаниям с конкретными критериями соответствия "годен/не годен" является неуместным методом устранения некоторых видов опасности в медицинских электрических изделиях.
Опасности, для которых критерии прохождения/несоответствия не могут быть разработаны, должны быть устранены с использованием других методов. Пример содержится в дополнительном стандарте для программируемых электронных систем. Как правило, нецелесообразно оценивать, обеспечивает ли программная система в элементе медицинского электрического изделия приемлемый уровень безопасности и производительности, подвергая оборудование или перечень программ указанному контролю или тестированию на соответствие критериям "годен/не годен". Лучшей альтернативой является установление того, что проектирование выполнялось в соответствии с положениями соответствующего стандарта системы качества (например, ИСО 9001).
Аналогичным образом определение процесса проектирования обеспечивает альтернативу проверке и испытаниям в таких областях, как оценка эргономической безопасности и влияния человеческих факторов. Эта альтернатива должна быть принята в МЭК 601-1 и его дополнительных и частных стандартах, если нецелесообразно указывать соответствующие критерии проверки или испытания.
Требования соответствия будут удовлетворены путем установления того, что проектирование было выполнено в соответствующей системе качества (например, путем демонстрации соответствующей регистрации в соответствии с ИСО 9001).
23 Клинические испытания и аналогичная оценка
Чтобы оценить его безопасность и производительность, некоторые органы власти могут требовать, чтобы некоторые типы медицинских электрических изделий подвергались контролируемым клиническим испытаниям.
Этот тип оценки выходит за рамки стандартов серии МЭК 601, которые должны быть ограничены вопросами, которые могут быть оценены с учетом конкретных требований соответствия, таких как требования к проверке или испытаниям или оценка процесса проектирования.
В приложении B обсуждается значение, которое имеют стандарты на продукцию в общей оценке/утверждении конкретного медицинского электрического изделия.
24 Требования к производительности
24.1 Общие положения
Требования к производительности варьируются в непрерывном спектре. На одном конце спектра находятся вопросы, которые важны для безопасного и правильного функционирования оборудования и которые должны быть включены в качестве требований безопасности и основных функциональных характеристик. На другом конце этого спектра некоторые эксплуатационные вопросы, которые не являются существенными для безопасного и правильного функционирования оборудования и не должны включаться в качестве требований безопасности или основных эксплуатационных характеристик.
Требования безопасности должны быть разработаны для вопросов, которые:
a) необходимы для обеспечения безопасной и эффективной работы продукта;
b) необходимы для многих пользователей, которые не смогут идентифицировать или указать их без ссылки на соответствующий стандарт или оценить их без специализированных лабораторных средств; а также
c) могут быть определены в полностью проверяемых технических условиях посредством воспроизводимых испытаний на соответствие или оценки процесса проектирования.
24.2 Требования к основным функциональным характеристикам
Некоторые требования к безопасности и основным функциональным характеристикам должны быть выражены в терминах минимальной производительности (например, частотная характеристика диагностического электрокардиографа).
Эти требования имеют решающее значение для правильного функционирования оборудования; пользователи должны полагаться на стандарты для руководства; и сертифицированное соответствие необходимо для подтверждения производительности оборудования.
24.3 Основные требования к раскрытию информации
В некоторых случаях целесообразно решать проблемы безопасности и установления основных функциональных характеристик, указав обязательные требования к раскрытию информации.
В тех случаях, когда существует согласие о том, что конкретное требование к производительности является существенным, но нет согласия относительно ограничений, которые должны применяться, изготовитель должен сообщить о характеристиках оборудования в соответствии со стандартной процедурой испытаний.
Такая ситуация может возникнуть, например, с требованиями к скорости потока для устройств управления инфузией с электропитанием.
24.4 "Необязательные" требования к функциональным характеристикам
Там, где нет полного согласия о том, что конкретное требование к производительности является существенным для безопасного и правильного функционирования оборудования, этот вопрос не следует включать в качестве основного требования.
Многие требования могут попадать в эту категорию, например, размер дисплея на мониторе ЭКГ. В тех случаях, когда согласовано, что международная стандартизация поможет медицинскому специалисту или изготовителю в определении пригодности для использования, в серии МЭК 601-3-xx должен быть подготовлен отдельный стандарт, содержащий необязательные требования к производительности или методам испытаний.
Основным вкладом этих стандартов должно быть установление методов испытаний, с помощью которых изготовители и потенциальные пользователи могут указывать и оценивать производительность.
Приложение А
Классификация рабочих частей
A.1 Введение
Определение и классификация рабочих частей в первом и втором изданиях МЭК 601-1 являются важной инициативой в международной стандартизации медицинских электрических изделий. Однако, чтобы эта система классификации была полностью эффективной в минимизации риска:
a) обоснование этих классификаций должно быть принято во внимание при разработке будущих стандартов на продукцию; а также
b) классификации должны быть понятны медицинскому персоналу, использующему медицинское электрическое изделие.
В настоящем приложении обсуждается обоснование этой системы классификации, и оно должно помочь в разработке будущих стандартов на продукцию и руководств по применению.
A.2 Применяемые части
А.2.1 Идентификация и маркировка
Детали оборудования, которые должны вступать в физический контакт с пациентом, называются рабочими частями. Общий стандарт требует, чтобы такие детали классифицировались как тип CF, тип BF или тип B. Рабочие части типов CF и BF также известны как рабочие части F-типа (плавающие).
Классификация обозначена на оборудовании следующими символами:
|
Некоторые частные стандарты определяют, какие части оборудования должны рассматриваться как рабочие.
А.2.2 Неэлектрические требования
МЭК 601-1 и некоторые частные стандарты требуют, чтобы рабочие части соответствовали определенным требованиям, которые более жесткие, чем требования к другим доступным частям (например, тепловые требования). В целом эти неэлектрические требования одинаковы для всех классификаций (типы CF, BF и B).
А.2.3 Электрические требования
Проводники, которые обеспечивают прохождение электричества между рабочей частью и пациентом, называются соединением(ями) пациента. Другие проводники в оборудовании, которые не изолированы или не отделены от соединения(ий) пациента до определенного уровня, называются контурами пациента.
МЭК 601-1 включает ряд электрических требований, которые применяются всякий раз, когда рабочая часть содержит какие-либо соединения с пациентом (например, испытания на диэлектрическую прочность, минимальные требования к утечке/зазору между цепью пациента и другими проводящими частями; ограничения утечки и вспомогательного тока пациента в нормальном состоянии и в определенных условиях нарушения).
Данные электрические требования различаются для рабочих частей типов CF, BF и B. Они определяют уровень защиты от поражения электрическим током через рабочую часть.
Некоторые частные стандарты запрещают рабочие части определенных классификаций, в то время как некоторые включают дополнительные электрические требования для рабочих частей.
A.3 Защита от поражения электрическим током
A.3.1 Введение
Медицинское электрическое изделие может быть в нормальном состоянии (например, все проводники защитного заземления и изоляция не повреждены, отсутствует попадание жидкости внутрь оборудования и т.д.), или оно может быть неисправно.
Рабочие части типов CF, BF и B обеспечивают разную степень защиты от поражения электрическим током. Необходимая защита зависит от состояния медицинского электрического изделия в непосредственной близости от пациента и от того, находится ли рабочая часть в непосредственном контакте с сердцем.
А.3.2 Защита в нормальных условиях
Если в электрическом оборудовании поблизости от пациента нет неисправностей, требования, приведенные в A.2.3, обеспечивают высокую степень безопасности (низкий уровень риска), когда рабочие части типов CF, BF или B используются в клинических применениях, которые не предполагают прямого контакта с сердцем. Тем не менее некоторые частные стандарты не допускают применения рабочих частей типа B (см. A.3.5).
Требования, приведенные в A.2.3 (включая пределы для тока утечки пациента и вспомогательного тока пациента), также обеспечивают разумную степень безопасности во время клинических процедур, в которых рабочие части находятся в прямом контакте с сердцем, при условии, что:
a) нет никаких неисправностей в каком-либо электрическом оборудовании поблизости от пациента в нормальном состоянии; а также
b) выравнивание потенциалов ограничивает напряжение, возникающее между доступными заземленными частями оборудования класса I в непосредственной близости от пациента, а также между этими точками и другими доступными объектами. (Это иногда называют работой в "эквипотенциальной зоне").
Тем не менее МЭК 601-1 и некоторые частные стандарты требуют применения рабочих частей типа CF в медицинских электрических изделиях, которые предназначены для непосредственного применения на сердце (см. А.3.4).
А.3.3 Защита в условиях неисправности
Вероятность и серьезность травм (например, от ожогов и поражения электрическим током) во время поражения электрическим током зависит:
a) от количества тока, который течет; а также
b) времени, за которое течет ток.
Напряжение на пациенте обычно ограничивается рядом мер предосторожности (например, защитное заземление в оборудовании класса I), и только в случае серьезной аварии пациент подключается непосредственно к сетевому напряжению. Тем не менее случайное заземление пациента является обычным явлением во многих клинических ситуациях и считается нормальным состоянием в МЭК 601-1.
В маловероятном случае непосредственного подключения пациента к сетевому напряжению опасность для пациента от электрического тока, протекающего через случайный путь к земле, не должна быть больше, чем при использовании другого электрического оборудования. Приемлемая безопасность обеспечивается основными требованиями к безопасности оборудования, а также мерами предосторожности пользователя, которые следует соблюдать при использовании любого электрического оборудования в медицинской практике.
Тем не менее использование медицинского электрического изделия с рабочими частями может увеличить опасность такой аварии из-за следующего:
a) некоторые применяемые детали могут обеспечивать очень низкий импеданс между пациентом и землей, и величина тока может быть намного больше, чем при случайном заземлении пациента;
b) физические отношения между рабочей частью и пациентом (например, фиксированное соединение физиологических записывающих электродов; пациент без сознания находится в контакте с операционным столом) могут быть такими, что ток протекает гораздо дольше, чем при случайном заземлении пациента;
c) рабочая часть может находиться в прямом электрическом контакте с сердцем, и в этом случае пороговое значение тока поражения электрическим током может быть до тысячи раз ниже, чем для тока, входящего и выходящего из пациента снаружи.
Одно из условий неисправности, указанных в A.2.3, требует, чтобы рабочие части F-типа выдерживали прямое подключение к сетевому напряжению. При этом условии ток утечки пациента ограничен 0,05 мА для типа CF и 0,50 мА для типа BF. Рабочие части типа B не должны выдерживать подключение к сетевому напряжению и могут обеспечивать прямое заземление пациента с помощью рабочих частей (см. также примечание к A.5.2).
Исходя из этих соображений, требования, указанные в A.2.3, означают, что если пациент находится на полном напряжении сети:
a) ток, который будет течь между пациентом и рабочей частью типа В, будет ограничен только сопротивлением пациента и может привести к поражению электрическим током;
b) максимальный ток, который может протекать между пациентом и рабочей частью типа BF, вряд ли приведет к поражению электрическим током, при условии, что рабочая часть не находится в прямом контакте с сердцем;
c) максимальный ток, который может протекать между пациентом и рабочей частью типа CF, вряд ли приведет к поражению электрическим током, даже если рабочая часть находится в прямом контакте с сердцем.
А.3.4 Непосредственное воздействие на сердце
МЭК 601-1 требует, чтобы все медицинские электрические изделия, предназначенные для непосредственного применения на сердце, имели рабочие части типа CF. При условии, что приняты меры предосторожности для обеспечения того, чтобы внутрисердечный контур (прямо или косвенно) не контактировал с каким-либо заземленным объектом, это обеспечивает высокую степень защиты от поражения электрическим током через рабочую часть, даже если:
a) произошел обрыв провода защитного заземления для другого элемента оборудования, чрезмерный ток утечки, утечка в корпусе или ток утечки пациента протекает через пациента от неисправного оборудования; или
b) напряжение между некоторыми заземленными объектами в непосредственной близости от пациента (включая электрическое оборудование класса I) приводит к чрезмерному току утечки, протекающему в пациенте из-за разности потенциалов в системе заземления.
Некоторые медицинские электрические изделия, которые предназначены для широкого спектра применений, также могут быть пригодными для использования в непосредственном контакте с сердцем. Требования к маркировке классификации МЭК 601-1 гарантируют, что пользователь может идентифицировать оборудование с рабочими частями типа CF для таких применений (см. также А.4).
А.3.5 Не контактирующие с сердцем (не внутрисердечные) применения
За исключением требования о том, что в оборудовании, предназначенном для непосредственного применения на сердце, требуются рабочие части типа CF, МЭК 601-1 не содержит каких-либо требований в отношении классификации, необходимой для конкретных типов оборудования. Подразумевается, что рабочие части типов CF, BF или B обеспечивают достаточную степень безопасности.
Тем не менее некоторые частные стандарты запрещают применять компоненты типа B в определенных видах оборудования. Обоснование заключается в том, что рабочие части типа B запрещены, когда соединения с пациентом создают особенно низкий импеданс или постоянные соединения с пациентом (например, электроды для записи или контроля ЭКГ).
Для не внутрисердечных применений существенное различие между рабочими частями F-типа и типа B состоит в том, что, если пациент случайно контактирует с сетевым напряжением, рабочая часть F-типа ограничивает ток, протекающий через него, до достаточно безопасного уровня, в то время как ток, протекающий в рабочей части типа B, может быть ограничен только сопротивлением пациента и может представлять серьезную опасность поражения электрическим током.
Следующие соображения должны быть приняты во внимание при разработке стандартов на продукцию при принятии решения о том, должны ли применяться рабочие части F-типа для конкретных типов оборудования, которые не предназначены для непосредственного применения на сердце:
a) обеспечивают ли соединения пациента особенно низкий импеданс или постоянное соединение с пациентом (например, электроды для записи или мониторинга ЭКГ); или они не приводят к большему риску, чем обычный, случайный контакт с любым другим предметом немедицинского электрического оборудования.
В первом случае рабочие части F-типа могут дать существенное преимущество. В последнем случае рабочая часть типа B может обеспечить адекватный уровень безопасности;
b) предназначено ли оборудование для использования в изоляции (например, в кабинете врача или в доме пациента), где пациент вряд ли напрямую подключен к другому медицинскому электрическому изделию; или в больничной среде, в которой ряд медицинских электрических изделий может использоваться одновременно.
В первом случае рабочие части типа B могут обеспечивать достаточный уровень безопасности. В последнем случае рабочие части F-типа могут дать значительное преимущество;
c) предназначено ли оборудование для использования в местах, где в электроустановке предусмотрены дополнительные средства защиты (например, устройства защитного отключения или изолированные источники питания). В этом случае рабочие части типа B могут обеспечивать достаточный уровень безопасности;
d) является ли оборудование оборудованием того типа, для которого могут быть предоставлены рабочие части F-типа без технических трудностей или чрезмерных затрат.
А.4 Пригодность рабочих частей для прямого соединения с сердцем
В соответствии с требованиями первого и второго изданий МЭК 601-1 все медицинские электрические изделия с рабочей частью, предназначенные изготовителем для непосредственного нанесения на сердце, должны иметь маркировку типа CF и должны соответствовать соответствующим требованиям.
Тем не менее маркировка типа CF также размещается на другом оборудовании, которое соответствует соответствующим требованиям, даже если оборудование указано изготовителем как неподходящее для подключения к сердцу.
Может быть аргумент в пользу ограничения использования знака типа CF на рабочих частях, которые предназначены изготовителем для прямого подключения к сердцу, но это не является требованием первого или второго издания МЭК 601-1.
А.5 Высокие уровни функционального тока и напряжения пациента
A.5.1 Общие положения
Некоторые медицинские электрические изделия подают существенное напряжение и ток через соединения пациента (например, кардиодефибрилляторы и высокочастотное хирургическое оборудование). Это обозначено в МЭК 601-1 как функциональный ток пациента.
A.5.2 Выдерживаемое напряжение дефибриллятора
Некоторые рабочие части имеют импедансы для ограничения потока функционального тока пациента от кардиодефибрилляторов. Тем не менее такие рабочие части все еще могут быть типов CF, BF или B, в зависимости только от способности выдерживать сетевое напряжение.
Следующие символы используются для обозначения классификации деталей с защитой от дефибриллятора:
|
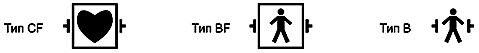
Примечание - В соответствии с требованиями МЭК 601-1 рабочие части, в которых ток утечки пациента превышает 0,50 мА при напряжении сети, подключенном к рабочей части, должны классифицироваться как тип B. Все рабочие части, которые обеспечивают прямое заземление на пациента, относятся к типу B. Однако рабочие части типа B не обязательно обеспечивают прямое заземление пациента.
A.5.3 Устойчивость к использованию совместно с высокочастотным (ВЧ) хирургическим оборудованием
Некоторые рабочие части предназначены для ограничения протекания функционального тока пациента от высокочастотного хирургического оборудования. Данные рабочие части могут быть типов CF, BF или B, в зависимости только от способности выдерживать сетевое напряжение. В первом или втором издании МЭК 601-1 отсутствуют символы для обозначения оборудования, которое будет продолжать работать при наличии высокочастотного хирургического оборудования.
A.5.4 Требования к изоляции выхода
Поскольку пациент может быть иным образом заземлен, источник функционального тока пациента от оборудования, такого как дефибрилляторы и высокочастотное хирургическое оборудование, должен быть изолирован от земли, чтобы предотвратить его прохождение по непредусмотренным путям. Однако эта изоляция должна соответствовать выходному напряжению и частоте, а не напряжению питающей сети, как это требуется для рабочих частей F-типа.
Приложение В
Роль стандартов в оценке и одобрении медицинских электрических изделий
Таблица В.1 - Опасности и причины
|
В таблице B.1 представлена часть матрицы, в которой опасности указаны в боковике, а причины - в головке таблицы.
МЭК 601-1 требует, чтобы опасности не могли возникнуть по какой-либо причине, то есть для каждого элемента всей матрицы должны быть приняты соответствующие меры для предотвращения конкретной опасности, возникающей в результате конкретной причины.
На практике некоторые опасности вряд ли являются следствием некоторых причин (например, сбой программного обеспечения вряд ли приведет к пожару). Для таких комбинаций никаких особых мер безопасности не требуется. Эта ситуация обозначена как N/A в соответствующем поле матрицы.
Другие комбинации требуют соответствующих мер безопасности (например, меры необходимы для предотвращения поражения электрическим током из-за отказа компонента и предотвращения функциональных опасностей из-за ошибки оператора). Ссылки в полях указывают соответствующие требования во втором издании МЭК 601-1. Для простоты в таблице B.1 не указаны все соответствующие номера разделов.
В некоторых случаях, в которых следует принимать меры для предотвращения конкретной опасности, возникающей из-за определенной причины, в серии стандартов МЭК 601 требования еще не определены (например, отказ компонента или сбой программного обеспечения могут вызвать функциональную опасность). Эта ситуация показана в таблице B.1 знаком "X" в поле.
Таблица B.2 - Роль стандартов в общей безопасности
|
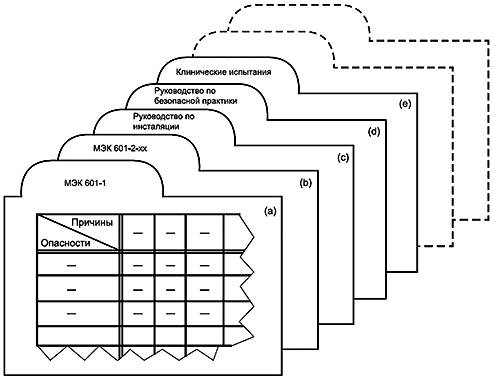
Таблица B.2 иллюстрирует, как общая безопасность включает в себя факторы, отличные от тех, которые рассматривают в серии МЭК 601. Как и в таблице B.1, опасности представлены вертикально, а причины представлены горизонтально на карте кэша.
Карта (а) представляет комбинации причин и опасностей, которые рассматриваются в МЭК 601-1.
Карта (b) представляет дополнительные причины и опасности, которые устраняются частными стандартами (например, функциональные опасности из-за отказов компонентов, которые приводят к несоблюдению основных требований к производительности). Такие опасности могут быть рассмотрены только в общих терминах в МЭК 601-1, но подробные требования к испытаниям должны быть включены в соответствующий частный стандарт.
Некоторые сочетания причин и опасностей рассматривают с помощью требований к установке, карты (c) или с помощью безопасной практики, карты (d).
Стандарты продукции, требования к установке и инструкции по безопасной практике не дают ответов на все вопросы при оценке клинической приемлемости новых технологий медицинского электронного оборудования. Дополнительные формы оценки, такие как предпродажные клинические испытания, могут быть сочтены необходимыми, карта (е).
Приложение С
Улучшение общей безопасности
С.1 Общая безопасность
Как уже говорилось во введении к настоящему стандарту, безопасное использование медицинского электрического изделия достигается за счет комбинации факторов, которые включают в себя:
a) функции, связанные с безопасностью, встроенные в оборудование;
b) осведомленность пользователей о потенциальных рисках, связанных с оборудованием, и, следовательно, о необходимости надлежащего обучения; а также
c) соответствующая установка, обслуживание и т.д.
Ранние стандарты на медицинские электрические изделия в основном касались безопасности конструкции оборудования, а разрыв между этим и общим желаемым уровнем безопасности определялся осведомленностью пользователей.
Однако практический эффект дальнейшего улучшения безопасности, связанной с оборудованием, может иметь неблагоприятные последствия в некоторых обстоятельствах из-за неправильной реакции людей. Эта проблема показана на рисунке С.1.
Например, медсестры могут привыкнуть полагаться на системы сигнализации, которые теперь включены в определенные устройства, вместо того, чтобы быть осторожными в том, что они делают. Когда им приходится использовать оборудование, которое не имеет таких сигналов тревоги, они гораздо чаще делают что-то не так.
C.2 Улучшенные стандарты продукта - непредвиденные последствия
Следовательно, одним из потенциально нежелательных побочных эффектов улучшения стандартов безопасности оборудования может быть снижение осведомленности о рисках и уровня ответственности, необходимого для пользователей.
Типичным примером является оборудование для диализа:
Несколько лет назад новые разработки в области почечного диализа привели к созданию дополнительных впускных отверстий для альтернативных концентратов в аппаратах для гемодиализа (бикарбонатных концентратов). Однако требуемое разбавление не было одинаковым во всех случаях (как в случае ацетатных концентратов), и дополнительные входные отверстия значительно увеличивали вероятность фатальной ошибки.
С первыми такими машинами было неясно, будет ли обнаружено неправильное соединение концентратов, и пользователи были обучены проверять окончательное решение перед диализом пациента. Пользователи были осведомлены о рисках, и было зарегистрировано очень мало несчастных случаев.
Более поздние машины включали функции безопасности (например, сигналы тревоги), чтобы предотвратить неправильное соединение концентратов. Тем не менее о происшествиях было сообщено, когда эти машины были введены и было обнаружено, что некоторые концентраты имеют слегка отличающиеся составы, которые могут помешать работе этих функций безопасности.
Проблема заключалась в том, что пользователи выработали привычку полагаться на сигналы тревоги на новых машинах и больше не проверяли окончательное решение перед набором номера. Это привело к несчастным случаям, которые не произошли бы с более ранними машинами, которые включали меньше функций безопасности.
Пользователи считали, что эти несчастные случаи были по вине изготовителей, потому что защитная система была неадекватной. Реальная проблема заключалась в том, что из-за улучшений продукта пользователи чувствовали себя менее ответственными за безопасность пациента.
Следует помнить, что новое медицинское электрическое изделие часто следует использовать вместе со старыми устройствами, которые не обеспечивают такой же уровень безопасности.
Ответом на эту проблему является соответствующее обучение пользователей и, возможно, необходимость ограничить использование более старых, менее безопасных устройств специально обученным персоналом.
Кто-то может спросить, почему эти "менее безопасные" устройства не следует выводить из эксплуатации. Практический ответ лежит в экономической реальности. Медицинские учреждения должны продолжать использовать старое оборудование, даже если оно не соответствует новым стандартам из-за ограничений по стоимости.
Продолжая на примере диализных аппаратов:
"Усовершенствования", которые были включены в современные машины, имели два последствия:
- появление нового класса аварий; а также
- снижение осведомленности о риске и, наконец, чувства ответственности пользователей.
Легко предвидеть следующий этап. Изготовители попытаются еще больше улучшить функции безопасности, и они, вероятно, будут включены в будущие стандарты.
Однако ошибочно полагать, что эту проблему можно решить только путем повышения безопасности оборудования. Существует большой риск того, что из-за поведения человека осведомленность пользователей будет продолжать снижаться, в то время как безопасность продукта возрастает (поскольку пользователи знают, что он увеличился), что приводит, в лучшем случае, к улучшению общей безопасности.
|
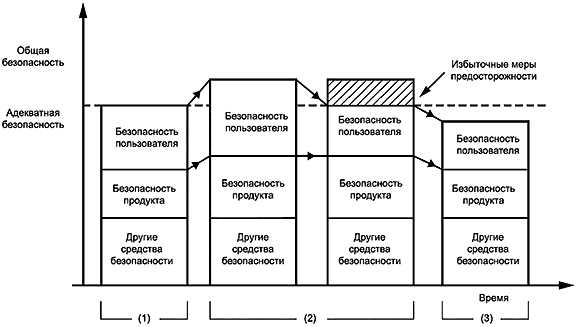
Рисунок С.1 - Общая безопасность
C.3 Потребность пользователей в осведомленности о риске
Включение функций безопасности в медицинское электрическое изделие не должно препятствовать выполнению пользователями соответствующих проверок (обозначенных как "избыточные меры предосторожности", см. C.1), так же, как если бы эти функции безопасности не существовали. Пользователи должны быть осведомлены о рисках и своих обязанностях, так как функции безопасности могут потерпеть неудачу, не становясь очевидными.
Это особенно относится к старому оборудованию, которое не соответствует основным требованиям к производительности. Такое оборудование может включать сигнализацию или защитные системы, отказ которых может быть не обнаружен пользователями.
Принцип отказоустойчивости должен применяться как к пользователям, так и к оборудованию. Избыточные меры предосторожности, принятые пользователями, должны обеспечивать, чтобы в случае "первой ошибки" пользователя при эксплуатации оборудования не было риска для пациента.
Такие меры предосторожности должны быть неотъемлемой частью обучения пользователей и должны быть четко указаны в эксплуатационной документации.
С.4 Вывод
Эти примеры иллюстрируют важность руководящих принципов и стандартов осведомленности пользователей для содействия пониманию необходимости обеспечения безопасности пользователя как одного из средств достижения общей безопасности. Это должен быть рекомендуемый подход для решения этой проблемы между пользователями и медицинским электрическим изделием.
При разработке или пересмотре стандартов безопасности продукции следует помнить, что общая безопасность требует общего глобального подхода, и адекватный уровень безопасности не может быть достигнут только путем повышения безопасности оборудования.
Приложение D
Факторы безопасности
МЭК 601-1 определяет и использует термин "коэффициент безопасности" в очень специфическом контексте, связанном с напряжениями, которые могут нести механически нагруженные элементы. Однако концепция факторов безопасности лежит в основе, без ссылки, допустимых пределов (пороговых предельных значений или TLV) для неизвестного количества параметров в стандартах серии МЭК 601.
При оценке риска опасные процессы могут быть стохастическими или нестохастическими. Стохастические процессы, такие как канцерогенез, являются результатом случайного возникновения биологических событий в клетках в ткани-мишени. В результате нельзя с уверенностью предсказать реакцию конкретного лица при определенных условиях воздействия. Использование факторов безопасности неуместно при работе со случайными процессами.
Как правило, правильное использование факторов безопасности при достижении приемлемых уровней воздействия на человека основывается на предположении, что существует пороговая доза, ниже которой не возникает никаких побочных эффектов. При условии, что изучаемое явление показывает порог, ниже которого не происходит никаких неблагоприятных воздействий, подход с использованием коэффициента безопасности устанавливает TLV ниже этого порога. Стохастические эффекты, для которых порог может не существовать, требуют других методов оценки риска.
Нестохастические эффекты, такие как электрически индуцированная тетания, более однородны по своей природе. Они происходят более предсказуемым образом после воздействия определенного критического уровня исследуемого стимула или токсиканта.
Для нестохастических эффектов один из подходов к установлению приемлемых уровней предельных значений воздействия на человека или пороговых значений заключается в применении подходящего фактора безопасности или коэффициента неопределенности к дозе или уровню воздействия, при которых в контролируемых исследованиях не наблюдалось побочных эффектов. Деление уровня ненаблюдаемого эффекта (NOEL) в контролируемых испытаниях на коэффициент безопасности (SF) дает пороговое предельное значение (TLV) для людей (TLV=NOEL/SF).
Следует выбрать TLV, для которого почти все люди не будут испытывать никаких побочных эффектов после повторного нормального воздействия. Наблюдения за профессионально незащищенным населением должны поддержать этот выбор.
Там, где NOEL был установлен, TLV часто включает 100-кратный коэффициент безопасности (SF=100). Это допускает 10-кратное изменение восприимчивости между животным и человеком, а также 10-кратное изменение индивидуальной восприимчивости в популяции людей. Когда уровень отсутствия эффекта отсутствует, коэффициент безопасности может быть применен к самому низкому уровню наблюдаемого эффекта (LOEL), с дополнительным коэффициентом 5- или 10-кратного включения, чтобы скорректировать отсутствие уровня отсутствия эффекта.
Опасные последствия большей степени тяжести могут потребовать более высоких факторов безопасности. И наоборот, в медицинской сфере пациент уже может подвергаться значительному риску из-за болезни и из-за терапевтической процедуры с использованием оборудования. В этом случае использование более низкого запаса прочности может быть уместным.
В некоторых случаях можно определить приемлемый уровень риска и соответствующий уровень воздействия. В таких случаях коэффициент безопасности должен быть равен единице (SF=1).
Правильное использование факторов безопасности является сложным и может быть только при наличии данных о воздействии опасности на человека или животное. Кроме того, использование коэффициента безопасности потребует оценки того, насколько приемлемы риски.
Коэффициенты безопасности не должны использоваться для установления пределов, если:
a) в сопроводительном обосновании прямо указано, как факторы безопасности повлияли на выбранные пределы;
b) обоснование указывает конкретные используемые факторы безопасности;
c) предоставляются ссылки на данные, которые комитет использовал для установления уровня бездействия или приемлемого уровня воздействия;
d) в обосновании изложены предположения относительно приемлемости рисков.
Приложение Е
История первого издания МЭК 513 и серии МЭК 601
Первое издание МЭК 513
Первое издание МЭК 513 было подготовлено подкомитетом 62А МЭК. Он был опубликован в 1976 году в виде отчета МЭК с заголовком "Основные аспекты философии безопасности электрооборудования, применяемого в медицинской практике".
В предисловии говорится, что отчет должен использоваться в качестве руководства для стандартов, которые должны быть выпущены техническим комитетом МЭК 62.
В отчете обсуждается клиническая среда, в которой используются медицинские электрические изделия, и многие опасности, характерные для данного типа электрооборудования. Он определяет необходимость стандартов безопасности во всех следующих областях для обеспечения безопасного использования медицинских электрических изделий:
a) нормы безопасности, касающиеся конструкции оборудования;
b) требования к установке для обеспечения безопасности электрооборудования в некоторых клинических применениях; а также
c) руководство пользователя, обеспечивающее безопасное обслуживание и эксплуатацию оборудования.
Отчет содержит общую философию для разработки серии МЭК 601 и обоснование некоторых требований, приведенных в первом издании МЭК 601-1. Отчет устанавливает следующее:
________________
МЭК 601-1 впоследствии не предусматривал оборудование типа H); МЭК 601-1:1977 "Безопасность медицинского электрического оборудования. Часть 1. Общие требования".
a) в течение срока службы оборудования может возникнуть ряд неисправностей, некоторые из которых могут быть предотвращены путем периодического осмотра и технического обслуживания. Эффект от неисправностей, которые могут остаться незамеченными, может быть предотвращен с помощью соответствующей конструкции и достаточных факторов безопасности. Вероятность возникновения двух независимых отказов одновременно считается очень малой, и возможна защитная система, в которой любой отдельный сбой может быть обнаружен до возникновения второго сбоя;
b) медицинские электрические изделия должны использовать защиту класса I, класса II или класса III для защиты от первого повреждения изоляции, но не класса 0 (впоследствии в МЭК 601-1 не предусматривались медицинские электрические изделия класса 0 или класса III). Чрезвычайно низкое напряжение должно быть доступно только на участках, которые вряд ли подключатся к пациенту;
c) ограниченные пределы тока утечки должны применяться к медицинскому электрическому оборудованию, которое должно быть классифицировано в новой системе как типы H, B, BF или CF, в зависимости от уровня тока утечки и изоляции от пациента;
d) наивысшая степень защиты (тип CF) требуется для всего оборудования, предназначенного для внутрисердечного применения. Другое оборудование может быть типов B, BF или CF.
Для оборудования, которое не предназначено для внутрисердечного применения, единственным обоснованием для отдельных классификаций типов BF и B является "описание степени и качества предусмотренных защитных мер". Однако, хотя МЭК 513 (и МЭК 601-1) заявляют, что для такого оборудования достаточно любого типа классификации, все большее число частных стандартов в настоящее время запрещает конкретные типы оборудования типа В, в которых "рабочая часть" устанавливает тесный контакт с пациентом.
За исключением внутрисердечных процедур, в отчете также не объясняется, как пользователь должен применять систему классификации тип CF/BF/B в клинической практике, чтобы определить, какой уровень защиты требуется для конкретной процедуры. Тем не менее он определяет необходимость применения руководств для решения этой и других проблем, в том числе необходимость дополнительных мер безопасности при электрической установке во время некоторых процедур. Тем не менее на момент подготовки отчета, лежащего в основе настоящего стандарта, МЭК разработало лишь несколько таких руководств по применению, и они не дают исчерпывающей информации об установке и/или о безопасном использовании медицинских электрических изделий.
В результате применения философии, изложенной в первом издании МЭК 513, первоначальный подход в техническом комитете 62 МЭК заключался в разделении стандартов на медицинские электрические изделия на отдельные требования "безопасности" и "функциональных характеристик". Общий стандарт и серия частных стандартов МЭК 601-2-xx должны были ограничиваться основными вопросами безопасности, чтобы гарантировать, что при нормальном использовании и при определенных условиях неисправности оборудование не будет представлять необоснованную опасность для пациента, оператора или окружающей среды. Там, где это необходимо, другие вопросы производительности, не связанные непосредственно с физической безопасностью, будут изложены в отдельной серии стандартов МЭК 601-3-xx.
Первое издание МЭК 601-1
Первое издание МЭК 601-1 было подготовлено подкомитетом 62А МЭК и опубликовано в 1977 году под названием "Безопасность медицинских электрических изделий. Часть 1. Общие требования".
Область применения первого издания МЭК 601-1 гласит:
"Настоящий стандарт применяется к МЕДИЦИНСКИМ ЭЛЕКТРИЧЕСКИМ ИЗДЕЛИЯМ, предназначенным для использования квалифицированным персоналом в ПАЦИЕНТНОЙ СРЕДЕ или под его надзором, или связанному с ПАЦИЕНТОМ таким образом, что это напрямую влияет на безопасность людей или животных в этой среде.
Настоящий стандарт устанавливает требования к транспортированию, хранению, установке, использованию и техническому обслуживанию такого ОБОРУДОВАНИЯ в условиях окружающей среды, указанных в подразделе 1.4 или изготовителем, или в соответствии с предписаниями в частных стандартах.
Хотя данный стандарт в первую очередь касается безопасности, он содержит некоторые требования, касающиеся надежной работы, когда это связано с безопасностью.
Опасности, возникающие в результате физиологических эффектов, вызванных предусмотренной функцией ОБОРУДОВАНИЯ, охватываемых настоящим стандартом, не рассматриваются.
(Неисчерпывающее) обследование МЕДИЦИНСКОГО ЭЛЕКТРИЧЕСКОГО ИЗДЕЛИЯ приведено в приложении А."
Целью первого издания МЭК 601-1 является:
"...установить требования к удовлетворительному уровню безопасности МЕДИЦИНСКОГО ЭЛЕКТРИЧЕСКОГО ИЗДЕЛИЯ, используемого в ПАЦИЕНТНОЙ СРЕДЕ, и послужить основой для требований безопасности частных стандартов для ОБОРУДОВАНИЯ".
Медицинское электрическое изделие определено в первом издании МЭК 601-1 как:
"Электрическое изделие, предназначенное для использования в среде ПАЦИЕНТА и связанное таким образом с ПАЦИЕНТОМ, что это может повлиять на безопасность ПАЦИЕНТА".
Второе издание МЭК 601-1
Второе издание МЭК 601-1 было также подготовлено подкомитетом 62А МЭК и опубликовано в 1988 году под названием "Изделия медицинские электрические. Часть 1. Общие требования безопасности".
Область распространения второго издания МЭК 601-1 гласит:
"Настоящий стандарт применяется к безопасности МЕДИЦИНСКОГО ЭЛЕКТРИЧЕСКОГО ИЗДЕЛИЯ (как определено в пункте 2.2.15).
Хотя данный стандарт в первую очередь касается безопасности, он содержит некоторые требования, касающиеся надежной работы, когда это связано с безопасностью.
УГРОЗЫ БЕЗОПАСНОСТИ, вытекающие из предполагаемой физиологической функции ОБОРУДОВАНИЯ, охватываемого настоящим стандартом, не рассматриваются.
Приложения в этом стандарте не являются обязательными, если они не сделаны явным заявлением в основном тексте".
Целью второго издания МЭК 601-1 является:
"...определить требования к безопасности МЕДИЦИНСКОГО ЭЛЕКТРИЧЕСКОГО ИЗДЕЛИЯ и послужить основой для требований безопасности частных стандартов".
Медицинское электрическое изделие определено во втором издании МЭК 601-1 как:
"Электрическое изделие, снабженное не более чем одним подключением к конкретной сети электропитания и предназначенное для диагностики, лечения или мониторинга ПАЦИЕНТА в условиях медицинского надзора и которое имеет физический или электрический контакт с ПАЦИЕНТОМ и/или передает энергию ПАЦИЕНТУ или от него, и/или обнаруживает такую передачу энергии ПАЦИЕНТУ или от него."
Основные различия между первым и вторым изданиями МЭК 601-1 описаны в A.1.2 приложения A "Общее руководство и обоснование", включение которого является нововведением во втором издании МЭК 601-1.
Частные стандарты
За публикацией первого издания МЭК 601-1 последовала первая серия стандартов МЭК 601-2-xx, устанавливающая частные требования для конкретных видов медицинских электрических изделий. Требования частных стандартов имеют приоритет над требованиями МЭК 601-1 для конкретного типа оборудования.
На момент публикации документа, послужившего основой настоящего стандарта, МЭК и ИСО (Международная организация по стандартизации) были опубликованы приблизительно тридцать частных стандартов в формате МЭК 601-2, а многие другие частные стандарты находились в стадии разработки.
Частные стандарты в серии МЭК 601-2-xx разработаны подкомитетом 62B "Оборудование для диагностической визуализации", подкомитетом 62C "Высокоэнергетическое оборудование и оборудование для ядерной медицины" и подкомитетом 62D "Электромедицинское оборудование".
Подкомитеты 62B и 62C разработали ряд частных стандартов безопасности и основных функциональных характеристик в серии МЭК 601-2-xx. Эти подкомитеты также подготовили ряд других стандартов производительности, но они не были опубликованы как части серии МЭК 601-3-xx.
Подкомитет 62D также разработал много частных стандартов в серии МЭК 601-2-xx. В каждом случае существенные требования к производительности были включены в форме минимальных требований с критериями прохождения/неуспешного испытания и/или требований об обязательном раскрытии. Потребность в других стандартах производительности еще не привела к публикации дополнительных стандартов.
Дополнительные стандарты
В дополнительных стандартах серии МЭК 601-1-хх определены общие требования безопасности, которые применимы:
a) к группам медицинских электрических изделий (например, радиологическое оборудование) или
b) специфической характеристике всех медицинских электрических изделий, которая не полностью учтена в общем стандарте (например, электромагнитная совместимость).
Первый дополнительный стандарт, МЭК 601-1-1, был опубликован в 1992 году под названием "Изделия медицинские электрические. Часть 1. Общие требования безопасности 1. Дополнительный стандарт. Требования безопасности для медицинских электрических систем".
Хотя это и не упоминается во втором издании МЭК 601-1, эти дополнительные стандарты являются частью общего стандарта. Таким образом, требования частного стандарта имеют приоритет перед требованиями общего стандарта.
Основные стандарты безопасности
Основные стандарты безопасности МЭК содержат требования, которые касаются конкретных аспектов (характеристик) безопасности, которые являются общими для большинства медицинских электрических изделий. Эти требования предназначены:
a) для обеспечения согласованности стандартов МЭК в областях, которые являются общими для ряда комитетов МЭК, избегая, таким образом, противоречивых требований;
b) способствования сокращению объема стандартов МЭК, тем самым экономя материал, затраты на печать и затраты для разработчиков стандартов и пользователей;
c) улучшения взаимопонимания инженеров разных технических дисциплин; а также
d) улучшения согласованности в стандартах МЭК.
Руководство МЭК 104 призывает технические комитеты, разрабатывающие стандарты на продукцию, ссылаться на основные стандарты безопасности везде, где это возможно.
________________
Руководство МЭК 104:1984 "Руководство по разработке стандартов безопастости".
Методы испытаний
Было предпринято согласованное действие для обеспечения того, чтобы требования в первом и втором изданиях МЭК 601-1 сопровождались воспроизводимыми методами испытаний с точно указанными критериями "годен/не годен". Намерение состояло в том, чтобы создать стандарт, который будет последовательно применяться различными пользователями.
Эта цель была в значительной степени достигнута. Предложения о новых требованиях безопасности часто отклонялись, если не могли быть разработаны воспроизводимые требования соответствия, даже когда существовала потребность в дополнительных требованиях. Однако некоторые требования МЭК 601-1 зависят от решений, касающихся возможности возникновения конкретных неисправностей, приводящих к опасности. Во многих случаях опасность, которая очевидна для одного человека с медицинской точки зрения, не определяется другим пользователем стандарта.
Эта проблема возникает из-за того, что в стандартах используются термины "условие единичного нарушения" и "угроза безопасности".
Условие единичного нарушения определяется во втором издании МЭК 601-1 как: "Условие, при котором одно средство защиты от УГРОЗЫ БЕЗОПАСНОСТИ в ОБОРУДОВАНИИ неисправно или имеет место внешнее ненормальное состояние (см. 3.6)".
Угроза безопасности определяется во втором издании следующим образом:
"Потенциально вредное воздействие на ПАЦИЕНТА, других людей, животных или окружающую среду, возникающее непосредственно от ОБОРУДОВАНИЯ".
Во втором издании МЭК 601-1 говорится, что каждый отдельный отказ компонента, который может быть разумно предвиден и который не связан с предполагаемым применением, должен быть исследован, чтобы определить, может ли это привести к угрозе безопасности. Более восьмидесяти требований касаются необходимости изучения условий неисправности, "если существует опасность". Поэтому существует некоторая неопределенность в отношении объема испытаний, требуемого МЭК 601-1, и это приводит к непоследовательному толкованию разработчиками оборудования и испытательными лабораториями.
УДК 615.841:006.354 | ОКС 11.040.01 | ||
Ключевые слова: изделия медицинские электрические, электрическое оборудование, безопасность, функциональные характеристики, требования, испытания, стандарты | |||
Электронный текст документа
и сверен по:
, 2020

{kind=link}