ГОСТ Р ИСО 14607-2017
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Имплантаты хирургические неактивные
ИМПЛАНТАТЫ МОЛОЧНЫХ ЖЕЛЕЗ
Частные требования
Non-active surgical implants. Mammary implants. Particular requirements
ОКС 11.040.40
ОКП 93 9300
Дата введения 2018-01-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "ЦИТОпроект" (ООО "ЦИТОпроект") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 453 "Имплантаты в хирургии"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 22 февраля 2017 г. N 64-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14607:2007* "Имплантаты хирургические неактивные. Имплантаты молочных желез. Частные требования" (ISO 14607:2007 "Non-active surgical implants - Mammary implants - Particular requirements", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
В настоящем стандарте в дополнение к требованиям, приведенным в стандарте уровня 1, представлен метод применения неактивных хирургических имплантатов, основанный на фундаментальных принципах ISO/TR 14283, а также метод, в котором согласованы соответствующие общие требования относительно использования медицинских изделий в клинической практике (см. приложение I директивы 93/42/ЕЕС от 14 июня 1993 г., а также изменения, внесенные директивой Комиссии 2003/12/CE).
Дополнительная информация по имплантатам молочных желез приведена в директиве 93/42/EEC, изданной информационным отделом Европейской комиссии по общественным и национальным вопросам применения имплантатов молочных желез.
Существует три уровня европейских стандартов, в которых рассматриваются требования, предъявляемые к пассивным хирургическим имплантатам (уровень 1 является наивысшим):
- уровень 1 - Общие требования к неактивным хирургическим имплантатам;
- уровень 2 - Частные требования к группам неактивных хирургических имплантатов;
- уровень 3 - Особые требования к отдельным типам неактивных хирургических имплантатов.
Настоящий стандарт является стандартом уровня 2 и содержит частные требования для группы имплантатов молочных желез.
Стандарт уровня 1, ИСО 14630, устанавливает требования, применимые ко всем неактивным хирургическим имплантатам. В ИСО 14630 также приведена информация о дополнительных требованиях, которые отражены в стандартах уровней 2 и 3.
Для соответствия всем требованиям необходимо начинать рассмотрение стандарта с наиболее низкого уровня.
1 Область применения
Настоящий стандарт устанавливает частные требования к имплантатам молочных желез в клинической практике.
В отношении безопасности настоящий стандарт устанавливает требования к показателям назначения, особенностям конструкции, материалам, оценке конструкции, производству, стерилизации, упаковке, а также информации, предоставляемой изготовителем.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты*, которые необходимо учитывать при использовании настоящего стандарта. В случае ссылок на документы, в которых приведена дата утверждения, необходимо пользоваться только указанной редакцией. В том случае, когда дата утверждения не приведена, следует пользоваться последней редакцией ссылочных документов, включая любые поправки и изменения к ним:
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ISO 34-1:2004 Rubber, vulcanized or thermoplastic - Determination of tear strength - Part 1: Trouser, angle and crescent test pieces (Резина вулканизированная или термопластичная. Определение прочности на разрыв. Часть 1. Раздвоенные, угловые и серповидные образцы)
ISO 37 Rubber, vulcanized or thermoplastic - Determination of tensile stress-strain properties (Резина вулканизированная или термопластичная. Определение упругопрочностных свойств при растяжении)
ISO 10993-1 Biological evaluation of medical devices - Part 1: Evaluation and testing (Биологическая оценка медицинских изделий. Часть 1. Оценка и испытания)
ISO 14155-1:2003 Clinical investigation of medical devices for human subjects - Part 1: General requirements ISO (Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования)
ISO 14155-2:2003 Clinical investigation of medical devices for human subjects - Part 2: Clinical investigation plans (Руководство no проведению клинических испытаний медицинских изделий. Часть 2. Планирование клинических испытаний)
ISO 14630 Non-active surgical implants - General requirements (Неактивные хирургические имплантаты. Общие требования)
NF S 99-401:1994 Medical devices - Silicone elastomer of medical grade (Медицинские устройства. Силиконовый эластомер медицинского класса)
Примечание - В библиографии приведены информативные ссылки на другие стандарты.
3 Термины и определения
В настоящем стандарте применены термины, приведенные в ИСО 10993-1, ИСО 14155-1, ИСО 14155-2 и ИСО 14630, а также следующие термины с соответствующими определениями:
3.1 передняя проекция: Максимальная высота имплантата при размещении его основания на плоской горизонтальной поверхности при его номинальном объеме.
3.2 основные размеры: Длина имплантата по большой оси и длина по малой оси при размещении его основания на плоской горизонтальной поверхности при его номинальном объеме.
3.3 диффузия: Перемещение материала внутрь и/или наружу имплантата через неповрежденную оболочку.
3.4 инъекционный порт: Часть имплантата, предназначенная для введения иглы с целью изменения объема имплантата.
3.5 имплантат молочной железы: Имплантат с оболочкой, разработанный с целью увеличения или возмещения объема молочной железы и наполняемый производителем либо хирургом.
3.6 клапан: Компонент оболочки, в который вставляется приспособление для накачивания имплантатов с регулируемым объемом.
3.7 оболочка: Оболочка имплантата.
3.8 средства ориентации: Пометка внутри или снаружи имплантата, помогающая хирургу позиционировать имплантат.
3.9 утечка: Вытекание из имплантата вещества из наполнителя или оболочки или вещества, образовавшегося в результате их взаимодействия.
3.10 шов: Уплотнение в месте соединения сплавленных или склеенных материалов.
4 Предполагаемое использование
Применяют требования раздела 4 ИСО 14630.
Особое внимание следует обратить на то, чтобы на клиническом состоянии и безопасности пациента не отразилось действие устройства в течение срока его службы и при условиях его нормального использования.
Примечание 1 - Информация о продолжительности предполагаемого функционирования представлена в 11.6.
Примечание 2 - Информация о пользе применения имплантатов молочной железы представлена в 7.2.
Примечание 3 - Информация о специфических рисках, связанных с имплантатами молочной железы, представлена в разделах 5, 6 и 7.
5 Особенности конструкции
Применяют требования раздела 5 ИСО 14630.
В целях удовлетворения требований, предъявляемых к предполагаемому функционированию, характеристики конструкции должны учитывать возможность обнаружения разрыва имплантата.
Кроме того, должно быть изучено влияние старения материалов.
6 Материалы
Применяют требования раздела 6 ИСО 14630.
Кроме того, при использовании силиконового эластомера применяют требования NF S 99- 401:1994. Особое внимание следует уделить:
- биологической оценке изделия и его компонентов в случае неисправности имплантата;
- стабильности материала (в особенности наполнителя).
7 Оценка конструкции
7.1 Общие требования
Применяют требования 7.1 ИСО 14630.
Имплантаты молочной железы должны быть сконструированы и изготовлены таким образом, что их использование в нормальных условиях и согласно предполагаемому назначению не поставило под угрозу клиническое состояние, безопасность или здоровье пациента. Любые остаточные явления или нежелательные побочные эффекты, которые могут быть связаны с их применением, должны представлять собой приемлемые риски в сравнении с пользой для пациента, принимая во внимание тот факт, что пользой считается прежде всего преимущества эстетического и психологического характера, будь то применение в реконструктивных и/или косметических целях.
Анализ рисков и оценка соответствия должны быть выполнены в отношении наполнителя, оболочки и имплантата молочной железы.
7.2 Доклиническая оценка
7.2.1 Общие требования
Доклиническая оценка имплантатов молочной железы должна соответствовать 7.2 ИСО 14630.
В тех случаях, когда испытание не приведено в настоящем стандарте или имеющееся испытание не применимо, производитель должен предоставить описание альтернативного утвержденного метода испытания и подготовки используемых образцов. Адекватность критериев соответствия/несоответствия, принятых для оценки, должна быть проверена перед проведением испытания.
Все испытания следует проводить на готовых стерилизованных изделиях или компонентах.
Выбранный размер образца основан на статистических расчетах, которые должны быть обоснованы и задокументированы.
Примечание - В отношении утвержденных методов испытаний, доступных для доклинической оценки, настоящий стандарт отражает современный технический уровень.
В случае уместности, для других материалов помимо силикона производитель должен рассмотреть и разработать испытания в соответствии с 7.2.2.
7.2.2 Механические испытания
7.2.2.1 Общие требования
Механические испытания следует проводить в соответствии с приложениями А, В, С, D и Е, результаты которых должны удовлетворять установленным требованиям.
Целью механических испытаний является подтверждение низкой частоты разрыва изделия при нормальных условиях эксплуатации.
7.2.2.2 Целостность оболочки
7.2.2.2.1 Общие требования
Следует оценить целостность оболочки.
Испытания характеристик оболочки из силиконового эластомера должны быть проведены в соответствии с приложением В и удовлетворять установленным требованиям. Кроме того, следует рассмотреть предположение о наихудшем сценарии.
Для других материалов помимо силиконового эластомера должны быть разработаны соответствующие испытания.
7.2.2.2.2 Удлинение
Испытание удлинения оболочки из силиконового эластомера следует выполнять в соответствии с В.1, результаты которого должны удовлетворять установленным требованиям.
7.2.2.2.3 Прочность на разрыв
Испытание прочности на разрыв следует проводить в соответствии с В.1.
7.2.2.2.4 Прочность соединений, швов или спаек
Испытание дефектоустойчивости соединений, швов и уплотнений следует проводить в соответствии с В.2, результаты которого должны удовлетворять установленным требованиям.
7.2.2.2.5 Дизайн оболочки
Следует тщательно подходить к выбору материалов, используемых для изготовления оболочки. Поверхности как внутри, так и снаружи оболочки должны минимизировать или предотвращать фрикционное истирание и между поверхностями оболочек, а также между поверхностью оболочки и местом установки имплантата. Если есть вероятность, что такое фрикционное истирание будет серьезной проблемой, изготовитель должен указать все соответствующие испытания, проведенные в целях подтверждения пригодности оболочки для имплантации.
7.2.2.3 Исправность клапана или инъекционного порта
Испытания на предмет исправности клапана или инъекционного порта следует проводить в соответствии с приложением С, результаты которых должны удовлетворять установленным требованиям.
7.2.2.4 Наполнитель
7.2.2.4.1 Общие требования
Физическая совместимость между наполнителем и оболочкой должна быть продемонстрирована путем предоставления долгосрочных данных о функционировании и целостности оболочки.
7.2.2.4.2 Испытание когезивных свойств силиконового геля
Если в качестве наполнителя использован силиконовый гель, следует проводить испытание в соответствии с приложением D на когезивные свойства для оценки как реологических свойств, так и чистоты геля. Испытания должны удовлетворять установленным требованиям в целях оптимизации клинической эффективности и безопасности.
Для других наполнителей помимо силиконового геля следует применять соответствующее и утвержденное испытание когезивных свойств.
7.2.2.5 Прочность имплантата
7.2.2.5.1 Общие требования
Статические испытания прочности на разрыв, усталостной прочности и ударопрочности должны проводить в соответствии с приложением Е и удовлетворять установленным требованиям.
7.2.2.5.2 Испытание усталостной прочности
Испытание усталостной прочности следует проводить в соответствии с Е.1. После испытания при осмотре с десятикратным увеличением на оболочке имплантата должны отсутствовать разрывы, трещины или разрезы.
7.2.2.5.3 Испытание ударопрочности
Испытание ударопрочности следует проводить в соответствии с Е.2, результаты которого должны удовлетворять установленным требованиям.
7.2.2.5.4 Статическое испытание прочности на разрыв
Статическое испытание прочности на разрыв должны проводить в соответствии с Е.3, его результаты следует документировать.
7.2.2.6 Обьем
Объем предварительно заполненного имплантата должен быть в пределах ±2,5% от объема, указанного на упаковке (см. 11.3). Объем должен быть указан в единицах СИ.
7.2.2.7 Размеры
Следует документировать основные размеры предполагаемого дизайна и передней проекции и их допустимые отклонения.
7.2.2.8 Поверхность
Если поверхность специально обработана, чтобы сформировать определенную текстуру, испытания поверхностных характеристик должны быть проведены в соответствии с приложением А, а их результаты зафиксированы.
7.2.3 Оценка химических свойств
7.2.3.1 Общие требования
Следует проводить оценку химических свойств оболочки и наполнителя.
7.2.3.2 Материал оболочки, силиконовый эластомер или материалы с покрытием
Для того чтобы оценить безопасность устройства, необходимо проводить анализ экстрагируемых или выделяемых химических веществ (особенно определение характеристик и количественный анализ материалов с низкой молекулярной массой).
7.2.3.3 Материалы для наполнителя
Должно быть выполнено подробное определение химических характеристик наполнителя.
Должны быть предоставлены долгосрочные данные о стабильности в физиологических условиях и результаты исследований ускоренного старения, чтобы продемонстрировать влияние времени и температуры на физические и химические характеристики устройства.
7.2.3.4 Испытание на утечку
Следует оценивать утечку из целого имплантата.
Примечание 1 - В настоящее время отсутствует утвержденный метод испытания. Для имплантатов с силиконовой оболочкой и силиконовым наполнителем неясно, какая доля утечки происходит из оболочки, а какая - из наполнителя. Методы исследований и требования по данному направлению находятся на стадии рассмотрения.
Примечание 2 - В настоящее время имеются два метода исследования, которые могут предоставить важную информацию относительно утечки: испытание на утечку в АСТМ Ф703-96 и испытание в приложении Н.
7.2.4 Оценка биологических свойств
Биологическая безопасность имплантата должна быть оценена в соответствии с требованиями ИСО 10993-1.
Следует оценить местную и системную токсичность любого вещества, попадающего в организм посредством имплантатов молочной железы. Токсикологическая оценка должна быть основана на химических характеристиках и токсикокинетике материалов, на имеющихся научных данных, касающихся токсикологических опасностей и рисков, и, в случае необходимости, на специфических испытаниях.
Кроме того, следует дать оценку возможных краткосрочных и долгосрочных эффектов, включая цитотоксичность, раздражение, гемосовместимость, генотоксичность, имплантацию, иммунотоксичность и другие формы системной токсичности, репродуктивной токсичности и канцерогенности. Кроме того, следует оценить влияние текстуры поверхности оболочки на окружающие ткани. Результаты оценки должны быть приняты во внимание при анализе рисков. Сведения о токсикокинетике потенциально токсичных или химически активных ингредиентов или продуктов разложения необходимы, так как есть вероятность, что после имплантации они могут попасть в организм в значительных количествах. Исходя из этого, требуется информация о распределении, преобразовании и выведении, применимая к пути воздействия.
Производитель должен определить и обосновать, есть ли необходимость в проведении испытаний in vivo.
Примечание - Оценка может включать исследование соответствующего опыта и/или испытание в реальных условиях. Подобного рода оценка позволяет сделать вывод о том, что проведение испытаний не требуется, если материал имплантата, изготовленный таким же образом, как и материал проектируемого устройства, имеет историю положительного использования в указанной роли (ИСО 10993-1:2003, раздел 6).
7.3 Клиническая оценка
Применяют требования 7.3 ИСО 14630.
Для клинических исследований применяют требования ИСО 14155-1 и ИСО 14155-2.
Примечание - Дополнительная информация по литературным данным представлена в ИСО 14155-1:2003, приложение А.
Целью клинической оценки является оценка количества и частоты местных осложнений, в частности капсулярная контрактура и разрывы/спадение имплантата молочной железы, после правильной имплантации.
Критерии приемлемости (т.е. безопасности и эффективности) клинической оценки должны быть четко определены для того, чтобы обеспечить проведение оценки риск/польза и предоставить доказательства безопасности и эффективности имплантата.
Для получения точного анализа результатов клинические данные должны быть основаны на определенной продолжительности последующего наблюдения за пациентами и на достаточном количестве репрезентативных пациентов.
Клинические данные, предоставляемые производителем, должны быть получены:
a) по результатам проспективных клинических исследований рассматриваемых имплантатов молочной железы, которые проводились по соответствующей программе;
b) на базе литературных данных по предшествующим клиническим исследованиям и данным, основанным на опыте применения имплантатов с теми же параметрами конструкции и функциональными характеристиками, как и оцениваемые имплантаты молочной железы.
Данные, полученные из литературных источников или практики применения других изделий, должны отвечать следующим критериям:
- идентичность оцениваемого изделия и изделий, которые являются предметом отчетов, должна быть продемонстрирована с точки зрения основных конструктивных параметров и эксплуатационных характеристик;
- все используемые данные должны быть получены из контролируемых клинических исследований, должным образом разработанных и проведенных когортных исследований, исследований типа "случай/контроль" или из грамотно заполненных историй болезни. Клинические данные должны быть получены, документированы и критически оценены специалистами, обладающими соответствующим опытом и знаниями. Причем данные должны быть опубликованы в рецензируемых журналах. Доказательства, полученные для научной оценки из разных отчетов об отдельных случаях или из необоснованных экспертных заключений, являются недостаточными.
7.4 Послепродажный контроль
Применяют требования 7.4 ИСО 14630.
В рамках предпродажных требований производитель должен также принять меры для будущей клинической оценки долгосрочного функционирования и частоты осложнений. Эти меры должны предусматривать анализ частоты случаев капсулярной контрактуры, разрыва/спадения и системных эффектов по истечении заранее установленных периодов времени.
8 Производство
Применяют требования раздела 8 ИСО 14630.
9 Стерилизация
Имплантаты должны поставляться в стерильном виде.
Применяют требования 9.1, 9.2 и 9.4 ИСО 14630.
10 Упаковка
Применяют требования раздела 10 ИСО 14630.
11 Информация, предоставляемая производителем
11.1 Общие требования
Применяют требования раздела 11 ИСО 14630. Приведенная ниже информация должна быть указана производителем на этикетке или в сборнике информации, предоставляемой производителем.
Примечание - Данные относительно вопросов отслеживаемости приведены в ИСО 16054 и CR 14060.
11.2 Повторная стерилизация
Если повторная стерилизация недопустима, это должно быть указано в информации, предоставляемой производителем. При возможности проведения повторной стерилизации, применяют требования 9.3 ИСО 14630.
Примечание - Изделие, требующее повторной стерилизации, расценивают как нестерильное.
11.3 Основные размеры
На этикетке должны быть указаны основные размеры, передняя проекция и номинальный объем.
11.4 Влияние диагностических методов
Должно быть указано влияние на имплантат диагностических методов, например маммографии или магнитно-резонансной томографии (МРТ).
11.5 Материалы для наполнителя
Для наполняемых имплантатов производитель должен указать рекомендуемый материал для наполнителя и инструкции по наполнению.
11.6 Информация об ожидаемом сроке службы
Производитель должен предоставить информацию об ожидаемой продолжительности исправного функционирования изделия, предпочтительно выраженную как относительная долговечность имплантата в течение 10 лет или менее, в соответствии с методом Каплана-Мейера или другим статистическим методом. Такая информация включает перечисление факторов, которые могут оказывать существенное влияние на фактический срок службы отдельного имплантата.
Примечание 1 - На практике не представляется возможным точно предсказать фактический срок службы отдельного имплантата. Понятно, что на некоторые факторы производитель повлиять не может. Эти факторы могут оказывать значительное влияние на срок службы отдельного изделия и включают конкретную процедуру имплантации, анатомические особенности и состояние здоровья пациента, его поведение и образ жизни (например, занятия спортом), а также предсказуемые и непредсказуемые внешние механические воздействия.
Примечание 2 - Производитель может выбрать предпочтительный метод отображения информации, касающейся ожидаемого срока службы при определенных условиях. Должна быть включена информация, основанная на статистических данных.
Примеры возможных методов включают указание:
- предполагаемого срока службы, достигающего ожидаемого значения;
- диапазона ожидаемых сроков службы;
- статистической информации, полученной на основе данных об уже имплантированных аналогичных изделиях.
Примечание 3 - Результаты испытаний, указанные в настоящем стандарте, предоставляют полезную информацию для производителя в плане процесса оценки того, каким образом представить данные об ожидаемом сроке службы.
11.7 Информация для пациента
Производитель должен предоставить пользователю информацию, адресованную пациенту, в соответствии с приложением F. Информационная поддержка пациента должна содержать форму информированного согласия. Эта форма должна быть подписана и датирована пациентом.
Примечание - Производитель не несет ответственности ни за передачу информации от пользователя пациенту, ни за подписание пациентом формы информированного согласия.
11.8 Этикетки
Упаковка должна включать не менее двух этикеток для использования в истории болезни пациента и/или карточке пациента. На этикетках должно быть перечислено следующее:
a) название или торговая марка и адрес производителя;
b) информация, необходимая для идентификации имплантата.
Примечание - Она должна включать следующее:
- коммерческое название имплантата;
- описание протеза;
- объем наполнения;
- имя пациента;
- левый или правый (отметить нужное);
c) серийный номер или номер партии.
11.9 Информация для пользователя
Производитель должен предоставить пользователю информацию в соответствии с приложением G.
11.10 Маркировка имплантатов
Применяют требования 11.3 ИСО 14630; кроме того, на имплантате должен быть указан номинальный объем или размер оболочки.
11.11 Карта производителя изделия
Карта производителя изделия должна быть заполнена врачом и выдана пациенту в целях отслеживаемости изделия. Эта карта/стикер должна(ен) включать как минимум следующую информацию:
- фирменное название имплантата;
- размер имплантата;
- серийный номер производителя или номер партии.
Примечание - Производитель не несет ответственности за передачу пациенту карты изделия с информацией.
Приложение А
(обязательное)
Испытание характеристик поверхности
А.1 Принцип
В этом испытании определяют средние характеристики поверхности имплантатов молочной железы.
А.2 Материалы
Имплантат молочной железы.
А.3 Процедура
Характеристики поверхности должны быть исследованы с помощью сканирующей электронной микроскопии (СЭМ) и задокументированы для того чтобы представить средние характеристики поверхности (стандартное отклонение). Характеристики поверхности (например, размер пор, пики и впадины) следует измерять на участке, равном приблизительно 4 мм.
Образцы должны включать не менее трех проб, взятых из основания, окружности и верхушки имплантата (всего девять). Они должны представлять поверхности в целом.
Примечание - Для производственного контроля (контроля качества) разрешается применение других методов (стилус, лазер и т.д.), откалиброванных с помощью СЭМ.
А.4 Отображение результатов
Должны быть записаны средние значения измерений и стандартное отклонение характеристик.
Данные предназначены для сбора информации с целью совершенствования знаний о взаимосвязи текстуры и функционирования.
Примечание - Данные, полученные в результате испытания на текущий момент, не могут быть связаны с функционированием или безопасностью изделия.
Приложение В
(обязательное)
Испытания целостности оболочки
В.1 Материал оболочки
В.1.1 Подготовка образца
Если иное не указано, все испытуемые образцы должны быть подготовлены с использованием заготовок Н2 в соответствии с ИСО 37. Если имплантат предварительно заполнен, силиконовый гель или другой материал должен быть убран. Испытания должны включать отметки на заготовке или средства ориентации, при их наличии на оболочке. Для очистки образцов рекомендуется использовать пропан-2-ол, если необходимо.
Испытания наиболее удобно проводить с помощью имеющейся в продаже машины для испытаний на растяжение. Во всех случаях образцы должны быть надежно зажаты с обоих концов, а затем растянуты с постоянной скоростью 500 мм/мин.
В.1.2 Удлинение
Удлинение следует определять в соответствии с требованиями ИСО 37. Удлинение должно составлять не менее 450%.
В.1.3 Остаточное удлинение
Испытание должны проводить в соответствии с требованиями ИСО 37.
Образец должен быть удлинен до 300%, удерживался при этом удлинении в течение 3 мин, а затем отпущен в исходное положение. После этого остаточное удлинение должно составлять не более 10%.
В.1.4 Прочность на разрыв
Прочность на разрыв следует определять в соответствии с методом С ИСО 34-1:2004. Результаты должны быть записаны.
В.2 Прочность соединений, швов и спаек
В.2.1 Принцип
Это испытание определяет прочность соединений, швов и спаек.
В.2.2 Процедура
В.2.2.1 Соединения, швы и спайки, которые имеют решающее значение для целостности оболочки, должны быть испытаны следующим образом. Образцы должны быть подготовлены и испытаны таким образом, как указано в В.1.1. Образец для испытаний должен быть взят из готового, стерилизованного изделия, как показано на рисунке В.1, таким образом, чтобы стык находился в пределах контрольной части образца.
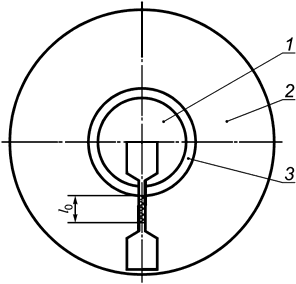
1 - накладка; 2 - оболочка; 3 - стык
Рисунок В.1 - Образец
Участок оболочки, примыкающий к скрепленной области, обозначенный l0 на рисунке В.1, должен выдерживать удлинение на 300% в течение 10 с.
Скрепления, швы, спайки и накладки, не имеющие решающего влияния на целостность оболочки, должны испытываться следующим образом. Образцы должны быть подготовлены в соответствии с В.1.1. Участок оболочки, примыкающий к скрепленной области, должен выдерживать удлинение на 100% в течение 10 с.
Приложение С
(обязательное)
Метод испытания исправности клапана и инъекционного порта
С.1 Исправность клапана
С.1.1 Принцип
Этот метод испытания определяет исправность клапана.
С.1.2 Материалы
Имплантат в сборе.
С.1.3 Процедура
Испытание клапана следует проводить на имплантате в сборе следующим образом.
Перед испытанием следует настроить клапан для имитации его использования с целью заполнения имплантата, как описано в инструкции по применению.
Применяют обратное давление (давление на внутреннюю сторону или просвет клапана), что эквивалентно 3 кПа (около 300 мм воды), используя воздух, воду или тестовую среду с продемонстрированной эквивалентностью. Поддерживают давление в течение 5 мин.
Проверяют клапан на наличие утечки. Если тестовой средой является воздух, погружают клапан в воду, чтобы проверить наличие утечки (пузырьков). Если использована жидкая тестовая среда, проверяют наличие капель, которые могут возникнуть на наружной поверхности клапана.
Уменьшают давление не более 0,3 кПа (около 30 мм воды). Поддерживают это давление в течение 5 мин и проверяют наличие утечки.
С.1.4 Требование
Никаких утечек не должно происходить во время испытания.
С.2 Исправность инъекционного порта
С.2.1 Принцип
Этот метод испытания определяет исправность инъекционного порта.
С.2.2 Материалы
Иглы, рекомендуемые производителем для нормального использования.
Вода или тестовая среда с продемонстрированной эквивалентностью.
С.2.3 Процедура
Испытание инъекционного порта на собранном имплантате следует проводить с помощью игл, рекомендуемых производителем для нормального использования.
Используя воду или тестовую среду с продемонстрированной эквивалентностью, применяют внутрипросветное давление 3 кПа (около 300 мм воды).
Делают проколы инъекционного порта не менее пяти раз с интервалом в 1 мин в пределах 1 мм рядом с центром участка.
Проверяют инъекционный порт на наличие утечки. Если тестовой средой является воздух, погружают накладку в воду, чтобы проверить на наличие утечки (пузырьков). Если использована жидкая тестовая среда, проверяют наличие капель, которые могут возникнуть на наружной поверхности порта.
С.2.4 Требования
Инъекционный порт считается негерметичным и не удовлетворяет требованиям испытания, если капли жидкости или пузырьки, которые могут появиться на проколотой поверхности, не статичны через 30 с.
Приложение D
(обязательное)
Испытание силиконового геля на когезивные свойства (только для силиконовых наполнителей)
D.1 Принцип
Этот метод испытания определяет когезивные свойства силиконового геля.
D.2 Материалы
Гель силиконовый.
D.3 Аппаратура
Испытательная машина, представленная на рисунке D.1, с внутренним объемом (100±5) мл.
D.4 Процедура
Испытание следует проводить при температуре (23±2)°С следующим образом:
a) используют машину для испытаний (см. рисунок D.1);
b) наполняют машину гелем.
Репрезентативный образец геля должен быть взят из одного имплантата достаточного размера, чтобы была возможность удалить гель одной сплошной массой. Следует проявлять осторожность при удалении геля и переносе его на испытательный стенд. Резкое перемешивание, неаккуратное обращение, образование пузырьков воздуха и т.д. приведет к ошибочным результатам;
c) в начале испытания гель должен быть на одном уровне с нижней поверхностью машины для испытаний и на одном уровне с верхней поверхностью или выше ее;
d) далее гель должен беспрепятственно течь через нижнее отверстие в течение 30 мин;
e) обращают внимание, если какая-то часть геля отделяется от испытуемого объема;
f) измеряют длину выступающей части геля.
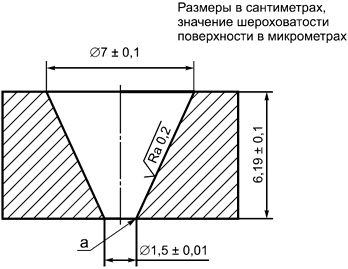
Примечание - Значением шероховатости поверхности Ra является средняя высота профиля ниже и выше линии, в соответствии с определением в ИСО 4287.
а - острый угол
Рисунок D.1 - Испытание когезивных свойств геля
D.5 Требования
Образец будет соответствовать требованиям испытания, если нет никакого разделения, и длина выступающей части геля менее или равна 30 мм.
Приложение E
(обязательное)
Механические испытания имплантата молочной железы в его имплантируемом состоянии
Е.1 Испытание на усталость
Е.1.1 Принцип
Этот метод испытания определяет усталостную прочность имплантата.
Е.1.2 Материалы
Имплантат молочной железы.
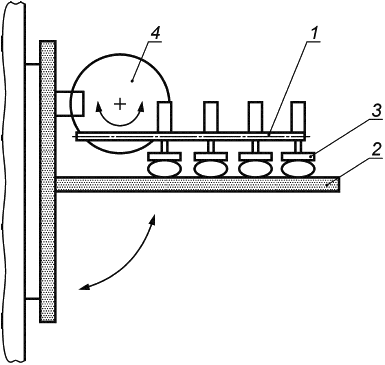
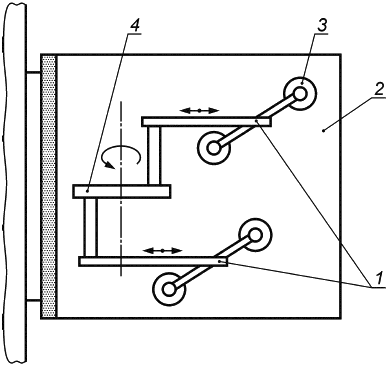
Е.1.3 Аппаратура
Испытательная машина схематически изображена на рисунке Е.1. Она состоит из неподвижной пластины и подвижной пластины, причем последняя с помощью соединительного рычага прикреплена к мотору, который генерирует возвратно-поступательное движение подвижной пластины. Подвижная пластина также включает в себя механизм регулировки, позволяющий изменять расстояние до неподвижной пластины. Таким образом, имплантат может быть сжат в соответствии с требованиями.
Общая длина перемещения подвижной пластины должна составлять 40 мм, что соответствует 20 мм в каждом направлении от центрального исходного положения. Мотор должен быть настроен на 200 циклов в минуту, что соответствует частоте 3,3 Гц.
Е.1.4 Процедура
Имплантат должен держаться с помощью силы сжатия между двумя противоположными вертикально расположенными опорными пластинами. Имплантат должен быть подвергнут деформации путем разнонаправленного перемещения одной из пластин. Сила сжатия удерживает протез между пластинами, тем самым подвергая имплантат воздействию сдвигающего усилия.
Наполняемые имплантаты должны быть заполнены в соответствии с инструкциями изготовителя до начала испытания. Испытание следует проводить при температуре (23±2)°С следующим образом:
a) определяют проекцию имплантата молочной железы;
b) помещают имплантат между двумя пластинами и регулируют расстояние между ними, чтобы оно соответствовало 80% от проекции;
c) проводят испытание в течение 2·10 циклов;
d) проводят осмотр имплантата в соответствии с Е.1.5.
Е.1.5 Требования
После этого испытания при визуальном осмотре с десятикратным увеличением на имплантате молочной железы должны отсутствовать любые разрывы, трещины или разрезы.
а)
b)
1 - соединительный рычаг; 2 - неподвижная пластина; 3 - подвижная пластина; 4 - мотор
Рисунок Е.1 - Машина для испытаний на усталость
Е.2 Испытание ударопрочности
Е.2.1 Принцип
Этот метод испытания определяет ударопрочность имплантата молочной железы. Испытание основано на вертикальном сбросе на имплантат груза определенной массы. Имплантат подвергается воздействию ударной силы, пропорциональной массе имплантата. Ударная сила изменяется за счет регулировки вертикального расстояния, с которого падает груз 4,4 кг.
Высоту падения рассчитывают следующим уравнением:
![]() ,
,
где Н - высота падения, мм;
м - масса имплантата, г.
Е.2.2 Аппаратура
Машина для испытаний схематически изображена на рисунке Е.2. Она состоит из рамы, снабженной подвижной балкой на двух опорах, к которой прикреплен груз общей массой 4,4 кг. При отсоединении от балки груз свободно движется по двум направляющим, что обеспечивает регулярное и воспроизводимое падение к основанию рамы. С имплантатом контактирует металлическая пластина диаметром 250 мм.
Балка содержит фиксирующий механизм, который может быть установлен на раме на разной высоте от основания. Для удобства на раме может быть установлен датчик высоты и ручная лебедка для позиционирования, а на балке - установлен механизм с электронным управлением для высвобождения груза.
Когда механизм, держащий груз, освобождает его, груз падает на имплантат. Образующаяся сила пропорциональна начальной высоте.
Е.2.3 Процедура
Наполняемые имплантаты должны быть заполнены в соответствии с инструкциями изготовителя до начала испытания. Испытание следует проводить следующим образом:
a) взвешивают имплантат;
b) вычисляют высоту падения, мм, в соответствии с массой имплантата и по уравнению, приведенному в Е.2.1;
c) отмечают проекцию имплантата и располагают балку таким образом, чтобы общее расстояние между сбрасываемым грузом и основанием рамы состояло из расчетной высоты сбрасывания и проекции имплантата;
d) располагают имплантат на основании рамы прямо по центру под ударной пластиной;
e) отпускают механизм, удерживающий груз;
f) проводят осмотр имплантата в соответствии с Е.2.1.
Е.2.4 Требование
Имплантат не должен быть поврежден.
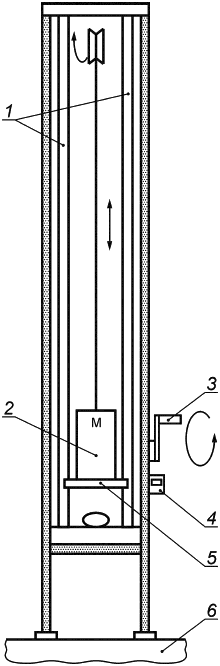
1 - направляющие; 2 - подвижная балка; 3 - ручная лебедка; 4 - датчик высоты; 5 - ударная пластина; 6 - основание рамы
Рисунок Е.2 - Машина для испытаний ударопрочности
Е.3 Статическое испытание прочности на разрыв
Е.3.1 Принцип
Это испытание определяет статическую прочность на разрыв имплантата молочной железы.
Е.3.2 Материалы
Имплантаты молочной железы.
Е.3.3 Аппаратура
Испытательная машина с компрессионным устройством с горизонтально закрепленной пластиной и подвижной пластиной.
Е.3.4 Процедура
Наполняемые имплантаты должны быть заполнены в соответствии с инструкциями изготовителя до начала испытания.
Помещают имплантат молочной железы в центре нижней пластины. На момент начала испытания сила, создающая повышенное давление, не будет действовать на имплантат молочной железы. Это означает, что на нулевую точку приложения силы не воздействует масса верхней пластины. Одновременно начинают запись сигнала приложения усилия и сигнала движения. Медленно повышают давление на имплантат (квазистатически) со скоростью 5 мм/мин или медленнее, пока он не разорвется. Во время этой процедуры сигналы расстояния и силы должны непрерывно фиксировать.
Во время испытания имплантат не должен выходить за пределы пластин. Если размер имплантата слишком большой, следует использовать пластины большего диаметра.
Проверяют имплантат на наличие повреждений и отмечают место разрыва, а также силу и высоту проекции в момент разрыва.
Приложение F
(обязательное)
Информация для пациента
Эту информацию пользователь должен предоставить пациенту задолго до операции. Что касается пункта с), то информация должна быть предоставлена после операции на адекватном носителе, таком как карта пациента.
Примечание 1 - В карте пациента также должна содержаться информация о локализации имплантата (слева или справа) и фамилия хирурга.
Информация должна быть представлена таким образом, чтобы ее было легко читать и понимать, на подходящих носителях, таких как справочник пациента, форма информированного согласия или карта пациента.
Примечание 2 - Производитель может использовать тот(те) носитель(и), какой(ие) считает наиболее подходящим(и) для данного изделия, но настоятельно рекомендуется использовать бумажный носитель, включающий всю следующую информацию.
Информация для пациента должна включать:
a) название или торговую марку и адрес производителя;
b) описание имплантата, включая тип, материалы и принципы устройства (например, предварительно заполненный или наполняемый имплантат), химические компоненты в общих терминах (например, силикон), характеристики (например, текстурированный или гладкий);
c) идентификационную информацию производителя для отслеживаемости (например, номер партии), размер (объем заполнения), форму и коммерческое название;
d) ожидаемый срок службы, выраженный в соответствии с 11.6;
e) следующее предупреждение: "Имплантаты молочной железы имеют ограниченный срок службы", и следующее сообщение: "Этот имплантат, возможно, придется удалить или заменить, что может означать повторную хирургическую операцию";
f) ожидаемую пользу;
g) ожидаемые риски: информация включает все возможные местные осложнения, такие как капсулярная контрактура, разрыв (упоминая о возможности "тихого" разрыва), утечка, спадение и сморщивание; а также потенциальное общее воздействие на здоровье;
h) нежелательные эффекты, например боль, инфекции, эстетические последствия (асимметрия, смещение, гипертрофированное рубцевание), изменения чувствительности сосков и груди;
i) данные о возможном влиянии имплантата на грудное вскармливание;
j) упоминание о необходимости консультации хирурга по поводу последующего медицинского наблюдения;
k) упоминание о необходимости консультации терапевта или фармацевта перед использованием препаратов для местного применения (например, стероидов) в области груди;
l) упоминание о необходимости продолжать посещать терапевта для проведения обычных обследований с целью выявления рака молочной железы;
m) упоминание о необходимости поставить в известность терапевта или хирурга о наличии имплантата, если запланировано хирургическое вмешательство в области груди;
n) влияние имплантата на диагностические методы, например маммография;
о) обязательное информирование рентгенолога при проведении маммографии для того, чтобы адаптировать сжатие молочной железы;
р) возможное влияние имплантата на самостоятельное обследование молочной железы.
Приложение G
(обязательное)
Информация для пользователя
Как минимум, должна быть предоставлена следующая информация:
a) показания к хирургическому вмешательству;
b) описание имплантата;
c) инструкция по применению;
d) противопоказания;
e) потенциальные осложнения и их возможное разрешение;
f) меры предосторожности при хирургической операции;
g) инструкции и меры предосторожности при удалении имплантата;
h) рекомендации относительно последующего медицинского наблюдения;
i) ожидаемый срок службы в соответствии с 11.6;
j) указание, требующее от хирурга предоставление гарантий, что пациент получит информацию, предоставленную производителем и описанную в 11.7;
k) вся доступная информация для выполнения 11.7.
Приложение H
(обязательное)
Оценка утечки силикона из имплантата молочной железы методом in vitro
Н.1 Принцип
Испытание состоит из погружения имплантатов при перемешивании в аналог биологической жидкости (SBF-раствор) при температуре (37±2)°С. Эта процедура in vitro должна имитировать pH- и ионную концентрацию плазмы крови человека (состав SBF приведен в Н.2.2) и оценивать количество силикона, выделяемое в течение 60 дней (+10 дней, если необходимо).
Определение количества силикона, выделяемого имплантатом, основано на измерении элементарного кремния в образцах SBF, регулярно берущихся из раствора. Анализы выполняют с помощью оптической эмиссионной спектрометрии с индуктивно-связанной плазмой (ОЭС-ИСП).
В конце исследования результаты обрабатывают, чтобы выразить общее количество силикона, выделенного имплантатом, как функцию следующих элементов:
- очный объем SBF-раствора, окружающего имплантат;
- точный объем, взятый из раствора для измерения количества кремния;
- теоретическое соотношение кремния/силикона.
Н.2 Материал и аппаратура
Шейкер орбитальный. Вращение должно быть достаточно эффективным, чтобы позволить перемешивать имплантаты должным образом с наименьшим возможным контактом со стенкой контейнера.
Спектрометр оптический эмиссионный с индуктивно-связанной плазмой.
Раствор кремния стандартный.
SBF-раствор.
Контейнеры пластиковые герметичные. Контейнеры, используемые для проведения анализа, не должны взаимодействовать с SBF-раствором и иметь форму, позволяющую полностью погрузить имплантат в раствор с наименьшим возможным контактом со стенкой контейнера (объемом в 7-9 раз большим, чем объем грудного имплантата).
Н.2.1 Подготовка образца
Для того чтобы убедиться в том, что все имплантаты занимают одинаковый объем и надлежащим образом погружены в раствор SBF, наполняемые имплантаты должны быть заполнены физиологическим раствором до их номинальных объемов.
Н.2.2 Аналог биологической жидкости SBF
Состав аналога биологической жидкости, ммоль/л, должен соответствовать составу, приведенному в таблице Н.1.
Таблица Н.1 - Состав аналога биологической жидкости
Na | 142,0 |
К+ | 5,0 |
Mg | 1,5 |
Са | 2,5 |
Cl | 147,8 |
НСО | 4,2 |
HPO | 1,0 |
SO | 0,5 |
Н.2.3 Приготовление раствора SBF
Растворяют каждую соль отдельно в 100 мл воды. Смешивают вместе все растворы, за исключением раствора хлорида кальция, который должен быть добавлен в последнюю очередь.
Н.2.4 Подготовка материала
Пластиковые контейнеры, применяемые для проведения исследования, должны быть очищены перед использованием, чтобы обеспечить удаление любой антиадгезивной силиконовой смазки, используемой при их изготовлении, а также других загрязняющих веществ. Заполняют контейнеры раствором моющего средства, плотно закрывают и перемешивают содержимое на орбитальном шейкере, затем тщательно промывают водой и высушивают тканью.
Н.3 Процедура
Проводят отбор восьми образцов в 0, 10, 20, 30, 40, 50, 60 и 70-й день (±2 дня).
Если один из образцов выходит за пределы диапазона, взятие другого осуществляется на следующий день (так Т может быть +3 дня).
Н.3.1 Условия определения выделения силикона и измерения количества кремния
Испытание следует проводить на трех имплантатах объемом (220±20) мл.
Имплантат (наполненный силиконовым гелем или физиологическим раствором) объемом V, мл, помещают в пластиковый контейнер. Добавляют SBF-раствор объемом (6±0,03) V.
Плотно закрывают контейнер и перемешивают на орбитальном шейкере в течение 1 ч. Берут на анализ 10 мл SBF и добавляют в контейнер тот же объем свежего раствора SBF. Первое взятие образцов соответствует данным Т=0 для каждого испытуемого имплантата.
Контейнер затем помещают в термостат с постоянной температурой (37±2)°С и каждый день перемешивают в течение 10 мин на орбитальном шейкере.
Каждые 10 дней берут по 10 мл SBF для анализа на ИСП-ОЭС и добавляют такой же объем свежего SBF. Также можно взвешивать контейнер с SBF и имплантатом каждый раз при взятии 10 мл для анализа и затем добавлять SBF до тех пор, пока не будет получена начальная масса. Эта процедура восполняет потерянный при работе с устройством объем SBF.
Каждые 10 дней берут по 10 мл SBF для анализа на ИСП-ОЭС и добавляют такой же объем свежего SBF. Таким образом, взятие образцов выполняют шесть раз на 10, 20, 30, 40, 50 и 60-й день (±2 дня). Если один из образцов выходит за пределы диапазона, взятие другого осуществляют на следующий день (так Т может быть +3 дня).
Анализ кремния следует осуществлять в течение 1 ч после забора образца из реакционной среды.
Перед каждой серией измерений спектрограф должен быть откалиброван по стандартным растворам кремния, приготовленным в SBF из 1000 мкг/мл стандартного раствора в воде. Концентрация этих стандартных растворов зависит от результатов предыдущих анализов.
Выбранный стандарт кремния должен быть сертифицирован в аккредитованной лаборатории.
Н.3.2 Отображение результатов
Каждый образец должен быть проанализирован несколько раз, чтобы получить среднюю концентрацию кремния и стандартное отклонение для каждого периода времени.
Для каждого типа имплантата результаты должны быть выражены в массе кремния и массе силикона, выделившегося из имплантата, в зависимости от времени.
Если
V - объем SBF-раетвора, добавленного в 0-й день;
- концентрация кремния, измеренная в 0-й день, мкг/мл;
- объем, взятый на 10-й день;
- концентрация кремния, измеренная на 10-й день, мкг/мл;
- объем, взятый на 20-й день;
- концентрация кремния, измеренная на 20-й день, мкг/мл.
Количество кремния X, мг, в растворе следует определять следующим образом:
в 0-й день Х, мг=(·V)/1000;
на 10-й день Х мг=(·V)/1000+(
·
)/1000;
на 20-й день Х мг=(·V)/1000+(
·
)/1000;
и т.д.
Наклон кривой соответствует среднему количеству кремния, выделившегося за день, в зависимости от времени.
Элементарный кремний, показанный ИСП-ОЭС, должен быть преобразован в общее количество силикона. Это преобразование делается для определения общего количества полидиметилсилоксана, выделившегося из отдельных имплантатов. Концентрацию, моль/мг, рассчитывают исходя из концентрации кремния (миллионная доля) к массе, мг, силикона.
Значение соотношения силикон/кремний вычисляют из теоретического коэффициента, указанного в 12-м издании Merck Index (часть Диметикон) с формулой [-(CH)
Si-О-]:кремний·2,64=силикон.
Примечание - Известно, что силикон является не единственным источником кремния в имплантате. Следовательно, данный расчет не отражает фактическое выделение силикона.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ISO 34-1:2004 | - | * |
ISO 34-1:2004 | - | * |
ISO 10993-1 | - | * |
ISO 14155-1:2008 | IDT | ГОСТ Р ИСО 14155-1-2008 "Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования" |
ISO 14155-2 | IDT | ГОСТ Р ИСО 14155-2-2008 "Руководство по проведению клинических испытаний медицинских изделий. Часть 2. Планирование клинических испытаний" |
ISO 14630 | IDT | ГОСТ Р ИСО 14630-2011 "Имплантаты хирургические неактивные. Общие требования" |
NF S 99-401:1994 | - | * |
* Соответствующий национальный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данных стандартов. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - IDT - идентичные стандарты. | ||
Библиография
[1] | ASTM D 3389-94, Standard test method for coated fabrics abrasion resistance |
[2] | ASTM F 703-96, Standard specification for implantable breast prostheses |
[3] | ASTM P 604-94, Standard classification for silicone elastomers used in medical applications |
[4] | ASTM D 412, Test method for rubber properties in tension |
[5] | ISO 14971, Medical devices - Application of risk management to medical devices |
[6] | ISO 4287, Geometrical Product Specifications (GPS) - Surface texture: Profile method - Terms, definitions and surface texture parameters |
[7] | ISO/TR 14283, Implants for surgery - Fundamental principles |
[8] | ISO 16054, Implants for surgery - Minimum data sets for surgical implants |
[9] | CR 14060, Medical device traceability [CEN Report] |
[10] | MEDDEV 2.7.1, Guidelines on Medical Devices - Evaluation of Clinical Data: A guide for manufacturers and notified bodies, April 2003 |
УДК 620.171.2.006.354 | ОКС 11.040.40 | ОКП 93 9300 | |
Ключевые слова: имплантаты для хирургии неактивные, молочные железы | |||
Электронный текст документа
и сверен по:
, 2017

{kind=link}