ГОСТ Р 53468-2009
(ИСО 16671:2003)
Группа П46
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Имплантаты офтальмологические
РАСТВОРЫ ИРРИГАЦИОННЫЕ ДЛЯ ОФТАЛЬМОЛОГИЧЕСКОЙ ХИРУРГИИ
Общие требования безопасности
Ophthalmic Implants. Irrigating solutions for ophthalmic surgery. General safety requirements
ОКС 11.040.40
ОКП 94 8100
Дата введения 2011-01-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Открытым акционерным обществом "ТКС-оптика" совместно с рабочей группой ПК 7 "Офтальмологическая оптика и приборы" Технического комитета ТК 296 "Оптика и оптические приборы" на основе собственного аутентичного перевода на русский язык стандарта, указанного в пункте 4
2 ВНЕСЕН Управлением технического регулирования и стандартизации Федерального агентства по техническому регулированию и метрологии
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 9 декабря 2009 г. N 625-ст
4 Настоящий стандарт является модифицированным по отношению к международному стандарту ИСО 16671:2003* "Имплантаты офтальмологические. Ирригационные растворы для офтальмологической хирургии" (ISO 16671:2003 "Ophthalmic implants. Irrigating solutions for ophthalmic surgery") путем:
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
- изменения наименования. Наименование настоящего стандарта изменено относительно наименования указанного международного стандарта для приведения его в соответствие с ГОСТ Р 1.5 (подраздел 3.6);
- изменения структуры. Сравнение структуры настоящего стандарта со структурой указанного международного стандарта приведено в дополнительном приложении И;
- введения дополнительных терминов с соответствующими определениями для лучшего понимания текста стандарта, которые выделены путем заключения их в рамки из тонких линий;
- изменения ссылочных нормативных документов;
- изменения отдельных фраз, которые в тексте стандарта также выделены курсивом*. Внесение указанных технических отклонений обусловлено особенностью объекта стандартизации, характерной для Российской Федерации
_______________
* В бумажном оригинале обозначения и номера стандартов и нормативных документов в разделе "Предисловие", подпункте 3.12 (Примечание), таблице Ж.1 приложения Ж и таблице И.1 приложения И приводятся обычным шрифтом, остальные по тексту документа выделены курсивом. - .
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
1 Область применения
Настоящий стандарт распространяется на офтальмологические ирригационные растворы (далее - ОИР) класса неактивных хирургических имплантатов, используемые во время хирургических операций на передней камере глаза субъекта. ОИР не оказывают принципиального иммунологического, фармакологического или метаболического действия.
Стандарт устанавливает требования безопасности в отношении предусмотренного применения, характеристик конструкции, доклинической и клинической оценок, стерилизации, упаковки и этикетирования ОИР и информации, предоставляемой их изготовителем.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ Р ИСО 10993-1-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования
ГОСТ Р ИСО 10993-5-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 5. Исследования на цитотоксичность: методы in vitro
ГОСТ Р ИСО 10993-6-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 6. Исследования местного действия после имплантации
ГОСТ Р ИСО 10993-7-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации
ГОСТ Р ИСО 10993-10-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 10. Исследования раздражающего и сенсибилизирующего действия
ГОСТ Р ИСО 10993-12-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 12. Приготовление проб и контрольные образцы
ГОСТ Р ИСО 14630-1999 Неактивные хирургические имплантаты. Общие технические требования
ГОСТ Р ИСО 14971-2009 Изделия медицинские. Применение менеджмента риска к медицинским изделиям
ГОСТ Р ИСО 15223-2002 Изделия медицинские. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации
ГОСТ Р 51148-1998 Изделия медицинские. Требования к образцам и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность
ГОСТ Р 51609-2000 Изделия медицинские. Классификация в зависимости от потенциального риска применения. Общие требования
ГОСТ Р 51892-2002 (ИСО 11979-1:99) Имплантаты офтальмологические. Интраокулярные линзы. Часть 1. Термины и определения
ГОСТ Р 52458-2005 (ИСО 11979-7:2001, ИСО 14155-1:2003) Имплантаты офтальмологические. Интраокулярные линзы. Клинические испытания
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодно издаваемому информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по соответствующим ежемесячно издаваемым информационным указателям, опубликованным в текущем году. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины по ГОСТ Р 51892, а также следующие термины с соответствующими определениями:
3.1 базовая окружность (reference circle): Окружности стандартного размера диаметром 10 или 25 мкм, используемые для измерения эквивалентного диаметра частицы. 3.2 диаметр эквивалентной площади (equivalent area diameter): Диаметр круга, площадь которого эквивалентна площади частицы. 3.3 динамический диапазон датчика (sensor dynamic range): Диапазон размеров микрочастиц, в котором они могут быть точно измерены и подсчитаны. 3.4 контейнер для хранения (storage container): Часть упаковки, предназначенная для защиты ОИР во время транспортирования и хранения и содержащая упаковочную вставку и герметичный стерильный пакет, внутри которого находятся ОИР и система доставки. 3.5 контрольная группа (control populations): Субъекты, которым имплантированы документированные ОИР. 3.6 клинический испытатель (clinical investigator): Лицо и/или организация, ответственные за проведение клинических испытаний и несущие клиническую ответственность за благополучие вовлеченных субъектов. 3.7 клиническое испытание (clinical investigation): Любое разработанное и запланированное систематическое изучение с целью проверки безопасности и/или эксплуатационных свойств ОИР с участием субъектов. 3.8 окончательный отчет (final report): Документ, заполняемый после завершения испытаний ОИР, в том числе клинических, содержащий описание и оценку результатов. |
3.9 офтальмологический ирригационный раствор, ОИР (ophthalmic irrigating solution): Водный раствор, который физиологически совместим с внутриглазной средой и функционирует преимущественно только механически.
3.10 план клинических испытаний, ПКИ (clinical investigation plan): Документ, определяющий цели, задачи, предполагаемую методологию, систему контроля, проведения анализа и хранения данных клинических испытаний. 3.11 поле зрения окулярной сетки, ПЗС (graticule field of view): Площадь внутри большой окружности, разделенная перекрестьем на квадранты. 3.12 серьезный неблагоприятный исход (adverse device effect): Послеоперационный исход, потенциально угрожающий зрению. Примечание - Примеры серьезных неблагоприятных исходов приведены в таблицах А.1 и А.2 ГОСТ Р 52458. |
3.13 система доставки (delivery system): Герметичный контейнер, в котором поставляется ОИР и дополнительные компоненты для введения данного ОИР в глаз.
3.14 стерильный барьер (sterile barrier): Пакет, в котором содержится ОИР и система доставки, которая поддерживает стерильность во время транспортирования и хранения. 3.15 субъект (subject): Лицо, участвующее в клинических испытаниях с применением испытуемого ОИР и/или в качестве субъекта контрольной группы. 3.16 объемный анализ (volumetry): Совокупность методов химического количественного анализа, основанных на измерении объемов растворов, осадков для установления концентрации (массы) определяемого вещества (включает титрометрический анализ). |
4 Общие требования безопасности ОИР
4.1 Требования к предусмотренным характеристикам
4.1.1 Предусмотренные характеристики ОИР должны быть определены изготовителем. Предусмотренные характеристики ОИР должны отвечать общим требованиям к предусмотренным характеристикам для неактивных хирургических имплантатов, установленных по ГОСТ Р ИСО 14630.
4.1.2 Предельные значения предусмотренных характеристик ОИР должны быть определены с учетом действующих нормативных документов, опубликованной клинической и научной литературы, идентифицированных результатов испытаний, доклинической оценки и клинических испытаний.
4.2 Требования к конструктивным особенностям
4.2.1 Общие требования
4.2.1.1 Должны применяться общие требования к предусмотренным характеристикам неактивных хирургических имплантатов по ГОСТ Р ИСО 14630.
4.2.2 Требования к концентрации компонентов
4.2.2.1 Концентрация каждого компонента вещества в готовом ОИР должна быть определена, документирована и представлена в виде массы вещества на единицу объема раствора.
4.2.2.2 Так как методика испытаний может оказать влияние на представленную фактическую концентрацию, в клиническом отчете должны быть приведены и описаны стандартные физико-химические методы. Нормативные документы на каждый компонент вещества, при их наличии, также должны быть указаны.
4.2.3 Требования к используемой воде
4.2.3.1 Чистота используемой воды должна соответствовать чистоте воды для инъекций в соответствии с [1], [2].
4.2.4 Требования к определению характеристик готового ОИР
4.2.4.1 Общие требования
Изготовитель должен описать и документировать физические характеристики, которые могут повлиять на эффективность использования ОИР в офтальмологической хирургии.
Примечание - Физические характеристики следует измерять в условиях, соответствующих времени использования ОИР.
4.2.4.2 Требования к показателю рН и буферной способности
Показатель рН готового ОИР следует измерять с помощью калиброванного рН-метра при температуре (25±2) °С. Значение показателя рН ОИР должно быть подтверждено документально.
Примечание - Значение показателя рН ОИР должно быть близким к значению показателя рН внутренней среды глазного яблока (рН 7,38) для предотвращения повреждения клеток роговичного эндотелия. Исследования in vitro показали, что диапазон значений показателя рН, переносимый эндотелием, сужается с увеличением времени воздействия.
Для определения буферной способности ОИР следует применять метод, пример которого приведен в приложении А.
Классификация ОИР по диапазону значений показателя рН и буферной способности раствора приведена в таблице 1.
Таблица 1 - Классификация ОИР по показателю рН и буферной способности
Группа | Буферная способность основания, моль/л на рН | Буферная способность кислоты, моль/л на рН | Диапазон значений показателя рН |
Без буферной добавки | Менее 0,0005 | Менее 0,004 | От 6,5 до 8,5 |
С умеренной буферной добавкой | От 0,0005 до 0,005 | От 0,004 до 0,04 | От 6,7 до 8,2 |
С буферной добавкой | Более 0,005 | Более 0,04 | От 7,2 до 7,6 |
4.2.4.3 Требования к химическим и биологическим загрязняющим примесям
Идентификация потенциально опасных химических и биологических загрязняющих примесей должна быть проведена посредством анализа рисков. В исходных материалах биологического происхождения эти примеси могут включать в себя протеины, нуклеиновые кислоты или другие биологические вещества. Примеси в готовом ОИР, представляющие потенциальную опасность для тканей глаза или системную опасность, должны быть идентифицированы, а их концентрация определена и представлена в окончательном отчете.
Примеси, загрязняющие ОИР, состоят из инородных, мобильных нерастворимых микрочастиц, кроме пузырьков газа, присутствующих в растворах.
Загрязняющие примеси должны быть определены с помощью стандартных аналитических методов, при их наличии, которые должны быть описаны. Предельные концентрации идентифицированных примесей должны быть установлены и описаны. Необходимость проведения испытания для проверки биологического действия этих примесей в процессе оценки биологической безопасности возникает в результате анализа рисков.
4.2.4.4 Требования к осмотическому давлению
Изготовитель должен определить и документировать диапазон осмотической концентрации ОИР. Осмотическое давление готового ОИР должно быть в диапазоне от 200 до 400 мОсм/кг. Осмотическое давление должно определяться с помощью парового или криоскопического осмометра при нормальных условиях.
4.2.4.5 Требования к спектральному коэффициенту пропускания
Коэффициент пропускания готового ОИР должен быть измерен в диапазоне от 300 до 1100 нм. Результаты должны быть представлены в графической форме: коэффициент пропускания, %, в зависимости от длины волны, нм.
4.2.4.6 Требования к микрочастицам
Присутствие в готовом ОИР микрочастиц определенных размеров, видимых и невидимых, приводит к потенциальной возможности возникновения в ОИР неблагоприятных явлений. При проведении оценки рисков необходимо определить потенциальную возможность загрязнения или образования частиц в ОИР в процессе изготовления в условиях, ожидаемых при транспортировании и хранении, а также при использовании ОИР.
Изготовитель должен охарактеризовать потенциальную опасность, связанную с частицами каждого вида, выявленными при проведении оценки рисков.
Общие требования к оценке потенциального риска применения готового ОИР должны соответствовать ГОСТ Р 51609.
В ОИР не должно содержаться видимых микрочастиц. Метод проверки загрязнения ОИР видимыми микрочастицами должен соответствовать методу, приведенному в приложении Б. Для определения в ОИР невидимых частиц следует применять методы по приложению В или Г.
Метод по приложению В следует применять при определении следующих уровней микрочастиц:
- не более 50 микрочастиц размерами не менее 10 мкм на 1 мл ОИР;
- не более 5 микрочастиц размерами не менее 25 мкм на 1 мл ОИР;
- не более 2 микрочастиц размерами не менее 50 мкм на 1 мл ОИР.
Метод по приложению Г следует применять при определении следующих уровней микрочастиц:
- не более 25 микрочастиц размером не менее 10 мкм на 1 мл ОИР;
- не более 2,5 микрочастиц размером не менее 25 мкм на 1 мл ОИР;
- не более 1 микрочастицы размером не менее 50 мкм на 1 мл ОИР.
Примечание - Метод с затенением света по приложению В основан на блокаде света. Аморфные, полужидкие или другие морфологически неразличимые микрочастицы способствуют затенению света и, следовательно, подсчету микрочастиц. В микроскопическом методе по приложению Г аморфные, полужидкие или другие морфологически неразличимые микрочастицы представляются в виде пятна или выцветшего места на поверхности мембранного фильтра и не считаются микрочастицами. Для компенсации этой разницы уровни микрочастиц данного метода составляют половину уровней микрочастиц метода по приложению В.
5 Требования к оценке конструкции ОИР
5.1 Общие требования
5.1.1 Оценка конструкции ОИР должна проводиться для подтверждения соответствия его предусмотренным характеристикам.
Требования к оценке конструкции ОИР должны соответствовать общим требованиям, приведенным в ГОСТ Р ИСО 14630 для неактивных хирургических имплантатов.
5.2 Требования к доклинической оценке биологической безопасности
5.2.1 Общие требования
5.2.1.1 Оценку биологической безопасности ОИР следует начинать с анализа рисков, проводимого и документированного в соответствии с ГОСТ Р ИСО 14971. Результаты анализа рисков должны определить испытания, необходимые для оценки биологической безопасности ОИР.
5.2.1.2 Для ОИР, содержащих материалы животного происхождения, должны быть применены анализ рисков и требования к менеджменту, приведенные в [3].
5.2.1.3 Для всех ОИР должны быть соблюдены требования, предъявляемые к оценке биологической безопасности по ГОСТ Р ИСО 10993-1, ГОСТ Р ИСО 10993-5, ГОСТ Р ИСО 10993-10, и требования, изложенные в настоящем подразделе.
Примечания
1 При проведении оценки риска изготовитель должен учитывать взаимодействие с другими офтальмологическими имплантатами.
2 На основании типичного клинического применения ОИР классифицируются как "Имплантируемые изделия; ткань/кость". Испытания изделий данной и других категорий, представленные в таблице 1 ГОСТ Р ИСО 10993-1, приведены только для руководства и не содержат максимальных или минимальных требований к испытаниям.
3 Допускается комбинировать испытания на биологическую совместимость, уменьшая число животных, необходимых для проведения испытания. Два испытания могут быть проведены одновременно на одном животном при условии, что испытуемое животное не подвергается чрезмерной боли или страданиям.
5.2.2 Требования к оценке на наличие бактериальных эндотоксинов
5.2.2.1 ОИР следует оценивать на наличие бактериальных эндотоксинов с помощью LAL-теста (на культуре limulus amoebocyte lysate) в соответствии с методикой, описанной в [4], или эквивалентной аттестованной методикой испытания.
5.2.2.2 ОИР, имеющий предел бактериальных эндотоксинов более 0,5 единиц эндотоксинов (ЕЭ) на миллилитр, считают не прошедшим испытание.
5.2.3 Требования к испытанию на послеоперационную воспалительную реакцию глаза
5.2.3.1 Если анализ риска указывает на необходимость проведения испытания на послеоперационную воспалительную реакцию глаза, то такое испытание ОИР следует проводить на животном в соответствии с приложением Е.
Выбор особи животного должен быть обоснован и оформлен документально. Следует применять требования, предъявляемые к содержанию животных, указанные в [5].
5.2.3.2 Испытание на животном должно отражать предполагаемое клиническое испытание. В процессе испытания должна быть проведена оценка на воспалительную реакцию глаза как в процессе офтальмологической хирургической операции, так и после нее, используя при этом оцениваемый ОИР и контрольный ОИР. Контрольный ОИР должен иметь подтверждение клинического использования его в качестве не оказывающего воспалительного действия на глаз в течение пяти лет. Объем испытуемого ОИР должен имитировать предполагаемое использование с учетом внутриглазного объема глаза человека и животного. Если испытуемый ОИР вызывает большее раздражение глаза или воспаление, чем ОИР, используемый как контрольный, необходимо провести оценку соотношения риск/польза.
5.2.3.3 Послеоперационную воспалительную реакцию глаза необходимо контролировать и оценивать в соответствии с приложением Д.
На основании анализа риска и требований к менеджменту соответствующая оценка в соответствующие моменты времени может включать в себя измерение толщины роговицы и биомикроскопию с использованием щелевой лампы. Все серьезные неблагоприятные воздействия следует документировать.
5.2.3.4 Результаты испытания раздражения и воспаления испытуемого ОИР должны быть меньше или равными результатам испытания контрольного ОИР. В противном случае испытуемый ОИР должен быть подвергнут клинической оценке.
5.3 Требования к клинической оценке
5.3.1 Общие требования
5.3.1.1 Клиническое испытание ОИР следует проводить по приложению Е. В дополнение к клиническому испытанию по приложению Е следует применять общие требования к клиническому испытанию субъектов по ГОСТ Р 52458.
6 Требования к стерильности ОИР
6.1 Готовый ОИР должен быть стерильным. При стерилизации должны применяться требования к стерилизации неактивных хирургических имплантатов, указанные в ГОСТ Р ИСО 14630, и требования по ГОСТ Р 51148.
6.2 Для стерилизации ОИР, а также первичного контейнера емкости не следует применять этиленоксид, если его применение не обосновано.
6.3 К ОИР или его первичному контейнеру, при стерилизации которых используется этиленоксид и связанные с ним загрязнения, проникающие в ОИР, должны быть применены следующие ограничения:
- этиленоксид: менее 20 мг/л;
- этиленхлоргидрин: менее 100 мг/л.
Примечание - Установлено, что требования, определяющие приемлемые уровни остаточного этиленоксида (ЭО), указанные в ГОСТ Р ИСО 10993-7, не подходят к изделиям, находящимся в контакте с высокочувствительными тканями, например, тканями глаза. Дополнительные указания по применению ГОСТ Р ИСО 10993-7 приведены в [6].
6.4 Для ОИР, которые периодически не стерилизуются, но подвергаются асептической обработке, должны применяться требования по [7]. Предельное значение нормы загрязнения для ОИР должно составлять 10, что должно быть подтверждено документально.
Примечание - Технический информационный отчет [4] не содержит требований по предельному содержанию этиленхлоргидрина (ЭХГ). Предполагается, что в будущей редакции международного стандарта будет установлено предельное содержание ЭХГ для интраокулярных изделий, содержащих хлорид, в четыре раза выше содержания ЭО. Это следует учитывать при оценке биологической безопасности ОИР и для соответствия будущей редакции стандарта.
6.5 К ОИР, которые не стерилизуют, но подвергают асептической обработке, должны быть применены требования [7]. Соответствие данному международному стандарту должно быть подтверждено аттестованным исследованием в среде с предельным значением нормы загрязнения 10.
Примечание - [7] устанавливает общие требования и содержит руководство по процессам, программам и методикам аттестации и контроля медицинских изделий, подвергаемых асептической обработке. Данный международный стандарт, в частности, применим к обработке физиологических (водных) растворов и, следовательно, имеет отношение к изготовлению ОИР. Будущие части данного международного стандарта будут посвящены специализированным процессам, таким как фильтрация и лиофилизация.
7 Требования к стабильности ОИР
7.1 Изготовитель должен определить и установить срок годности ОИР при хранении в складских помещениях и системах доставки. Определение срока годности при хранении должно быть проведено в реальном масштабе времени или проверено по ускоренной методике испытания для подтверждения, что основные характеристики по безопасности и эффективные эксплуатационные качества готового ОИР и системы доставки находятся в установленных пределах в течение обозначенного срока годности при ожидаемых условиях транспортирования и хранения. Испытание в реальном масштабе времени должно быть проведено при нормальных условиях хранения и изменений температуры, а относительная влажность воздуха должна составлять (60±20)%.
При испытании по ускоренной методике температура должна быть не более 45 °С, а относительная влажность воздуха должна составлять 40%.
7.2 Параметры, которые необходимо контролировать во время исследования срока годности при хранении: рН, осмоляльность раствора, уровни микрочастиц, стерильность, цвет, чистота и другие параметры, установленные при анализе рисков как критические для безопасного применения ОИР.
7.3 Установленный срок службы ОИР при хранении должен быть пересмотрен, если при анализе рисков выявлено изменение технологии, что может повлиять на стабильность ОИР.
Примечание - Изменения состава ОИР, исходных материалов и их поставщиков, условий изготовления, включая процесс стерилизации, тип упаковки или упаковочные материалы, могут оказать неблагоприятное влияние на срок службы ОИР при хранении.
8 Требования к упаковке ОИР
8.1 Требования к защите от повреждения
8.1.1 ОИР должны быть защищены от повреждения при хранении и транспортировании.
8.1.2 Должны быть применены требования к упаковке медицинских изделий, указанные в ГОСТ Р ИСО 14630 и [8].
8.2 Требования к защите стерильности
ОИР при транспортировании должны быть упакованы таким образом, чтобы оставаться стерильными в пределах или ниже норм, указанных для условий транспортирования, хранения и погрузочно-разгрузочных работ. Должны соблюдаться требования к стерильному барьеру, указанные в [9].
9 Требования к информации, предоставляемой изготовителем ОИР
9.1 Должны соблюдаться общие требования к информации, предоставляемой изготовителем медицинских изделий, указанные в [10], и частные требования к ОИР, приведенные в настоящем разделе.
9.2 По применимости допускается использовать символы вместо текста. Если используются символы, должны быть применены требования ГОСТ Р ИСО 15223.
На этикетке должны быть указаны присутствие в ОИР группы буферной добавки и, при ее наличии, буферная способность.
9.3 Если ОИР подвержен повреждению под действием факторов окружающей среды, на транспортном контейнере должны быть нанесены четкие предупредительные знаки.
Номер партии и конечный срок действия могут быть указаны на самоклеящейся этикетке.
9.4 Вкладыш должен быть вложен в упаковку так, чтобы его можно было взять и прочитать, не повреждая стерильного барьера.
9.5 Минимальная информация, наносимая на контейнер для хранения, вкладыш в упаковку, стерильный барьер и первичный контейнер, приведена в таблице 2.
Таблица 2 - Информация, предоставляемая изготовителем
Информация | Контейнер для хранения | Вкладыш в упаковку | Стерильный барьер (при наличии) | Первичный контейнер |
Наименование изготовителя или уполномоченного представителя | + | + | + | + |
Адрес изготовителя | + | + | - | - |
Торговое наименование изделия | + | + | + | + |
Описание комплекта поставки и инструкция по использованию | - | + | - | - |
Краткое описание химического состава изделия и объема поставки | + | + | - | - |
Условия хранения | + | + | - | - |
Показания к применению | - | + | - | - |
Противопоказания к применению | - | + | - | - |
Меры предосторожности и предупреждения об известном взаимодействии с другими имплантатами | - | + | - | - |
Сообщение о том, что содержимое предназначено для разового использования | + | + | + | + |
Надпись: "Стерильно" и метод(ы) стерилизации изделия и первичного контейнера | + | + | + | + |
Надпись: "Не использовать, если стерильный барьер нарушен" | + | + | + | |
Конечный срок годности | + | - | + | + |
Сообщение о наличии в ОИР буферной добавки, буферная способность (при наличии) согласно таблице 1 | - | + | - | - |
Номер партии | + | - | + | + |
Примечание - Номер партии, срок годности и дату стерилизации допускается не указывать на стерильном барьере, если он прозрачный и необходимую информацию можно прочитать на первичном контейнере, не нарушая его герметичность. | ||||
Приложение А
(справочное)
Методы измерения рН и определения буферной способности ОИР
А.1 Общие требования
А.1.1 В настоящем приложении приведен пример методов измерения рН и определения буферной способности ОИР.
А.2 Требования к оборудованию и вспомогательным устройствам
А.2.1 Стеклянная колба.
А.2.2 Бюретка емкостью 10 мл для подачи титрированного раствора с ценой деления 0,1 мл.
А.2.3 Чашка для размешивания.
А.2.4 Палочка для помешивания.
А.2.5 Гидроокись натрия (NaOH), 0,005-нормальный стандартный объемный раствор.
А.2.6 Соляная кислота (HCI), 0,01-нормальный стандартный объемный раствор.
А.2.7 Калиброванный рН-метр.
А.3 Порядок подготовки, проведения измерения и правила обработки результатов измерения
А.3.1 Растворы гидроокиси натрия и соляной кислоты приготовить общепринятыми методами и перед применением разбавить до нужной концентрации.
А.3.2 В стеклянную колбу влить 25 мл образца (объем ) и измерить
с помощью калиброванного рН-метра.
А.3.3 С помощью бюретки добавить достаточное количество титрированного стандартного объемного раствора HCI, чтобы обеспечить изменение показателя рН на 1,0 единицу рН (). Записать объем добавленного титрированного раствора (
).
А.3.4 Повторить операции со второй аликвотной пробой образца, используя титрированный стандартный объемный раствор NaOH.
А.3.5 Рассчитать буферную способность по формуле
![]() ,
,
где - объем добавленного титрированного раствора;
- объем раствора образца, мл;
- нормальность титрированного раствора.
А.3.6 В окончательном отчете записать буферную способность по кислоте и буферную способность по основанию, моль/л на рН (или моль-экв./л).
Для растворов со значительной буферной способностью объем образца допускается регулировать до получения нужного объема титрированного раствора.
Приложение Б
(обязательное)
Метод проверки загрязнения ОИР видимыми микрочастицами
Б.1 Общие требования
Метод проверки заключается в проведении визуальной оценки ОИР на наличие видимых микрочастиц с помощью устройства наблюдения.
Б.2 Требования к оборудованию и вспомогательным устройствам
Б.2.1 Устройство наблюдения, в состав которого входят:
Б.2.1.1 Матовая черная панель, установленная вертикально.
Б.2.1.2 Матовая белая панель, установленная вертикально рядом с черной панелью.
Б.2.1.3 Регулируемый патрон, снабженный источником света и рассеивателем.
Б.2.1.4 Источник света в точке наблюдения должен создавать освещенность не менее 2000 лк. Для емкостей из цветного стекла и пластмассы освещенность должна быть более 2000 лк.
Б.3 Порядок подготовки, проведения проверки и правила обработки результатов проверки
Б.3.1 Снять все этикетки с емкости, содержащей ОИР, промыть и высушить ее наружные стороны.
Б.3.2 Осторожно поворачивая емкость, следить за появлением микрочастиц перед белой поверхностью. Воздушные пузыри в емкости недопустимы.
Б.3.3 Появление микрочастиц считать в течение 5 с.
Б.3.4 Провести операции по Б.3.2-Б.3.3 перед черной панелью.
Б.3.5 Сумму микрочастиц, появившихся перед белой и черной панелями, считают уровнем загрязнения ОИР видимыми микрочастицами и документируют.
Приложение В
(справочное)
Метод проверки загрязнения ОИР невидимыми микрочастицами*
_______________
* В бумажном оригинале наименование приложения В выделено курсивом. - .
В.1 Общие требования
Метод проверки заключается в проведении оценки ОИР на наличие невидимых микрочастиц с помощью устройства электронной системы подсчета микрочастиц по ослаблению света.
В.2 Требования к оборудованию и вспомогательным устройствам
В.2.1 Общие требования
В.2.1.1 Устройство представляет собой электронную систему подсчета микрочастиц, взвешенных в растворе, обеспечивающую соответствующую подачу ОИР, в которой должен быть установлен датчик ослабления света, например, типа KL-04, фирма RION.
В.2.2 Требования к пределу измерения концентрации датчика
В.2.2.1 Используемое устройство должно иметь предел измерения концентрации максимального числа определяемых микрочастиц на миллилитр, который превышал бы предполагаемую концентрацию микрочастиц в испытуемом ОИР.
Предел измерения концентрации устройства указывается уровнем подсчета микрочастиц, при котором совпадающий счет вследствие одновременного присутствия двух или более микрочастиц в просматриваемом объеме датчика ослабления света составляет менее 10% подсчетов, суммированных для микрочастиц размерами 10 мкм. Предел измерения концентрации микрочастиц должен быть указан в паспорте устройства.
В.2.3 Требования к динамическому диапазону датчика
В.2.3.1 Динамический диапазон датчика должен охватывать наименьший размер микрочастиц, которые подлежат подсчету в испытуемом ОИР.
В.2.4 Требования к воде
В.2.4.1 Используемая вода должна быть пропущена через пористый фильтр для очистки ее от микрочастиц. Размеры пор фильтра должны быть не более 1,2 мкм.
В.2.5 Требования к калибровке
В.2.5.1 Устройство калибруют по набору сферических микрочастиц размерами от 10 до 25 мкм в монодисперсной суспензии. Набор сферических микрочастиц диспергируют в воде по В.2.4.1 до получения соответствующей концентрации микрочастиц. В процессе диспергирования не допускаются агрегатные состояния микрочастиц.
В.3 Порядок подготовки, проведения проверки и правила обработки результатов проверки
В.3.1 Общие требования
В.3.1.1 Проверку проводят в условиях ограниченного загрязнения микрочастицами с использованием вытяжного шкафа с ламинарным потоком воздуха через высокоэффективный сухой воздушный фильтр (НЕРА).
В.3.1.2 Для обеспечения соответствия рабочих параметров устройства требуемой точности проверки оператор, проводящий проверку, должен пройти соответствующее обучение.
В.3.1.3 Перед контрольной проверкой используемая стеклянная посуда должна быть чисто вымыта с применением теплого моющего средства и тщательно ополоснута большим количеством воды по В.2.4.1.
В.3.1.4 Во время переноса исследуемого ОИР в емкость, подлежащую контролю, не допускается проникновение воздушных пузырьков.
В.3.2 Порядок подготовки к проведению контрольной проверки
В.3.2.1 Контрольная проверка заключается в определении загрязнения микрочастицами пяти образцов воды по В.2.4.1 объемом 5 мл каждый в соответствии с В.3.3.
В.3.2.2 Если число микрочастиц размерами более 10 мкм превышает 25 в совокупном объеме образцов воды 25 мл, то общие требования безопасности проведения проверки не являются достаточными для проведения основной проверки ОИР. Следует повторить подготовительные этапы и провести повторную контрольную проверку.
В.3.2.3 ОИР, прошедшие контрольную проверку, подвергают процедуре определения загрязнения.
В.3.3 Порядок проведения проверки и оценка результатов
В.3.3.1 Для многокомпонентных ОИР требуется тщательное смешивание отдельных компонентов в соответствии с указаниями на этикетке.
В.3.3.2 Перемешивают содержимое ОИР, медленно переворачивая емкость не менее 25 раз. В случае необходимости, осторожно удаляют герметичную упаковку.
В.3.3.3 Очищают внешние поверхности емкости с помощью струи воды по В.2.4.1, снимают крышку, не допуская загрязнения содержимого емкости. Дают выйти пузырькам газа в течение 2 мин.
В.3.3.4 Берут четыре порции ОИР, каждая из которых содержит (5±0,2) мл, и подсчитывают число микрочастиц, размеры которых равны или более 10 и 25 мкм.
В.3.3.5 Подсчитывают и документально оформляют среднее число микрочастиц в ОИР, подлежащем исследованию.
Приложение Г
(рекомендуемое)
Микроскопический анализ твердых (невидимых) микрочастиц
Г.1 Общие требования
Г.1.1 Микроскопический анализ предусматривает подсчет числа невидимых твердых микрочастиц в ОИР на единицу объема после того, как эти частицы соберутся на пористом мембранном фильтре.
Г.1.2 При выполнении микроскопического анализа не предусмотрен подсчет аморфных, полужидких или других морфологически неотчетливых частиц, которые имеют вид пятна или изменения цвета на поверхности мембранного фильтра. Такие частицы имеют небольшой рельеф или не имеют рельефа поверхности и имеют студенистый или пленкообразный вид.
Г.2 Требования к оборудованию и вспомогательным устройствам
Г.2.1 Микроскоп
Г.2.1.1 Микроскоп (сложный бинокулярный), комбинация объектива и окуляра обеспечивает увеличение (100±10). Апохроматический или лучший объектив должен иметь номинальное увеличение 10
и минимальную числовую апертуру 0,25. Объектив должен быть совместим с эпископическим осветителем. Увеличение окуляров - 10
.
Г.2.2.1 Микроскоп снабжен механическим столиком, который может фиксировать мембранный фильтр диаметром 25 или 47 мм и сканировать всю его площадь.
Г.2.2 Осветители
Г.2.2.1 Два осветителя, оснащенные голубыми светофильтрами дневного света для снижения усталости глаза оператора:
- внешний осветитель, регулируемый для обеспечения косого освещения, падающего под углом от 10° до 20° к горизонтали;
- эпископический осветитель светлого поля, встроенный в микроскоп.
Г.2.3 Окулярная сетка с окружностями
Г.2.3.1 Окулярная сетка с окружностями (см. рисунок Г.1), откалиброванная таким образом, чтобы измерительные окружности занимали 2% номинального поля зрения в плоскости столика для данных объектива и окуляра.
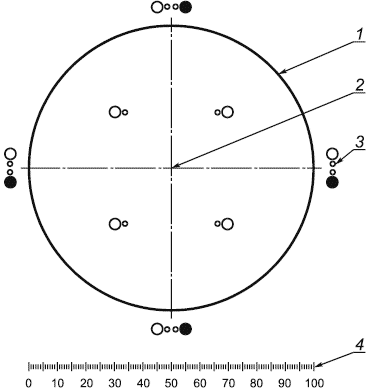
1 - поле зрения окулярной сетки; 2 - перекрестье; 3 - окружности сравнения; 4 - линейная шкала
Рисунок Г.1 - Окулярная сетка с окружностями
Г.2.3.2 Прозрачные и черные измерительные окружности диаметрами 10 и 25 мкм при увеличении 100 являются сравнительными шкалами для измерения частиц.
Г.2.4 Объект-микрометр
Г.2.4.1 Объект-микрометр с ценой деления 10 мкм, имеющий сертификат соответствия, выдаваемый в установленном порядке аккредитованным органом по сертификации.
Г.2.5 Устройство фильтрации и вспомогательные принадлежности
Г.2.5.1 Фильтровальная воронка объемом, подходящим для испытания, минимальным диаметром 21 мм. Воронка должна быть изготовлена из пластмассы, стекла или нержавеющей стали.
Г.2.5.2 Фильтровальный диффузор, выполненный в виде подставки под фильтр, изготовленный из нержавеющей стали или спеченного стекла.
Г.2.5.3 Источник вакуума.
Г.2.5.4 Пинцет с закругленными концами.
Г.2.5.5 Дозаторы растворителя с фильтрующим соплом, обеспечивающим подачу растворителя, отфильтрованного от частиц толщиной не более 1,2 мкм при давлении от 70 до 550 кПа.
Г.2.5.6 Мембранные фильтры, 25 или 47 мм, с сеткой или без сетки, черного или темно-серого цвета, изготовленные из целлюлозно-эфирной смеси, размерами пор не более 1,0 мкм.
Г.2.5.7 Чашка Петри.
Г.2.5.8 Вытяжной шкаф с ламинарным потоком воздуха через высокоэффективный сухой воздушный фильтр (НЕРА) с содержанием не более 3500 частиц/м.
Г.2.5.9 Стерильные перчатки.
Г.2.5.10 Деионизированная вода.
Г.2.5.11 Изопропиловый спирт.
Г.3 Порядок подготовки к проведению анализа
Г.3.1 Общие требования
Г.3.1.1 Определяют слепую пробу путем следующих операций:
- подают 50 мл деионизированной или фильтрованной дистиллированной воды;
- проводят откачку воздуха и пропускают всю воду через мембранный фильтр;
- вынимают мембрану из основания фильтровальной воронки и кладут поверх полоски двухсторонней ленты в чашке Петри;
- высушивают мембрану и проводят ее исследование под микроскопом при увеличении 100.
Г.3.1.2 Если в пределах отфильтрованного участка пробы имеется не более 20 микрочастиц размерами, более или равными 10 мкм, и 5 микрочастиц размерами, более или равными 25 мкм, то фоновый уровень микрочастиц считают достаточно низким для проведения микроскопического анализа.
Г.3.1.3 При проведении анализа следует использовать обеспыленные стерильные перчатки, устройство фильтрации и принадлежности, тщательно вымытые с использованием теплого раствора моющего средства, смываемого деионизированной водой или изопропиловым спиртом. Перед использованием следует отфильтровать дистиллированную или деионизированную воду фильтрами с отверстиями не более 1,2 мкм.
Г.3.1.4 Окончательную сушку устройства фильтрации и принадлежностей проводят в вытяжном шкафу с ламинарным потоком воздуха.
Рекомендуется устанавливать вытяжной шкаф в отдельном помещении с подачей отфильтрованного воздуха.
Г.3.2 Подготовка микроскопа
Г.3.2.1 Включают внешний вспомогательный осветитель падающего косого освещения и устанавливают его вблизи столика микроскопа.
Г.3.2.2 Фокусируют осветитель так, чтобы получить концентрированный участок света на мембране фильтра, расположенного на столике.
Г.3.2.3 Регулируют вспомогательный осветитель падающего косого освещения по высоте так, чтобы угол падающего света составлял от 10° до 20° к горизонтали; микрочастицы на мембранном фильтре должны иметь отчетливые темные тени.
Г.3.2.4 Регулируют внутренний эпископический осветитель светлого поля, полностью открыв полевую и апертурную диафрагмы. Центрируют нить накала лампы и фокусируют микроскоп на фильтр, содержащий микрочастицы.
Г.3.2.5 Регулируют интенсивность отраженного света до тех пор, пока микрочастицы не станут четко различимы и не обнаружат отчетливые видимые тени.
Г.3.2.6 Устанавливают самое низкое значение интенсивности эпископического освещения, затем повышают его до тех пор, пока тени, отбрасываемые частицами, не покажут минимально заметное снижение контраста.
Г.3.3 Подготовка устройства фильтрации
Г.3.3.1 Моют фильтровальную воронку (Г.2.5.1), подставку и диффузор в растворе жидкого моющего средства и в горячей воде, промывают горячей проточной водопроводной водой, потом промывают фильтрованной деионизированной водой под давлением. Повторяют процедуру промывания с использованием фильтрованного изопропилового спирта. Промывают устройство фильтрации фильтрованной деионизированной водой, подаваемой под давлением.
Г.3.3.2 С помощью чистого пинцета с закругленными концами вынимают мембрану из основания воронки фильтра.
Г.3.3.3 Струей фильтрованной очищенной воды, подаваемой под низким давлением, тщательно промывают обе стороны фильтра, начиная сверху, поворачивая взад и вперед к основанию.
Г.3.3.4 Собирают чистое устройство так, чтобы диффузор находился в верхней части основания, а чистый мембранный фильтр - в верхней части диффузора.
Г.3.3.5 Устанавливают узел воронки в верхней части основания и закрепляют.
Г.3.4 Подготовка к анализу испытуемых ОИР
Готовят испытуемые ОИР в следующей последовательности.
Г.3.4.1 Вне вытяжного шкафа снимают все наружные крышки, герметизирующие полосы и незакрепленные или отстающие бумажные ярлыки. Промывают внешние поверхности контейнера фильтрованной деионизированной водой, предусмотрев защиту емкости от попадания загрязнений из окружающей среды.
Г.3.4.2 Изымают содержимое контейнеров. Содержимое контейнеров со съемными пробками можно изъять непосредственным снятием крышек, также допускается использовать устройство для отбора проб с иглой для проникновения в крышку контейнера. Пробы ОИР, упакованные в пластиковые контейнеры, можно изъять, сделав отверстие в колпачке контейнера или отрезав уголок чистым лезвием или ножницами.
Г.3.4.3 Число проб испытуемых ОИР должно быть достаточным для обеспечения статистически обоснованной оценки того, соответствует ли партия или другая крупная группа представленных ОИР заданным пределам или превышает их.
Г.3.4.4 Если объем контейнера менее 25 мл, следует испытать ОИР из не менее чем 10 контейнеров. Допускается проверять единичные контейнеры, если объем ОИР отдельного контейнера составляет не менее 25 мл. В случае, если вместимость контейнера более 25 мл, проверяют одиночные контейнеры. Проверку ОИР в контейнере более 25 мл проводят на основании соответствующего плана выборочного контроля.
Г.4 Порядок проведения анализа
Г.4.1 Порядок анализа испытуемых ОИР
Г.4.1.1 Перемешивают контейнеры, содержащие испытуемые ОИР, 20-кратным опрокидыванием.
Г.4.1.2 Вскрывают контейнеры таким способом, который создаст наименьшее возможное число фоновых частиц. Если объем ОИР в контейнере менее 25 мл, следует вскрыть и смешать содержимое не менее 10 контейнеров в чистой емкости.
Г.4.1.3 Переливают весь объем ОИР в фильтровальную воронку и включают вытяжку.
Г.4.1.4 Если объем ОИР, подлежащего фильтрации, превышает вместимость фильтровальной воронки, постепенно добавляют раствор порциями, пока весь объем ОИР не будет отфильтрован.
Г.4.1.5 После последней порции ОИР промывают стенки воронки, направляя под низким давлением по кругу на стенки воронки поток фильтрованной деионизированной воды. Прекращают промывание воронки прежде, чем объем снизится до 1/4 уровня наполнения. Поддерживают вакуум, пока вся жидкость не пройдет в воронку.
Г.4.1.6 Отключив вытяжку, с помощью пинцета вынимают мембрану из основания фильтровальной воронки и помещают в чашку Петри, закрепив ее с помощью двусторонней ленты и нанеся идентификационную маркировку испытуемого ОИР.
Г.4.1.7 Высушивают фильтровальную мембрану в вытяжном шкафу со слегка приоткрытой крышкой чашки Петри.
Г.4.1.8 Рассматривают под микроскопом с увеличением 100 фильтровальную мембрану при снятой крышке чашки Петри.
Г.4.1.9 Если в зоне фильтрации имеется не более 20 частиц размерами, более или равными 10 мкм, и не более 5 частиц, размерами, более или равными 25 мкм, то фоновый уровень частиц достаточно низок для проведения микроскопического анализа.
Г.4.1.10 Если число частиц превышает эти требования, фоновый уровень частиц не подходит для проведения микроскопического анализа. Следует повторять подготовительные этапы, пока фоновый уровень частиц не станет подходящим для проведения данного испытания.
Г.4.2 Порядок проведения работ с окулярной сеткой и подсчет микрочастиц
Г.4.2.1 Относительную погрешность окулярной сетки определяют с помощью объект-микрометра, проводя следующие операции:
- совмещают линейную шкалу сетки со шкалой объект-микрометра, чтобы они были параллельны;
- сравнивают шкалы, используя как можно больше делений на каждой шкале;
- записывают число делений на линейной шкале сетки - и число делений на шкале объект-микрометра -
;
- рассчитывают относительную погрешность окулярной сетки по следующей формуле:
![]() . (Г.1)
. (Г.1)
Допустима относительная погрешность в пределах ±2%.
Г.4.2.2 Основным методом измерения с использованием окулярной сетки с окружностями является мысленный перенос изображения каждой микрочастицы в поле зрения окулярной сетки, а затем сравнение их с окружностями сравнения диаметрами 10 и 25 мкм. Процесс измерения выполняется без наложения микрочастиц на окружности сравнения, микрочастицы не сдвигаются со своего места в пределах поля зрения окулярной сетки.
Г.4.2.3 Для измерения белых и прозрачных микрочастиц следует использовать внутренний диаметр светлых опорных окружностей, для измерения темных микрочастиц - наружный диаметр черных непрозрачных окружностей сравнения.
Г.4.2.4 Фокусируя окулярную сетку регулированием диоптрийного кольца правого окуляра, поворачивают ее в правом окуляре так, чтобы линейная шкала находилась внизу поля зрения, наблюдая исследуемый образец не в фокусе.
Г.4.2.5 Фокусируют микроскоп на исследуемый ОИР. Наблюдают ОИР только через правый окуляр. Наблюдая через левый окуляр и регулируя диоптрийное кольцо, добиваются фокусировки исследуемого ОИР.
Г.4.2.6 При выполнении полного подсчета не принимают во внимание поле зрения сетки, ограниченное большой окружностью. Используют вертикальное перекрестье.
Г.4.2.7 Сканируют всю мембрану справа налево по траектории, которая примыкает, но не перекрывает первую траекторию сканирования. Повторяют процедуру, перемещаясь вправо-влево, пока все частицы на мембране не будут подсчитаны.
Г.4.2.8 Записывают общее число частиц, размеры которых составляют 10 мкм или более, и общее число частиц, размеры которых составляют 25 мкм или более.
Г.4.3 Правила обработки результатов анализа
Г.4.3.1 Рассчитывают число частиц на контейнер по формуле
, (Г.2)
где - общее число подсчитанных частиц;
- объем испытуемого ОИР, мл.
Г.4.3.2 Корректируют степень разбавления для получения числа микрочастиц при подготовке ОИР, т.е. рассчитывают число выявленных микрочастиц на объем исследуемого ОИР.
Приложение Д
(обязательное)
Испытание ОИР методом внутриглазной имплантации*
_______________
* В бумажном оригинале наименование приложения Д выделено курсивом. - .
Д.1 Общие требования
Д.1.1 При проведении испытания внутриглазной имплантации проводят оценку местного действия на живую ткань образца ОИР, имплантированного хирургическим способом в место, соответствующее предусмотренному применению, маршруту и длительности контакта как на микро-, так и макроуровнях.
Д.1.2 Общие требования к испытаниям на имплантацию соответствуют указанным в ГОСТ Р ИСО 10993-6 и ГОСТ Р ИСО 10993-12.
Д.1.3 В качестве места для имплантации может быть использована передняя камера или стекловидная полость глаза животного, подходящего для испытания.
Д.1.4 В соответствии с требованиями [5] испытание на животном должно быть сокращено до оправданного минимума.
Д.2 Порядок проведения испытания
Д.2.1 Необходимый объем ОИР, соответствующий предполагаемому использованию, вводят в переднюю камеру или в переднюю часть стекловидной полости глаза.
Д.2.2 Имплантацию осуществляют с минимально возможной травмой глаза для того, чтобы хирургическая травма глаза не маскировала повреждения, полученные в результате воздействия испытуемого или контрольного ОИР.
Д.2.3 В качестве образца сравнения используют другой зарегистрированный ОИР, присутствующий на рынке не менее пяти лет и предназначенный для тех же целей.
Д.3 Правила обработки результатов испытания
Д.3.1 Воспалительная реакция глаза, возникающая после введения ОИР, должна постоянно контролироваться и классифицироваться согласно стандартизованной системе количественных показателей для биомикроскопического исследования с использованием щелевой лампы через 4-6 ч и биомикроскопического исследования и измерения толщины роговицы через 24, 48, 72 ч и 1 нед после введения ОИР.
Д.3.2 Внутриглазное давление измеряют в моменты времени, указанные в оценке риска. Допускается приводить дополнительные оценки в зависимости от длительности изучения имплантации.
Д.3.3 Результаты испытаний должны быть занесены в окончательный отчет.
Приложение Е
(обязательное)
Требования к клиническому испытанию ОИР*
_______________
* В бумажном оригинале наименование приложения Е выделено курсивом. - .
Е.1 Общие требования
Должны быть применены общие требования к клиническим испытаниям по ГОСТ Р 52458 и частные требования подразделов Е.2-Е.4 настоящего приложения.
Е.2 План клинического испытания
Е.2.1 Должно быть проведено статистическое клиническое испытание в контролируемых условиях. Целью исследования должна быть документально подтвержденная безопасность нового ОИР во время операции по сравнению с контрольным ОИР. Анализ рисков должен определить исходную гипотезу, а для расчета необходимого числа субъектов в каждой контрольной группе должны быть использованы стандартные биостатистические формулы.
Е.2.2 Контрольным раствором должен быть документированный ОИР, широко продаваемый в течение последних пяти лет, предназначенный и утвержденный для того же применения. Число не доведенных до конца субъектов в каждой контрольной группе должно составлять не более 10% общего числа вовлеченных в исследование субъектов.
Примечание - Исследования, проводимые в одной стране, могут привести к появлению дополнительных требований по выполнению правовых норм в других странах.
Е.2.3 Каждый клинический испытатель должен проводить одинаковую хирургическую операцию для всех субъектов.
Е.2.4 Наблюдатели не должны принимать участие в клиническом испытании. Если нельзя достигнуть сравнительного исследования нового и контрольного ОИР, послеоперационные исследования может провести независимый наблюдатель, который не должен знать, какой ОИР был использован.
Е.2.5 Длительность использования ОИР и объем, применяемый для каждого субъекта, должны быть подтверждены документально.
Е.2.6 Серьезные неблагоприятные операционные и послеоперационные исходы, а также вредные воздействия ОИР должны быть документально подтверждены и занесены в окончательный отчет в соответствии с ГОСТ Р 52458.
Е.2.7 Субъектов наблюдают до операции и через следующие промежутки времени после операции: (6±2) ч (только для измерения внутриглазного давления); (24±4) ч; 1 нед ±2 сут; 1 мес ±7 сут; 3 мес ±2 нед.
Е.2.8 В процессе клинических испытаний должны быть оценены следующие переменные параметры:
а) послеоперационное изменение внутриглазного давления,
б) толщина роговицы,
в) число клеток эндотелия роговицы,
г) острота зрения,
д) другие параметры, идентифицируемые оценкой риска.
Е.3 Оценка параметров
Е.3.1 Послеоперационное изменение внутриглазного давления
Если оценка риска указывает на необходимость мониторинга внутриглазного давления, то его следует измерять с помощью аппланационного тонометра Гольдмана. Давление следует измерять при каждом посещении субъекта.
Е.3.2 Толщина роговицы
Если оценка риска указывает на необходимость измерения толщины роговицы, то измерения проводят до операции и через 24 ч, 1 нед и 1 мес после операции.
Е.3.3 Послеоперационная воспалительная реакция глаза
Степень реакции воспаления глаза оценивают методом биомикроскопии с использованием щелевой лампы и клинически классифицируют при каждом посещении за исключением посещения через 6 ч.
Е.3.4 Состояние эндотелия роговицы
Состояние эндотелия роговицы оценивают путем подсчета числа клеток эндотелия роговицы до операции и через 3 мес после операции.
Е.3.5 Острота зрения
Скорректированную остроту зрения измеряют до операции и через 1 нед, 1 и 3 мес после операции.
Е.4 Примеры расчета числа субъектов, необходимого для проведения клинических испытаний
Е.4.1 Расчет числа субъектов для измерения толщины роговицы
Е.4.1.1 Пример основан на изменении толщины роговицы в качестве основного критерия. За нулевую гипотезу () принимают то обстоятельство, когда разница в изменении толщины роговицы испытуемого и контрольного ОИР до операции и через 24 ч после операции больше или равна заданному пределу клинической эквивалентности (
, мм). Альтернативной гипотезой (
) является то обстоятельство, когда разница в изменении толщины роговицы испытуемого и контрольного ОИР до операции и через 24 ч после операции меньше заданного предела клинической эквивалентности (
, мм).
Таким образом
![]()
![]() , (Е.1)
, (Е.1)
где и
![]() - средние значения числа субъектов для испытуемой и контрольной групп.
- средние значения числа субъектов для испытуемой и контрольной групп.
Е.4.1.2 Чтобы построить двухсторонний доверительный интервал для разницы в изменении толщины роговицы, который находится в пределах ±, мм, когда истинная разница равна нулю, при мощности 80% и стандартном отклонении для изменения
, мм, необходимое число субъектов в группе рассчитывается по следующей формуле:
![]() , (Е.2)
, (Е.2)
где и
- стандартные нормальные отклонения, соответствующие
и
.
Е.4.1.3 Испытуемый ОИР считают эквивалентным контрольному ОИР, если двухсторонний 95%-ный доверительный интервал для разницы в изменении толщины роговицы находится в соответствующих пределах клинической эквивалентности ±, мм.
Если стандартное отклонение при изменении толщины роговицы до операции и через 24 ч после операции составляет 0,05 мм, а предел клинической эквивалентности установлен на ±0,025 мм, в испытуемой группе должно быть 86 субъектов. Дополнительные субъекты, не менее 10%, должны быть включены в испытания, чтобы предусмотреть отсев.
Е.4.2 Расчет числа субъектов для подсчета клеток эндотелия
Е.4.2.1 Пример основан на процентном изменении клеток эндотелия в качестве основного конечного критерия. За нулевую гипотезу () принимают то обстоятельство, когда разница в процентном изменении клеток эндотелия испытуемого и контрольного ОИР до операции и спустя 3 мес после операции больше или равна заданному пределу клинической эквивалентности (
, мм). Альтернативной гипотезой (
) является то обстоятельство, когда разница в процентном изменении клеток эндотелия испытуемого и контрольного ОИР до операции и спустя 24 ч после операции меньше заданного предела клинической эквивалентности (
, мм).
Таким образом:
![]()
![]() , (E.3)
, (E.3)
где и
![]() - средние значения числа субъектов для испытуемой и контрольной групп.
- средние значения числа субъектов для испытуемой и контрольной групп.
Е.4.2.2 Чтобы построить двухсторонний доверительный интервал для разницы в процентном изменении клеток эндотелия, который находится в пределах ±, мм, когда истинная разница равна нулю, при мощности 80% и стандартном отклонении для изменения
, мм, необходимое число субъектов в группе рассчитывается по следующей формуле:
![]() , (E.4)
, (E.4)
где и
- стандартные нормальные отклонения, соответствующие
и
.
Е.4.2.3 Испытуемый ОИР считают эквивалентным контрольному ОИР, если двухсторонний 95%-ный доверительный интервал для разницы в процентном изменении клеток эндотелия находится в соответствующих пределах клинической эквивалентности ±, мм.
Если стандартное отклонение при процентном изменении клеток эндотелия до операции и через 3 мес после операции составляет 15%, а предел клинической эквивалентности установлен на ±7,5%, в испытуемой группе должно быть 86 субъектов. Дополнительные субъекты, не менее 10%, должны быть включены в испытания, чтобы предусмотреть отсев.
Приложение Ж
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации, использованным в настоящем стандарте в качестве нормативных ссылок
Ж.1 Сравнение ссылочных международных (региональных) стандартов с национальными стандартами Российской Федерации, использованными в настоящем стандарте в качестве нормативных ссылок, приведено в таблице Ж.1.
Таблица Ж.1 - Сравнение ссылочных международных (региональных) стандартов с национальными стандартами Российской Федерации, использованными в настоящем стандарте в качестве нормативных ссылок
Обозначение ссылочного национального стандарта Российской Федерации | Обозначение и наименование ссылочного международного стандарта и условное обозначение степени соответствия ссылочному национальному стандарту |
ГОСТ Р ИСО 10993-1-2009 | ИСО 10993-1:1997 Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования (IDT) |
ГОСТ Р ИСО 10993-5-2009 | ИСО 10993-5:1999 Оценка биологического действия медицинских изделий. Часть 5. Исследования на цитотоксичность: методы in vitro (IDT) |
ГОСТ Р ИСО 10993-6-2009 | ИСО 10993-6:1994 Оценка биологического действия медицинских изделий. Часть 6. Исследования местного действия после имплантации (IDT) |
ГОСТ Р ИСО 10993-7-2009 | ИСО 10993-7:1995 Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации (IDT) |
ГОСТ Р ИСО 10993-9-2009 | ИСО 10993-9:1999 Оценка биологического действия медицинских изделий. Часть 9. Основные принципы идентификации и количественного определения потенциальных продуктов деградации (IDT) |
ГОСТ Р ИСО 10993-10-2009 | ИСО 10993-10:2002 Оценка биологического действия медицинских изделий. Часть 10. Исследования раздражающего и сенсибилизирующего действия (IDT) |
ГОСТ Р ИСО 10993-12-2009 | ИСО 10993-12:2007 Оценка биологического действия медицинских изделий. Часть 12. Приготовление проб и контрольные образцы (IDT) |
ГОСТ Р ИСО 14630-1999 | ИСО 14630:1997 Неактивные хирургические имплантаты. Общие требования (IDT) |
ГОСТ Р ИСО 14971-2009 | ИСО 14971:2007 Медицинские изделия. Применение менеджмента риска к медицинским изделиям (IDT) |
ГОСТ Р ИСО 15223-2002 | ИСО 15223:2000 Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и сопроводительной документации (IDT) |
ГОСТ Р 51148-1998 | - |
ГОСТ Р 51609-2000 | - |
ГОСТ Р 51892-2002 | - |
ГОСТ Р 52458-2005 | ИСО 11979-7:2001 Имплантаты офтальмологические. Внутриглазные линзы. Часть 7. Клинические исследования (MOD) |
Приложение И
(справочное)
Сопоставление структуры настоящего стандарта со структурой примененного международного стандарта
И.1 Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта представлено в таблице И.1. Указанное в таблице изменение структуры межгосударственного стандарта относительно структуры примененного международного стандарта обусловлено приведением его в соответствие с требованиями ГОСТ Р 1.5.
Таблица И.1 - Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта
Структура международного стандарта | Структура настоящего стандарта |
1 Область распространения | 1 Область применения |
2 Ссылки на нормативные документы | 2 Нормативные ссылки |
3 Термины и определения | 3 Термины и определения* |
4 Предполагаемые характеристики | 4 Общие требования безопасности ОИР 4.1 Требования к предусмотренным характеристикам |
5 Конструктивные особенности | 4.2 Требования к конструктивным особенностям** |
5.1 Общие положения | 4 2 1 Общие требования |
5.2 Концентрация компонентов | 4.2.2 Требования к концентрации компонентов |
5.3 Используемая вода | 4.2.3 Требования к используемой воде |
5.4 Характеристики готового изделия | 4.2.4 Требования к определению характеристик готового ОИР |
5.4.1 Общие положения | |
5.4.2 Показатель рН и буферная емкость | 4.2.4.1 Общие требования |
5.4.3 Химические и биологические (загрязняющие) примеси | 4.2.4.2 Требования к показателю рН и буферной способности |
5.4.4 Осмотичность | 4.2.4.3 Требования к химическим и биологическим загрязняющим примесям |
5.4.5 Спектральный коэффициент пропускания | |
5.4.6 Микрочастицы | 4.2.4.4 Требования к осмотическому давлению |
5.4.6.1 Общие сведения (Учтено в 4.2.4.6) | 4.2.4.5 Требования к спектральному коэффициенту пропускания |
5.4.6.2 Видимые частицы (Учтено в 4.2.4.6) | 4.2.4.6 Требования к микрочастицам |
6 Оценка структуры | 5 Требования к оценке конструкции ОИР** |
6.1 Общие положения | 5.1 Общие требования |
6.2 Доклиническая оценка биологической безопасности | 5.2 Требования к доклинической оценке биологической безопасности |
6.2.1 Общие положения | 5.2.1 Общие требования |
6.2.2 Испытание на бактериальные эндотоксины | 5.2.2 Требования к оценке на наличие бактериальных эндотоксинов |
6.2.3 Послеоперационная воспалительная реакция глаза | 5.2.3 Требования к испытанию на послеоперационную воспалительную реакцию глаза |
6.3 Клиническая оценка | 5.3 Требования к клинической оценке |
5.3.1 Общие требования | |
7 Стерилизация | 6 Требования к стерильности ОИР |
8 Стабильность изделия | 7 Требования к стабильности ОИР |
9 Упаковка | 8 Требования к упаковке ОИР |
9.1 Защита от повреждения во время хранения и транспортирования | 8.1 Требования к защите от повреждения |
9.2 Поддержание стерильности при транспортировке | 8.2 Требования к защите стерильности |
10 Информация, предоставляемая изготовителем | 9 Требования к информации, предоставляемой изготовителем ОИР |
Приложение А (справочное) Пример метода измерения показателя рН и определения буферной емкости | Приложение А (справочное) Методы измерения рН и определения буферной способности ОИР** |
А.1 Общие сведения | А.1 Общие требования |
А.2 Оборудование и реактивы | А.2 Требования к оборудованию и вспомогательным устройствам |
А.3 Методика | А.3 Порядок подготовки, проведения измерений и правила обработки результатов измерения |
Приложение В (обязательное) Загрязнение микрочастицами (видимые микрочастицы) | Приложение Б (обязательное) Метод проверки загрязнения ОИР видимыми микрочастицами** |
В.1 Общие сведения | Б.1 Общие требования |
В.2 Устройство | Б.2 Требования к оборудованию и вспомогательным устройствам |
В.3 Методика | Б.3 Порядок подготовки, проведения проверки и правила обработки результатов проверки |
Приложение С (справочное) Микроскопический анализ твердых частиц | Приложение В (справочное) Метод проверки загрязнения ОИР невидимыми микрочастицами** |
С.1 Общие сведения | В.1 Общие требования |
С.2 Устройство | В.2 Требования к оборудованию и вспомогательным устройствам |
С.2.1 Общие положения | В.2.1 Общие требования |
С.2.2 Предел концентрации | В.2.2 Требования к пределу измерения концентрации датчика |
С.2.3 Динамический диапазон | В.2.3 Требования к динамическому диапазону датчика |
С.2.4 Вода, очищенная от частиц | В.2.4 Требования к воде |
С.2.5 Калибровка | В.2.5 Требования к калибровке |
С.3 Методика | В.3 Порядок подготовки, проведение проверки и правила обработки результатов проверки |
С.3.1 Контрольное испытание | В.3.1 Общие требования |
С.3.2 Метод | В.3.2 Порядок подготовки к проведению контрольной проверки |
В.3.3 Порядок проведения проверки и оценка результатов | |
Приложение D (справочное) Метод испытания с загораживанием света при загрязнении микрочастицами (субвидимые частицы) | Приложение Г (рекомендуемое) Микроскопический анализ твердых (невидимых) микрочастиц |
D.1 Введение | Г.1 Общие требования |
D.2 Испытательная установка | Г.2 Требования к оборудованию и вспомогательным устройствам |
D.2.1 Микроскоп | Г.2.1 Микроскоп |
D.2.2 Осветители | Г.2.2 Осветители |
D.2.3 Окулярная сетка | Г.2.3 Окулярная сетка с окружностями |
D.2.4 Микрометр | Г.2.4 Объект-микрометр |
D.2.5 Устройство фильтрации | Г.2.5 Устройство фильтрации и вспомогательные принадлежности |
D.3 Метод | Г.3 Порядок подготовки к проведению анализа |
D.3.1 Подготовка | Г.3.1 Общие требования |
D.3.1.1 Внешние условия при испытании | |
D.3.1.2 Подготовка микроскопа | Г.3.2 Подготовка микроскопа |
D.3.1.3 Работа окулярной сетки (учтено в Г.4.2) | |
D.3.1.4 Подготовка устройства фильтрации | Г.3.3 Подготовка устройства фильтрации |
D.3.2 Методика испытаний | Г.3.4 Подготовка к анализу испытуемых ОИР |
D.3.2.1 Препараты для испытания | |
D.3.2.2 Определение продукта (жидкие препараты) | Г.4 Порядок проведения анализа |
D.3.3 Подсчет частиц | Г.4.1 Порядок анализа испытуемых ОИР |
D.3.3.1 Общие положения | Г.4.2 Порядок проведения работ с окулярной сеткой и подсчет микрочастиц |
D.3.3.2 Методика полного подсчета | Г.4.3 Правила обработки результатов анализа |
Приложение Е (обязательное) Испытание на имплантацию внутрь глаза | Приложение Д (обязательное) Испытание ОИР методом внутриглазной имплантации** |
Е.1 Общие сведения | Д.1 Общие требования |
Е.2 Методика испытания | Д.2 Порядок проведения испытания |
Е.3 Оценка испытания | Д.3 Правила обработки результатов испытания |
Приложение F (справочное) Клиническое испытание | Приложение Е (обязательное) Требования к клиническому испытанию ОИР |
F.1 Общие положения | Е.1 Общие требования |
F.2 План клинического испытания | Е 2 План клинического испытания |
F.3 Конечные результаты | Е.3 Оценка параметров |
F.3.1 Послеоперационное изменение давления | Е.3.1 Послеоперационное изменение внутриглазного давления |
F.3.2 Толщина роговицы | Е.3.2 Толщина роговицы |
F.3.3 Послеоперационное воспаление | Е.3.3 Послеоперационная воспалительная реакция глаза |
F.3.4 Подсчет клеток эндотелия | Е.3.4 Состояние эндотелия роговицы |
F.3.5 Острота зрения | Е.3.5 Острота зрения |
F.3.6 Дополнительные конечные пункты (учтено в Е.1) | |
F.4 Примеры вычисления количества пациентов | Е.4 Примеры расчета числа субъектов, необходимого для проведения клинических испытаний |
F.4.1 Вычисление количества пациентов, основанное на изменении толщины роговицы | Е.4.1 Расчет числа субъектов для измерения толщины роговицы |
F.4.2 Вычисление количества пациентов, основанное на потере клеток эндотелия | Е.4.2 Расчет числа субъектов для подсчета клеток эндотелия |
- | Приложение Ж (справочное) Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации, использованным в настоящем стандарте в качестве нормативных ссылок |
Библиография | Библиография |
* В настоящий стандарт включены термины из ГОСТ Р 51892 и термины, необходимые для лучшего понимания текста стандарта. ** Данные разделы и приложения в настоящем стандарте разбиты на пункты (подпункты), что обусловлено необходимостью приведения их в соответствие с ГОСТ Р 1.5. *** Изменение статуса приложений в настоящем стандарте обусловлено необходимостью учета потребностей национальной экономики Российской Федерации. | |
Библиография
[1] | USP 24 <788> | Фармакопея США, 24-е издание. Пятое Дополнение 1996, <788>, Частичное вещество в инъекциях. Испытание на подсчет микроскопических частиц |
USP 24 <788> | United States Pharmacopoeia, 24th revision. Fifth Supplement 1996. <788>, Particulate matter in injections-Microscopic particle count test | |
[2] | ГФ II* | Государственная фармакопея. Издание 11 (в части воды) |
________________ * Документ в информационных продуктах не содержится. За информацией о документе Вы можете обратиться в Службу поддержки пользователей. - . | ||
[3] | ЕН 12442-1:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 1. Анализ и менеджмент риска |
EN 12442-1:2000 | Animal tissues and their derivates utilized in the manufacture of medical devices - Part 1: Analysis and management of risk | |
EH 12442-2:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 2. Регулирование источника, сбора и обработки | |
EN 12442-2:2000 | Animal tissues and their derivates utilized in the manufacture of medical devices - Part 2: Controls on sourcing, collection and handling | |
EH 12442-3:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 3. Аттестация уничтожения и/или дезактивации вирусов и других передаваемых агентов | |
EN 12442-3:2000 | Animal tissues and their derivates utilized in the manufacture of medical devices - Part 3: Validation of elimination and/or inactivation of viruses and other transmissible agents | |
[4] | 1USP 24 <85> | Фармакопея Соединенных Штатов, 24-е издание, <85> Определение бактериальных эндотоксинов |
USP 24 <85> | United States Pharmacopoeia, 24th revision, <85> Bacterial endotoxins test | |
[5] | ИСО 10993-2:2006 | Оценка биологического действия медицинских изделий. Часть 2. Требования к условиям содержания животных (ISO 10993-2:2006) (Biological evaluation of medical devices - Part 2: Animal welfare requirements) |
[6] | AAMI TIR No. 19:1998 | Технический информационный отчет по ИСО 10993-7 AAMI TIR No. 19:1998 Technical Information Report for ISO 10993-7 |
[7] | ИСО 13408-1:1998 | Асептическая обработка изделий для охраны здоровья. Часть 1. Общие требования |
ISO 13408-1:1998 | Aseptic processing of health care products - Part 1: General requirements | |
[8] | ИСО 11607-1:2006 | Упаковка окончательно стерилизованных медицинских изделий. Часть 1. Требования к материалам, системам стерильных барьеров и упаковочным системам |
ISO 11607-1:2006 | Packaging for terminally sterilized medical devices - Part 1: Requirements for materials, sterile barrier systems and packaging systems | |
ИСО 11607-2:2006 | Упаковка окончательно стерилизованных медицинских изделий. Часть 2. Требования к аттестации процессов формирования, герметизации и сборки | |
ISO 11607-2:2006 | Packaging for terminally sterilized medical devices - Part 2: Validation requirements for forming, sealing and assembly processes | |
[9] | ЕН 868-1:1997 | Упаковочные материалы и системы для медицинских изделий, подлежащих стерилизации. Часть 1. Общие требования и методы испытания |
EN 868-1:1997 | Packaging materials and systems for medical devices which are to be sterilized - Part 1: General requirements and test methods | |
[10] | ЕН 1041:1998 | Информация, предоставляемая изготовителем медицинской продукции |
EN 1041:1998 | Information supplied by the manufacturer with medical devices | |
Электронный текст документа
и сверен по:
, 2010

{kind=link}