ГОСТ 31579-2012
(ISO 15798:2001)
Группа П46
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
Имплантаты офтальмологические
ИЗДЕЛИЯ ОФТАЛЬМОЛОГИЧЕСКИЕ ВИСКОХИРУРГИЧЕСКИЕ
Общие требования безопасности
Ophthalmic implants. Ophthalmic viscosurgical devices. General safety requirements
МКС 11.040.70
Дата введения 2015-01-01
Предисловие
Цели, основные принципы и порядок проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0-92 "Межгосударственная система стандартизации. Основные положения" и ГОСТ 1.2-2009 "Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, применения, обновления и отмены"
Сведения о стандарте
1 ПОДГОТОВЛЕН Федеральным государственным унитарным предприятием "Всероссийский научно-исследовательский институт стандартизации и сертификации в машиностроении" (ВНИИНМАШ)
2 ВНЕСЕН Федеральным агентством по техническому регулированию и метрологии (Росстандарт)
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 24 мая 2012 г. N 41)
За принятие проголосовали:
Краткое наименование страны по МК (ИСО 3166) 004-97 | Код страны по | Сокращенное наименование национального органа по стандартизации |
Азербайджан | AZ | Азстандарт |
Беларусь | BY | Госстандарт Республики Беларусь |
Казахстан | KZ | Госстандарт Республики Казахстан |
Киргизия | KG | Кыргызстандарт |
Россия | RU | Росстандарт |
Таджикистан | TJ | Таджикстандарт |
Узбекистан | UZ | Узстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 1 ноября 2012 г. N 644-ст межгосударственный стандарт ГОСТ 31579-2012 (ISO 15798:2001) введен в действие в качестве национального стандарта Российской Федерации с 1 января 2015 г.
5 Настоящий стандарт модифицирован по отношению к международному стандарту ISO 15798:2001* Ophthalmic implants - Ophthalmic viscosurgical devices (Имплантаты офтальмологические. Офтальмологические вискохирургические изделия) путем изменения структуры.
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Сравнение структуры международного стандарта со структурой настоящего стандарта приведено в приложении ДБ.
Дополнительные и измененные фразы, слова, показатели и/или их значения выделены в тексте стандарта курсивом*
________________
* В бумажном оригинале обозначения и номера стандартов и нормативных документов в разделе "Предисловие", приложении ДА и по тексту документа отмеченные знаком "**", приводятся обычным шрифтом, остальные по тексту документа выделены курсивом. - .
Степень соответствия - модифицированная (MOD).
Стандарт подготовлен на основе применения ГОСТ Р 53078-2008 (ИСО 15798:2001)
6 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодном информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты." В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячном информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
1 Область применения
Настоящий стандарт распространяется на офтальмологические вискохирургические изделия (далее - ОВИ) класса неактивных хирургических имплантатов, используемые во время хирургических операций в передней камере глаза субъекта. ОВИ предназначены для формирования и сохранения объема, защиты внутриглазных тканей и манипулирования ими при хирургическом вмешательстве. Фармакологическое действие ОВИ не рассматривается.
Стандарт устанавливает требования безопасности в отношении предусмотренного применения, характеристик конструкции, доклинической и клинической оценки, стерилизации, упаковки и этикетирования ОВИ и информации, предоставляемой изготовителем этих изделий.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие межгосударственные стандарты*:
_______________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ГОСТ ISO 10993-1-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования
ГОСТ ISO 10993-5-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 5. Исследование на цитотоксичность: методы in vitro
ГОСТ ISO 10993-6-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 6. Исследование местного действия после имплантации
ГОСТ ISO 10993-7-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации
ГОСТ ISO 10993-9-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 9. Основные принципы идентификации и количественного определения потенциальных продуктов деструкции
ГОСТ ISO 10993-10-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 10. Исследование раздражающего и сенсибилизирующего действия
ГОСТ ISO 10993-12-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 12. Приготовление проб и стандартные образцы
ГОСТ ISO 10993-16-2011 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 16. Моделирование и исследование токсикокинетики продуктов деструкции и вымывания
ГОСТ ISO 13485-2011 Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования
ГОСТ ИСО 14630-2002* Неактивные хирургические имплантаты. Общие технические требования
________________
* На территории Российской Федерации действует ГОСТ Р ИСО 14630-2011, здесь и далее по тексту. - .
ГОСТ ISO 14971-2011 Изделия медицинские. Применение менеджмента риска к медицинским изделиям
ГОСТ 31214-2003 Изделия медицинские. Требования к образцам и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность
ГОСТ 31508-2012 Изделия медицинские. Классификация в зависимости от потенциального риска применения. Общие требования
ГОСТ 31580.7-2012 (ISO 11979-7:2001) Имплантаты офтальмологические. Интраокулярные линзы. Часть 7. Клинические испытания
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя "Национальные стандарты" за текущий год. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применен термин "не доведенный до конца" по национальному стандарту государств, упомянутых в предисловии, как проголосовавших за принятие настоящего межгосударственного стандарта, а также следующие термины с соответствующими определениями:
3.1 базовая окружность (reference circle): Окружность стандартного размера диаметром 10 или 25 мкм, используемая для измерения эквивалентного диаметра частицы.
3.2 вязкоупругое изделие (viscoelastic): Изделие, обладающее свойствами вязкости и упругости.
3.3 диаметр эквивалентной площади (equivalent area diameter): Диаметр круга, площадь которого эквивалентна площади частицы.
3.4 контейнер для хранения (storage container): Часть упаковки, предназначенная для защиты изделия во время транспортирования и хранения и содержащая упаковочную вставку и стерильный барьер.
3.5 контрольная группа (control populations): Субъекты, которым имплантированы документированные офтальмологические вискохирургические изделия.
3.6 клинический испытатель (clinical investigator): Лицо и/или организация, ответственное(ая) за проведение клинических испытаний и несущее(ая) клиническую ответственность за благополучие вовлеченных субъектов.
3.7 клиническое испытание (clinical investigation): Любое разработанное и запланированное систематическое изучение в целях проверки безопасности и/или эксплуатационных свойств офтальмологических вискохирургических изделий с участием субъектов.
3.8 наблюдатель (monitor): Лицо, назначенное организатором для наблюдения за соответствием действий клинического испытателя протоколу клинических испытаний и проверки исходных данных.
3.9 нулевая сдвиговая вязкость (zero shear viscosity): Установившаяся сдвиговая вязкость при скорости сдвига, приближающейся к нулю.
3.10 организатор (sponsor): Лицо и/или организация, которые несут ответственность за начало и/или проведение клинических испытаний ОВИ.
3.11 окончательный отчет (final report): Документ, заполняемый после окончания испытаний ОВИ, в том числе клинического, содержащий описание и оценку результатов.
3.12 офтальмологическое вискохирургическое изделие; ОВИ (ophthalmic viscosurgical device): Кратковременный имплантат, применяемый при хирургическом вмешательстве в передней камере глаза субъекта.
3.13 первичный контейнер (primary container): Ампула или шприц, содержащая(ий) офтальмологическое вискохирургическое изделие.
Примечание - Первичный контейнер является частью системы доставки.
3.14 план клинических испытаний; ПКИ (clinical investigation plan): Документ, определяющий цели, задачи, предполагаемую методологию, систему контроля, проведения анализа и хранения данных клинических испытаний.
3.15 поле зрения окулярной сетки; ПЗС: Площадь внутри большой окружности.
3.16 реологически активный компонент (rheologically active component): Соединение или смесь соединений в готовом офтальмологическом вискохирургическом изделии, придающее(ая) изделию вязкие или вязкоупругие свойства.
3.17 сдвиговая вязкость (shear viscosity): Способность вещества сопротивляться деформации под воздействием нагрузки.
Примечания
1 Значение сдвиговой вязкости определяют как частное от деления значения касательного напряжения на значение скорости сдвига в установившемся сдвиговом потоке.
2 Сдвиговую вязкость выражают в миллипаскаль-секундах (МПа·с). Ранее сдвиговую вязкость выражали в сантипуазах (сР).
3.18 серьезный неблагоприятный исход (adverse devise effect): Послеоперационный исход, потенциально угрожающий зрению.
Примечание - Примеры серьезных неблагоприятных исходов приведены в ГОСТ 31580.7, таблицы А.1 и А.2 (приложение А).
3.19 система доставки (delivery system): Герметичный контейнер, в котором поставляют изделие и дополнительные компоненты для введения данного изделия в глаз.
3.20 стерильный барьер (sterile barrier): Пакет, содержащий офтальмологическое вискохирургическое изделие и систему доставки, поддерживающую стерильность во время транспортирования и хранения.
3.21 субъект (subject): Лицо, участвующее в клинических испытаниях с применением испытуемого офтальмологического вискохирургического изделия и/или в качестве лица контрольной группы.
3.22 упругость (elasticity): Свойство физического тела восстанавливать первоначальную форму после деформации.
Примечание - Значение упругости определяют как отношение значения нагрузки (силы, возникающей внутри тела) к размеру деформации (изменению размеров тела).
4 Общие требования безопасности офтальмологических вискохирургических изделий
4.1 Требования к предусмотренным характеристикам
4.1.1 Общие требования к предусмотренным характеристикам ОВИ должны соответствовать требованиям для неактивных хирургических имплантатов по ГОСТ ИСО 14630. Дополнительно к ним изготовитель должен описать и документировать функциональные характеристики ОВИ в отношении:
a) химического состава;
b) реологических свойств;
c) эффективности защиты роговичного эндотелия.
4.2 Требования к конструктивным особенностям
4.2.1 Общие требования
4.2.1.1 Общие требования к предусмотренным характеристикам неактивных хирургических имплантатов - по ГОСТ ИСО 14630.
4.2.1.2 Все требования, предъявляемые к определениям характеристик, описанные в подразделе, должны быть соблюдены при испытании готового стерильного ОВИ.
Примечание - Указанные требования предназначены для применения при аттестации материалов, но необязательны к применению в стандартной программе обеспечения/контроля качества.
4.2.1.3 Чистота используемой воды должна соответствовать чистоте воды для инъекций в соответствии с [1]*.
________________
* См. раздел Библиография. - .
4.2.2 Требования к определению характеристик реологически активных компонентов
4.2.2.1 Характеристика химических свойств
Изготовитель должен предоставить описание каждого реологически активного компонента, содержащегося в ОВИ. Необходимо перечислить исходные материалы, используемые при его изготовлении, а также требования к качеству продукции. Исходные материалы должны соответствовать нормативным документам при их наличии. К реологически активным компонентам, извлеченным из источников животного происхождения, применяют требования региональных стандартов [2], [3] и [4].
Если реологически активным компонентом является высокомолекулярный органический полимер, все повторяющиеся подзвенья, составляющие полимер, должны быть идентифицированы в химическом отношении, все связи между ними должны быть описаны. Также должны быть описаны перекрестные связи.
Природа смеси реологически активного компонента в готовом ОВИ должна быть описана (например, растворенная, диспергированная). Если представлен раствор, должна быть определена растворимость реологически активного компонента в растворителе при температуре его хранения и при температуре (25±2) °С.
4.2.2.2 Концентрация
Концентрация вещества каждого реологически активного компонента в готовом ОВИ должна быть представлена в виде массы вещества на единицу объема раствора. Поскольку методика испытаний может оказать влияние на представленную фактическую концентрацию, должны быть указаны стандартные физические и химические методы.
4.2.2.3 Распределение молекулярной массы
Если реологически активным компонентом ОВИ является высокомолекулярный органический полимер, должна быть представлена средняя молекулярная масса.
Признано, что многие ОВИ содержат высокомолекулярные полидисперсные полимеры и что распределение молекулярной массы может быть комплексным. В таких случаях изготовитель должен провести дополнительные испытания с целью представить адекватное описание распределения молекулярной массы компонентов в готовом ОВИ и соответствующий отчет. Дополнительные методы, по возможности, должны быть стандартизованы.
4.2.3 Требования к определению характеристик готового офтальмологического вискохирургического изделия
4.2.3.1 Общие требования
Реологические и оптические свойства ОВИ являются физическими характеристиками, которые определяют их эксплуатационные качества в офтальмологической хирургии, поэтому физические свойства ОВИ, идентифицированные ниже, должны быть полно и точно описаны. Реологические свойства должны быть измерены в условиях, предполагаемых в момент применения ОВИ.
4.2.3.2 Сдвиговая вязкость
Сдвиговая вязкость ОВИ, поставляемого конечному потребителю, должна быть измерена в диапазоне скоростей сдвига, которые возникают в процессе обычного использования ОВИ. Измерения следует проводить при температуре (25±2) °С. Испытательное оборудование и другие условия измерения должны быть документированы.
Примечание - Предполагаемый диапазон скоростей сдвига расположен от одного крайнего значения 0,001 с, приближаясь к нулевому сдвигу, когда вязкоупругий материал находится в неподвижном состоянии в передней камере, до другого крайнего значения около 1000 с
, приближаясь к условиям, когда вязкоупругий материал впрыскивают в глаз через канюлю. Для ОВИ с низкой вязкостью измерить сдвиговую вязкость при очень низких скоростях сдвига невозможно. В таком случае вязкость может быть измерена при скорости сдвига от 1000 с
до самого низкого практически определяемого значения. Для определения нулевой сдвиговой вязкости ОВИ с очень высокой вязкостью (более 2·10
мПа·с) могут потребоваться скорости сдвига ниже 0,001 с
.
Соотношение вязкость/скорость сдвига должно быть представлено в виде стандартного графика зависимости логарифма вязкости от логарифма скорости сдвига. Вязкость должна быть измерена с помощью вращательного вискозиметра при нормальных условиях. Нулевую сдвиговую вязкость определяют как установившуюся сдвиговую вязкость при приближающейся к нулю скорости сдвига. Для растворов с высокой вязкостью измерения проводят с помощью реометра с постоянной нагрузкой.
4.2.3.3 Упругость
Упругость ОВИ должна быть измерена на частотах от 0,01 до 20 Гц. Измерения следует проводить при температуре (25±2) °С. Испытательное оборудование и другие условия измерения должны быть документированы. Логарифмы модуля вязкости и модуля упругости должны быть представлены на графике в зависимости от логарифма частоты. Данные также могут быть представлены в виде графика зависимости упругости от логарифма частоты в процентном выражении.
4.2.3.4 Химическое описание компонентов
Изготовитель должен документально подтвердить общую природу растворителя, а также представить подробный список всех компонентов, обоснование их включения в состав и молярную концентрацию в готовом ОВИ. Компоненты должны соответствовать требованиям нормативных документов при их наличии.
4.2.3.5 Значение рН
Значение рН готового ОВИ следует измерять посредством калиброванного рН-метра при температуре (25±2) °С. Значение рН ОВИ должно находиться в диапазоне от 6,8 до 7,6. Значение рН ОВИ должно быть близким к значению рН стекловидного вещества (рН 7,38), чтобы предотвратить повреждение клеток роговичного эндотелия. Исследования in vitro показали, что диапазон рН, переносимый эндотелием, сужается с увеличением времени воздействия.
4.2.3.6 Химические и биологические загрязняющие примеси
Определение потенциально опасных химических и биологических загрязняющих примесей должно быть проведено посредством анализа рисков. В исходных материалах биологического происхождения эти примеси могут включать в себя белки, нуклеиновые кислоты и другие вещества. Примеси в готовом ОВИ, полученные из исходных материалов или появляющиеся в процессе изготовления (например, вещества, образующие перекрестные связи, и антиоксиданты), представляющие собой потенциальную опасность для тканей глаза или системную опасность, должны быть идентифицированы, а их концентрация в готовом ОВИ должна быть определена.
Примечание - Частыми загрязнителями являются капли кремниевой смазки, вытекающие из шприца, ошибочно принимаемые за воздушные пузырьки или частицы. Этот источник загрязнения ОВИ необходимо рассмотреть при оценке рисков.
Загрязняющие примеси должны быть определены с помощью стандартных аналитических методов (при их наличии), которые должны быть описаны. Предельные концентрации идентифицированных примесей должны быть установлены и описаны. Необходимость проведения испытания для проверки биологического действия этих примесей в процессе оценки биологической безопасности определяют по результатам анализа рисков.
4.2.3.6* Осмотическая концентрация
__________________
* Нумерация соответствует оригиналу. - .
Изготовитель должен определить и документировать диапазон осмотической концентрации ОВИ. Осмоляльность готового ОВИ должна быть от 200 до 400 мОсм/кг. Осмоляльность должна быть определена с помощью парового или криоскопического осмометра при нормальных условиях.
4.2.3.8 Коэффициент пропускания
Коэффициент пропускания готового ОВИ должен быть измерен в диапазоне от 300 до 1100 нм. Результаты должны быть представлены в графической форме: коэффициент пропускания, % в зависимости от длины волны, нм.
4.2.3.9 Твердые частицы
При наличии в готовом ОВИ частиц определенного размера и характеристик потенциально возможно возникновение неблагоприятных явлений: например, избыточное или длительное по времени повышение внутриглазного давления (далее - ВГД).
При проведении оценки рисков необходимо определить потенциальную возможность загрязнения или образования частиц в ОВИ в процессе изготовления, в условиях, ожидаемых при транспортировании и хранении, а также при использовании ОВИ. В частности, необходимо учитывать потенциальную возможность агрегирования, полимеризации и прилипания частиц к внутриглазным тканям.
Примечание - ОВИ, содержащие синтетические полимеры, подвержены значительно более высокому риску образования микрогелей, идентификация которых и определение их количественного состава методом рассеянного света или микроскопическими методами затруднительны.
Изготовитель должен привести характеристику потенциальной опасности, связанной с частицами каждого вида, выявленного при проведении оценки рисков.
Изготовитель должен охарактеризовать виды, размеры и количество частиц, имеющихся в готовом ОВИ. Должны быть установлены предельные значения по частицам каждого вида, подтвержденные соответствующим обоснованием.
Примечание - Метод определения количества частиц представлен в приложении С.
4.2.3.10 Показатель преломления офтальмологических вискохирургических изделий
Показатель преломления ОВИ должен быть измерен с помощью рефрактометра при температуре (25±2) °С при фиксированной длине волны.
Примечание - Диапазон рекомендуемых значений показателей преломления должен быть приведен в технических условиях на ОВИ конкретного типа.
4.3 Требования к оценке свойств готового офтальмологического вискохирургического изделия
4.3.1 Общие требования
4.3.1.1 Требования к оценке свойств ОВИ должны соответствовать общим требованиям, приведенным в ГОСТ ИСО 14630 для неактивных хирургических имплантатов, и системным требованиям ГОСТ ISO 13485.
4.3.1.2 Требования к ОВИ и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность, должны соответствовать ГОСТ 31214.
4.3.1.3 Общие требования к оценке потенциального риска применения ОВИ должны соответствовать ГОСТ 31508.
4.3.2 Требования к оценке биологической безопасности
4.3.2.1 Общие требования
Оценку биологической безопасности ОВИ следует начинать с анализа рисков и проводить и документировать в соответствии с ГОСТ ISO 14971. По результатам анализа рисков должны быть определены испытания, необходимые для оценки биологической безопасности ОВИ.
Для ОВИ, содержащих материалы животного происхождения, должны быть применены анализ рисков и требования к менеджменту, приведенные в [2], [3] и [4].
Для всех ОВИ должны быть соблюдены требования, предъявляемые к оценке биологической безопасности по ГОСТ ISO 10993-1, ГОСТ ISO 10993-5, ГОСТ ISO 10993-10, и требования, изложенные в настоящем подразделе.
Примечания
1 На основании типичного клинического применения в передней камере глаза ОВИ классифицируют как "имплантируемые изделия; ткань/кость". Испытания изделий данной и других категорий, представленные в таблице 1 ГОСТ ISO 10993-1, приведены только для руководства без указания максимальных или минимальных требований.
2 Допускается комбинировать испытания на биологическую совместимость, уменьшая число животных, необходимых для проведения испытания. Два испытания могут быть проведены одновременно на одном животном при условии, что испытуемое животное не подвергают чрезмерной боли или страданиям.
4.3.2.2 Испытание имплантацией внутрь глаза
Для проведения данного испытания должна быть использована позиция имплантации: в передней камере глаза или стекловидном теле. Должны быть применены частные требования к испытаниям имплантацией внутрь глаза, указанные в приложении А.
Если испытуемое ОВИ вызывает большую реакцию глаза или воспаление, чем ОВИ, используемое как контрольное, необходимо провести оценку соотношения риск/польза.
4.3.2.3 Испытание на бактериальные эндотоксины
ОВИ должно быть оценено на наличие бактериальных эндотоксинов с помощью LAL-теста (на культуре limulus amoebocyte lysate) в соответствии с методикой, описанной в [5], или эквивалентной аттестованной методикой испытания. ОВИ, в котором предел бактериальных эндотоксинов превышает 0,5 единицы эндотоксинов (ЕЭ) на миллилитр, считают не прошедшим испытание.
4.3.2.4 Испытание на внутриглазное давление
Испытание на внутриглазное давление должно быть проведено в соответствии с методикой, представленной в приложении В.
Если испытуемое ОВИ вызывает более высокое давление или более длительное повышение давления, чем ОВИ, используемое как контрольное, необходимо провести оценку соотношения риск/польза.
Результаты испытания должны быть использованы для определения вероятного значения и длительности послеоперационного повышения внутриглазного давления. Это повлияет на план клинического испытания и может потребовать послеоперационных измерений внутриглазного давления дополнительно к приведенным в 4.4.3 периодам времени.
4.3.2.5 Удаление остаточного офтальмологического вискохирургического изделия из передней камеры
При отсутствии литературных данных по удалению остаточного ОВИ скорость, с которой остаточное ОВИ вымывается из передней камеры через трабекулярную зону, должна быть определена с использованием соответствующего метода испытания, например с помощью люминесцентных или радиоизотопных маркеров, и представлена в отчете.
4.3.2.6 Деградация и токсикокинетика
При отсутствии литературных данных о поведении ОВИ изготовитель должен представить доказательства: пути удаления, биотрансформацию и катаболические продукты компонентов. Идентификацию и количественное определение продуктов деструкции, исследование токсикокинетики продуктов деструкции и вымывания проводят по ГОСТ ISO 10993-9 и ГОСТ ISO 10993-16**.
4.4 Требования к клинической оценке
4.4.1 Общие требования
Настоящий подраздел устанавливает требования к клинической оценке ОВИ в передней камере глаза. Должны быть применены общие требования к неактивным хирургическим имплантатам по национальному стандарту государств, упомянутых в предисловии, как проголосовавших за принятие настоящего межгосударственного стандарта и частные требования 4.4.2-4.4.6.
4.4.2 План клинического испытания
4.4.2.1 Должно быть проведено статистическое клиническое испытание в контролируемых условиях. Целью исследования должна быть документально подтвержденная безопасность нового вязкоупругого ОВИ во время операции в передней камере по сравнению с контрольным ОВИ. Анализ рисков должен определить исходную гипотезу, а для расчета необходимого числа субъектов в каждой контрольной группе должны быть использованы стандартные биостатистические формулы.
4.4.2.2 Контрольным изделием должно быть документированное ОВИ, широко продаваемое в течение последних пяти лет и предназначенное для того же применения. Клинический испытатель должен задействовать не менее 20 субъектов или более 25% общего числа субъектов, вовлеченных в исследование. Число не доведенных до конца субъектов в каждой контрольной группе должно составлять не более 10% общего числа вовлеченных в исследование субъектов.
Примечание - Исследования, проводимые в одной стране, могут вызвать появление дополнительных требований по выполнению правовых норм в других странах.
4.4.2.3 Каждый клинический испытатель должен использовать при исследовании линзы одного и того же типа и одинаковую хирургическую операцию для всех субъектов.
4.4.2.4 Наблюдатели не должны принимать участие в клиническом исследовании. При невозможности сравнения результатов исследований нового и контрольного ОВИ послеоперационные исследования разрешается провести независимому наблюдателю, который не должен знать, какое ОВИ было использовано.
4.4.2.5 Если изготовитель предполагает сделать дополнительные заявления, например в отношении послеоперационного действия ОВИ, для поддержания таких заявлений должны быть определены дополнительные ориентиры, а также выполнен соответствующий пересчет необходимого числа субъектов.
4.4.2.6 В процессе клинических испытаний должны быть оценены следующие переменные параметры:
a) значение и длительность послеоперационного повышения внутриглазного давления;
b) число клеток эндотелия роговицы;
c) воспалительная реакция глаза или другие неблагоприятные межоперационные или послеоперационные явления.
4.4.2.7 Расчет числа субъектов, необходимых для проведения клинических испытаний ОВИ в испытуемых и контрольной группах, представлен в приложении D.
4.4.3 Послеоперационная оценка повышения внутриглазного давления
4.4.3.1 Внутриглазное давление должно быть измерено с помощью аппланационного тонометра Гольдмана перед операцией и после операции в следующие периоды времени:
(6±2) ч; (24±4) ч; 1 неделя ± 2 сут; 1 мес ± 7 сут; 3 мес ± 2 недели.
4.4.3.2 В случае если внутриглазное давление отдельного испытуемого субъекта остается повышенным в течение более 24 ч, должны быть проведены дополнительные измерения, пока значение внутриглазного давления не достигнет уровня нормы.
4.4.3.3 Назначение медикаментов для понижения давления в любые периоды времени должно быть оформлено документально, и данные должны быть предоставлены отдельно по каждому субъекту. Если официально опубликованные источники, результаты испытаний на животных или клинический опыт доказывают, что максимальное внутриглазное давление появляется вне послеоперационного диапазона от 4 до 8 ч, изготовитель должен изменить план клинического испытания и внести в него дополнительное измерение внутриглазного давления в момент, близкий к максимальному значению внутриглазного давления.
4.4.3.4 Мониторинг испытуемых субъектов с внутриглазным давлением, более или равным 30 мм рт.ст., должен быть проведен через неделю и позже в течение всего исследования. Если частотность мониторинга статистически превышает ожидаемое нулевое значение, следует рассмотреть возможность преждевременного прекращения клинического исследования. Данный анализ должен быть проведен во время исследования по всему числу испытуемых субъектов, а в конце исследования - по каждой испытуемой группе. По завершении исследования анализ измерений внутриглазного давления должен включать в себя расчет средних значений, а также частотности значений внутриглазного давления, более или равного 30 мм рт.ст.
4.4.4 Подсчет числа клеток эндотелия роговицы
Состояние эндотелия роговицы оценивают путем подсчета числа клеток эндотелия роговицы до операции и через (3±0,5) мес после операции.
4.4.5 Послеоперационная воспалительная реакция глаза
Степень реакции воспаления глаза оценивают методом биомикроскопии с использованием щелевой лампы и клинически классифицируют при каждом измерении внутриглазного давления.
4.4.6 Серьезный неблагоприятный исход
Клинические исследователи должны предоставлять отчеты о серьезных межоперационных и послеоперационных исходах заказчику немедленно, как только о них стало известно. Все другие неблагоприятные исходы должны быть документированы в историях болезни.
5 Требования к стерилизации
5.1 ОВИ должно быть стерилизовано в соответствии с требованиями к стерилизации, указанными в ГОСТ ИСО 14630 для неактивных хирургических имплантатов.
5.2 К ОВИ или его компонентам, при стерилизации которых используют влажное тепло, должны быть применены требования по [6].
5.3 К ОВИ или его компонентам, при стерилизации которых используют этиленоксид, должны быть применены требования по [7].
Примечание - Установлено, что требования, определяющие приемлемые уровни остаточного этиленоксида, указанные в ГОСТ ISO 10993-7, неприменимы к ОВИ, находящимся в контакте с высокочувствительными тканями, например тканями глаза. Дополнительные указания по применению требований ГОСТ ISO 10993-7 приведены в [8]. Для интраокулярных ОВИ специальных категорий предельное содержание этиленоксида для интраокулярной линзы установлено на уровне 0,5 мкг в день, но не более 1,25 мкг в целом. Пределы для других интраокулярных ОВИ могут быть предварительно заданы исходя из массы ОВИ.
Предельное содержание этиленоксида для ОВИ должно составлять 60 мкг/мл (дополнительная информация по методике измерения приведена в [8]).
Примечание - В техническом информационном отчете [8] отсутствуют требования по предельному содержанию этиленхлоргидрина. Предполагается, что в новой редакции [8] будет установлено предельное содержание этиленхлоргидрина для интраокулярных ОВИ, содержащих хпорид, в четыре раза превышающее содержание этиленоксида. Это следует учитывать при оценке биологической безопасности ОВИ.
5.4 К ОВИ, при стерилизации которых используют облучение, должны быть применены требования по [9].
Примечание - Известно, что многие ОВИ содержат высокомолекулярные полимеры, не являющиеся термостойкими, и что стерилизация влажным теплом может оказать отрицательное воздействие на их реологические свойства. Если ОВИ нельзя стерилизовать влажным теплом, применяют асептическую обработку.
5.5 К ОВИ, которые не стерилизуют, но подвергают асептической обработке, должны быть применены требования [10]. Соответствие данному международному стандарту должно быть подтверждено аттестованным исследованием в среде с предельно допустимым уровнем загрязнения - 10.
Примечание - Стандарт [10] устанавливает общие требования и содержит руководство по процессам, программам и методикам аттестации и контроля медицинских изделий, подвергаемых асептической обработке. Стандарт [10], в частности, применим к обработке физиологических (водных) растворов и, следовательно, имеет отношение к изготовлению ОВИ. Следующие части стандарта [10] будут посвящены специализированным процессам, таким как фильтрация и лиофилизация.
6 Требования к стабильности офтальмологических вискохирургических изделий
6.1 Изготовитель должен определить и установить срок годности ОВИ при хранении в складских помещениях и систему доставки. Определение срока годности при хранении должно быть проведено в реальном масштабе времени или по ускоренной методике при температуре не более 45 °С с целью подтвердить, что основные характеристики по безопасности и эффективные эксплуатационные качества готового ОВИ и системы доставки находятся в установленных пределах в течение обозначенного срока годности при ожидаемых условиях транспортирования и хранения.
6.2 Параметры и характеристики, которые необходимо контролировать во время исследований срока годности при хранении: реологический профиль, рН, стерильность и другие факторы, установленные при анализе рисков как критические для безопасного применения ОВИ.
6.3 Изменения состава ОВИ, исходных материалов и их поставщиков, условий изготовления, включая процесс стерилизации, тип упаковки или упаковочные материалы, могут оказать неблагоприятное влияние на срок службы ОВИ при хранении. Установленный срок службы ОВИ при хранении должен быть пересмотрен, если при анализе рисков выявлено изменение технологии изготовления, что может повлиять на стабильность ОВИ.
7 Требования к целостности и характеристике комплекта доставки
7.1 ОВИ поставляют в герметичных контейнерах вместе с канюлей для введения ОВИ в глаз. Эти два компонента являются комплектом доставки.
7.2 Отсутствие механического повреждения комплекта доставки должно быть подтверждено испытаниями для применения ОВИ по назначению.
7.3 Физико-химическая совместимость ОВИ и комплекта доставки должны быть также оценены.
8 Требования к упаковке
8.1 Защита от повреждения при хранении и транспортировании
Должны быть применены требования к упаковке медицинских изделий, указанные в ГОСТ ИСО 14630 и [11].
8.2 Защита стерильности при транспортировании
ОВИ должны быть упакованы таким образом, чтобы оставаться стерильными в пределах, указанных для условий транспортирования, хранения и обращения. Должны быть соблюдены требования к стерильной упаковке, указанные в [12].
9 Требования к информации, предоставляемой изготовителем
9.1 Должны быть соблюдены общие требования к информации, предоставляемой изготовителем медицинских изделий, указанные в [13], и частные требования к ОВИ, приведенные в настоящем разделе.
9.2 Допускается использовать символы вместо текста. При использовании символов должны быть соблюдены требования по национальному стандарту государств, упомянутых в предисловии, как проголосовавших за принятие настоящего межгосударственного стандарта.
9.3 Если ОВИ подвержено повреждению под действием факторов окружающей среды, на контейнере для хранения должны быть нанесены четкие предупредительные знаки.
9.4 Номер партии и конечный срок действия могут быть указаны на самоклеящейся этикетке.
9.5 Вкладыш должен быть вложен в упаковку так, чтобы его можно было взять и прочитать, не повреждая стерильного барьера.
9.6 Минимальная информация, наносимая на упаковочный контейнер, вкладыш в упаковку, стерильный барьер и первичный контейнер, приведена в таблице 1.
Таблица 1 - Информация, предоставляемая изготовителем
Информация | Контейнер для хранения | Вкладыш в упаковку | Стерильный барьер | Первичный контейнер |
Наименование изготовителя или уполномоченного представителя | + | + | + | - |
Адрес изготовителя | + | + | - | - |
Торговое наименование ОВИ | + | + | + | - |
Описание комплекта поставки и инструкция по его использованию | - | + | - | - |
Краткое описание химического состава ОВИ и объема поставки | + | + | - | - |
Описание соответствующих конструктивных параметров, которые могут повлиять на безопасность и характеристики ОВИ, в том числе: концентрация; распределение молекулярной массы; рН; осмомоляльность и др. | - | + | - | - |
Графическое представление реологического профиля: график логарифмической зависимости вязкости (МПа·с) от сдвига (с | - | + | - | - |
Условия хранения | + | + | - | - |
Показания к применению | - | + | - | - |
Противопоказания к применению | - | + | - | - |
Инструкция по применению, включая рекомендации по удалению ОВИ (при необходимости) | + | + | - | - |
Меры предосторожности и предупреждения | - | + | - | - |
Сообщение о том, что содержимое предназначено для разового использования | - | + | + | - |
Надпись: "Стерильно" и метод(ы) стерилизации ОВИ и системы доставки | + | + | + | - |
Надпись: "Не использовать, если стерильный барьер нарушен" | - | - | + | - |
Конечный срок годности | + | - | + | + |
Номер партии после слова "ЛОТ" | + | - | + | + |
Примечание - Номер партии, срок годности и дату стерилизации допускается не указывать на стерильном барьере, если он прозрачный и необходимую информацию можно прочесть на первичном контейнере, не нарушая его герметичности. | ||||
Приложение А
(обязательное)
Методика испытания имплантацией внутрь глаза
А.1 Общие требования
А.1.1 При испытании имплантацией внутрь глаза проводят оценку местного действия на живую ткань образца ОВИ, имплантированного хирургическим способом в месте, соответствующем предусмотренному применению, маршруту и длительности контакта как на микроуровнях, так и макроуровнях. Общие требования к испытаниям на имплантацию соответствуют указанным в ГОСТ ISO 10993-6, ГОСТ ISO 10993-12.
А.1.2 В качестве места для имплантации может быть использована передняя камера или стекловидная полость глаза животного, подходящего для испытания.
В соответствии с требованиями [14] испытание на животном должно быть сокращено до оправданного минимума.
А.2 Порядок проведения испытания
А.2.1 Соответствующий объем ОВИ вводят в переднюю камеру или в переднюю часть стекловидной полости глаза. Перед введением определенного объема испытуемого или контрольного ОВИ в область передней камеры или стекловидного тела допускается удалить равный объем влаги из соответствующей камеры.
А.2.2 Имплантацию осуществляют с минимально возможной травмой глаза, не маскирующей повреждения, полученные в результате воздействия испытуемого или контрольного материала.
А.2.3 В качестве контроля используют другое зарегистрированное ОВИ, присутствующее на рынке не менее пяти лет и предназначенное для тех же целей. Контрольный ОВИ имплантируют одновременно в то же место в другой глаз.
Примечание - Предпочтительна двусторонняя имплантация, но допускается и односторонняя имплантация, если того требуют местные предписания.
А.3 Правила обработки результатов испытания
А.3.1 Воспалительную реакцию, возникающую после введения материала, постоянно контролируют и классифицируют согласно стандартизованной системе количественных показателей для биомикроскопического исследования с использованием щелевой лампы с интервалами времени: 4-6, 24, 48, 72 ч и 1 неделя после введения.
А.3.2 Допускается вводить дополнительные сроки контроля в зависимости от продолжительности исследования.
А.3.3 Результаты испытаний должны быть внесены в окончательный отчет.
Приложение В
(обязательное)
Методика испытания на внутриглазное давление *
_________________
* Слова "Методика испытания" в наименовании приложения B в бумажном оригинале выделены курсивом. - .
В.1 Общие требования
В.1.1 Хирургическая операция в передней камере глаза с использованием ОВИ может сопровождаться повышением внутриглазного давления. Это является приемлемым последствием использования ОВИ и не должно существенно повлиять на зрительную функцию или восстановление глазных тканей. Значительное или продолжительное повышение внутриглазного давления может вызвать боль или дискомфорт и привести к повреждению тканей глаза.
В.1.2 Методика испытания позволяет контролировать повышение внутриглазного давления после замены влаги на ОВИ такого же объема в передней камере глаза испытуемого животного. ОВИ остается в глазу, следовательно, испытание не имитирует клиническое использование, при котором хирург удаляет как можно больше ОВИ, перед тем как закрыть надрез. Длительность и значение изменения внутриглазного давления во время предклинического испытания могут быть больше, чем длительность и значение внутриглазного давления во время клинического использования.
В.1.3 Испытание проводят только для сравнения ОВИ с контрольным ОВИ, утвержденным для такого же использования.
В.2 Порядок проведения испытания
В.2.1 Объем водянистой влаги, эквивалентный 25% объема передней камеры глаза испытуемого животного, удаляют и заменяют на ОВИ равного объема.
В.2.2 В качестве контроля используют другое зарегистрированное ОВИ, присутствующее на рынке не менее пяти лет и предназначенное для тех же целей. Контрольный ОВИ имплантируют одновременно в то же место в другой глаз.
Примечание - Предпочтительна двусторонняя имплантация, так как число животных, необходимое для обеспечения достоверности испытания, будет меньше, чем при односторонней имплантации. Допускается и односторонняя имплантация, если того требуют местные предписания.
В.3 Правила обработки результатов испытания
В.3.1 Внутриглазное давление измеряют методом аппланационной тонометрии перед операцией и в следующие периоды времени после операции: 1 ч ± 15 мин; 2 ч ± 30 мин; (4±1) ч; (8±2) ч; (12±2)ч; (24±4) ч; 1 неделя ± 2 сут; 1 мес ± 7 сут. Скорость и длительность повышения внутриглазного давления существенно зависят от природы ОВИ и его вязкости. Как только эти показатели стабилизируются, число измерений внутриглазного давления может быть изменено, чтобы более точно следовать его изменению. Могут потребоваться дополнительные проверки внутриглазного давления, если оно остается повышенным в течение более 24 ч после испытания.
В.3.2 Результаты испытаний должны быть внесены в окончательный отчет.
Приложение С
(рекомендуемое)
Микроскопический анализ твердых частиц
С.1 Общие требования
С.1.1 Микроскопический анализ предусматривает подготовку образцов ОВИ для количественного определения твердых частиц на основе метода по [15]. Перед фильтрацией и микроскопическим анализом ОВИ должны быть разбавлены из-за вязкости.
С.1.2 Допускается использовать альтернативные методы определения слепой пробы и подготовки образца. Необходимо тщательно оценивать изменения, которые могут повлиять на подсчет частиц.
С.2 Требования к оборудованию и вспомогательным устройствам
С.2.1 Микроскопы и принадлежности
С.2.1.1 Микроскоп (сложный бинокулярный), комбинация объектива и окуляра обеспечивает увеличение (100±10). Апохроматический объектив или лучший должен иметь номинальное увеличение 10
и минимальную числовую апертуру 0,25. Для исследования в отраженном свете должны быть предусмотрены эпископические осветители.
С.2.1.2 Два осветителя, оснащенных синими светофильтрами дневного света для снижения усталости глаза оператора:
- внешний осветитель, регулируемый для обеспечения косого освещения, падающего под углом 10°-20° к горизонтали;
- эпископический осветитель светлого поля, встроенный в микроскоп.
С.2.1.3 Сетка для улучшенного микроскопического анализа (далее - сетка IMA), калиброванная таким образом, что измерительные окружности в плоскости предметного столика микроскопа составляют 10 или 25 мкм с точностью 2%. Допускается использовать альтернативную сетку с требуемой точностью калибровки.
С.2.1.4 Микрометр ценой деления 10 мкм (100 дел./мм).
С.2.2 Устройство фильтрации и вспомогательные принадлежности
С.2.2.1 Фильтровальная воронка объемом, подходящим для испытания, и минимальным диаметром 21 мм. Воронка должна быть изготовлена из пластмассы, стекла или нержавеющей стали.
С.2.2.2 Воронка для фильтра Гельмана, 25 мм/200 мл (деталь N 4203).
С.2.2.3 Воронка для ультрафиолетового фильтра, 47 мм/300 мл (деталь N ХХ10 047 24).
С.2.2.4 Фильтровальный диффузор, выполненный в виде подставки под фильтр, изготовленный из нержавеющей стали или спеченного стекла.
С.2.2.5 Источник вакуума.
С.2.2.6 Пинцет с закругленными концами.
С.2.2.7 Дозаторы растворителя с фильтрующим соплом, обеспечивающим подачу растворителя, отфильтрованного от частиц размером не более 1,2 мкм при давлении от 689,5 до 5561 мбар (10-80 фунт/дюйм).
С.2.2.8 Мембранные фильтры диаметром 25 или 47 мм с сеткой или без сетки черного или темно-серого цвета, изготовленные из целлюлозно-эфирной смеси размером пор 0,8 мкм.
С.2.2.9 Чашка Петри.
С.2.2.10 Вытяжной шкаф с ламинарным потоком через высокоэффективный сухой воздушный фильтр (НЕРА) с содержанием не более 100 частиц размером от 0,5 мкм на кубический фут (приблизительно 3531 частиц/м).
С.2.2.11 Стерильные перчатки.
С.2.2.12 Одежда с подобранными рукавами.
С.2.2.13 Деионизированная вода.
С.2.2.14 Изопропанол.
С.2.2.15 Химический стакан вместимостью 600 мл с тефлоновой палочкой для перемешивания.
С.3 Порядок подготовки к проведению анализа
С.3.1 Общие требования
С.3.1.1 Первоначальное испытание: испытывают вместе 10 единиц ОВИ.
С.3.1.2 Повторное испытание: испытывают вместе 20 единиц ОВИ.
С.3.1.3 Перед испытанием сетка IMA должна быть измерена, ее относительная погрешность должна быть сертифицирована на соответствие требованиям Фармакопеи США [15]
С.3.2 Настройка микроскопа
С.3.2.1 Включают оба осветителя (С.2.1.2) (внешний вспомогательный осветитель падающего косого освещения и внутренний эпископический осветитель светлого поля).
С.3.2.2 Устанавливают объектив с увеличением 10 и регулируют межзрачковое расстояние до появления слитного изображения.
С.3.2.3 Фокусируют правый окуляр на сетку (С.2.1.3) при установленном образце перефокусировкой микроскопа (С.2.1.1) до появления светлого поля с невидимыми деталями образца. Закрывают левый глаз и вращают диоптрийное кольцо правого окуляра до появления изображения сетки в фокусе.
С.3.2.4 Фокусируют микроскоп на мелкую деталь образца.
С.3.2.5 Фокусируют левый парфокальный окуляр, закрывают правый глаз и вращают диоптрийное кольцо левого окуляра до тех пор, пока изображение мелкой детали образца не станет четким.
Открыв оба глаза, убеждаются, что изображение образца является четким в обоих глазах.
С.3.2.6 Регулируют вспомогательный осветитель падающего косого освещения по высоте так, чтобы угол падающего света составлял от 10° до 20° к горизонтали: частицы на мембранном фильтре (С.2.2.8) должны иметь отчетливые темные тени.
С.3.2.7 Регулируют внутренний эпископический осветитель светлого поля, полностью открыв полевую и апертурную диафрагмы. Центрируют нить накала лампы и фокусируют микроскоп на фильтр, содержащий частицы. Регулируют интенсивность отраженного света до тех пор, пока частицы не станут четко различимыми и не обнаружат отчетливые видимые тени.
С.3.2.8 Устанавливают самое низкое значение интенсивности эпископического освещения, затем повышают его до тех пор, пока тени, отбрасываемые частицами, не покажут минимально заметное снижение контраста.
С.3.3 Подготовка фильтровального устройства
С.3.3.1 Надевают перчатки (С.2.2.11) и одежду (С.2.2.12), моют руки фильтрованной до 0,22 мкм деионизированной водой (С.2.2.13).
С.3.3.2 Моют вручную химический стакан вместимостью 600 мл, палочку для перемешивания (С.2.2.15), фильтровальную воронку (С.2.2.1), подставку и диффузор в горячем, не содержащим ионов, растворе детергента. Промывают их горячей проточной водопроводной водой, затем промывают фильтрованной деионизированной водой под давлением.
С.3.3.3 Переносят вымытое устройство и другое необходимое оборудование в вытяжной шкаф с ламинарным потоком (С.2.2.10), защищенный фильтрованным воздухом НЕРА.
С.3.3.4 Промывают наружные и внутренние поверхности фильтровального устройства фильтрованным до 1,2 мкм изопропанолом под давлением.
С.3.3.5 Окончательно промывают наружные и внутренние поверхности фильтровального устройства и другого необходимого оборудования фильтрованной до 1,2 мкм деионизированной водой.
С.3.3.6 С помощью пинцета (С.2.2.6) тщательно вымывают в потоке фильтрованной деионизированной воды под низким давлением обе стороны фильтра сверху донизу, поворачивая вперед и назад, и устанавливают чистый фильтр по центру подставки диффузора.
С.3.3.7 Помещают фильтровальную воронку сверху подготовленной подставки с фильтром и закрепляют.
С.4 Порядок проведения анализа
С.4.1 Порядок проверки фонового загрязнения
С.4.1.1 С помощью дозатора растворителя (С.2.2.7) наполняют химический стакан 500 мл фильтрованной деионизированной воды.
Выливают все содержимое химического стакана в фильтровальное устройство.
С.4.1.2 После последней порции раствора промывают стенки воронки, направляя под низким давлением по кругу на стенки воронки поток фильтрованной деионизированной воды. Прекращают промывание воронки, прежде чем объем снизится до одной четвертой уровня наполнения. Поддерживают вакуум, пока вся жидкость не пройдет в воронку.
С.4.1.3 Ослабив вакуум, пинцетом вынимают мембрану из подставки фильтровальной воронки и помещают в чашку Петри (С.2.2.9).
С.4.1.4 Высушивают фильтровальную мембрану в шкафу со слегка приоткрытой крышкой чашки Петри.
С.4.1.5 Рассматривают под микроскопом с увеличением 100 фильтровальную мембрану при снятой крышке чашки Петри.
С.4.1.6 Если в зоне фильтрации имеется не более 20 частиц размером, более или равным 10 мкм, и не более пяти частиц размером, более или равным 25 мкм, то фоновый уровень частиц достаточно низок для проведения микроскопического анализа.
С.4.1.7 Если число частиц превышает эти требования, фоновый уровень частиц не подходит для проведения микроскопического анализа. Следует повторять подготовительные этапы, пока фоновый уровень частиц не подойдет для проведения данного испытания.
С.4.2 Порядок проведения фильтрации офтальмологических вискохирургических изделий
С.4.2.1 Снимают все наружные крышки, герметизирующие ленты и свободные или отстающие бумажные ярлыки. Вымывают наружные поверхности контейнеров вручную теплым моющим раствором, не содержащим ионов, и промывают в теплой проточной водопроводной воде, после чего окончательно промывают фильтрованной деионизированной водой под давлением.
С.4.2.2 Просушивают наружные поверхности контейнеров фильтрованным до 0,2 мкм сжатым воздухом.
Переносят вымытые и просушенные образцы в вытяжной шкаф, защищенный фильтрованным воздухом НЕРА.
С.4.2.3 Подготовляют чистое фильтровальное устройство, как указано в С.3.3.
С.4.2.4 Перемешивают тефлоновой палочкой в заранее вымытом химическом стакане все содержимое 10 единиц ОВИ, разбавляют до получения объема не более 500 мл.
С.4.2.5 В вытяжном шкафу размешивают содержимое стакана в течение не менее 30 мин.
С.4.2.6 Вливают все содержимое в фильтровальное устройство.
С.4.2.7 Проводят операции по С.4.1.2-С.4.1.3.
С.4.2.8 Прикрепляют к чашке Петри ярлык с идентификацией образца.
С.4.2.9 Проводят операции по С.4.1.4.
С.4.3 Порядок проведения работ с сеткой IMA и подсчета частиц
С.4.3.1 Основной метод анализа по сетке IMA заключается в мысленной трансформации изображения каждой частицы в окружность и сравнении ее с эталонными окружностями сетки диаметром 10 и 25 мкм. Процесс подсчета выполняют без наложения частицы на эталонные окружности; при этом частицы не должны перемещаться со своего места в пределах поля зрения сетки (большая окружность). Для подсчета светлых и прозрачных частиц используют внутренний диаметр прозрачных эталонных окружностей сетки. Для подсчета темных частиц используют наружный диаметр черных непрозрачных эталонных окружностей сетки.
С.4.3.2 При выполнении данного анализа не допускается подсчитывать аморфные, полужидкие или иные морфологически неотчетливые материалы, которые имеют вид пятен или имеют изменения цвета на поверхности мембраны.
С.4.3.3 При выполнении полного подсчета не принимают во внимание поле зрения сетки, ограниченное большой окружностью. Используют вертикальную визирную нить.
С.4.3.4 Проводят операции по С.4.1.5.
С.4.3.5 Сканируют всю мембрану справа налево по траектории, которая примыкает, но не перекрывает первую траекторию сканирования. Повторяют процедуру, перемещаясь вправо-влево, пока все частицы на мембране не будут подсчитаны.
С.4.3.6 Записывают общее число частиц, размер которых составляет 10 мкм или более, и общее число частиц, размер которых составляет 25 мкм или более. Оценивают размер частицы, не изменяя увеличения микроскопа и не меняя освещенность.
С.5 Правила обработки результатов анализа
С.5.1 Рассчитывают число частиц на контейнер по формуле
, (С.1)
где - общее число подсчитанных частиц;
- общее число ОВИ, участвовавших в испытании.
Примечание - Микроскопический анализ твердых частиц может быть проведен другими доступными для конкретной лаборатории методами, дающими идентичные результаты.
Приложение D
(справочное)
Расчет числа субъектов, необходимых для проведения клинических испытаний*
_______________
* В бумажном оригинале наименование приложения D выделено курсивом. - .
D.1 При клинических испытаниях основным критерием при расчете размера выборки субъектов является повышение внутриглазного давления. Повышение внутриглазного давления является основным вопросом безопасности используемых в настоящее время ОВИ. Клинические испытания предназначены для того, чтобы показать, что испытуемое ОВИ не хуже контрольного в отношении встречаемости пиков внутриглазного давления выше 30 мм рт.ст.
D.2 За нулевую гипотезу () принимают то обстоятельство, когда встречаемость пиков внутриглазного давления свыше 30 мм рт.ст.
за вычетом контрольной встречаемости пиков внутриглазного давления свыше 30 мм рт.ст.
превышает минимально обнаруживаемую разность между двумя величинами
.
Таким образом:
![]() ;
;
![]() .
.
D.3 Минимальное число оцениваемых субъектов , необходимых для каждой испытуемой группы, определяют по формуле
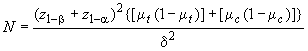 . (D.1)
. (D.1)
D.4 При контрольной встречаемости 0,3, минимально обнаруживаемой разности
0,13, показателях
![]() 0,80 и
0,80 и 0,10 минимальное число субъектов в каждой исследуемой группе
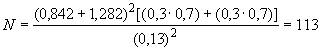 .
.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов межгосударственным стандартам, использованным в настоящем стандарте в качестве нормативных ссылок
Таблица ДА.1
Обозначение ссылочного межгосударственного стандарта | Обозначение и наименование ссылочного международного стандарта и условное обозначение степени его соответствия ссылочному межгосударственному стандарту |
ГОСТ ISO 10993-1-2011 | ИСО 10993-1:1997 Оценка биологического действия медицинских изделий. Часть 1. Оценка и испытание (IDT) |
ГОСТ ISO 10993-5-2011 | - |
ГОСТ ISO 10993-6-2011 | ИСО 10993-6:1994 Оценка биологического действия медицинских изделий. Часть 6. Испытания на местное действие после имплантации (IDT) |
ГОСТ ISO 10993-7-2011 | ИСО 10993-7:1995 Оценка биологического действия медицинских изделий. Часть 7. Остатки стерилизации этиленоксидом (IDT) |
ГОСТ ISO 10993-9-2011 | ИСО 10993-9:1999 Оценка биологического действия медицинских изделий. Часть 9. Основные принципы идентификации и количественного определения потенциальных продуктов деструкции (IDT) |
ГОСТ ISO 10993-10-2011 | - |
ГОСТ ISO 10993-12-2011 | - |
ГОСТ ISO 10993-16-2011 | ИСО 10993-16:1999 Оценка биологического действия медицинских изделий. Часть 16. Моделирование и исследование токсикокинетики продуктов деструкции и вымывания (IDT) |
ГОСТ ISO 13485-2011 | - |
ГОСТ ИСО 14630-2002 | ИСО 14630:1997 Неактивные хирургические имплантаты. Общие требования (IDT) |
ГОСТ ISO 14971-2011 | ИСО 14971-1:1998 Медицинские изделия. Менеджмент риска. Часть 1. Применение анализа рисков (IDT) |
- | ИСО 15223:2000 Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и сопроводительной документации (IDT) |
ГОСТ 31214-2003 | - |
ГОСТ 31508-2012 | - |
ГОСТ 31580.7-2012 | ИСО 14155-1:2003 Клиническое исследование медицинских изделий для людей. Часть 1. Общие требования (NEQ) |
Примечание - В настоящей таблице использованы следующие условные обозначения степени соответствия стандартов: | |
Приложение ДБ
(справочное)
Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта
ДБ.1 Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта представлено в таблице ДБ.1. Указанное в таблице изменение структуры межгосударственного относительно структуры примененного международного стандарта обусловлено приведением его в соответствие с требованиями ГОСТ 1.5-2004**.
Таблица ДБ.1 - Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта
Структура международного стандарта | Структура настоящего стандарта |
1 Область распространения | 1 Область применения |
2 Нормативные ссылки | 2 Нормативные ссылки |
3 Термины и определения | 3 Термины и определения |
4 Предусмотренные характеристики | 4 Общие требования безопасности офтальмологических вискохирургических изделий |
4.1 Требования к предусмотренным характеристикам | |
5 Конструктивные особенности | 4.2 Требования к конструктивным особенностям |
5.1 Общие положения | 4.2.1 Общие требования |
5.2 Определение характеристик реологически активных компонентов | 4.2.2 Требования к определению характеристик реологически активных компонентов |
5.2.1 Характеристика химических свойств | 4.2.2.1 Характеристика химических свойств |
5.2.2 Концентрация | 4.2.2.2 Концентрация |
5.2.3 Распределение молекулярной массы | 4.2.2.3 Распределение молекулярной массы |
5.3 Определение характеристик готового изделия | 4.2.3 Требования к определению характеристик готового офтальмологического вискохирургического изделия |
5.3.1 Общие положения | 4.2.3.1 Общие требования |
5.3.2 Сдвиговая вязкость | 4.2.3.2 Сдвиговая вязкость |
5.3.3 Упругость | 4.2.3.3 Упругость |
5.3.4 Химическое описание компонентов | 4.2.3.4 Химическое описание компонентов |
5.3.5 рН | 4.2.3.5 Значение рН |
5.3.6 Химические и биологические загрязняющие примеси | 4.2.3.6 Химические и биологические загрязняющие примеси |
5.3.7 Осмотичность | 4.2.3.7 Осмотическая концентрация |
5.3.8 Спектральный коэффициент пропускания | 4.2.3.8 Коэффициент пропускания |
5.3.9 Твердые частицы | 4.2.3.9 Твердые частицы |
5.3.10 Показатель преломления | 4.2.3.10 Показатель преломления офтальмологических вискохирургических изделий |
6 Параметры конструкции | 4.3 Требования к оценке свойств готового офтальмологического вискохирургического изделия |
6.1 Общие положения | 4.3.1 Общие требования |
6.2 Оценка биологической безопасности | 4.3.2 Требования к оценке биологической безопасности |
6.2.1 Общие положения | 4.3.2.1 Общие требования |
6.2.2 Испытание на внутриглазную имплантацию | 4.3.2.2 Испытание имплантацией внутрь глаза |
6.2.3 Испытание на бактериальные эндотоксины | 4.3.2.3 Испытание на бактериальные эндотоксины |
6.2.4 Оценка повышения внутриглазного давления | 4.3.2.4 Испытание на внутриглазное давление |
6.2.5 Удаление остаточного ОВИ из передней камеры | 4.3.2.5 Удаление остаточного офтальмологического вискохирургического изделия из передней камеры |
6.2.6 Деградация и токсикокинетика | 4.3.2.6 Деградация и токсикокинетика |
6.3 Клиническая оценка | 4.4 Требования к клинической оценке |
6.3.1 Общие положения | 4.4.1 Общие требования |
6.3.2 План клинического испытания | 4.4.2 План клинического испытания |
6.3.3 Послеоперационное повышение внутриглазного давления | 4.4.3 Послеоперационная оценка повышения внутриглазного давления |
6.3.4 Подсчет числа клеток эндотелия роговицы | 4.4.4 Подсчет числа клеток эндотелия роговицы |
6.3.5 Послеоперационное воспаление | 4.4.5 Послеоперационная воспалительная реакция глаза |
6.3.6 Неблагоприятные события | 4.4.6 Серьезный неблагоприятный исход |
7 Стерилизация | 5 Требования к стерилизации |
8 Стабильность продукции | 6 Требования к стабильности офтальмологических вискохирургических изделий |
9 Целостность и характеристика системы подачи | 7 Требования к целостности и характеристике комплекта поставки |
10 Упаковка | 8 Требования к упаковке |
10.1 Защита от повреждения при хранении и транспортировке | 8.1 Защита от повреждения при хранении и транспортировании |
10.2 Поддержание стерильности при транспортировке | 8.2 Защита стерильности при транспортировании |
11 Информация, предоставляемая изготовителем | 9 Требования к информации, предоставляемой изготовителем |
Приложение А (обязательное) Испытание глазной имплантацией | Приложение А (обязательное) Методика испытания имплантацией внутрь глаза |
А.1 Общие сведения | А.1 Общие требования |
А.2 Методика испытания | А.2 Порядок проведения испытания |
А.3 Оценка результатов | А.3 Правила обработки результатов испытания |
Приложение В (обязательное) Испытание на внутриглазное давление | Приложение В (обязательное) Методика испытания на внутриглазное давление |
В.1 Общие сведения | В.1 Общие требования |
В.2 Методика испытания | В.2 Порядок проведения испытания |
В.3 Оценка результатов | В.3 Правила обработки результатов испытания |
Приложение С (справочное) Микроскопический анализ твердых частиц | Приложение С (рекомендуемое) Микроскопический анализ твердых частиц |
С.1 Основные сведения | С.1 Общие требования |
С.2 Термины и определения | - |
С.3 Оборудование | С.2 Требования к оборудованию и вспомогательным устройствам |
С.4 Образцы | С.3.1.1 |
С.5 Процедура испытания | С.3 Порядок подготовки к проведению анализа |
С.5.1 Общие сведения | С.3.1 Общие требования |
С.5.2 Настройка микроскопа | С.3.2 Настройка микроскопа |
С.5.3 Подготовка фильтровальной установки | С.3.3 Подготовка фильтровального устройства |
С.5.4 Проверка фонового загрязнения | С.4 Порядок проведения анализа |
С.5.5 Подготовка образца | С.4.2 Порядок проведения фильтрации офтальмологического вискохирургического изделия |
С.5.6 Работа с сеткой IMA и подсчет частиц | С.4.3 Порядок проведения работ с сеткой IMA и подсчета частиц |
С.6 Результаты испытания | С.5 Правила обработки результатов анализа |
Приложение D (справочное) Число пациентов для клинических испытаний | Приложение D (справочное) Расчет числа субъектов, необходимых для проведения клинических испытаний |
Библиография | Библиография |
| |
Библиография
[1] | ГФ 11* | Государственная фармакопея. Издание 11 (в части воды) |
________________ * Документ в информационных продуктах не содержится. За информацией о документе Вы можете обратиться в Службу поддержки пользователей. - . | ||
[2] | ЕН 12442-1:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 1. Анализ и менеджмент риска |
(EN 12442-1:2000) | (Animal tissues and their derivates utilized in the manufacture of medical devices - Part 1: Analysis and management of risk) | |
[3] | ЕН 12442-2:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 2. Регулирование источника, сбора и обработки |
(EN 12442-2:2000) | (Animal tissues and their derivates utilized in the manufacture of medical devices - Part 2: Controls on sourcing, collection and handling) | |
[4] | ЕН 12442-3:2000 | Животные ткани и их производные, используемые при изготовлении медицинских изделий. Часть 3. Аттестация уничтожения и/или дезактивации вирусов и других передаваемых агентов |
(EN 12442-3:2000) | (Animal tissues and their derivates utilized in the manufacture of medical devices - Part 3: Validation of elimination and/or inactivation of viruses and other transmissible agents) | |
[5] | USP 24 <85> | Фармакопея Соединенных Штатов, 24-е издание, <85> Определение бактериальных эндотоксинов |
(USP 24 <85>) | (United States Pharmacopoeia, 24th revision, <85> Bacterial endotoxins test) | |
[6] | ИСО 17665-1:2006 | Стерилизация изделий для охраны здоровья. Влажное тепло. Часть 1. Требования к разработке, аттестации и текущему контролю процесса стерилизации медицинских изделий (Взамен ИСО 11134:1994) |
(ISO 17665-1:2006) | (Sterilization of health care products - Moist heat - Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices) | |
[7] | ИСО 11135:1994 | Медицинские изделия. Аттестация и текущий контроль стерилизации этиленоксидом |
(ISO 11135:1994) | (Medical devices -Validation and routine control of ethylene oxide sterilization) | |
[8] | AAMI TIR No.19:1998 | Технический информационный отчет по ИСО 10993-7 |
(AAMI TIR No.19:1998) | (Technical Information Report for ISO 10993-7) | |
[9] | ИСО 11137-1:2006 | Стерилизация изделий для охраны здоровья. Радиация. Часть 1. Требования к разработке, аттестации и текущему контролю процесса стерилизации медицинских изделий |
(ISO 11137-1:2006) | (Sterilization of health care products. Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices) | |
ИСО 11137-2:2006 | Стерилизация изделий для охраны здоровья. Радиация. Часть 2. Определение стерилизационной дозы облучения | |
(ISO 11137-2:2006) | (Sterilization of health care products. Radiation - Part 2: Establishing the sterilization dose) | |
ИСО 11137-3:2006 | Стерилизация изделий для охраны здоровья. Радиация. Часть 3. Руководство по дозиметрическим аспектам | |
(ISO 11137-3:2006) | (Sterilization of health care products. Radiation - Part 3: Guidance on dosimetric aspects) | |
[10] | ИСО 13408-1:1998 | Асептическая обработка изделий для охраны здоровья. Часть 1. Общие требования |
(ISO 13408-1:1998) | (Aseptic processing of health care products - Part 1: General requirements) | |
[11] | ИСО 11607-1:2006 | Упаковка окончательно стерилизованных медицинских изделий. Часть 1. Требования к материалам, системам стерильных барьеров и упаковочным системам |
(ISO 11607-1:2006) | (Packaging for terminally sterilized medical devices - Part 1: Requirements for materials, sterile barrier systems and packaging systems) | |
ИСО 11607-2:2006 | Упаковка окончательно стерилизованных медицинских изделий. Часть 2. Требования к аттестации процессов формирования, герметизации и сборки | |
(ISO 11607-2:2006) | (Packaging for terminally sterilized medical devices - Part 2: Validation requirements for forming, sealing and assembly processes) | |
[12] | EH 868-1:1997 | Упаковочные материалы и системы для медицинских изделий, подлежащих стерилизации. Часть 1. Общие требования и методы испытания |
(EN 868-1:1997) | (Packaging materials and systems for medical devices which are to be sterilized - Part 1: General requirements and test methods) | |
[13] | EH 1041:1998 | Информация, предоставляемая изготовителем медицинской продукции |
(EN 1041:1998) | (Information supplied by the manufacturer with medical devices) | |
[14] | ИСО 10993-2:2006 | Оценка биологического действия медицинских изделий. Часть 2. Требования к условиям содержания животных |
(ISO 10993-2:2006) | (Biological evaluation of medical devices - Part 2: Animal welfare requirements) | |
[15] | Фармакопея США, 24-е издание. Пятое Дополнение 1996, <788>, Частичное вещество в инъекциях. Испытание на подсчет микроскопических частиц | |
(United States Pharmacopoeia, 24th revision. Fifth Supplement 1996. <788>, Particulate matter in injections-Microscopic particle count test) | ||
_________________________________________________________________________________________
УДК 616.7:006.354 МКС 11.040.70 П46 MOD
Ключевые слова: офтальмологические имплантаты, вискохирургические изделия, требования безопасности, характеристики, оценка свойств, клиническая оценка, стерилизация, упаковка, информация, методы оценки
_____________________________________________________________________________________________
Электронный текст документа
и сверен по:
, 2014

{kind=link}