ГОСТ Р ИСО 15378-2017
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПЕРВИЧНЫЕ УПАКОВОЧНЫЕ МАТЕРИАЛЫ ДЛЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Частные требования по применению ИСО 9001:2008 с учетом надлежащей производственной практики (GMP)
Primery packaging materials for medicinal products. Particular requirements for the application of ISO 9001:2008 with reference to good manufacturing practice (GMP)
ОКС 03.120.10
11.040.01
Дата введения 2018-07-01
Предисловие
1 ПОДГОТОВЛЕН Государственным бюджетным образовательным учреждением высшего профессионального образования "Первый Московский государственный медицинский университет имени И.М.Сеченова" Министерства здравоохранения Российской Федерации (Первый МГМУ имени И.М.Сеченова) на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 458 "Разработка, производство и контроль качества лекарственных средств"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 12 сентября 2017 г. N 1076-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 15378:2015* "Первичные упаковочные материалы для лекарственных средств. Особые требования по применению ИСО 9001:2008 с учетом надлежащей производственной практики (GMP)" (ISO 15378:2015 "Primary packaging materials for medicinal products - Particular requirements for the application of ISO 9001:2008, with reference to Good Manufacturing Practice (GMP)", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВЗАМЕН ГОСТ Р 53699-2009 (ИСО 15378:2006)
6 ПЕРЕИЗДАНИЕ. Март 2020 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт определяет принципы надлежащей производственной практики (GMP) и устанавливает требования к системе менеджмента качества, применимой к первичным упаковочным материалам для лекарственных средств. Реализация принципов GMP при производстве и контроле качества первичных упаковочных материалов в рамках организаций-производителей имеет чрезвычайно важное значение для обеспечения безопасности пациентов, получающих лекарственные препараты, из-за непосредственного контакта первичной упаковки с препаратом. Применение GMP в отношении фармацевтических упаковочных материалов помогает обеспечить соответствие этих материалов требованиям и потребностям фармацевтической промышленности.
Настоящий стандарт применяется к первичным упаковочным материалам и содержит основные положения стандарта ИСО 9001:2008.
В отношении построения настоящего стандарта действуют следующие правила:
- разделы или подразделы, цитируемые полностью и без изменений из ИСО 9001:2008, заключены в рамку;
- текст, выделенный курсивом*, содержит дополнительную информацию по GMP, касающуюся первичных упаковочных материалов (в соответствии с ИСО 15378).
________________
* В оригинале обозначения и номера стандартов и нормативных документов приводятся обычным шрифтом. - .
Термины и определения по GMP содержатся в разделе 3. При включении в перечень источник приводится в скобках.
ИСО 9001:2008, Системы менеджмента качества - Требования 01 Общие положения Для создания системы менеджмента качества необходимо стратегическое решение организации. На разработку и внедрение системы менеджмента качества организации влияют: a) ее внешняя среда, изменения или риски, связанные с этой средой; b) изменяющиеся потребности; c) конкретные цели; d) выпускаемая продукция; e) применяемые процессы; f) размер и структура организации. Настоящий стандарт не предполагает единообразия в структуре систем менеджмента качества или их документации. Требования к системе менеджмента качества, установленные настоящим стандартом, являются дополняющими по отношению к требованиям к продукции. Информация, обозначенная как "Примечание", носит характер методических указаний для понимания или разъяснения соответствующего требования. Настоящий стандарт может использоваться внутренними и внешними сторонами, включая органы по сертификации, в целях оценки способности организации выполнять требования потребителей, требования к продукции, являющиеся обязательными к исполнению в соответствии с действующим законодательством (далее - обязательные требования), и собственные требования. При разработке настоящего стандарта были учтены принципы менеджмента качества, установленные ИСО 9000 и ИСО 9004. |
Основной задачей настоящего стандарта является определение гармонизированных требований к первичным упаковочным материалам. Он содержит некоторые частные требования к первичным упаковочным материалам, заимствованные из правил GMP и касающиеся производства, контроля и т.п. лекарственных средств.
Процессный подход
ИСО 9001:2008, Системы менеджмента качества - Требования 02 Процессный подход Настоящий стандарт направлен на применение "процессного подхода" при разработке, внедрении и улучшении результативности системы менеджмента качества в целях повышения удовлетворенности потребителей путем выполнения их требований. Для успешного функционирования организация должна определить и осуществлять менеджмент многочисленных взаимосвязанных видов деятельности. Деятельность, использующая ресурсы и управляемая в целях преобразования входов в выходы, может рассматриваться как процесс. Часто выход одного процесса образует непосредственно вход следующего. Применение в организации системы процессов наряду с их идентификацией и взаимодействием, а также менеджмент процессов, направленный на получение желаемого результата, могут быть определены как "процессный подход". Преимущество процессного подхода состоит в непрерывности управления, которое он обеспечивает на стыке отдельных процессов в рамках их системы, а также при их комбинации и взаимодействии. При применении в системе менеджмента качества такой подход подчеркивает важность: a) понимания и выполнения требований; b) необходимости рассмотрения процессов с точки зрения добавляемой ими ценности; c) достижения запланированных результатов выполнения процессов и обеспечения их результативности; d) постоянного улучшения процессов, основанного на объективном измерении. Приведенная на рисунке 1 модель системы менеджмента качества, основанной на процессном подходе, иллюстрирует связи между процессами, представленными в разделах 4-8. Эта модель показывает, что потребители играют существенную роль в установлении требований, рассматриваемых в качестве входов. Мониторинг удовлетворенности потребителей требует оценки информации о восприятии потребителями выполнения их требований. Приведенная на рисунке 1 модель охватывает все основные требования настоящего стандарта, но не показывает процессы на детальном уровне. Примечание 1 - Кроме того, ко всем процессам может быть применен цикл "Plan - Do - Check - Act" (PDCA). Цикл PDCA можно кратко описать так: - планирование (plan) - разработка целей и процессов, необходимых для достижения результатов в соответствии с требованиями потребителей и политикой организации; - осуществление (do) - внедрение процессов; - проверка (check) - постоянные контроль и измерение процессов и продукции в сравнении с политикой, целями и требованиями на продукцию и сообщение о результатах; - действие (act) - принятие действий по постоянному улучшению показателей процессов. |
|
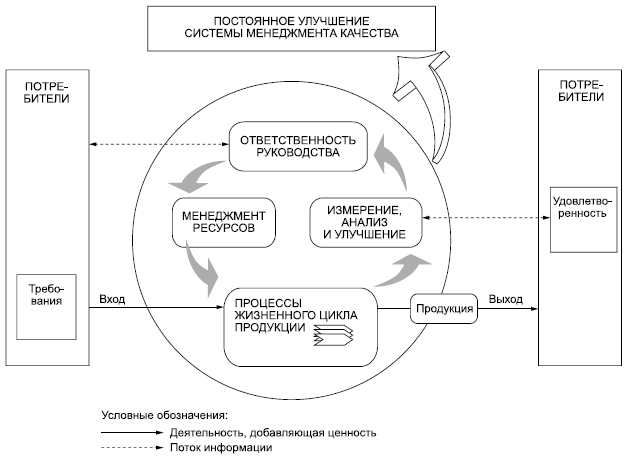
ИСО 9001:2008, Системы менеджмента качества - Требования 0.3 Связь с ИСО 9004 ИСО 9001 и ИСО 9004 являются стандартами на системы менеджмента качества, которые дополняют друг друга, но их можно применять также независимо. ИСО 9001 устанавливает требования к системе менеджмента качества, которые могут быть использованы для внутреннего применения организациями, а также в целях сертификации или заключения контрактов. Стандарт направлен на результативность системы менеджмента качества при выполнении требований потребителей и соответствующих законодательных и других обязательных требований. Ко времени публикации стандарта ИСО 9001:2008 стандарт ИСО 9004 находился на стадии пересмотра. Новая версия ИСО 9004 будет содержать рекомендации для менеджмента по достижению устойчивого успеха любой организации в сложной, требовательной и постоянно изменяющейся среде. ИСО 9004 представляет более широкий взгляд на менеджмент качества, чем ИСО 9001; он нацеливает на удовлетворение потребностей и ожиданий всех заинтересованных сторон на основе систематического и постоянного улучшения деятельности организации. Однако этот стандарт не предназначен для целей сертификации, заключения контрактов и выполнения обязательных требований. |
Совместимость с другими системами управления
Настоящий стандарт содержит требования ИСО 9001:2008 и дополнительно частные требования к первичным упаковочным материалам, которые позаимствованы из правил GMP по организации производства и контроля качества лекарственных средств и адаптированы, если это целесообразно.
ИСО 9001:2008, Системы менеджмента качества - Требования 0.4 Совместимость с другими системами управления При разработке настоящего стандарта должное внимание было уделено положениям ИСО 14001:2004 для улучшения совместимости этих двух стандартов в интересах сообщества пользователей. Приложение A показывает соответствие между ИСО 9001:2008 и ИСО 14001:2004. Настоящий стандарт не содержит конкретных требований к другим системам менеджмента, таким как менеджмент охраны окружающей среды, менеджмент профессионального здоровья и безопасности, финансовый менеджмент или менеджмент рисков. Однако стандарт позволяет организации согласовать или интегрировать свою собственную систему менеджмента качества с другими системами менеджмента с соответствующими требованиями. Организация может адаптировать действующую(ие) систему(ы) менеджмента для создания системы менеджмента качества, соответствующей требованиям настоящего стандарта. |
Примечание - ИСО 9001:2008, приложение A не включено в настоящий стандарт.
1 Область применения
1.1 Общие положения
Настоящий стандарт устанавливает требования, предъявляемые к системе менеджмента качества в тех случаях, когда организации требуется продемонстрировать способность производить первичные упаковочные материалы для лекарственных средств, которые стабильно соответствуют требованиям потребителя, в том числе регуляторным требованиям и международным стандартам, применимым к первичной упаковке.
В настоящем стандарте неоднократно используется выражение "если это целесообразно". В том случае, если определенное требование сопровождается данной фразой, это означает, что данное требование является целесообразным, за исключением случаев, когда организация может документально обосновать иное.
ИСО 9001:2008, Системы менеджмента качества - Требования 1.1 Общие положения Настоящий стандарт устанавливает требования, предъявляемые к системе менеджмента качества в том случае, когда организация: a) нуждается в демонстрации своей способности всегда поставлять продукцию, отвечающую требованиям потребителей и соответствующим обязательным требованиям, и b) ставит своей целью повышение удовлетворенности потребителей посредством эффективного применения системы менеджмента качества, включая процессы постоянного ее улучшения, и обеспечение соответствия требованиям потребителей и соответствующим обязательным требованиям. Примечание 1 - В настоящем стандарте термин "продукция" применим только: a) к предназначенной для потребителя или затребованной им продукции; b) любым заданным результатам процессов жизненного цикла. Примечание 2 - Законодательные и регуляторные требования могут быть сформулированы в форме правовых требований. |
1.2 Применение
Настоящий стандарт применяется при проектировании, производстве и поставке первичных упаковочных материалов для лекарственных средств. Он также применим в целях сертификации.
ИСО 9001:2008, Системы менеджмента качества - Требования 1.2 Применение Все требования настоящего стандарта являются общими и предназначены для применения всеми организациями, независимо от их вида, размера и поставляемой продукции. В том случае, если какое-либо требование (требования) настоящего стандарта нельзя применить вследствие специфики организации и ее продукции, допускается его (их) исключение. При допущенных исключениях заявления о соответствии настоящему стандарту приемлемы в том случае, если данные исключения подпадают под требования раздела 7 и не влияют на способность или ответственность организации предоставлять продукцию, соответствующую требованиям потребителей и применимым законодательным и регуляторным требованиям. |
2 Нормативные ссылки
В тексте настоящего стандарта приводятся ссылки на документы, перечисленные ниже. Данные документы являются обязательными для целей настоящего стандарта. Для датированных документов применима только процитированная редакция. Для недатированных документов применима последняя редакция (включая любые дополнения) цитируемого документа.
ИСО 9001:2008, Системы менеджмента качества - Требования 2 Нормативные ссылки Нижеприведенные документы являются обязательными для целей настоящего стандарта. Для датированных документов применима только процитированная редакция. Для недатированных документов применима последняя редакция (включая любые дополнения) цитируемого документа. ISO 9000:2005, Системы менеджмента качества - Основные положения и словарь ISO 14698-1, Cleanrooms and associated controlled environments - Biocontamination control - Part 1: General principles and methods (Чистые помещения и связанные с ними контролируемые среды. Контроль биозагрязнений. Часть 1. Общие принципы и методы). ISO 14698-2, Cleanrooms and associated controlled environments - Biocontamination control - Part 2: Evaluation and interpretation of biocontamination data (Чистые помещения и связанные с ними контролируемые среды. Контроль биозагрязнений. Часть 2. Оценка и анализ данных по биологическому загрязнению) |
3 Термины и определения
В рамках настоящего стандарта используются термины и определения, приведенные в стандарте ИСО 9000, а также следующие термины и определения.
ИСО 9001:2008, Системы менеджмента качества - Требования 3 Термины и определения В настоящем документе используются термины и определения, приведенные в стандарте ИСО 9000. В тексте настоящего стандарта под термином "продукция" может также пониматься "услуга". |
Прочие термины и определения, используемые в настоящем стандарте, относятся непосредственно к правилам надлежащей производственной практики (GMP), применимым к производству первичных упаковочных материалов для лекарственных средств.
________________
Способ упорядочения и группировки терминов и определений в настоящем стандарте соответствует способу, использованному в ИСО 9000.
3.1 Термины, связанные с качеством
3.1.1 претензия потребителя: Информация, предоставленная потребителем о недостатках и (или) несоответствиях.
Примечание 1 - Информация может быть передана в устной или письменной форме.
Примечание 2 - Предметом претензии может быть качество, количество или условия поставки первичного упаковочного материала (3.4.18.1).
3.2 Термины, связанные с менеджментом
3.2.1 надлежащая производственная практика (GMP): Управление качеством (3.2.2) и обеспечение качества в процессе производства (3.4.20).
Примечание 1 - Определения терминов "управление качеством" (3.2.2) и "обеспечение качества" приведены в стандарте ИСО 9000:2005, 3.2.10 и 3.2.11.
Примечание 2 - Требования, предъявляемые к надлежащей производственной практике (GMP) в фармацевтической промышленности, установлены в стандарте по обеспечению качества (см. [32]).
Примечание 3 - Требования надлежащей производственной практики (GMP) для первичных упаковочных материалов (3.4.18.1) включают, помимо наличия подходящего персонала, помещений и оборудования, наличие системы менеджмента качества, которая включает контроль поступающих исходных материалов (3.4.28), производство (3.4.20), наличие соответствующей документации, выполнение санитарно-гигиенических требований на производстве, контроль готовой продукции (3.8.5), записи о реализации, рассмотрение жалоб и самоконтроль.
Примечание 4 - Понятия "GMP" и "современная надлежащая производственная практика" (cGMP) являются равнозначными. Руководства по GMP постоянно актуализируются в соответствии с меняющимися требованиями современного уровня развития науки и производства. Это и привело к появлению термина "cGMP", который иногда используется. Фармацевтическая промышленность предполагает, что организации (3.3.1) принимают во внимание современные требования GMP в рамках своих программ постоянного улучшения качества.
3.2.2 управление качеством: Часть менеджмента качества, направленная на выполнение требований, предъявляемых к качеству.
Примечание 1 - Управление качеством включается* в себя проверку или испытания, удостоверяющие, что продукция соответствует требованиям спецификаций (3.7.3).
___________________
* Текст документа соответствует оригиналу. - .
[ИСО 9000:2005, 3.2.10]
3.3 Термины, связанные с организацией
3.3.1 организация: Группа лиц и ресурсов с установленными обязанностями, полномочиями и взаимосвязями.
Примечание 1 - В рамках настоящего стандарта под организацией понимается компания, осуществляющая производство (3.4.20) первичного упаковочного материала (3.4.18.1).
[ИСО 9000:2005, 3.3.1, измененный путем добавления примечания 1]
3.3.2 аутсорсинг: Выполнение части процесса или всего процесса другой организацией (3.3.1).
Примечание 1 - Зачастую аутсорсинг называют субподрядом.
3.3.3 служба качества: Подразделение организации, которое отвечает как за обеспечение качества (QA), так и за управление качеством (QC) (3.2.2).
Примечание 1 - Служба (службы) качества могут представлять собой отдельные подразделения, занимающиеся обеспечением качества и контролем качества соответственно, или же единичное лицо (группу лиц) в зависимости от размера и структуры организации (3.3.1).
3.4 Термины, связанные с процессами и продукцией
3.4.1 воздушный шлюз: Закрытое пространство, предназначенное для контролирования воздушного потока.
Примечание 1 - Данное пространство, как правило, снабжено по меньшей мере двумя взаимосвязанными дверями, установленными между двумя и более комнатами, и используется для перехода персонала или перемещения материалов при условии соблюдения различных условий, например чистоты потока воздуха при входе.
3.4.2 сборка: Соединение вместе первичных упаковочных материалов (3.4.18.1) и (или) их компонентов.
Примечание 1 - Примерами могут служить сборка частей пипеток, компонентов инъекционных систем или установка защитных колпачков для игл на предварительно наполненных шприцах.
3.4.3 чистое помещение: Помещение, в котором контролируется концентрация взвешенных в воздухе частиц и которое построено и используется таким образом, чтобы минимизировать попадание, образование и сохранение частиц внутри помещения, и в котором при необходимости контролируются прочие требуемые параметры, такие как температура, влажность и давление.
[ИСО 14644-1:1999, 2.1.1]
3.4.4 чистая зона: Специальное пространство, в котором контролируется концентрация взвешенных в воздухе частиц и которое построено и используется таким образом, чтобы минимизировать попадание, образование и сохранение частиц внутри зоны, и в котором при необходимости контролируются прочие требуемые параметры, такие как температура, влажность и давление.
Примечание 1 - Данная зона может быть открытой или закрытой, а также располагаться внутри или вне чистого помещения (3.4.3).
[ИСО 14644-1:1999, 2.1.2]
3.4.5 контаминация: Попадание любого нежелательного материала в первичный упаковочный материал (3.4.18.1).
Примечание 1 - Готовая продукция (3.4.11) может быть контаминирована физически (твердые частицы), химически или биологически (биотоксины и эндотоксины).
Примечание 2 - Контаминация может произойти в процессе производства (3.4.20), упаковки, хранения и (или) распространения вследствие контаминированных систем вентиляции, оборудования для отбора проб, материалов, помещений, тары или по вине персонала.
3.4.6 контролируемая среда: Среда, созданная и эксплуатируемая таким образом, чтобы контролировать возможное попадание потенциальных контаминантов.
3.4.7 перекрестная контаминация: Контаминация (3.4.5) материала или продукции другим материалом или продукцией.
Примечание 1 - Перекрестную контаминацию также иногда называют смешиванием.
Примечание 2 - См. [31].
3.4.8 дата изготовления: Дата наступления одной из первых стадий процесса производства первичного упаковочного материала (3.4.18.1), или процесса упаковки, или процесса выпуска готовой продукции, которая может служить предметом согласования с потребителем.
3.4.9 документированная процедура: Разработанная, документированная, утвержденная, внедренная и поддерживаемая процедура.
3.4.10 срок годности: Предполагаемый предельный срок использования.
Примечание 1 - См. также определение срока хранения (3.4.26).
Примечание 2 - Обычно срок годности представляет собой период, в течение которого ожидается, что первичный упаковочный материал (3.4.18.1) остается пригодным для использования при условии хранения в установленных условиях, и по истечении которого он не должен использоваться.
3.4.11 готовая продукция: Первичный упаковочный материал (3.4.18.1), которой прошел все стадии производства (3.4.20).
3.4.12 однородность (гомогенность): Единообразие характеристик материала и их значений для его определенного количества.
Примечание 1 - Понятие "однородность" также может включать в себя единообразие материалов или определенных характеристик материалов, представляющих особое значение.
3.4.13 промежуточная продукция: Первичный упаковочный материал (3.4.18.1), прошедший некоторые, но не все стадии производства (3.4.20).
Примечание 1 - Промежуточная продукция требует дальнейшей обработки, прежде чем станет готовой продукцией (3.4.11).
3.4.14 очистка линии: Удаление (промывка/продувка линии) всего, что связано с предыдущим производственным циклом.
Примечание 1 - Как правило, очистка линии производится до начала очередного производственного цикла для предотвращения любой ошибки или перекрестной контаминации (3.4.7). Обычно требуется, чтобы производственное оборудование (линия) и соответствующее рабочее пространство (3.4.31) были полностью очищены от всех материалов, отходов, продукции, образцов, документов и т.п., которые использовались в ходе предыдущего производственного цикла, до внесения материалов, образцов продукции, документов и т.п., необходимых для начала следующего производственного цикла.
3.4.15 изготовление: Все операции, включающие в себя закупку и получение материалов для производства (3.4.20), упаковку, маркировку, управление качеством (3.2.2), выпуск, хранение, реализацию продукции, а также соответствующие средства управления.
3.4.16 лекарственное средство: Любое вещество или комбинация веществ, предназначенные для лечения или профилактики заболевания у людей или животных.
Примечание 1 - Любое вещество или сочетание веществ, вводимое человеку или животным с диагностическими целями или для восстановления, корректировки или изменения физиологических функций человека или животных, также рассматривается как лекарственное средство.
Примечание 2 - См. [31].
Примечание 3 - Фармацевтические или лекарственные препараты, в том числе используемые в клинических исследованиях, также могут называться лекарственными средствами.
3.4.17 подготовка печатного оригинала: Вся подготовительная деятельность, предшествующая печати.
Примечание 1 - Сюда относятся концептуальное решение, дизайн, графическое исполнение, репрография, пленка, изготовление печатной формы, шелкотрафаретная печать, цифровые файлы и шрифтоносители.
3.4.18 Упаковочные материалы
3.4.18.1 первичные упаковочные материалы: Материалы, используемые в фармацевтической упаковке, которая будет содержать, герметизировать или использоваться для дозированного применения лекарственного средства (3.4.16) и которая будет находиться в непосредственном контакте с лекарственным средством.
3.4.18.2 вторичные упаковочные материалы: Упаковочные материалы, не вступающие в контакт с лекарственным средством, такие как картонные коробки с текстом и без текста, этикетки, брошюры или листки-вкладыши (или рекламные листки), обертки и контейнеры для транзитных перевозок, например складные коробки.
3.4.19 вспомогательные средства: Материалы, используемые для реализации процесса.
Примечание 1 - Материал не включается в спецификацию (3.7.3) и может быть удален на финальной стадии производства или до нее.
Пример - Сжатый воздух, прокатная смазка, разделительная смазка для пресс-форм.
3.4.20 производство: Процессы, оканчивающиеся получением первичных упаковочных материалов (3.4.18.1).
Примечание 1 - Данные процессы образуют полный производственный цикл, начиная от приемки исходных материалов (3.4.28) с последующей обработкой, упаковкой и заканчивая получением готовой продукции (3.4.11).
3.4.21 критический для качества: Параметр, влияющий на качество первичного упаковочного материала (3.4.18.1).
Примечание 1 - Материал, операция или условия процесса, требования к испытаниям или любые другие соответствующие параметры могут считаться критическими для качества в том случае, если невыполнение требований, предъявляемых к ним, может иметь существенные пагубные последствия.
3.4.22 карантин: Статус материалов или продукции, изолированных до вынесения решения об их последующем одобрении или последующей выбраковки.
Примечание 1 - Подкарантинный материал, как правило, изолируется физическим или иным эффективным способом.
3.4.23 реализация: Общий термин, охватывающий все процессы, требуемые для достижения необходимого результата от проектирования до поставки продукции.
3.4.24 восстановление: Обработка или повторная обработка первичных упаковочных материалов (3.4.18.1) для их соответствия требованиям спецификаций (3.7.3).
3.4.25 повторная обработка: Повторение части процесса производства.
Примечание 1 - Продолжение части технологического процесса после того, как контроль в процессе производства показал, что данная часть процесса не завершена, является нормальной практикой и не относится к повторной обработке.
3.4.26 срок хранения: Период времени, в течение которого ожидается, что первичные упаковочные материалы (3.4.18.1) будут соответствовать требованиям (спецификациям) при хранении в определенных условиях и по истечении которого материалы не должны использоваться.
Примечание 1 - См. также срок годности (3.4.10).
3.4.27 стандартная операционная процедура (СОП): Утвержденная, документированная процедура (3.4.9) или ряд процедур, рабочих инструкций и инструкций по испытанию продукции и управлению.
3.4.28 исходный материал: Сырье/компоненты/субстанции, используемые при производстве первичных упаковочных материалов (3.4.18.1).
3.4.29 стерильность: Состояние, характеризующееся отсутствием жизнеспособных микроорганизмов.
[ИСО 14937:2009, 3.26]
3.4.30 обработка поверхности: Процесс, направленный на улучшение поверхности первичных упаковочных материалов (3.4.18.1).
Пример - Покрытие силиконом или другая обработка внутренних стеклянных поверхностей, покрытие внутренних или внешних поверхностей стеклянных контейнеров или резиновых.
3.4.31 рабочее пространство: Определенное пространство, где проводятся операции по подготовке, производству, упаковке, испытанию или приемочному контролю и где после данных операций обычной* проводится очистка линии.
___________________
* Текст документа соответствует оригиналу. - .
Примечание 1 - Подобное пространство обычно обозначено при помощи барьеров, разметки на полу или аналогичных средств и может содержать оборудование, например, производственное, испытательное, компьютеры, рабочие столы, пробопечатные станки.
3.5 Термины, относящиеся к характеристикам
3.5.1 серия, партия: Определенное количество первичных упаковочных материалов (3.4.18.1), произведенных в ходе одного или нескольких процессов с запланированными однородными характеристиками и стабильным качеством.
Примечание 1 - Для удовлетворения производственных требований или требований потребителя серия может быть разделена на ряд подсерий, объединяемых позднее в единую однородную серию.
Примечание 2 - При непрерывном производстве серией считается доля продукции, определяемая либо заданным количеством, либо количеством, произведенным за установленный промежуток времени.
3.5.2 прослеживаемость: Возможность проследить предысторию, применение или местонахождение рассматриваемого объекта.
Примечание 1 - Применительно к продукции прослеживаемость может относиться к:
- источнику материалов и частей,
- предыстории обработки и
- распространению и местонахождению продукции после поставки.
Примечание 2 - В области метрологии принято определение, приведенное в Руководстве ИСО/IEC 99:2007, 2.41 (третье издание Международного словаря по метрологии).
[ИСО 9000:2005, 3.5.4, модифицированный путем обновления ссылки в примечании 2]
3.6 Термины, связанные с соответствием
3.6.1 утвержденный: Имеющий подтвержденный статус соответствия.
Примечание 1 - Соответствие может подтверждаться на любом этапе процесса [для исходных материалов (3.4.28), вспомогательных средств (3.4.19), упаковочных материалов или готовой продукции (3.4.11)].
3.6.2 выпуск серии: Решение о выпуске серии (3.5.1) для продажи или поставки после официального рассмотрения досье на серию (3.7.1), осуществляемого службой качества (3.3.3) или лицом, назначенным службой качества.
3.6.3 отклонение: Отступление от утвержденной (3.6.1) стандартной операционной процедуры (3.4.27) или действующего стандарта.
3.6.4 несоответствие спецификации: Результаты испытаний, не соответствующие требованиям спецификации (3.7.3).
3.6.5 отбракованный: Статус исходных материалов (3.4.28), вспомогательных средств (3.4.19), промежуточной продукции (3.4.13) или готовой продукции (3.4.11), результаты испытания которых не удовлетворяют одному или нескольким требованиям спецификации (3.7.3) и которые признаны [обычно службой качества (3.3.3)] непригодными для использования.
3.6.6 отбраковка: Процесс установления, выполняемый обычно службой качества (3.3.3), того, что исходные материалы (3.4.28), вспомогательные средства (3.4.19), промежуточная продукция (3.4.13) или готовая продукция (3.4.11) непригодны для использования.
3.6.7 архивные образцы: Материалы или готовая продукция (3.4.11), хранимые для будущего сравнения.
Примечание 1 - Данные образцы обычно отбираются в достаточном количестве и хранятся в определенных условиях в течение заданного периода времени для последующего сравнения.
3.6.8 возврат: Процесс отправки первичных упаковочных материалов (3.4.18.1) обратно в организацию (3.3.1).
3.6.9 переработка: Действие, предпринятое в отношении несоответствующей продукции для того, чтобы она соответствовала требованиям.
Примечание 1 - Сортировка может рассматриваться как переработка.
[ИСО 9000:2005, 3.6.7, модифицированный путем добавления примечания 1]
3.7 Термины, связанные с документацией
3.7.1 досье на серию: Документы и записи, дающие представление о предыстории серии (3.5.1), включая информацию, относящуюся к производству и контролю, и облегчающие ее прослеживаемость (3.5.2).
3.7.2 номер серии, номер партии: Уникальный идентификатор серии (3.5.1).
Примечание 1 - Номер серии может представлять собой сочетание цифр, букв и (или) символов, идентифицирующих серию (3.5.1) (или партию) и способствующих определению предыстории производства и распределения.
[ИСО 9000:2005, 3.7.3]
3.7.3 спецификация: Документ, в котором указаны требования.
3.7.4 спецификация требований пользователя: Утвержденный (3.6.1) документ, содержащий спецификации (3.7.3) продукции, производящейся из материала на данном оборудовании, а также функциональные, операционные и (или) технические аспекты оборудования или процесса, требуемые для производства желаемой продукции.
3.8 Термины, связанные с испытаниями
3.8.1 автоматизированный контроль: Оценка соответствия, осуществляемая контрольным оборудованием без человеческого вмешательства.
Примечание 1 - Контрольное оборудование может включать в себя оптоэлектронные компоненты (камеры), лазерные системы, ультразвуковые системы и связанные с ними системы обработки данных, а также прочее оборудование.
3.8.2 калибровка (поверка): Процесс проверки или регулирования [путем сопоставления со стандартным образцом (эталоном)] точности средства измерения.
Примечание 1 - Калибровка (поверка) может быть также описана как ряд операций, устанавливающих при заданных условиях зависимость между значениями, полученными с помощью средства измерения, или значениями, представляемыми вещественной мерой, и соответствующими известными значениями стандартного образца (эталона).
3.8.3 управление изменениями: Документированное управление изменениями.
Примечание 1 - К изменениям могут, в частности, относиться изменения в сырье, спецификациях (3.7.3), производственной базе, оборудовании, производственных процессах и методах испытаний.
3.8.4 двойная проверка: Документированная верификация (3.8.13) действия, результата или записи вторым лицом или системой.
Примечание 1 - Частью такого процесса подтверждения может быть вторая подтверждающая подпись в ходе внутрипроизводственного контроля, производственная документация и записи о качестве для серии (3.5.1), подписанные вторым лицом, или контроль с помощью электронных приборов. Как правило, результаты двойной проверки подписываются вторым лицом.
3.8.5 контроль готовой продукции: Испытания, проводящиеся на готовой продукции (3.4.11) для определения ее соответствия требованиям спецификации (3.7.3).
3.8.6 внутрипроизводственный контроль: Действия, предпринимаемые в процессе производства (3.4.20), с целью проверки соответствия продукции требованиям спецификации (3.7.3).
Примечание 1 - Для того чтобы продукция соответствовала требованиям, может потребоваться мониторинг процессов и корректирование средств производства (3.4.20).
Примечание 2 - Контроль за окружающей средой или оборудованием также может считаться частью внутрипроизводственного контроля.
3.8.7 квалификация монтажа (IQ): Процесс получения и документирования свидетельства того, что оборудование было поставлено и смонтировано в соответствии с его спецификацией (3.7.3).
[ISO/TS 11139:2006, 2.22]
3.8.8 квалификация функционирования (OQ): Процесс получения и документирования свидетельства того, что смонтированное оборудование функционирует в рамках предварительно установленных пределов при его использовании в соответствии с операционными процедурами.
[ISO/TS 11139:2006,2.27]
3.8.9 квалификация эксплуатации (PQ): Верификация (3.8.13) того факта, что предложенная спецификация (3.7.3) на производственную базу, оборудование или систему пригодна для предполагаемого использования.
[ISO/TS 11139:2006, 2.30]
3.8.10 процесс квалификации; квалификация: Процесс, подтверждающий способность выполнять установленные требования.
Примечание 1 - Термин "квалифицирован" используется для обозначения соответствующего статуса.
Примечание 2 - Квалификация включает в себя квалификацию проекта (DQ), квалификацию монтажа (IQ) (3.8.7), квалификацию функционирования (OQ) (3.8.8) и может включать в себя квалификацию эксплуатации (PQ) (3.8.9), а также реквалификацию.
Примечание 3 - Квалификация может быть применена к производственной базе, оборудованию и вспомогательным средствам.
[ISO 9000:2005, 3.8.6, модифицированный путем добавления термина "квалификация" и примечаний 1 и 2]
3.8.11 сопоставление: Сравнение между теоретическим и фактическим объемом выпущенной или используемой готовой продукции (3.4.11) с учетом нормальной вариации.
Примечание 1 - Сравнение проводится с учетом отходов, образцов и других потерь, присущих процессу.
3.8.12 валидация: Подтверждение посредством представления объективных свидетельств того, что требования, предъявляемые к конкретному предполагаемому использованию или применению, выполнены.
Примечание 1 - Термин "валидирован" используется для обозначения соответствующего статуса.
Примечание 2 - Валидация может быть проведена в отношении процессов, продукции и программного обеспечения.
[ИСО 9000:2005, 3.8.5, модифицированный путем добавления примечания 2]
3.8.13 верификация: Подтверждение (на основе представления объективных свидетельств) того, что установленные требования были выполнены.
Примечание 1 - Термин "верифицирован" используется для обозначения соответствующего статуса.
Примечание 2 - При разработке и проектировании верификация представляет собой процесс изучения результатов рассматриваемой деятельности с целью установления соответствия упомянутой деятельности заданным требованиям.
Примечание 3 - В настоящем стандарте термин "верификация" используется с целью обеспечения того, что производственные системы надлежащим образом смонтированы и функционируют корректно; этого также можно достичь посредством квалификации монтажа (3.8.7) и квалификации функционирования (3.8.8).
[ИСО 9000:2005, 3.8.4, модифицированный путем добавления примечания 3]
3.9 Термины, связанные с управлением рисками
3.9.1 анализ риска: Процесс, направленный на осмысление природы риска и определение его уровня.
Примечание 1 - Анализ риска предоставляет основу для сравнительной оценки риска (3.9.3) и принятия решения об обработке риска.
Примечание 2 - Анализ риска включает в себя количественную оценку риска (3.9.2).
[ИСО 73:2009, 3.6.1]
3.9.2 оценка риска: Совокупный процесс идентификации риска (3.9.4), анализа риска (3.9.1) и сравнительной оценки риска (3.9.3).
[ИСО 73:2009, 3.4.1]
3.9.3 сравнительная оценка риска: Процесс сравнения результатов анализа риска (3.9.1) с критериями риска с целью определения, являются ли риск и (или) его величина приемлемыми или допустимыми.
Примечание 1 - Сравнительная оценка риска способствует принятию решения об инициировании работ по снижению риска.
[ИСО 73:2009, 3.7.1]
3.9.4 идентификация риска: Процесс выявления, признания и описания рисков.
Примечание 1 - Идентификация риска включает в себя идентификацию источников риска, событий, их причин и их потенциальных последствий.
Примечание 2 - Идентификация риска может включать в себя ретроспективные данные, теоретический анализ, обоснованные и экспертные точки зрения, а также потребности заинтересованных сторон.
[ИСО 73:2009, 3.5.1]
3.9.5 управление рисками: Скоординированные действия, направленные на руководство и управление организацией (3.3.1) с учетом риска.
[ИСО 73:2009, 2.1]
4 Система менеджмента качества
4.1 Общие требования
ИСО 9001:2008, Системы менеджмента качества - Требования 4 Системы менеджмента качества 4.1 Общие требования Организация должна разработать, документировать, внедрить и поддерживать в рабочем состоянии систему менеджмента качества, а также постоянно улучшать ее эффективность в соответствии с требованиями настоящего стандарта. Организация должна: a) определять процессы, необходимые для системы менеджмента качества, и их применение во всей организации (см. 1.2); b) определять последовательность и взаимодействие данных процессов; c) определять критерии и методы, необходимые для обеспечения результативности как при осуществлении этих процессов, так и при управлении ими; d) обеспечивать наличие ресурсов и информации, необходимых для поддержания этих процессов и их мониторинга; e) осуществлять мониторинг, измерение там, где это возможно, и анализ данных процессов и f) принимать меры, необходимые для достижения запланированных результатов и постоянного улучшения этих процессов. |
g) охарактеризовать свою общую политику, намерения и подход к обеспечению качества продукции.
ИСО 9001:2008, Системы менеджмента качества - Требования Организация должна управлять данными процессами в соответствии с требованиями настоящего стандарта. В том случае, если организация решает передать сторонней организации выполнение какого-либо процесса, влияющего на соответствие продукции требованиям, она должна обеспечить со своей стороны управление таким процессом. Вид и степень управления процессами, переданными сторонним организациям, должны быть определены в системе менеджмента качества. Примечание 1 - Вышеупомянутые процессы, необходимые для системы менеджмента качества, включают в себя процессы управленческой деятельности руководства, обеспечения ресурсами, процессы жизненного цикла продукции, измерения, анализа и улучшения. Примечание 2 - Процесс, переданный другой организации, является процессом, необходимым для системы менеджмента качества организации и который по выбору организации выполняется сторонней организацией. Примечание 3 - Обеспечение управления процессами, переданными сторонним организациям, не освобождает организацию от ответственности за соответствие всем требованиям потребителей, законодательным и регуляторным требованиям. Выбор вида и степени управления процессом, переданным сторонней организации, зависит от таких факторов, как: a) возможное влияние переданного сторонним организациям процесса на способность организации поставлять продукцию, соответствующую требованиям; b) степень участия в управлении процессом, переданным сторонней организации; c) возможность обеспечения необходимого управления посредством применения требований 7.4. |
4.1.1 Управление риском
Организация должна обеспечить применение системы управления рисками в процессах, связанных с проектированием/разработкой, производством и поставкой первичных упаковочных материалов в части, касающейся качества первичной упаковки; следует поддерживать соответствующие записи (см. 4.2.4).
Управление риском должно применяться, помимо прочего, в отношении нижеперечисленных аспектов:
- управление изменениями,
- очистка,
- претензии,
- контаминация,
- управление проектированием (новая продукция/новые процессы),
- гигиена и здоровье,
- маркировка,
- техническое обслуживание,
- управление материалами,
- несоответствия, дефекты качества,
- защита от насекомых и животных,
- цепочки закупки и поставок,
- переработка,
- прослеживаемость,
- валидация, верификация и квалификация.
Примечание - Принципы и руководства по управлению рисками приведены, например, в руководствах ИСО 31000, ИСО 14971 или ICH Q9 [35] GAMP5 [33]. Руководство по различным методам идентификации и оценки рисков, а также степени и управлению опасностями, связанными с определенными процессами или действиями, приведено в МЭК 31010.
4.2 Требования, предъявляемые к документации
4.2.1 Общие положения
ИСО 9001:2008, Системы менеджмента качества - Требования 4.2.1 Общие положения Документация системы менеджмента качества должна включать в себя: a) документально оформленные заявления о политике и целях в области качества; b) руководство по качеству; c) документированные процедуры и записи, требуемые настоящим стандартом; d) документы, включая записи, определенные организацией как необходимые ей для обеспечения эффективного планирования, осуществления процессов и управления ими. Примечание 1 - Использованный в настоящем стандарте термин "документированная процедура" означает, что процедура разработана, документально оформлена, внедрена и поддерживается в рабочем состоянии. Один документ может содержать требования одной или более процедуры. Требование о наличии документированной процедуры может быть реализовано более чем одним документом. Примечание 2 - Степень документированности системы менеджмента качества в одной организации может отличаться от степени документированности в другой в зависимости от: a) размера организации и вида деятельности, b) сложности процессов и их взаимодействия и c) компетенции персонала. Примечание 3 - Документация может существовать в любой форме или на любом носителе. |
Примечание 4 - Документированные процедуры, рабочие инструкции, инструкции по проведению испытаний в целях производства и управления, требуемые настоящим стандартом, могут называться стандартными операционными процедурами (СОП).
Организации необходимо документально оформить общую политику, намерения и подход к управлению риском, валидации и управлению изменениями.
4.2.1.1 Администрирование компьютеризированными системами и данными
Должны существовать документированные процедуры:
a) для присвоения ответственности с целью обеспечения безопасности и поддержания информационных технологий и самих данных;
b) для обеспечения безопасности сети и файлов и обеспечения положения, при котором только авторизованный персонал имеет доступ к системам и файлам;
c) для обеспечения целостности файлов в том случае, если файлы хранятся в общем доступе, например на файловом сервере, доступном с нескольких рабочих станций;
d) охватывающие управление паролями и алгоритмы безопасности, включая "спящий режим", который должен существовать на случай отсутствия персонала за компьютером, и
e) для резервного копирования и восстановления электронных данных о продукции, при этом процедуры определяют частоту резервного копирования, метод и носитель, а также физический процесс безопасного хранения файлов с данными; носители с резервными данными должны быть идентифицированы и прослеживаемы.
Организация должна иметь документированный план восстановления информационных технологий (ИТ), в котором детально описана система для частичного и полного восстановления данных в случае отказа ИТ-системы. Система должна верифицироваться через установленные промежутки времени с целью обеспечения возможности восстановления данных.
При замене ИТ-систем в рамках управления изменениями должен быть определен доступ к ранее использованным системам и данным (см. 7.5.1.3).
4.2.2 Руководство по качеству
ИСО 9001:2008, Системы менеджмента качества - Требования 4.2.2 Руководство по качеству Организация должна разработать и поддерживать в рабочем состоянии руководство по качеству, содержащее: a) область применения системы менеджмента качества, включая подробности и обоснование любых исключений (см. 1.2); b) документированные процедуры, разработанные для системы менеджмента качества, или ссылки на них и c) описание взаимодействия процессов системы менеджмента качества. |
4.2.2.1 Организация должна четко определить, в какой степени настоящий стандарт применим к ее процессам.
Примечание 1 - Организация может определить, относится ли настоящий стандарт ко всей выпускаемой продукции (для фармацевтических и иных целей) или исключительно к продукции для фармацевтических целей.
4.2.2.2 Руководство по качеству должно описывать структуру документации, используемой в системе менеджмента качества.
4.2.3 Управление документацией
ИСО 9001:2008, Система менеджмента качества - Требования 4.2.3 Управление документацией Документация системы менеджмента качества должна быть управляемой. Записи, представляющие собой специальный вид документов, должны быть управляемыми согласно требованиям 4.2.4. Для определения необходимых средств управления должна быть разработана документированная процедура, предусматривающая: a) официальное утверждение документов с точки зрения их достаточности до выпуска; b) анализ и актуализацию по мере необходимости и повторное официальное утверждение документов; c) обеспечение идентификации изменений и статуса пересмотра документов; d) обеспечение наличия соответствующих версий документов в местах их применения; e) обеспечение сохранения документов четкими и легко идентифицируемыми; f) обеспечение идентификации и управление рассылкой документов внешнего происхождения, определенных организацией как необходимые для планирования и функционирования системы менеджмента качества, и g) предотвращение непреднамеренного использования устаревших документов и применение соответствующей идентификации таких документов, сохраненных для каких-либо целей. |
4.2.3.1 Организация должна убедиться в том, что изменения в документах анализируются и утверждаются либо первичной согласительной инстанцией, либо другой назначаемой инстанцией, имеющей доступ к соответствующей исходной информации для обоснования своих решений.
4.2.3.2 Организация должна установить период, в течение которого будет храниться по меньшей мере одна копия устаревшего контролируемого документа (см. также 4.2.4.8).
4.2.3.3 Если для подписания документов используются электронные подписи, они должны контролироваться для обеспечения безопасности, эквивалентной безопасности использования рукописной подписи.
4.2.3.4 Контролируемые документы должны включать в себя уникальный идентификатор (например, название/номер документа, выпуск и номер страницы).
4.2.4 Управление записями
ИСО 9001:2008, Системы менеджмента качества - Требования 4.2.4 Управление записями Записи, разработанные и утвержденные для представления свидетельств соответствия требованиям и эффективного функционирования системы менеджмента качества, должны находиться под управлением. Организация должна разработать и внедрить документированную процедуру для определения средств управления, необходимых для идентификации, хранения, защиты, извлечение*, сохранения и удаления записей. Записи должны оставаться четкими, легко идентифицируемыми и восстанавливаемыми. |
___________________
* Текст документа соответствует оригиналу. - .
Примечание 1 - К записям относятся данные о производстве серии продукции, а также прочие записи о качестве, такие как отчеты об отклонениях и расследовании.
4.2.4.1 Электронные записи подлежат такому же управлению, которое требуется для других записей (см. 4.2.4 и 7.5.2.9).
4.2.4.2 Отдельные записи должны быть четкими, несмываемыми, выполненными сразу же после выполнения деятельности (в порядке выполнения работ), датированными и подписанными лицом, делающим запись. Исправления в записях должны быть датированными, подписанными и сопровождаемыми разъяснениями по мере необходимости, оставляя разборчивой первоначальную запись.
4.2.4.3 Организация должна определять критические процессы и показатели, в которых для выпуска серии требуется двойная проверка. В записях должны быть четко обозначены идентифицированная проверка и стадии. Если одна из проверок проводится электронно, это должно быть четко определено.
4.2.4.4 Для каждой серии первичного упаковочного материала организация должна составлять и вести запись, которая обеспечивает прослеживаемость (см. 7.5.3) и определяет количество произведенной продукции и количество, утвержденное к реализации.
4.2.4.5 Организация должна определять те показатели документации по серии, которые подлежат верификации.
4.2.4.6 Документация по серии должна быть верифицирована и утверждена.
4.2.4.7 Все записи, касающиеся производства, управления, испытаний, распределения и расследования, должны сохраняться в течение по меньшей мере пяти лет с даты производства первичного упаковочного материала или по договоренности с потребителем.
Примечание - Может потребоваться сохранение записей о первичных упаковочных материалах до конца срока хранения лекарственного средства, как оговорено потребителем.
5 Ответственность руководства
5.1 Обязательства руководства
ИСО 9001:2008, Системы менеджмента качества - Требования 5.1 Обязательства руководства Высшее руководство должно обеспечивать наличие свидетельств принятия своих обязательств по разработке и внедрению системы менеджмента качества, а также постоянному улучшению ее эффективности посредством: a) доведения до сведения персонала организации важности выполнения требований потребителей, а также законодательных и регуляторных требований; b) разработки политики в области качества; c) обеспечения разработки целей в области качества; d) проведения анализа со стороны руководства и e) обеспечения необходимыми ресурсами. |
5.2 Ориентация на потребителя
ИСО 9001:2008, Системы менеджмента качества - Требования 5.2 Ориентация на потребителя Высшее руководство должно обеспечивать определение и выполнение требований потребителей для повышения их удовлетворенности (см. 7.2.1 и 8.2.1). |
Примечание - Основные требования потребителя к организации связаны с приемлемой производственной базой, компетентным и подготовленным персоналом, процессами, предназначенными для обеспечения безопасности продукции и отсутствия перекрестной контаминации, а также со способностью постоянно выпускать продукцию, отвечающую техническим требованиям потребителя.
5.2.1 Аудиты со стороны потребителя
Организация должна разрешать существующему/будущему потребителю или его представителям (по взаимному согласию) проводить аудиты качества для анализа и оценки своей системы менеджмента качества.
5.3 Политика в области качества
ИСО 9001:2008, Системы менеджмента качества - Требования 5.3 Политика в области качества Высшее руководство должно обеспечивать положение, при котором политика в области качества: a) соответствует целям организации; b) включает в себя обязательство соответствовать требованиям и постоянно повышать эффективность системы менеджмента качества; c) создает основы для постановки и анализа целей в области качества; d) доводится до сведения персонала организации и понятна ему; e) анализируется на соответствие современным требованиям. |
5.4 Планирование
5.4.1 Цели в области качества
ИСО 9001:2008, Системы менеджмента качества - Требования 5.4.1 Цели в области качества Высшее руководство организации должно обеспечить положение, при котором цели в области качества, включая необходимые для выполнения требований к продукции [см. 7.1, а)], были разработаны и утверждены в соответствующих подразделениях и на соответствующих уровнях организации. Цели в области качества должны быть измеримыми и согласуемыми с политикой в области качества. |
5.4.2 Планирование системы менеджмента качества
ИСО 9001:2008, Системы менеджмента качества - Требования 5.4.2 Планирование системы менеджмента качества Высшее руководство должно обеспечивать: a) планирование системы менеджмента качества для выполнения требований, приведенных в 4.1, а также для достижения целей в области качества; b) поддержание целостности системы менеджмента качества при планировании и внедрении в нее изменений. |
5.5 Ответственность, полномочия и обмен информацией
5.5.1 Ответственность и полномочия
ИСО 9001:2008, Системы менеджмента качества - Требования 5.5.1 Ответственность и полномочия Высшее руководство должно обеспечивать определение и доведение до сведения персонала организации ответственности и полномочий. |
5.5.1.1 Организация должна поддерживать в актуальном состоянии текущие образцы (см. 4.2.4) подписей ответственных лиц. Рекомендуется иметь перечни подписей и (или) идентификационные перечни всего персонала, проводящего проверки или двойные проверки этапов процесса, осуществляющего внутрипроцессный контроль и т.п.
5.5.1.2 Служба качества, отвечающая за принятие решений, критических для обеспечения качества, должна обладать полномочиями по принятию таких решений независимо от производственных процессов.
5.5.2 Представитель руководства
ИСО 9001:2008, Системы менеджмента качества - Требования 5.5.2 Представитель руководства Высшее руководство должно назначить представителя из состава руководства организации, который независимо от других обязанностей должен нести ответственность и иметь полномочия, распространяющиеся: a) на обеспечение разработки, утверждения, внедрения и поддержания в рабочем состоянии процессов, необходимых для системы менеджмента качества; b) на представление отчетов высшему руководству о продуктивности системы менеджмента качества и необходимости ее улучшения; c) на содействие распространению понимания требований потребителей по всей организации. Примечание - Ответственность представителя руководства может включать в себя поддержание связи с внешними сторонами по вопросам, касающимся системы менеджмента качества. |
5.5.3 Внутренний обмен информацией
ИСО 9001:2008, Системы менеджмента качества - Требования 5.5.3 Внутренний обмен информацией Высшее руководство должно обеспечивать разработку и утверждение в организации соответствующих процессов обмена информацией, включая информацию, относящуюся к эффективности системы менеджмента качества. |
5.5.3.1 Правила GMP, описанные в настоящем стандарте, и регуляторные требования должны по мере необходимости доводиться до сведения каждого уровня организации.
5.5.3.2 Высшее руководство должно быть своевременно уведомлено о ситуациях, критичных для обеспечения качества.
Примечание - К примерам процессов обмена информацией относятся те, которые касаются обмена информацией о политике в области качества, анализов со стороны руководства, результатов внутренних проверок качества и корректирующих и предупреждающих действий.
5.6 Анализ со стороны руководства
5.6.1 Общие положения
ИСО 9001:2008, Системы менеджмента качества - Требования 5.6.1 Общие положения Высшее руководство должно анализировать через запланированные интервалы времени систему менеджмента качества организации в целях обеспечения ее постоянной пригодности, достаточности и эффективности. Данный анализ должен включать в себя оценку возможностей для улучшения и потребности в изменениях в системе менеджмента качества организации, в том числе в политике и целях в области качества. Записи об анализе со стороны руководства должны поддерживаться в актуальном состоянии (см. 4.2.4). |
5.6.2 Входные данные для анализа
ИСО 9001:2008, Системы менеджмента качества - Требования 5.6.2 Входные данные для анализа Входные данные для анализа со стороны руководства должны включать в себя следующую информацию: a) результаты аудитов (проверок); b) отзывы потребителей; c) результативность процессов и соответствие продукции; d) статус предупреждающих и корректирующих действий; e) последующие действия, вытекающие из предыдущих анализов со стороны руководства; f) изменения, которые могли бы повлиять на систему менеджмента качества; g) рекомендации по улучшению. |
h) эффективность подготовки.
5.6.3 Выходные данные анализа
ИСО 9001:2008, Системы менеджмента качества - Требования 5.6.3 Выходные данные анализа Выходные данные анализа со стороны руководства должны включать в себя все решения и действия, относящиеся к: a) улучшению эффективности системы менеджмента качества и ее процессов; b) улучшению продукции в соответствии с требованиями потребителей; c) потребности в ресурсах. |
d) потребности в обучении.
6 Управление ресурсами
6.1 Обеспечение ресурсами
ИСО 9001:2008, Системы менеджмента качества - Требования 6.1 Обеспечение ресурсами Организация должна определить и предоставить ресурсы, необходимые для: a) внедрения и поддержания в рабочем состоянии системы менеджмента качества, а также постоянного улучшения ее эффективности; b) повышения удовлетворенности потребителей путем удовлетворения их требований. |
6.2 Персонал
6.2.1 Общие положения
ИСО 9001:2008, Системы менеджмента качества - Требования 6.2.1 Общие положения Персонал, выполняющий работу, влияющую на соответствие продукции требованиям, должен быть компетентным на основе полученного образования, подготовки, навыков и опыта. Примечание - На соответствие продукции требованиям непосредственно или косвенно может влиять персонал, выполняющий любую работу в рамках системы менеджмента качества. |
6.2.2 Компетентность, осведомленность и подготовка
ИСО 9001:2008, Системы менеджмента качества - Требования 6.2.2 Компетентность, осведомленность и подготовка Организация должна: a) определять необходимую компетентность персонала, выполняющего работу, которая влияет на соответствие требованиям к качеству продукции; b) где это возможно, обеспечивать подготовку или предпринимать другие действия в целях достижения необходимой компетентности; c) оценивать эффективность принятых мер; d) обеспечивать осведомленность своего персонала об актуальности и важности его деятельности и вкладе в достижение целей в области качества; e) поддерживать в актуальном состоянии соответствующие записи об образовании, подготовке, навыках и опыте (см. 4.2.4). |
6.2.2.1 Обучение GMP
6.2.2.1.1 Должно регулярно проводиться дополнительное обучение, которое включает осведомленность о применимых правилах GMP и всех процедурах и методах, которые оказывают влияние на качество продукции и систему менеджмента качества. Такое обучение должно охватывать:
a) риск контаминации и перекрестной контаминации;
b) потенциальную опасность для конечного пользователя/пациента в случае контаминации продукции;
c) влияние любых отклонений от установленных процедур, процессов или спецификаций на качество продукции потребителя или конечного пользователя.
6.2.2.1.2 Особое внимание должно уделяться подготовке персонала, занятого производством стерильных компонентов или компонентов, подвергающихся последующей стерилизации.
6.2.2.1.3 Должно быть организовано специальное обучение, касающееся микробиологической контаминации и контаминации механическими частицами, а также потенциального риска подобной контаминации для пациента.
6.2.2.1.4 С определенной периодичностью должно проводиться повторное обучение (повторный инструктаж).
6.2.2.1.5 Должна быть организована подготовка временного персонала, или он должен находиться под наблюдением подготовленного лица.
6.2.2.1.6 При найме консультантов для консультирования по вопросам качества должны вестись записи об их квалификации и видах оказываемых услуг.
6.2.2.1.7 Контрагенты и посетители должны надлежащим образом инструктироваться до входа в помещения, где осуществляется производство.
6.3 Инфраструктура
ИСО 9001:2008, Системы менеджмента качества - Требования 6.3 Инфраструктура Организация должна определять, обеспечивать и поддерживать инфраструктуру, необходимую для достижения соответствия требованиям, предъявляемым к продукции. Инфраструктура может включать в себя, если применимо: a) здания, рабочее пространство и связанные с ним системы; b) технологическое оборудование (как технические, так и программные средства); c) вспомогательные службы (такие как транспортная, служба связи или информационная система). |
6.3.1 Инфраструктура должна управляться, эксплуатироваться и обслуживаться таким образом, чтобы предупредить контаминацию продукции, включая, но не ограничиваясь, следующие требования:
- производственные объекты должны быть защищены от входа персонала, не имеющего прав доступа в них;
- персонал должен входить в определенные области производственных и складских помещений, а также помещений контроля/обеспечения качества в надлежащей одежде;
- планировка, проектирование и эксплуатация должны сводить к минимуму риск возникновения ошибок и позволять осуществлять эффективную уборку и техническое обслуживание во избежание перекрестной контаминации и любых нежелательных воздействий на качество продукции, основываясь на оценке риска;
- помещения для переодевания, мытья рук и туалеты должны располагаться рядом с зонами, где осуществляются манипуляции с продукцией и ее обработка; в том случае, если это может повлиять на качество продукции, данные помещения должны быть отделены от производственной зоны, а их вентиляционные системы не должны сообщаться напрямую.
6.3.2 Складские помещения должны:
- иметь подходящую вместимость, позволяющую упорядоченно хранить исходные материалы и продукцию, и
- соответствовать качеству материалов и продукции.
6.4 Производственная зона
ИСО 9001:2008, Системы менеджмента качества - Требования 6.4 Производственная зона Организация должна создать производственную зону, необходимую для достижения соответствия требованиям, предъявляемым к продукции, и управлять ею. Примечание - Термин "производственная зона" относится к условиям, в которых выполняется работа, в том числе физическим, экологическим и другим факторам (таким как шум, температура, влажность, освещенность или погодные условия). |
6.4.1 Требования, предъявляемые к производственной зоне
6.4.1.1 Организация должна разработать и утвердить документированные требования по охране здоровья, чистоте, одежде и контролю доступа персонала, если контакт между таким персоналом и первичным упаковочным материалам или производственной зоной может негативно отразиться на качестве первичной упаковки.
6.4.1.2 Если условия производственной зоны могут отрицательно повлиять на качество первичных упаковочных материалов, организация должна определить соответствующие условия производственной зоны и разработать систему по их результативному мониторингу и контролю.
6.4.1.3 В случае необходимости должны быть созданы и документированы специальные условия по контролированию контаминированных или потенциально контаминированных первичных упаковочных материалов, чтобы препятствовать контаминации других первичных упаковочных материалов, производственной зоны или персонала.
6.4.1.4 Необходимо использовать покрытие, закрывающее открытые первичные упаковочные материалы, если посредством документированной оценки риска не обосновано иное.
6.4.2 Классификация чистых зон/помещений
Чистые зоны/помещения должны быть классифицированы и управляться/мониторироваться.
См. ИСО 14644-1, ИСО 14644-2, ИСО 14644-3, ИСО 14644-5.
Касательно проектирования, строительства и ввода в действие чистых помещений см. ИСО 14644-2 и ИСО 14644-4.
Если это уместно, мониторинг биоконтаминации (загрязнения) должен проводиться в соответствии с требованиями ИСО 14698-1 и ИСО 14698-2.
6.4.3 Управление риском контаминации
Организация должна определить и управлять рисками возможной контаминации первичных упаковочных материалов, например:
a) личная гигиена и состояние здоровья;
b) личная одежда, ювелирные изделия, включая пирсинги, и косметика;
c) курение, употребление еды, питья, личных медикаментов и жевание;
d) переработка и утилизация отходов;
e) микробиологическая контаминация;
f) защитная одежда, подходящая для класса производственного помещения.
Примечание - Для снижения риска загрязнения могут использоваться дверные доводчики, защитные воздушные или пластиковые завесы.
6.4.4 Защита от насекомых и животных
Должна быть реализована и поддерживаться документированная, эффективная программа по защите от насекомых и животных (грызунов).
6.4.5 Материалы и технические системы (вспомогательные службы)
6.4.5.1 Все технические системы (например, воздухо-, газо-, паро-, водоснабжение) должны быть оценены на предмет возможного влияния на качество первичных упаковочных материалов и любых связанных с ними рисков. Должны вестись записи проводимой оценки (см. 4.2.4).
Оценка должна включать в себя прочие жидкости (например, смазочные жидкости, охлаждающие жидкости, гидравлические масла и т.п.), которые могут случайно вступить в контакт с первичным упаковочным материалом.
В зависимости от рисков должна быть рассмотрена возможность использования жидкостей пищевого качества.
6.4.5.2 Там, где это необходимо, должны быть предусмотрены соответствующие вентиляционные и вытяжные системы для сведения к минимуму риска контаминации. Особое внимание следует уделять системам рециркуляции воздуха.
6.4.5.3 Если вода вступает в непосредственный контакт с первичным упаковочным материалом или исходным материалом или используется для мытья оборудования, контактирующего с продукцией, необходимо определять и контролировать ее качество.
6.4.5.4 Вспомогательные средства, используемые в производственном процессе, должны быть определены, подвергаться документированной оценке риска на предмет их потенциального влияния на качество первичных упаковочных материалов, а их использование должно контролироваться.
6.5 Работы по техническому обслуживанию и уборке
6.5.1 Организация должна разработать и утвердить документированные требования к техническому обслуживанию (например, в отношении технологических процессов, систем и оборудования), когда проводимые работы или их отсутствие могут отразиться на качестве продукции.
6.5.2 Следует вести записи о работах по техническому обслуживанию (см. 4.2.4).
6.5.3 Работы по ремонту и техническому обслуживанию не должны представлять опасности для качества продукции. Работы по техническому обслуживанию не должны приводить к контаминации, а по их окончании должна проводиться документированная проверка чистоты.
6.5.4 Организация должна обеспечивать использование, эксплуатацию и там, где это уместно, поддержание в надлежащем состоянии инфраструктуры в соответствии с правилами GMP и во избежание контаминации продукции (включая контроль механических частиц и микробиологический контроль в случае необходимости).
6.5.5 Организация должна составить и задокументировать план уборки, в котором принимается во внимание риск контаминации.
Примечание - Документированные процедуры и план уборки должны содержать, где это уместно, нижеприведенную информацию:
- методы уборки;
- используемые материалы, например, чистящие/дезинфицирующие средства;
- зоны/оборудование, подлежащие уборке (очистке);
- меры предосторожности и порядок уборки на случай пролива;
- требуемые записи.
После очистки обычной практикой является хранение оборудования в чистом и высушенном состоянии, отдельно от контаминированного оборудования.
6.5.6 Необходимо вести комплект технической документации по оборудованию и монтажу, имеющим критическое значение для обеспечения качества.
6.5.7 Оборудование, критическое для обеспечения качества, имеющее дефекты, должно быть выведено из эксплуатации и (или) четко промаркировано соответствующим образом, а произведенная на нем продукция проверена (см. 8.2.4). До повторного ввода в эксплуатацию оборудование должно быть верифицировано как пригодное к использованию.
7 Процессы жизненного цикла продукции
7.1 Планирование процессов жизненного цикла продукции
ИСО 9001:2008, системы менеджмента качества - Требования 7.1 Планирование процессов жизненного цикла продукции Организация должна планировать и разрабатывать процессы, необходимые для обеспечения жизненного цикла продукции. Планирование процессов жизненного цикла продукции должно быть согласовано с требованиями к другим процессам системы менеджмента качества (см. 4.1). При планировании процессов жизненного цикла продукции организация должна определить в установленном порядке: а) цели в области качества и требования к продукции; b) потребность в разработке процессов и документов, а также в обеспечении ресурсами для конкретной продукции; c) необходимую деятельность по верификации и валидации, мониторингу, измерению, контролю и испытаниям для конкретной продукции, а также критерии приемки продукции; d) записи, необходимые для обеспечения свидетельства того, что процессы жизненного цикла продукции и продукция соответствуют требованиям (см. 4.2.4). Результат этого планирования должен быть представлен в форме, соответствующей практике организации. Примечание 1 - Документ, определяющий процессы системы менеджмента качества (включая процессы жизненного цикла продукции) и ресурсы, которые предстоит направить на конкретную продукцию, проект или контракт, может рассматриваться как план качества. Примечание 2 - При разработке процессов жизненного цикла продукции организация может также применять требования 7.3. |
7.1.1 При планировании процессов жизненного цикла продукции следует учитывать требование к единообразной обработке первичных упаковочных материалов. При планировании следует также учитывать необходимость отбора и сохранения образцов в надлежащем состоянии.
7.1.2 Организация должна обеспечить положение, при котором в планирование и реализацию на протяжении всего жизненного цикла продукции включаются процессы управления риском; следует вести соответствующие записи (см. 4.2.4).
7.2 Процессы, связанные с потребителем
7.2.1 Определение требований, относящихся к продукции
ИСО 9001:2008, Системы менеджмента качества - Требования 7.2.1 Определение требований, относящихся к продукции Организация должна определить: a) требования, установленные потребителями, включая требования к поставке и деятельности после поставки; b) требования, не определенные потребителем, но необходимые для конкретного или предполагаемого использования, когда оно известно; c) законодательные и регуляторные требования, применимые к продукции; d) любые дополнительные требования, рассматриваемые организацией как необходимые. Примечание - Деятельность после поставки может включать в себя, например, действия по гарантийному обеспечению, контрактным обязательствам, таким как услуги по техническому обслуживанию, и дополнительные услуги, такие как утилизация или полное уничтожение. |
7.2.1.1 Должны быть определены и документированы требования, относящиеся к продукции, включая изменения, требующие уведомления.
7.2.1.2 Должны быть определены и документированы требования потребителей, касающиеся предотвращения несанкционированного использования отходов производства первичных упаковочных материалов (включая образцы, печатные материалы, ярлыки).
7.2.2 Анализ требований, относящихся к продукции
ИСО 9001:2008, Системы менеджмента качества - Требования 7.2.2 Анализ требований, относящихся к продукции Организация должна анализировать требования, относящиеся к продукции. Данный анализ должен проводиться до принятия организацией обязательства поставлять продукцию потребителю (например, участие в тендерах, принятие контрактов или заказов, принятие изменений к контрактам или заказам) и должен обеспечивать: a) определение требований к продукции; b) согласование требований контракта или заказа, отличающихся от ранее сформулированных; c) способность организации выполнять определенные требования. Записи результатов анализа и последующих действий, вытекающих из анализа, должны поддерживаться в рабочем состоянии (см. 4.2.4). Если потребители не выдвигают документированных требований, организация должна подтвердить их у потребителя до принятия к исполнению. Если требования к продукции изменены, организация должна обеспечить, чтобы соответствующие документы были исправлены, а соответствующий персонал был поставлен в известность об изменившихся требованиях. Примечание - В некоторых ситуациях, таких как продажи, осуществляемые через Интернет, практически нецелесообразно проводить официальный анализ каждого заказа. Вместо этого анализ может распространяться на соответствующую информацию о продукции, такую как каталоги или другие рекламные материалы. |
7.2.3 Связь с потребителем
ИСО 9001:2008, Системы менеджмента качества - Требования 7.2.3 Связь с потребителем Организация должна определять и осуществлять эффективные мероприятия по поддержанию связи с потребителями в отношении: a) информации о продукции; b) оформления запросов, контракта или заказа, включая поправки; c) обратной связи от потребителей, включая претензии потребителей. |
7.2.3.1 Организация должна разработать, внедрить и поддерживать в рабочем состоянии систему обратной связи для заблаговременного оповещения о потенциальных и реальных проблемах качества и содействия вкладу потребителей в систему корректирующих и предупреждающих действий.
7.2.3.2 Если этого требует потребитель, организация должна согласовывать с ним, какие изменения требуют письменного подтверждения до утверждения, а какие - только уведомления. О предлагаемых изменениях необходимо своевременно информировать и согласовывать процесс внесения изменений (см. 7.2.1).
Между организацией и потребителем рекомендуется иметь документированное техническое соглашение об обеспечении качества, в котором рассматриваются меры, принимаемые в отношении несоответствий (см. 8.3).
7.2.3.3 Если это оговорено потребителем, образцы и сертификационные документы, предоставляемые для сравнительной оценки, испытания на стабильность или клинических исследований для целей подачи заявления на регистрацию лекарственных средств, должны соответствовать правилам и нормам GMP.
В рамках подачи заявления на регистрацию лекарственных средств потребитель может затребовать от организации указанную информацию. Представитель организации, обладающий достаточной квалификацией в данном вопросе, должен утвердить подобную информацию.
Пример - Информация о составе, данные испытаний, спецификации, информация о методах контроля и условиях обработки.
Изменения, которые оказывают влияние на любые данные, предоставленные организацией, должны быть переданы потребителю или регуляторным органам в установленном порядке (см. 7.2.3 и 7.5.1.3).
Примечание - Для сохранения конфиденциальности данных организации научная и техническая информация может быть передана в виде досье непосредственно соответствующим инстанциям, например в виде регистрационного досье или сертификата соответствия требованиям Европейской фармакопеи в связи с подачей заявления на регистрацию лекарственного средства, должны вестись соответствующие записи.
7.3 Проектирование и разработка
7.3.1 Планирование проектирования и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.1 Планирование проектирования и разработки Организация должна планировать проектирование и разработку продукции и управлять этими процессами. В ходе планирования проектирования и разработки организация должна определить: а) стадии проектирования и разработки; b) проведение анализа, верификации и валидации, соответствующих каждой стадии проектирования и разработки; c) ответственность и полномочия в области проектирования и разработки. Организация должна управлять взаимодействием различных групп, занятых проектированием и разработкой, в целях обеспечения эффективной связи и четкого распределения ответственности. Результаты планирования должны актуализироваться, если это необходимо, в процессе проектирования и разработки. Примечание - Анализ, верификация и валидация проектирования и разработки имеют разные цели, поэтому их можно проводить и записи по ним вести как отдельно, так и в любых сочетаниях, подходящих для продукции и организации. |
7.3.1.1 Организация должна внедрять документированные процедуры проектирования и разработки. Данные процедуры должны включать в себя оценку риска, определение соответствующих аспектов GMP и любого потенциального влияния на потребителя и в конечном счете на пациента.
7.3.1.2 Ответственность за проектирование и оценку риска должна быть согласована между потребителем и организацией.
7.3.1.3 В ходе процесса проектирования и разработки следует предусмотреть верификацию пригодности выходных данных проектирования и разработки до окончательного согласования спецификаций на производство.
7.3.2 Входные данные для проектирования и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.2 Входные данные для проектирования и разработки Входные данные, относящиеся к требованиям к продукции, должны быть определены, а записи должны поддерживаться в актуальном состоянии (см. 4.2.4). Входные данные должны включать в себя: a) функциональные и эксплуатационные требования; b) применимые законодательные и регуляторные требования; c) там, где это возможно, информацию, заимствованную из предыдущих аналогичных проектов; d) прочие требования, необходимые для проектирования и разработки. Входные данные должны анализироваться на достаточность. Требования должны быть полными, недвусмысленными и непротиворечивыми. |
7.3.3 Выходные данные проектирования и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.3 Выходные данные проектирования и разработки Выходные данные проектирования и разработки должны быть представлены в форме, подходящей для проведения верификации относительно входных требований к проектированию и разработке, а также должны быть официально утверждены до их последующего использования. Выходные данные проектирования и разработки должны: a) соответствовать входным требованиям к проектированию и разработке; b) обеспечивать соответствующей информацией по закупкам, производству и обслуживанию; c) содержать критерии приемлемости продукции или ссылки на них; d) определять характеристики продукции, существенные для ее безопасного и правильного использования. Примечание - Информация по производству и обслуживанию может включать в себя подробные данные о сохранении продукции. |
Примечание - Приветствуется совместная работа организации и потребителя, направленная на верификацию соответствия первичных упаковочных материалов их предполагаемому применению.
7.3.4 Анализ проекта и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.4 Анализ проекта и разработки На соответствующих стадиях должен проводиться систематический анализ проекта и разработки в соответствии с запланированными мероприятиями (см. 7.3.1) с целью: а) оценки способности результатов проектирования и разработки удовлетворять требованиям; b) выявления любых проблем и внесения предложений по необходимым действиям. В состав участников подобного анализа должны включаться представители подразделений, имеющих отношение к анализируемой(ым) стадии(ям) проектирования и разработки. Записи результатов анализа и всех необходимых действий должны поддерживаться в актуальном состоянии (см. 4.2.4). |
7.3.5 Верификация проекта и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.5 Верификация проекта и разработки Верификация должна осуществляться в соответствии с запланированными мероприятиями (см. 7.3.1) с целью удостоверения, что выходные данные проектирования и разработки соответствуют входным требованиям. Записи результатов верификации и всех необходимых действий должны поддерживаться в актуальном состоянии (см. 4.2.4). |
7.3.6 Валидация проекта и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.6 Валидация проекта и разработки Валидация проекта и разработки должна осуществляться в соответствии с запланированными мероприятиями (см. 7.3.1) с целью удостоверения, что полученная в результате продукция соответствует требованиям к установленному или предполагаемому использованию, если оно известно. Где это практически возможно, валидация должна быть завершена до поставки или применения продукции. Записи результатов валидации и всех необходимых действий должны поддерживаться в актуальном состоянии (см. 4.2.4). |
7.3.7 Управление изменениями проекта и разработки
ИСО 9001:2008, Системы менеджмента качества - Требования 7.3.7 Управление изменениями проекта и разработки Изменения проекта и разработки должны быть идентифицированы, а записи должны поддерживаться в актуальном состоянии. Изменения должны быть проанализированы, верифицированы и валидированы соответствующим образом, а также одобрены до внесения. Анализ изменений проекта и разработки должен включать в себя оценку влияния изменений на составные части и уже поставленную продукцию. Записи результатов анализа изменений и любых необходимых действий должны поддерживаться в актуальном состоянии (см. 4.2.4). |
7.3.7.1 Уведомление
Об изменениях, которые влияют на какие-либо предоставляемые данные, следует сообщать потребителю, а в случае предоставления организацией технического досье/регистрационного досье - непосредственно регуляторным органам.
7.3.7.2 Изменение проекта
При осуществлении изменения действующая валидация и документы, на которые повлияло изменение, должны быть проанализированы и пересмотрены, а персонал должен пройти соответствующую переподготовку.
Примечание 1 - К выходным данным проектирования и разработки относятся записи (спецификации, технология производства, технические чертежи, технические или исследовательские протоколы) и образцы.
Примечание 2 - Конфиденциальная научная и техническая информация (организации) может быть представлена в виде досье непосредственно в регуляторные органы [например, техническое досье и (или) регистрационное досье].
7.4 Закупки
7.4.1 Процесс закупок
ИСО 9001:2008, Системы менеджмента качества - Требования 7.4.1 Процесс закупок Организация должна обеспечивать соответствие закупленной продукции установленным требованиям к закупкам. Тип и степень управления, применяемые по отношению к поставщику и закупленной продукции, должны зависеть от ее влияния на последующие стадии жизненного цикла продукции или готовую продукцию. Организация должна оценить и выбрать поставщиков на основе их способности поставлять продукцию в соответствии с требованиями организации. Должны быть разработаны критерии отбора, оценки и повторной оценки. Записи результатов оценивания и любых необходимых действий, вытекающих из оценки, должны поддерживаться в актуальном состоянии (см. 4.2.4). |
7.4.1.1 Организация должна утвердить поставщиков:
a) исходных материалов,
b) вспомогательных средств, критических для обеспечения качества, и
c) упаковочных материалов для использования в чистых помещениях.
7.4.1.2 Организация должна уведомить потребителя до аутсорсинга сторонней организации любой части производственного процесса.
7.4.1.3 Все услуги, переданные на аутсорсинг сторонним организациям, которые могут повлиять на качество продукции, должны контролироваться, включая подготовку печатного оригинала, услуги лабораторий, стерилизацию, услуги по калибровке и квалификации, техническое обслуживание, уборку (очистку), перевозку, борьбу с вредителями и подрядчиков по переработке отходов в зависимости от рисков.
7.4.1.4 Консультанты, предоставляющие рекомендации по производству и контролю первичных упаковочных материалов, должны считаться поставщиками.
7.4.1.5 Поставщики материалов и услуг, критических для обеспечения качества, должны утверждаться службой качества или лицом, назначенным службой качества.
7.4.1.6 Организация должна оценивать, а также вести записи относительно компетентности лабораторий, выполняющих работы, критические для обеспечения качества. Для выполнения работ, критических для обеспечения качества, организация должна использовать только лаборатории, одобренные с точки зрения их компетентности.
7.4.1.7 Если выполнение процесса стерилизации передано на аутсорсинг сторонней организации, организация должна убедиться в том, что процесс отвечает требованиям 7.5.1 и 7.5.2.
7.4.1.8 Изменение источника получения сырья, критического для обеспечения качества, подлежит процессу управления изменениями.
7.4.2 Информация о закупках
ИСО 9001:2008, Системы менеджмента качества - Требования 7.4.2 Информация о закупках Информация о закупках должна описывать заказанную продукцию, включая, где это необходимо, требования, предъявляемые к: a) официальному утверждению продукции, процедур, процессов и оборудования, b) квалификации персонала и c) системе менеджмента качества. Организация должна обеспечивать адекватность установленных требований к закупкам до их сообщения поставщику. |
7.4.2.1 Организация должна сохранять соответствующую информацию по закупкам, то есть документы (см. 4.2.3) и записи (см. 4.2.4), в объеме, требуемом для обеспечения прослеживаемости согласно 7.5.3.
7.4.3 Верификация закупленной продукции
ИСО 9001:2008, Системы менеджмента качества - Требования 7.4.3 Верификация закупленной продукции Организация должна разработать, внедрить и осуществлять контроль или другую деятельность, необходимую для обеспечения соответствия закупленной продукции установленным требованиям к закупкам. В том случае, если организация или ее потребитель предполагает осуществить верификацию у поставщика, то организация должна указать предполагаемую договоренность относительно верификации и порядок выпуска продукции в информации по закупкам. |
7.4.3.1 Поступающие материалы подлежат физическому или административному карантину до их утверждения и выпуска для использования.
Примечание - В исключительных случаях может использоваться испытываемый материал, при условии наличия безотказных процедур, не допускающих выпуска первичного упаковочного материала до подтверждения статуса подобных материалов.
7.4.3.2 Для материалов, критических для обеспечения качества, организация должна периодически верифицировать релевантную и (или) критическую информацию, полученную от поставщиков данных материалов и содержащуюся в сертификате анализа, сертификате соответствия или актах испытаний.
Примечание 1 - Данная верификация может подразумевать сходные испытания на месте производства, или же проводимые независимым подрядчиком, или плановый аудит производственных мощностей с целью обеспечения высокого уровня достоверности информации, предоставленной поставщиком. В качестве альтернативы при надлежащем обосновании аудит может быть заменен сертифицированной системой менеджмента.
Примечание 2 - Вместо подобного испытания, проводимого самой организацией, отчет об анализе (например, сертификат анализа, соответствия, акт испытания) может быть принят от поставщика при условии, что по меньшей мере одно специфичное определение подлинности проводится для данного материала или компонента изделия самой организацией.
7.4.3.3 Должны вестись записи по верификации (см. 4.2.4).
7.4.3.4 Отбор проб должен осуществляться в соответствии с методикой отбора проб, с использованием процедур, средств и оборудования, не допускающих контаминацию.
7.5 Производство и обслуживание
7.5.1 Управление производством и обслуживанием
ИСО 9001:2008, Системы менеджмента качества - Требования 7.5.1 Управление производством и обслуживанием Организация должна планировать и осуществлять производство и обслуживание в управляемых условиях. Управляемые условия должны включать в себя там, где это применимо: a) наличие информации, описывающей характеристики продукции; b) наличие рабочих инструкций при необходимости; c) использование подходящего оборудования; d) наличие и применение оборудования для мониторинга и измерений; e) проведение мониторинга и измерений; f) осуществление выпуска, поставки и действий после поставки продукции. |
g) определение даты производства с учетом задействованных процессов;
h) уделение особого внимания маркировке, этикетированию и упаковке для обеспечения результативного контроля и предотвращения ошибок;
i) документированную процедуру с определением управления отклонениями в процессе. Должны быть расследованы отклонения, критические для обеспечения качества, и зарегистрированы полученные результаты (см. 4.2.4).
7.5.1.1 Управление чистотой продукции и контаминацией
7.5.1.1.1 Организация должна разработать, утвердить и соблюдать документированные требования к чистоте первичных упаковочных материалов и процедуры по предотвращению контаминации продукции и оборудования.
Должны оцениваться потенциальные риски, связанные с любыми материалами или вспомогательными средствами, которые могут нести угрозу безопасности пациента, например, трансмиссивная губчатая энцефалопатия.
7.5.1.1.2 Все технологические процессы, осуществляемые в чистых или контролируемых зонах, включая меры по охране окружающей среды, производство, внутрипроизводственный контроль и упаковку первичных упаковочных материалов, должны отвечать установленным условиям и функциональным критериям конкретной зоны. Чистые помещения должны быть оборудованы воздушными шлюзами.
7.5.1.1.3 Технологические процессы в контролируемых рабочих условиях должны быть согласованы между потребителем и организацией.
7.5.1.1.4 Организация должна также разработать и утвердить документированные требования к чистоте первичных упаковочных материалов, когда:
a) первичная упаковка очищается организацией до проведения стерилизации и (или) его использования, или
b) первичные упаковочные материалы будут поставляться нестерилизованными и их чистота важна для использования, или
c) технологические реагенты должны быть удалены с продукции в процессе производства.
7.5.1.1.5 Должны быть определены контейнеры для хранения, их соответствующая оснастка и линии загрузки и разгрузки.
7.5.1.1.6 Особое внимание (например, при идентификации, обеспечении безопасности и чистоты) следует уделять в первую очередь загрузке и разгрузке контейнеров для насыпных грузов/бункеров.
7.5.1.1.7 Манипуляции с контейнерами и их перемещение должны осуществляться в чистоте и не приводить к контаминации механическими частицами. Для продукции, контактирующей с упаковочными материалами, они должны быть закрыты или надлежащим образом герметизированы (опечатаны).
7.5.1.1.8 Для очистки оборудования, используемого при производстве первичных упаковочных материалов, должны быть разработаны письменные процедуры. Должны вестись записи об очистке оборудования, критического для обеспечения качества первичных упаковочных материалов (см. 4.2.4).
7.5.1.1.9 Следует определять содержание и уровень очистки производственного оборудования/производственных участков.
7.5.1.1.10 При производстве некоторых материалов свойственно использование переработанных материалов (например, стекла, алюминия, бумаги, термопластиков). Параметры переработки должны быть определены и согласованы с потребителем.
7.5.1.1.11 Если это не согласовано с потребителем, термопластичные материалы не должны подвергаться повторному измельчению или не подлежат повторному использованию в первичных упаковочных материалах.
7.5.1.1.12 Должен быть организован контроль очистки линии между производством различных серий для удаления всех материалов и документов, не требуемых для последующей операции. Должны вестись записи работ по очистке линий (см. 4.2.4).
Примечание - В качестве примера можно привести документированную проверку чистоты контейнеров многоразового использования, применяемых для хранения исходных материалов в процессе обработки, проводимую до их загрузки другим материалом, во избежание перекрестной контаминации.
7.5.1.1.13 Частично документированной оценке риска должны подвергаться системы, предназначенные для снижения времени простоя оборудования и не позволяющие проводить полную очистку линии (сокращенная очистка линии или система автоматизированной переналадки); подобные системы должны функционировать в контролируемом режиме для обеспечения качества продукции.
7.5.1.1.14 Палеты должны быть собраны из материалов, пригодных для конкретной продукции; их источник должен прослеживаться, а сами палеты - контролироваться для снижения риска контаминации.
Примечание - Деревянные палеты могут быть контаминированы в результате переноса любых реактивов, использованных для обработки прочих палет.
7.5.1.2 Управление разделением
Для снижения риска перекрестной контаминации/смешивания материалов, промежуточной и готовой продукции, они должны разделяться подходящими способами, основанными на оценке риска (например, физическое разделение, маркировка, штрихкоды, электронное местоположение).
7.5.1.3 Управление изменениями
7.5.1.3.1 Организация должна разработать, утвердить и использовать документированную процедуру для эффективной системы управления изменениями, включающей сравнительную оценку риска (рисков), связанных с предлагаемыми изменениями, которые могут оказать потенциальное влияние на качество поставляемой продукции.
7.5.1.3.2 В процессе сравнительной оценки влияния изменения на качество продукции должна быть определена необходимость валидации или повторной валидации.
7.5.1.3.3 Процедура управления изменениями организации должна обеспечивать формирование дополнительных данных, демонстрирующих, что изменения приведут к производству продукции желаемого качества и безопасности, соответствующих утвержденной спецификации.
7.5.1.3.4 Определенные подразделения должны иметь полномочия и нести ответственность за утверждения изменений. Утвержденные изменения должны внедряться не бесконтрольно.
7.5.1.4 Конкретные требования к стерильности первичных упаковочных материалов
Организация должна вести записи (см. 4.2.4) о параметрах процесса стерилизации, который использовался для стерилизации каждой серии. Записи о стерилизации должны быть прослеживаемыми для каждой серии первичного упаковочного материала.
7.5.2 Валидация процессов производства и обслуживания
ИСО 9001:2008, Системы менеджмента качества - Требования 7.5.2 Валидация процессов производства и обслуживания Организация должна валидировать любые процессы производства и обслуживания, результаты которых не могут быть верифицированы последующим мониторингом или измерениями, из-за чего недостатки становятся очевидными только после начала использования продукции или после предоставления услуги. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов. Организация должна разработать мероприятия по этим процессам, в том числе, если это применимо: a) определенные критерии для анализа и утверждения процессов; b) утверждение соответствующего оборудования и квалификации персонала; c) использование конкретных методов и процедур; d) требования к записям (см. 4.2.4); e) повторную валидацию. |
7.5.2.1 Организация должна идентифицировать процессы, критические для обеспечения качества, в рамках своей деятельности, в частности те, которые влияют на качество первичных упаковочных материалов. Контроль за любыми из этих процессов, результат которых не может быть верифицирован посредством последующего мониторинга или измерения, должен демонстрироваться путем валидации и документироваться.
7.5.2.2 Для определения того, какие процессы критичны для обеспечения качества, и степени валидации, необходимой для демонстрации управления этими процессами, используется оценка риска. Анализ риска должен быть связан со свойствами продукции, определяющими ее качество.
7.5.2.3 Оборудование, инженерные системы и средства производства, используемые для производства первичных упаковочных материалов, должны проходить верификацию и (или) квалификацию/валидацию в соответствии с документированной оценкой риска.
7.5.2.4 Верификация и (или) квалификация/валидация должны проводиться при возникновении существенных изменений в средствах производства, оборудовании и процессах, которые могут повлиять на качество продукции.
Примечание - Управление изменениями процесса валидации является частью политики управления изменениями организации.
7.5.2.5 Насколько это целесообразно, валидация той или иной продукции должна проводиться по согласованию с потребителем.
7.5.2.6 Результаты валидации должны регистрироваться (см. 4.2.4). Записи о валидации должны сохраняться на протяжении всего срока службы оборудования и процесса и еще в течение двух лет после списания оборудования или по договоренности с потребителем.
7.5.2.7 В отношении программного обеспечения, используемого в процессах, критических для обеспечения качества, должны проводиться функциональные испытания в достаточном количестве и в соответствующих условиях для верификации прослеживаемости, точности передачи и сохранения данных. Система должна проверяться, например, путем ввода правильных и неправильных данных для определения прослеживаемости, точности передачи и сохранения данных или записей.
7.5.2.8 Результаты данных испытаний и проверок должны регистрироваться (см. 4.2.4).
7.5.2.9 Электронные записи должны сохраняться и защищаться от потери и случайного повреждения и должны храниться в форме, допускающей восстановление; если это невозможно, должны сохраняться бумажные экземпляры в течение двух лет после списания оборудования или по договоренности с потребителем (см. 4.2.4.1).
Примечание - Дополнительные данные, касающиеся защиты данных, менеджмента и валидации программного обеспечения, приведены в [27], [32] и [33].
7.5.2.10 При аутсорсинге любого процесса, критического для обеспечения качества, организация должна убедиться в том, что процесс отвечает требованиям настоящего стандарта.
7.5.2.11 Если стерилизация является обязательной, организация должна разработать и утвердить документированные процедуры по валидации процессов стерилизации. Процессы стерилизации должны пройти валидацию до первоначального использования и периодически проходить повторную валидацию. Должны сохраняться записи результатов валидации процессов стерилизации (см. 4.2.4).
См. ИСО 11135, ИСО 11137-1 или ИСО 11137-2.
7.5.2.12 В том случае, если стерилизация является обязательной, организация должна подвергнуть первичные упаковочные материалы валидированному процессу стерилизации и зарегистрировать все контрольные параметры процесса стерилизации. При аутсорсинге процесса стерилизации организация должна убедиться в том, что процесс отвечает требованиям настоящего стандарта.
См. ИСО 14937.
7.5.3 Идентификация и прослеживаемость
ИСО 9001:2008, Системы менеджмента качества - Требования 7.5.3 Идентификация и прослеживаемость Если это целесообразно, организация должна идентифицировать продукцию с помощью соответствующих средств на всех стадиях ее жизненного цикла. Организация должна идентифицировать статус продукции по отношению к требованиям мониторинга и измерений на всех стадиях ее жизненного цикла. Если прослеживаемость является обязательной, то организация должна управлять уникальной идентификацией продукции и поддерживать записи в рабочем состоянии (см. 4.2.4). Примечание - В ряде отраслей промышленности менеджмент конфигурации является средством поддержания идентификации и прослеживаемости. |
7.5.3.1 Организация должна разработать и поддерживать систему прослеживания всех производственных материалов от источника получения до жизненного цикла продукции с определением масштаба прослеживания и требуемых записей на основе оценки риска (см. 4.2.4, 8.3 и 8.5).
7.5.3.2 Записи о производстве серии должны быть обозначены уникальным номером серии или идентификационным номером.
7.5.3.3 Должны сохраняться записи об использовании оборудования, критического для обеспечения качества (см. 4.2.4). Данные записи должны также включать информацию об очистке и техническом обслуживании наряду с производственными операциями. Работы по техническому обслуживанию должны документироваться и прослеживаться до конкретной производственной операции или единицы оборудования.
7.5.3.4 Организация должна разработать, утвердить и поддерживать документированные процедуры с целью обеспечения идентификации и отделения от нормальной продукции возвращаемых организации первичных упаковочных материалов (например, для повторной обработки в соответствии с установленными требованиями).
7.5.4 Собственность потребителя
ИСО 9001:2008, Системы менеджмента качества - Требования 7.5.4 Собственность потребителя Организация должна проявлять заботу о собственности потребителя, пока она находится под управлением организации или используется ею. Организация должна идентифицировать, верифицировать, защищать и сохранять собственность потребителя, предоставленную для использования или включения в продукцию. Если собственность потребителя утеряна, повреждена или признана непригодной для использования, организация должна известить об этом потребителя и поддерживать записи в актуальном состоянии (см. 4.2.4). Примечание - Собственность потребителя может включать в себя интеллектуальную собственность и сведения личного характера. |
7.5.5 Сохранение продукции
ИСО 9001:2008, Системы менеджмента качества - Требования 7.5.5 Сохранение продукции Организация должна сохранять продукцию в ходе внутренней обработки и в процессе поставки к месту назначения в целях поддержания ее соответствия установленным требованиям. Если это применимо, сохранение продукции должно включать в себя идентификацию, погрузочно-разгрузочные работы, упаковку, хранение и защиту. Требование сохранения должно быть также применено и к составным частям продукции. |
7.5.5.1 Организация должна разработать и поддерживать систему контроля продукции с ограниченным сроком хранения или требующую особых условий хранения. Такие особые условия хранения должны контролироваться и регистрироваться (см. 4.2.4). Сроки хранения должны быть обоснованными.
7.5.5.2 Продукция должна быть четко обозначена, разделена и должна подлежать ответственному хранению, а также должна быть защищена от попадания посторонних веществ или контаминации. Упаковка, используемая для получения и содержания продукции, должна быть чистой и пригодной для применения. Поставки должны сопровождаться соответствующей документацией. Документация на поставку должна соответствовать конкретной серии.
7.5.5.3 В случае повторного использования контейнеров для упаковки предшествующие ярлыки должны быть удалены или надписи на них стерты. Контейнеры должны быть чистыми или проверенными на чистоту в соответствии с документированной процедурой.
7.5.5.4 В случае необходимости на ярлыке должны быть указаны и должны выполняться особые условия транспортировки или хранения первичных упаковочных материалов.
7.6 Управление оборудованием для мониторинга и измерения
ИСО 9001:2008, Системы менеджмента качества - Требования 7.6 Управление оборудованием для мониторинга и измерения Организация должна определить мониторинг и измерения, которые предстоит осуществлять, а также оборудование для мониторинга и измерений, необходимое для обеспечения свидетельства соответствия продукции установленным требованиям. Организация должна установить процессы для обеспечения того, чтобы мониторинг и измерения могли быть выполнены и в действительности были выполнены в соответствии с требованиями к ним. При необходимости обеспечения получения достоверных результатов измерительное оборудование должно: a) быть откалибровано и (или) поверено в установленные периоды или перед его применением с использованием эталонов, прослеживаемых до международных или национальных эталонов. При отсутствии таких эталонов основа, использованная для калибровки или поверки, должна быть зарегистрирована (см. 4.2.4); b) быть отрегулировано или повторно отрегулировано по мере необходимости; c) иметь идентификатор в целях установления статуса калибровки; d) иметь защиту от регулировок, которые сделали бы недостоверными результаты измерения; e) быть защищенным от повреждения и порчи в ходе обращения, технического обслуживания и хранения. Кроме того, организация должна оценить и зарегистрировать достоверность предыдущих результатов измерения, если выявлено, что оборудование не соответствует требованиям. Организация должна предпринять соответствующее действие в отношении такого оборудования и любой затронутой продукции. Записи результатов калибровки и поверки должны поддерживаться в рабочем состоянии (см. 4.2.4). Если при мониторинге и измерении установленных требований используют программное обеспечение, его способность удовлетворять предполагаемому применению предварительно должна быть подтверждена и повторно подтверждена по мере необходимости. Примечание - Подтверждение соответствия компьютерного программного обеспечения предполагаемому применению обычно предусматривает его верификацию и менеджмент конфигурации в целях поддержания его пригодности для использования. |
7.6.1 Должны проводиться регулярные регистрируемые испытания автоматического контрольного оборудования с нагрузкой (например, испытания 100% фотокамерных инспекционных систем и считывателей штрихкодов) для проверки постоянной функциональной пригодности.
7.6.2 Испытательное оборудование, используемое для определения приемлемости исходных материалов, критических для обеспечения качества, промежуточной/обрабатываемой или готовой продукции, должно калиброваться или должны проводиться дополнительные квалификационные испытания по мере необходимости.
8 Измерение, анализ и улучшение
8.1 Общие положения
ИСО 9001:2008, Системы менеджмента качества - Требования 8.1 Общие положения Организация должна планировать и внедрять процессы мониторинга, измерения, анализа и улучшения, необходимые для: a) демонстрации соответствия требованиям, предъявляемым к продукции; |
Данное требование относится как к промежуточной продукции, так и к первичным упаковочным материалам.
b) обеспечения соответствия системы менеджмента качества; c) постоянного повышения эффективности системы менеджмента качества. Указанная деятельность должна включать в себя определение применимых методов, в том числе статистических, и степень их использования. |
8.2 Мониторинг и измерение
8.2.1 Удовлетворенность потребителей
ИСО 9001:2008, Системы менеджмента качества - Требования 8.2.1 Удовлетворенность потребителей В качестве одного из способов измерения работы системы менеджмента качества организация должна проводить мониторинг информации, касающейся восприятия потребителем выполнения организацией его требований. Должны быть определены методы получения и использования данной информации. Примечание - Мониторинг восприятия потребителями может включать в себя получение информации из таких источников, как исследования удовлетворенности потребителей, данные от потребителей о качестве поставленной продукции, исследования мнений пользователей, анализ оттока клиентов, благодарности, претензии по гарантийным обязательствам и отчеты распространителей. |
8.2.2 Внутренний аудит
ИСО 9001:2008, Системы менеджмента качества - Требования 8.2.2 Внутренний аудит Организация должна проводить внутренние аудиты (проверки) через запланированные интервалы времени в целях установления того, что система менеджмента качества: a) соответствует запланированным мероприятиям (7.1), требованиям настоящего стандарта и требованиям к системе менеджмента качества, разработанным организацией, и b) эффективно внедрена и поддерживается в рабочем состоянии. Программа аудитов (проверок) должна планироваться с учетом статуса и важности процессов и участков, подлежащих аудиту, а также результатов предыдущих аудитов. Критерии, область применения, частота и методы аудитов должны быть определены. Выбор аудиторов и проведение аудитов должны обеспечивать объективность и беспристрастность процесса аудита. Аудиторы не должны проверять свою собственную работу. Должна быть разработана и утверждена документированная процедура для определения ответственности и требований, связанных с планированием и проведением аудитов, ведением записей и составлением отчетов о результатах. Записи об аудитах и их результатах должны поддерживаться в актуальном состоянии (см. 4.2.4). Руководство, ответственное за проверяемые области деятельности, должно обеспечить, чтобы все необходимые коррекции и корректирующие действия предпринимались без излишней отсрочки для устранения обнаруженных несоответствий и вызвавших их причин. Последующие действия должны включать в себя верификацию принятых мер и отчет о результатах верификации (см. 8.5.2). Примечание - См. ИСО 19011. |
8.2.3 Мониторинг и измерение процессов
ИСО 9001:2008, Системы менеджмента качества - Требования 8.2.3 Мониторинг и измерение процессов Организация должна использовать подходящие методы мониторинга и, где это применимо, измерения процессов системы менеджмента качества. Эти методы должны демонстрировать способность процессов достигать запланированных результатов. Если запланированные результаты не достигаются, то должны предприниматься необходимые коррекции и корректирующие действия. Примечание - При определении подходящих методов организация должна учитывать тип и объем мониторинга или измерений, подходящих для каждого из таких процессов, в отношении их влияния на соответствие требованиям к продукции и на результативность системы менеджмента качества. |
Служба качества должна обеспечивать проведение расследования, устранения и документирования отклонений, критических для обеспечения качества.
8.2.4 Мониторинг и измерение продукции
ИСО 9001:2008, Системы менеджмента качества - Требования 8.2.4 Мониторинг и измерение продукции Организация должна осуществлять мониторинг и измерять характеристики продукции в целях верификации соблюдения требований, предъявляемых к продукции. Это должно осуществляться на соответствующих стадиях процесса жизненного цикла продукции согласно запланированным мероприятиям (см. 7.1). Свидетельства соответствия критериям приемлемости должны поддерживаться в рабочем состоянии. В записях должно(ы) быть указано(ы) лицо(а), утвердившее(ие) выпуск продукции для доставки потребителю (см. 4.2.4). Выпуск продукции и предоставление услуги потребителю не должны осуществляться до тех пор, пока все запланированные действия (см. 7.1) не будут удовлетворительно завершены, если соответствующим полномочным лицом или органом и, где это применимо, потребителем не утверждено иное. |
8.2.4.1 Результаты за пределами спецификаций
Любой результат за пределами спецификаций должен расследоваться в соответствии с документированной процедурой, а полученные данные должны регистрироваться (см. 4.2.4).
8.2.4.2 Входной контроль и входные испытания
Должны быть разработаны и утверждены и должны выполняться требования для всех используемых материалов. Поступающие материалы должны контролироваться или иным образом проверяться на соответствие установленным требованиям.
8.2.4.3 Внутрипроизводственный контроль
8.2.4.3.1 Организация должна в соответствии с требованиями документированных процедур контролировать и испытывать продукцию в процессе обработки.
8.2.4.3.2 Должны быть определены процедуры отбора проб, обеспечивающие репрезентативность образцов для оцениваемого процесса. Образцы не следует возвращать в производственную зону, если они были направлены на отдельную испытательную площадку.
8.2.4.3.3 После выхода из строя оборудования или незапланированной остановки процесса должен быть проведен дополнительный внутрипроизводственный контроль.
8.2.4.4 Выпуск серии
Организация должна внедрить процесс утверждения выпуска серии продукции.
Примечание 1 - Поставка до выпуска серии может осуществляться по соглашению с потребителем.
Если контроль готовой продукции является обязательным, то он должен быть завершен до выпуска серии. Должны быть определены процедуры отбора проб с целью обеспечения репрезентативности образцов по отношению к оцениваемой серии. Образцы не следует возвращать в производственную зону, если они были направлены на отдельную испытательную площадку.
Для выпуска серии должен проводиться анализ документации на серию.
Примечание 2 - Контроль готовой продукции может не включать в себя все параметры, приведенные в спецификации, в зависимости от системы и стратегии управления.
8.2.4.5 Архивные образцы
Архивные образцы должны быть отобраны в соответствии с требованиями организации и (или) потребителя.
8.2.4.6 Данные о производстве и контроле
По запросу потребителя или его представителя данные о производстве и контроле, относящиеся к продукции (за исключением конфиденциальной интеллектуальной собственности организации), должны быть предоставлены с целью верификации того, что производственный процесс, внутрипроизводственный контроль и испытательное оборудование функционируют надлежащим образом.
8.3 Управление несоответствующей продукцией
ИСО 9001:2008, Системы менеджмента качества - Требования 8.3 Управление несоответствующей продукцией Организация должна обеспечивать идентификацию продукции, не соответствующей требованиям, и управление ею в целях предотвращения непреднамеренного использования или поставки такой продукции. Должна быть разработана и утверждена документированная процедура для определения средств управления и соответствующей ответственности и полномочий для действий с несоответствующей продукцией. Если применимо, организация должна предпринимать в отношении несоответствующей продукции одно или несколько нижеперечисленных действий: a) устранение обнаруженного несоответствия; b) санкционирование использования, выпуска или приемки продукции, если получено разрешение от соответствующего полномочного лица или органа и, где это применимо, потребителя; c) предотвращение ее первоначального предполагаемого использования или применения; d) предпринятие шагов, соответствующих потенциальным или имеющимся эффектам несоответствия, в том случае, если несоответствующая продукция была выявлена после поставки или начала использования. После того как несоответствующая продукция исправлена, она должна быть подвергнута повторной верификации для подтверждения соответствия требованиям. Записи о характере несоответствий и любых последующих предпринятых действиях, включая полученные разрешения на использование, выпуск и т.п., должны поддерживаться в актуальном состоянии (см. 4.2.4). |
8.3.1 Несоответствующая продукция или несоответствующие материалы должны подвергаться карантину в ожидании принятия решения о корректирующих или иных действиях. Когда идет речь об исправлении путем переработки или модификации, следует провести и зарегистрировать оценку риска любого отрицательного влияния переработки на продукцию (см. 4.2.4 и 7.5.1).
8.3.2 Переработка и (или) модификация должны проводиться в соответствии с документированной процедурой, которая была утверждена службой качества. Процедура переработки должна быть согласована с потребителем в том случае, если это является оговоренным требованием.
8.3.3 Если материал для первичной упаковки был произведен в условиях чистых помещений, любая переработка должна осуществляться в тех же условиях.
8.3.4 Любое предложение о выпуске несоответствующей продукции должно осуществляться посредством документального соглашения, утвержденного потребителем.
8.3.5 В случае забраковки первичные упаковочные материалы должны утилизироваться или уничтожаться в соответствии с документированной процедурой.
8.4 Анализ данных
ИСО 9001:2008, Системы менеджмента качества - Требования 8.4 Анализ данных Организация должна определять, собирать и анализировать соответствующие данные для демонстрации пригодности и результативности системы менеджмента качества, а также оценивания, в какой области возможно постоянное улучшение системы менеджмента качества. Данные должны включать в себя информацию, полученную в результате мониторинга и измерения и из других подходящих источников. Анализ данных должен представлять информацию, касающуюся: a) удовлетворенности потребителей (см. 8.2.1); b) соответствия требованиям к продукции (см. 8.2.4); c) характеристик и тенденций процессов и продукции, включая возможности проведения предупреждающих действий (см. 8.2.3 и 8.2.4); d) поставщиков (см. 7.4). |
Организация должна разработать, внедрить и поддерживать документированные процедуры, включая требования к анализу данных, для выявления существующих или потенциальных причин выпуска несоответствующей продукции или других проблем качества.
8.5 Улучшение
8.5.1 Постоянное улучшение
ИСО 9001:2008, Системы менеджмента качества - Требования 8.5.1 Постоянное улучшение Организация должна постоянно повышать эффективность системы менеджмента качества посредством использования политики и целей в области качества, результатов аудитов, анализа данных, корректирующих и предупреждающих действий, а также анализа со стороны руководства. |
Изменения, предлагаемые в качестве части постоянного улучшения, подлежат менеджменту риска.
8.5.2 Корректирующие действия
ИСО 9001:2008, Системы менеджмента качества - Требования 8.5.2 Корректирующие действия Организация должна предпринимать корректирующие действия в целях устранения причин несоответствий для предупреждения повторного их возникновения. Корректирующие действия должны быть адекватны по отношению к последствиям выявленных несоответствий. Должна быть разработана документированная процедура для определения требований: a) к анализу несоответствий (включая претензии потребителей); b) к установлению причин несоответствий; c) к оцениванию необходимости действий, требуемых для предупреждения повторения несоответствий; d) к определению и осуществлению необходимых действий; e) к записям результатов предпринятых действий (см. 4.2.4); f) к анализу результативности предпринятых корректирующих действий. |
8.5.2.1 Организация должна своевременно расследовать все претензии потребителей и сообщить о проводимых корректирующих действиях на все производственные участки и участки, связанные с производством. Действия должны осуществляться как можно быстрее, и в соответствии с согласованным графиком следует вести записи расследования (см. 4.2.4).
8.5.2.2 Должно быть обосновано и зарегистрировано отсутствие корректирующих и (или) предупреждающих действий по результатам расследования претензий потребителей (см. 4.2.4).
8.5.3 Предупреждающие действия
ИСО 9001:2008, Системы менеджмента качества - Требования 8.5.3 Предупреждающие действия Организация должна определять действия в целях устранения причин потенциальных несоответствий для предупреждения их появления. Предупреждающие действия должны соответствовать возможным последствиям потенциальных проблем. Должна быть разработана и утверждена документированная процедура для определения требований: a) к установлению потенциальных несоответствий и их причин; b) к оцениванию необходимости действий в целях предупреждения появления несоответствий; c) к определению и осуществлению необходимых действий; d) к записям результатов предпринятых действий (см. 4.2.4); e) к анализу результативности предпринятых предупреждающих действий. |
Приложение A
(обязательное)
Требования GMP к печатным материалам для первичной упаковки
А.1 Средства получения печатных оттисков
А.1.1 Общие положения
А.1.1.1 Файлы, содержащие иллюстрации/исходные материалы, должны быть поименованы и храниться в соответствии с документированной процедурой, которая позволяет легко их идентифицировать, контролируемо выпускать и прослеживать.
А.1.1.2 Все средства получения печатных оттисков должны:
a) иметь четкое и однозначное обозначение, чтобы обеспечить прослеживаемость до исходного материала;
b) быть произведены на основе основного исходного материала, находящегося у потребителя, и быть прослеживаемыми до данного материала;
c) проверяться на соответствие бумажному носителю или электронным данным, одобренным потребителем, и регистрироваться (см. 4.2.4);
d) храниться в защищенной зоне, имеющей четкую систему санкционированного выпуска и возврата на хранение.
А.1.2 Сочетаемые печатные формы
Когда требуется больше одной печатной формы, должна существовать документированная система, обеспечивающая использование всех форм в комплекте. В том случае, когда комплект форм содержит типовой проект для нескольких работ, каждая отдельная форма в комплекте должна иметь четкое, однозначное обозначение и быть документирована.
А.1.3 Изменение текста/проекта
Когда проект требует использования нескольких форм и некоторые из них должны быть заменены из-за изменения в тексте/проекте, должна существовать документированная процедура, обеспечивающая замену затронутых форм и сохранение других средств в комплекте. Исходные формы подлежат повторной идентификации.
А.1.4 Верификация
А.1.4.1 Общие положения
Верификация проекта посредством получения печатных оттисков должна проводиться в период подготовки к работе печатного станка и до получения разрешения на выпуск продукции.
А.1.4.2 Карантин и уничтожение
Организация должна иметь:
a) документированную процедуру, обеспечивающую надлежаще оформленную изоляцию средств подготовки печатного оригинала и печатных материалов проекта, подвергающегося пересмотру;
b) документированную систему, детально описывающую методику утилизации ненужных средств подготовки печатного оригинала и получения печатных оттисков; они должны приводиться в непригодное состояние и утилизироваться контролируемым и надежным способом.
А.2 Процессы печати и преобразования
А.2.1 Наладка (подготовка к работе) печатного станка
А.2.1.1 Первоначальную предпечатную подготовку следует проводить с использованием чистых компонентов или материалов.
А.2.1.2 При подготовке к последующим процессам можно использовать материал из изначального процесса печати той же серии.
А.2.1.3 Изначальный приправочный материал может повторно использоваться в ходе подготовительного процесса для получения правильного цвета.
А.2.1.4 Материал, используемый для приправки, должен быть изолирован, а затем утилизирован как производственные отходы.
А.2.2 Системы переналадки
Системы переналадки, предназначенные для сокращения времени, затрачиваемого на подготовку (например, автоматизированная смена формы), и не позволяющие проводить полную очистку линий, подлежат документированной оценке рисков и должны использовать средства управления для обеспечения защиты продукции. Все печатные издания, полученные в ходе предыдущей операции, должны удаляться с линии до получения официального разрешения на выпуск тиража. Все средства управления должны регистрироваться (см. 4.2.4).
А.2.3 Сохраняемые образцы
А.2.3.1 Все сохраняемые печатные образцы должны быть четко обозначены и должны подлежать надежному хранению.
А.2.3.2 Образцы, используемые для иных целей (например, администрирование/продажа), должны уничтожаться, если они более не контролируются организацией.
А.2.4 Запасные печатные средства
А.2.4.1 В ходе производства, если запасные формы изготавливают на основе существующих фиксированных утвержденных исходных материалов (например, негативов или пошаговых снимков с использованием технологии "компьютер - печатная форма"), операция может быть продолжена после проведения новой проверки первой партии.
А.2.4.2 В ходе производства, если формы создают на основе новых материалов, операция должна быть отменена; заменяемые формы необходимо рассматривать как новые оригиналы. Последующую продукцию необходимо рассматривать как новую серию. Введение всех заменяемых печатных средств необходимо регистрировать (см. 4.2.4).
А.2.5 Печать комплектов
Печать комплектов (процесс печати более одного макета на подложке в ходе одного производственного цикла) признается и относится к риску, связанному с высокой вероятностью контаминации. Соответственно, подобная печать должна быть разрешена исключительно после согласования с потребителем и после завершения документированной оценки риска, предназначенной для определения степени и возможности уменьшить риск перекрестной контаминации (см. 7.2.3).
А.2.6 Серийное производство и хранение запасов
А.2.6.1 Серийное производство и хранение продукции на складе осуществляется только в том случае, если это оговорено контрактом.
А.2.6.2 Организация должна управлять хранением для обеспечения безопасности и целостности продукции и сохранять ее прослеживаемость до производства и используемых материалов.
А.2.7 Цифровая печать
А.2.7.1 Гибкие возможности цифровой печати являются источником новых видов деятельности, которыми необходимо управлять и документировать их для обеспечения точности и защищенности печатной продукции.
А.2.7.2 Применение цифровой печати и других специальных требований к продукции должно быть согласовано с потребителем.
А.2.7.3 Организация должна разработать надежную систему доступа к файлам, предназначенную для предотвращения непреднамеренного использования несоответствующих исходных файлов.
А.2.7.4 В случае, если альтернативная система безопасности не разработана, контролирующий компьютер, связанный с цифровой печатной машиной, должен содержать в своей памяти только один исходный файл для текущего производственного цикла, и удаление этого файла должно быть частью документированной очистки линии.
А.2.7.5 Рабочие параметры для достижения приемлемого цвета должны быть установлены в ходе официального процесса и зарегистрированы (см. 4.2.4).
А.2.7.6 Для рулонной печати, осуществляемой на непрерывной основе, должен существовать подходящий метод обеспечения разделения продукции; последующие процессы должны верифицироваться с целью обеспечения корректного разделения продукции и удаления разделителя.
Пример - Определенная перенастройка или интервал между каждой печатью, например напечатанный замещающий текст или пустой материал, считывание и верификация кодов.
А.3 Система безопасности на основе штрихкодов
А.3.1 Общие положения
Для обеспечения безопасности продукции и предотвращения перекрестной контаминации штрихкоды безопасности могут быть включены в макет печатных упаковочных материалов с целью верификации организацией в процессе производства и (или) потребителем в процессе операции упаковывания.
Примечание - Наиболее распространены двухмерные матричные штрихкоды, система меток.
Организация может добавить собственные идентификационные коды в макет продукции, если это предусмотрено контрактом.
В том случае, если организация ответственна за определение штрихкода безопасности, каждый цвет макета должен быть включен в штрихкод. Для всех штрихкодов, если цвет несовместим с требованиями сканирующего оборудования, необходимо уведомить об этом потребителя.
А.3.2 Верификация методов/оборудования
А.3.2.1 По возможности каждую единицу продукции со штрихкодом безопасности необходимо подвергать верификации посредством использования онлайн-сканирующего оборудования для обеспечения того, чтобы коды были читаемы и была произведена необходимая продукция. Сканирование штрихкода безопасности должно выполняться в течение последнего допустимого производственного процесса.
А.3.2.2 Конфигурация программного обеспечения/контроля сканирующего оборудования должна контролироваться для предотвращения несанкционированного доступа. Там, где это возможно, коды должны загружаться из независимого источника (например, спецификация или официальное подтверждение).
А.3.2.3 Должна быть разработана эффективная система забраковки любой продукции, не прошедшей процесс сканирования. Любая продукция, забракованная онлайн-сиcmемой сканирования, должна быть проверена для определения причины забраковки, а непригодные компоненты впоследствии должны быть утилизированы. Данные события должны быть зарегистрированы и проанализированы до выпуска продукции.
А.3.2.4 Сканирующее онлайн-оборудование и связанный с ним механизм забраковки должны подвергаться испытаниям в ходе производства для проверки того, эффективны ли они в обнаружении и удалении материалов с несоответствующими штрихкодами. Проведение такого мониторинга должно осуществляться в начале процесса через запланированные интервалы времени и быть зарегистрировано (см. 4.2.4).
А.3.2.5 Любая продукция, для производства которой было указано применение электронного сканирования, но фактически оно не осуществлялось, должна быть надлежащим образом одобрена и отражена в записях о качестве. Потребитель должен быть уведомлен об этом, и от него должно быть получено документальное одобрение до выпуска продукции.
А.3.2.6 Для рулонного многополосного производства все линии должны подлежать верификации при помощи штрихкода. Там, где это выполнить невозможно, при согласовании с потребителем только одна линия может подвергаться верификации.
А.3.2.7 Должно быть предусмотрено выполнение измерений вне технологического процесса и (или) верификации образцов штрихкодов на всех линиях.
А.3.3 Товарные штрихкоды
В том случае, если товарный штрихкод включен в макет (EAN, Code 39, PZN и т.д.), в течение всего процесса производства необходимо выполнять документированную проверку образца.
А.3.4 Рулонные материалы и продукция
А.3.4.1 Если иное не указано потребителем:
a) склейка должна быть произведена с использованием склеивающей ленты ярких цветов на обеих сторонах рулонного материала;
b) должны быть проверены обе стороны склейки для обеспечения того, что идентичные материалы соединены и совмещены с точной приводкой.
Может быть определено предельное количество склеек.
А.3.4.2 Количество материала (длина, вес или численность) для производства каждого рулона должно быть определено в точных пределах, согласованных с потребителем, и указано на рулоне.
А.3.4.3 Номер серии, номер рулона и дата производства должны быть указаны на внутренней поверхности втулки каждого рулона.
А.3.4.4 Для предотвращения перекрестной контаминации полотно должно быть выработано до появления чистого материала в конце производственного цикла, чтобы гарантировать отсутствие материала в печатной машине.
А.3.4.5 В том случае, если необходимо оставить печатный материал в оборудовании по преобразованию (конвертингу) в связи с трудностью восстановления целостности рулонного материала (например, устройства для продольной резки), должна быть разработана документированная процедура для удаления и утилизации материала, используемого для проведения нового макета через машину.
А.3.4.6 Если существует вероятность производства материала с пропусками печати по причине, связанной с проектом или работой печатного оборудования, организация должна иметь безопасную систему для обнаружения, удаления и отделения продукция*, произведенной с пропущенными цветом или текстом.
___________________
* Текст документа соответствует оригиналу. - .
Приложение B
(справочное)
Руководство по требованиям, предъявляемым к верификации, квалификации и валидации первичных упаковочных материалов
В.1 Общие положения
В настоящем информационном приложении описан подход при необходимости проведения верификации или квалификации и валидации. Руководство относится к требованиям по валидации, приведенным в 7.5.2, а также к требованиям по проектированию и разработке, приведенным в 7.3.
|
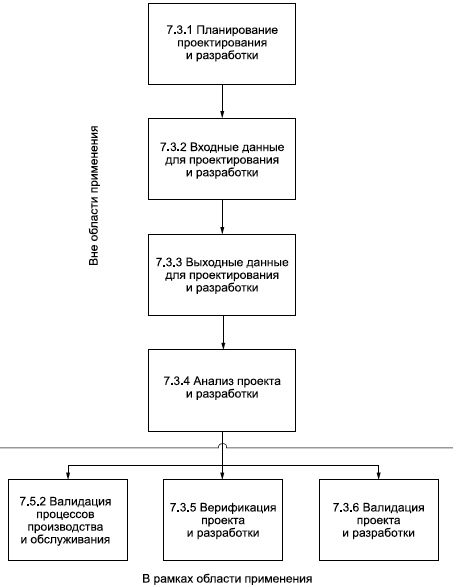
Рисунок В.1 - Область применения настоящего приложения
В тексте настоящего приложения будут упоминаться только верификация/квалификация оборудования и валидация процессов/продукции.
В том случае, если проектирование и разработка относятся к оборудованию, верификация дизайна и разработки равноценна верификации/квалификации оборудования.
В том случае, если проектирование и разработка относятся к процессам или продукции, валидация проектирования и разработки равноценна валидации процесса/продукции.
К объектам, для которых может потребоваться верификация/квалификация, относятся:
- оборудование, использующееся для производства первичных упаковочных материалов, вспомогательные и технические средства;
- испытательное оборудование, использующееся для определения пригодности исходных материалов, критических для качества продукции, промежуточной и готовой продукции;
К объектам, для которых может потребоваться валидация, относятся:
- процессы;
- индивидуальная продукция, если это необходимо или согласовано с потребителем.
Целью верификации/квалификации/валидации является подтверждение путем предоставления документальных свидетельств того, что предопределенные требования спецификаций стабильно выполняются.
В.2 Факторы, которые необходимо учитывать
B.2.1 Общие положения
Документированная оценка риска используется для определения того, какое оборудование и какие процессы подлежат верификации/квалификации/валидации. Валидация продукции является необязательной и проводится либо в соответствии с внутренним решением организации, или по запросу потребителя.
Верификация/квалификация оборудования и валидация процессов/продукции могут быть проведены независимо друг от друга, например валидация нового процесса/продукции не требует повторной верификации/квалификации существующего оборудования.
B.2.2 Факторы, которые необходимо учитывать до проведения верификации/квалификации/валидации
Необходимые условия верификации/квалификации оборудования перечислены ниже:
- одобренные/согласованные требования спецификаций;
- определение функций и согласованных обязанностей (организация и поставщик);
- определение критических параметров процесса;
- обучение GMP и верификации/квалификации.
Необходимые условия валидации процесса/продукции перечислены ниже:
- одобренные/согласованные спецификации на процесс/продукцию;
- верифицированное/квалифицированное оборудование;
- определение функций и согласованных обязанностей (организация и потребитель);
- определение критических параметров процесса;
- обучение (операторы, персонал, отвечающий за качество, инженеры и т.д.);
- стандартные операционные процедуры, детально описывающие процедуры работы (функционирования) и очистки оборудования.
B.2.3 Валидация программного обеспечения
Программное обеспечение может быть валидировано/верифицировано (см. 7.5.2.7 и 7.5.2.8) при помощи функциональных испытаний. Программное обеспечение, являющееся частью оборудования, может быть включено в верификацию/квалификацию оборудования.
Примечание - В руководстве по надлежащей практике автоматизированного производства (GAMP) [33] могут быть приведены указания по проведению функциональных испытаний.
В.3 Мастер-план валидации (VMP)
В.3.1 Общие положения
Мастер-план валидации (VMP) описывает работы по валидации (что в данном случае также включает в себя верификацию/квалификацию), а также порядок их выполнения, согласующийся с общим подходом к валидации. Рекомендуется регулярный (например, ежегодный) пересмотр плана.
План, как правило, включает следующие элементы:
- планирование валидации (см. В.3.2) и составление графика;
- организационную структуру работ по валидации - функции и обязанности (см. В.3.3);
- сводную информацию об оборудовании и процессах, критических для качества (см. В.3.4 и В.3.5);
- ссылки на существующую документацию (например, на существующий отчет по валидации), если применимо;
- ссылки на применимые процедуры (например, общий подход к валидации, формат документации).
Для крупных проектов (различные работы по верификации/квалификации/валидации объединены в один проект) целесообразной может являться разработка отдельных мастер-планов валидации.
В.3.2 Планирование валидации
B.3.2.1 Общие положения
Рекомендуемым подходом является перспективная валидация, однако в некоторых условиях приемлемыми являются сопутствующая или ретроспективная валидация (данное утверждение неприменимо в отношении процессов стерилизации, см. 7.5.2.11).
B.3.2.2 Перспективная валидация
Перспективная валидация осуществляется до начала промышленного производства.
B.3.2.3 Сопутствующая валидация
Сопутствующая валидация должна применяться только для квалификации технологических характеристик и валидации процессов.
Сопутствующая валидация состоит из работ, проводимых параллельно с выпуском промышленной продукции, когда промышленная продукция выпускается до завершения валидации. Сопутствующая валидация должна использовать принципы и процедуры, связанные с перспективной валидацией.
Валидация, осуществляемая в процессе периодического производства является отличительной чертой производства первичных упаковочных материалов. В некоторых случаях станки/линии используются для периодического производства. В подобных случаях сопутствующая валидация может быть прервана (также на более длительный срок) и продолжена с началом следующего периода.
B.3.2.4 Ретроспективная валидация
Ретроспективная валидация подразумевает, что промышленная продукция была выпущена до завершения процесса валидации.
Данный подход включает в себя получение документированного свидетельства того, что смонтированное и эксплуатируемое оборудование выпускает продукцию стабильного качества, путем проведения ретроспективного анализа данных, полученных по целому ряду функциональных параметров и сырьевых материалов. Такой анализ может распространяться на записи о техническом обслуживании и инженерные записи, записи о качестве и претензии потребителей.
B.3.2.5 Метод крайних вариантов/матричный метод
В общем случае может применяться метод крайних вариантов/матричный метод, основываясь на оценке риска. В том случае, если была доказана эквивалентность оборудования, метод крайних вариантов/матричный метод может быть использован для:
- уменьшения числа монтажных и функциональных испытаний в процессе верификации/квалификации оборудования;
- уменьшения числа серий для квалификации эксплуатации в процессе квалификации оборудования;
- уменьшения числа серий для валидации процесса в ходе валидации продукции или процесса;
- увеличения периода до повторной валидации отдельного оборудования и поочередного осуществления повторной валидации на эквивалентном оборудовании.
Нижеприведенная таблица демонстрирует указанный подход.
Таблица В.1 - Пример использования матричного метода
Тип валидации | Станок 1 | Станок 2 | Станок 3 |
Первичная валидация | Три серии | Одна серия | Одна серия |
Повторная валидация | Одна серия | Не проводится | Не проводится |
Повторная валидация | Не проводится | Одна серия | Не проводится |
Повторная валидация | Не проводится | Не проводится | Одна серия |
Повторная валидация | Одна серия | Не проводится | Не проводится |
В том случае, если несколько видов продукции производятся по одному и тому же технологическому процессу, для уменьшения количества серий для валидации может использоваться экспериментальный план, заключающийся в испытании только крайних вариантов, например продукции наибольшего и наименьшего размера. План подразумевает, что крайние варианты будут репрезентативны по отношению ко всем образцам, находящимся в диапазоне между ними. В случае возможности определения "наихудшего случая" достаточным является испытание только данного варианта вместо крайних вариантов.
В любом случае должно иметься достаточно знаний о процессе и продукции, на которых будет основываться мотивация принятия решения.
Должны быть задокументированы эквивалентность оборудования, техническое обоснование и доводы в пользу принятия того или иного решения.
B.3.3 Функции и обязанности
Детализированная информация о функциях и обязанностях (с указанием лиц, ответственных за осуществление этапов валидации и одобрение результатов) должна являться частью соответствующей документации по проведению валидации и должна быть обозначена до начала работы, за исключением случаев, когда данная информация приводится в других документах или процедурах.
B.3.4 Оборудование, критическое для качества
Оценка риска является неотъемлемой частью процесса принятия решений и может быть использована для определения необходимости проведения верификации или квалификации оборудования. Таким образом, все оборудование должно быть оценено на предмет потенциального влияния на качество продукции.
Оборудование является критическим для качества в том случае, если на один из нижеперечисленных вопросов дается положительный ответ.
- Повлияет ли отказ оборудования непосредственно на качество продукции, как это определено для типа продукции, которая на нем производится?
- Используется ли оборудование для гарантирования стерильности продукции?
- Используется ли оборудование для управления или измерения технологических этапов или параметров, критических для качества?
- Получают ли с помощью оборудования данные/записи для приемки или забраковки?
- Находится ли оборудование в непосредственном контакте с продукцией?
- Используется ли оборудование для предотвращения или устранения контаминации или для очистки?
В том случае, если оборудование считается критическим, должна быть проведена верификация или квалификация, и оборудование включается в мастер-план валидации.
В том случае, если оборудование не считается критическим, применяются принципы надлежащей инженерной практики, и оно не включается в мастер-план валидации.
B.3.5 Процессы, критические для качества
Валидация процессов производится в том случае, когда результат процесса невозможно верифицировать при помощи последующего мониторинга или измерения. Это также относится к процессам, недостатки которых могут быть выявлены только после начала использования продукции (см. 7.5.2.1).
Нижеприведенное дерево принятия решений может быть использовано для определения того, является ли процесс критическим или нет, на основании риска, что будет выпущена продукция, не соответствующая установленным заранее требованиям спецификации.
|
Рисунок В.2 - Дерево принятия решений для валидации процессов/процессов, критических для качества
В.4 Выполнение верификации/квалификации/валидации
В.4.1 Этапы верификации или квалификации
В.4.1.1 Общие положения
Верификация или квалификация должны основываться на оценке риска.
Верификация/квалификация оборудования состоит из следующих этапов: оценка риска, установление требований спецификации и верификация/квалификация.
Решение о виде необходимой документации должно приниматься в начале процесса и документироваться, за исключением случаев, когда одно приведено в описании процедур валидации.
|
DS - спецификация проекта;
FAT - заводские приемочные испытания;
FDS - функциональная спецификация проекта;
FS - функциональная спецификация;
SAT - приемочные испытания на месте;
URS - спецификация требований пользователя.
Рисунок В.3 - Этапы верификации/квалификации
В.4.1.2 Оценка рисков
В основе спецификации и процесса верификации/квалификации должна лежать оценка риска. Оценка риска также должна надлежащим образом применяться на каждом этапе, документация должна основываться на риске для качества продукции.
B.4.1.3 Спецификация требований
B.4.1.3.1 Общие положения
Спецификации требований могут быть объединены в один или более спецификационный документ в зависимости от степени критичности оборудования (как стандартного, так и произведенного на заказ) или в зависимости от ответственности (организации или поставщика). Каждая спецификация должна содержать четко определенные, конкретные, проверяемые и достижимые критерии приемлемости.
B.4.1.3.2 Спецификация требований пользователя
В 3.7.4 спецификации требований пользователя определены как утвержденный документ, в котором указаны спецификации продукции, производящейся из материала на данном оборудовании, а также функциональные, операционные и (или) технические аспекты оборудования или процесса, требуемые для производства желаемой продукции. Спецификации требований пользователя могут включать в себя рекомендации по расположению оборудования с предусмотрением достаточного места для перемещения материалов и персонала, доступность запасных частей и удобство уборки и очистки линии. В качестве альтернативы подобная информация может быть предоставлена в другой документации.
B.4.1.3.3 Спецификация оборудования
Важно, чтобы все критические аспекты были включены в спецификацию.
Критическими аспектами производственных систем обычно являются функции, свойства, возможности и эксплуатационные характеристики или характеристики, необходимые для производственного процесса и всей системы для обеспечения стабильного качества продукции.
B.4.1.4 Верификация/квалификация оборудования
Для верификации того, что все оборудование пригодно для предполагаемого применения, было надлежащим образом смонтировано и функционирует корректно, в соответствии со спецификациями требований, должен использоваться системный подход. Степень верификации/квалификации и детализация документации должны основываться на рисках, включая риски, связанные с качеством продукции, а также сложностью и новизной оборудования.
Верификация/квалификация может быть разделена на два этапа: квалификация монтажа и квалификация эксплуатации, однако данные этапы могут быть объединены в один, как это обычно бывает при испытании у поставщика и при испытании на производственной площадке.
Разрешается использование имеющихся данных, например, данные, полученные в ходе испытания у поставщика, не обязательно должны быть воспроизведены на производственной площадке, если только транспортирование оборудования не влияет на испытания.
Монтажные испытания должны включать в себя верификацию того, что оборудование смонтировано в соответствии со спецификациями, откалибровано (если применимо).
Эксплуатационные испытания должны включать в себя верификацию надлежащего функционирования оборудования при верхних и нижних границах требуемого эксплуатационного предела.
B.4.1.5 Квалификация эксплуатации
При проведении квалификации эксплуатации производственные материалы используются для верификации того, что оборудование является надежным, а первичный упаковочный материал может стабильно производиться в обычных условиях эксплуатации. Квалификация эксплуатации оценивает общую работоспособность линии для обеспечения того, что она может стабильно производить продукцию, соответствующего, установленного стандартом качества. Процесс проведения испытания и результаты производства подходящего числа последовательно произведенных серий (обычно трех) официально документируют и утверждают.
На основании управления риском (например, оборудование является стандартным и все спецификации продукции были испытаны на каждой серии до выпуска) квалификация эксплуатации может быть пропущена или объединена с валидацией процесса или продукции.
Для использования трех последовательных серий продукции может быть сделано исключение в том случае, если производственный процесс является длительным, например, когда одна серия материала может потребовать нескольких недель непрерывного производства. В подобном случае квалификация эксплуатации может быть проведена на трех подсериях, производство каждой из которых осуществляется на протяжении не менее одного дня.
В.4.2 Валидация процессов и продукции
В.4.2.1 Валидация процессов
Валидация процессов состоит из определенного числа последовательных серий (обычно трех), произведенных в нормальных условиях, однако с большим числом отбора проб и дополнительными испытаниями по сравнению с обычным производством. Валидация процессов может быть объединена с квалификацией эксплуатации. В случае длительного процесса производства для серий, используемых при валидации процессов, действует то же исключение (см. В.4.1.5).
Валидация процессов должна быть основана на надежных знаниях о процессах и их вариациях/изменчивости. Валидация процесса не зависит от продукции. Может быть использован метод крайних вариантов/матричный метод.
В.4.2.2 Валидация продукции
В целом согласуется с валидацией процессов, однако могут быть учтены конкретные требования потребителя.
В.5 Документация для верификации, квалификации и валидации
В.5.1 Общие положения
Все документы должны быть проанализированы и утверждены до начала верификации/квалификации/валидации/выпуска.
Ответственность за любое решение о выпуске должна лежать на службе качества. Любой пересмотр документации должен отслеживаться при помощи управления версиями.
|
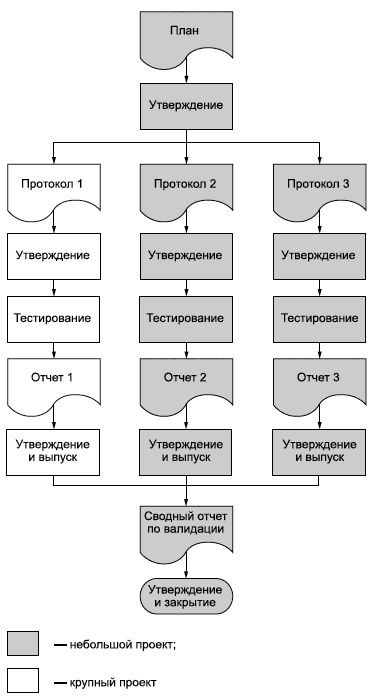
Рисунок В.4 - Пример структуры документации
B.5.2 План (факультативно)
Для уточнения действий, предпринимаемых на различных этапах верификации/квалификации/валидации проекта, может быть разработан один или несколько планов, за исключением случая, когда данные действия описаны в процедуре валидации.
В план могут быть включены следующие позиции:
- описание проекта/область применения;
- функции и обязанности;
- планирование;
- анализ риска;
- суммарные критерии приемлемости;
- валидационный подход;
- документы, которые будут созданы;
- ссылки на процедуры и управление изменениями;
- определение условий статуса карантина для серий, используемых в квалификации эксплуатации или валидации.
План и протокол могут быть объединены в единый документ.
B.5.3 Протокол
Должен быть разработан и утвержден письменный протокол, в котором указывается, как будет проводиться верификация/квалификация/валидация.
В протокол могут быть включены следующие позиции:
- описание проекта/область применения;
- функции и обязанности;
- обоснование выбранного подхода;
- испытания, которые будут проведены, с указанием методов и условий;
- детализированные критерии приемлемости для каждого испытания;
- план отбора проб;
- критические параметры процесса;
- ссылки на процедуры, управление изменениями и спецификации требований;
- необходимые условия.
B.5.4 Отчет
Должен быть подготовлен отчет, ссылающийся на протокол верификации/квалификации/валидации.
В отчет могут быть включены следующие позиции:
- сводка результатов испытаний;
- исходные данные;
- выявленные отклонения и корректирующие действия, которые были или будут предприняты;
- выводы;
- изменения в плане, как это определено в протоколе, с надлежащим обоснованием.
После удовлетворительного завершения испытаний должен быть осуществлен официальный переход на следующий этап верификации/квалификации/валидации, оформленный в виде письменного одобрения.
B.5.5 Сводный отчет по валидации (факультативно)
Сводный отчет о валидации готовится после завершения всех этапов верификации/квалификации/валидации и завершает проект.
Сводный отчет о валидации может включать в себя следующие позиции:
- ссылки на протоколы и отчеты;
- изменения в планах и обоснования любых изменений;
- официальное одобрение и завершение проекта.
B.5.6 Архивирование
Записи о верификации, или квалификации и валидации, должны сохраняться согласно 4.2.4.7.
В.6 Поддержание валидации
B.6.1 Общие положения
Должна поддерживаться верификация/квалификация оборудования и валидация процессов/продукции. Любые изменения должны быть оценены с точки зрения риска их влияния на статус валидации. При необходимости следует провести надлежащую верификацию/квалификацию/валидацию или повторную квалификацию/валидацию.
Изменения/улучшения валидационных процедур также должны оцениваться с точки зрения риска для установления необходимости повторной валидации.
B.6.2 Управление изменениями
Для управления изменениями в процессе верификации/квалификации/валидации должны быть внедрены конкретные средства управления. Официальное, документированное управление изменениями должно быть реализовано после выпуска оборудования/процесса/продукции и сохраняться в течение всего жизненного цикла оборудования/процесса/продукции.
B.6.3 Последующие мероприятия
По окончании верификации/квалификации/валидации необходимо предпринять надлежащие действия для устранения любых нарушений, например недостатков.
Все действия должны быть задокументированы.
B.6.4 Техническая профилактика
Для гарантирования надлежащего функционирования оборудования после его выпуска должен быть разработан и утвержден план технической профилактики (включающий калибровку).
B.6.5 Повторная валидация/квалификация
Оборудование не требует повторной квалификации, а процесс - повторной валидации до тех пор, пока они функционируют в управляемом режиме и в них или в результирующую продукцию не вносятся изменения. Определение того, что оборудование/процесс функционируют в управляемом режиме, осуществляется посредством анализа данных об управлении процессом, получаемых ежедневно, и анализа любых данных об испытании продукции на соответствие спецификациям и возможным изменениям.
В случае внесения изменений или возникновения отклонений, оборудование/процесс должны быть проанализированы и оценены. При необходимости проводится повторная валидация/квалификация.
Периодическая квалификация/валидация может потребоваться, например, для чистых помещений, процессов стерилизации.
Приложение C
(справочное)
Взаимосвязь между пунктами настоящего стандарта и структурами более высокого уровня
В настоящем приложении описана взаимосвязь между пунктами/подпунктами настоящего стандарта и пунктами/подпунктами, приведенным в структурах более высокого уровня, используемых в стандартах систем менеджмента, согласно директивам ИСО/IEC, часть 1, приложение SL, дополнение 2. Оно предназначено для связи структуры стандарта ИСО 15378 со структурой более высокого уровня, приведенной в директиве ИСО/IEC, часть 1, после того, как завершится пересмотр ИСО 9001, лежащего в основе настоящего стандарта.
Таблица С.1 - Взаимосвязь между настоящим стандартом и структурами более высокого уровня для использования в стандартах системы менеджмента (см. директивы ИСО/IEC, часть 1, приложение SL, дополнение 2) (1 из 4)
Пункт/подпункт настоящего стандарта | Пункты/подпункты структуры более высокого уровня для использования в стандартах системы менеджмента согласно директивам ИСО/IEC, часть 1, приложение SL, дополнение 2 | Примечания |
1 Область применения | 1 Область применения | |
2 Нормативные ссылки | 2 Нормативные ссылки | |
3 Термины и определения | 3 Термины и определения | |
5.2 Ориентация на потребителя | 4 Среда организации | |
5.6 Анализ со стороны руководства | 4.1 Понимание организации и ее среды | |
5.2 Ориентация на потребителя | 4 Среда организации | |
4.2.2 Руководство по качеству | 4 Среда организации | |
4 Система менеджмента качества | 4 Среда организации | |
5 Ответственность руководства | 5 Лидерство | |
5.1 Обязательства руководства | 5.1 Лидерство и приверженность | |
5.3 Политика в области качества | 5 Лидерство | |
5.5 Ответственность, полномочия и обмен информацией | 5 Лидерство | |
5.4.2 Планирование системы менеджмента качества | 6 Планирование | |
5.4 Планирование | 6 Планирование | |
5.4.1 Цели в области качества | 6.1 Цели в области качества и планирование их достижения | |
6.1 Обеспечение ресурсами | 7 Средства обеспечения | |
6.3 Инфраструктура | 7.1 Ресурсы | |
6.2 Персонал | 7 Средства обеспечения | |
6.2.2 d) | 7 Средства обеспечения | |
5.5.1 Ответственность и полномочия | 7 Средства обеспечения | |
5.5.3 Внутренний обмен информацией | 7.4 Обмен информацией | |
4.2 Требования, предъявляемые к документации | 7 Средства обеспечения | |
7 Процессы жизненного цикла продукции | 8 Деятельность на стадиях жизненного цикла продукции и услуг | |
7.1 Планирование процессов жизненного цикла продукции | 8.1 Планирование и управление деятельностью на стадиях жизненного цикла продукции и услуг | |
8.1 Общие положения | 9 Оценка результатов деятельности | |
8.2 Мониторинг и измерение | 9.1 Мониторинг, измерение, анализ и оценка | |
8.2.2 Внутренний аудит | 9 Оценка результатов деятельности | |
5.6 Анализ со стороны руководства | 9 Оценка результатов деятельности | |
8.5 Улучшение | 10 Улучшение | |
8.3 Управление несоответствующей продукцией | 10.1 Несоответствия и корректирующие действия | |
8.5 Улучшение | 10 Улучшение | |
8.5.1 Постоянное улучшение | 10.2 Постоянное улучшение |
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего межгосударственного стандарта |
ISO 14698-1 | IDT | ГОСТ ИСО 14698-1-2005 "Чистые помещения и связанные с ними контролируемые среды. Контроль биозагрязнений. Часть 1. Общие принципы и методы" |
ISO 14698-2 | IDT | ГОСТ ИСО 14698-2-2005 "Чистые помещения и связанные с ними контролируемые среды. Контроль биозагрязнений. Часть 2. Анализ данных о биозагрязнениях" |
Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: | ||
Библиография
[1] | ISO 9001 | Quality management systems - Requirements | |
________________
| |||
[2] | ISO 9004 | Managing for the sustained success of an organization - A quality management approach | |
[3] | ISO 10007 | Quality management systems - Guidelines for configuration management | |
[4] | ISO 10012 | Measurement management systems - Requirements for measurement processes and measuring equipment | |
[5] | ISO 11135 | Sterilization of health-care products - Ethylene oxide - Requirements for the development, validation and routine control of a sterilization process for medical devices | |
[6] | ISO 11137-1 | Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices | |
[7] | ISO 11137-2 | Sterilization of health care products - Radiation - Part 2: Establishing the sterilization dose | |
[8] | ISO/TS 11139 | Sterilization of health care products - Vocabulary | |
___________________ | |||
[9] | ISO 13485 | Medical devices - Quality management systems - Requirements for regulatory purposes | |
[10] | ISO 14001:2004 | Environmental management systems - Requirements with guidance for use | |
___________________ | |||
[11] | ISO 14644-1 | Cleanrooms and associated controlled environments - Part 1: Classification of air cleanliness by particle concentration | |
[12] | ISO 14644-2 | Cleanrooms and associated controlled environments - Part 2: Specifications for monitoring and periodic testing to prove continued compliance with ISO 14644-1 | |
[13] | ISO 14644-3 | Cleanrooms and associated controlled environments - Part 3: Test methods | |
[14] | ISO 14644-4 | Cleanrooms and associated controlled environments - Part 4: Design | |
[15] | ISO 14644-5 | Cleanrooms and associated controlled environments - Part 5: Operations construction and start-up | |
[16] | ISO 14644-7 | Cleanrooms and associated controlled environments - Part 7: Separative devices (clean air hoods, gloveboxes, ISOIators and mini-environments) | |
[17] | ISO 14644-8 | Cleanrooms and associated controlled environments - Part 8: Classification of air cleanliness by chemical concentration (ACC) | |
[18] | ИСО 14937:2009 | Sterilization of health care products - General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process for medical devices | |
[19] | ISO 14971 | Medical devices - Application of risk management to medical devices | |
[20] | ISO 19011 | Guidelines for auditing management systems | |
[21] | ISO 31000 | Risk management - Principles and guidelines | |
[22] | IEC 31010 | Risk management - Risk assessment techniques | |
[23] | ISO/IEC 90003 | Software engineering - Guidelines for the application of ISO 9001:2008 to computer software Under revision | |
___________________ | |||
[24] | ISO/IEC Guide 2 | Standardization and related activities - General vocabulary | |
[25] | ISO Guide 73 | Risk management - Vocabulary | |
[26] | ISO/IEC | Internation al* vocabulary of metrology - Basic and general concepts and associated terms (VIM) | |
________________ * Текст документа соответствует оригиналу. - . | |||
[27] | IEC 60601-1 | Medical electrical equipment - Part 1: General requirements for basic safety and essential performance | |
[28] | IEC 60812 | Analysis techniques for system reliability - Procedure for failure mode and effects analysis (FMEA) | |
[29] | IEC 61025 | Fault tree analysis (FTA) | |
[30] | IEC 61882 | Hazard and operability studies (HAZOP studies) - Application guide | |
[31] | EU Guidelines to Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, http:/ ec.europa.eu/health/documents | ||
[32] | US/FDA Code of Federal Regulations. Title 21, Part 11, www.fda.gov | ||
[33] | GAMP 5, A Risk-Based Approach to Compliant GxP Computerized Systems, http://www.ispe.org | ||
[34] | PIC/S Guide PE 008-3, Explanatory Notes for Industry on the Preparation of a Site Master File, http://www.picscheme.org/pics.php | ||
[35] | ICH Q9. Quality risk management, http://www.ich.org | ||
[36] | ICH Q10. Pharmaceutical quality system, http://www.ich.org | ||
[37] | EU Guidelines to Good Manufacturing Practice for Medical Products for Human and Veterinary Use. Volume 4, Annex 15 Qualification and Validation and Annex 18 Good Manufacturing Practice for Active Pharmaceutical Ingredients Requirements, http://ec.europa.eu/health/documents/ | ||
[38] | The Pharmaceutical Quality Group. http://www.pqg.org/pharma/ | ||
[39] | ASTM E 2500, Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment | ||
[40] | Приказ Минпромторга России от 14 июня 2013 г. N 916 "Об утверждении правил надлежащей производственной практики" | ||
УДК 658.562.014:006.354 | ОКС 03.120.10 |
Ключевые слова: первичные упаковочные материалы, лекарственные средства, система менеджмента качества, ответственность руководства, менеджмент рисков, процессы жизненного цикла продукции | |
Электронный текст документа
и сверен по:
, 2020

{kind=link}