ГОСТ Р ИСО 15378-2014
Группа Т59
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
МАТЕРИАЛЫ ДЛЯ ПЕРВИЧНОЙ УПАКОВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Особые требования по применению ИСО 9001:2008 с учетом правил надлежащей производственной практики (GMP)
Primary packaging materials for medicinal products. Particular requirements for the application of ISO 9001:2008, with reference to good manufacturing practice (GMP)
ОКС 03.120.10
11.040.01
ОКСТУ 0025
Дата введения 2015-06-01
Предисловие
1 ПОДГОТОВЛЕН Открытым акционерным обществом "Всероссийский научно-исследовательский институт сертификации" (ОАО "ВНИИС") на основе собственного аутентичного перевода на русский язык международного стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 076 "Системы менеджмента"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 1 августа 2014 г. N 839-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 15378:2011* "Материалы для первичной упаковки лекарственных средств. Особые требования по применению ИСО 9001:2008 с учетом правил надлежащей производственной практики (GMP)" (ISO 15378:2011 "Primary packaging materials for medicinal products - Particular requirements for the application of ISO 9001:2008, with reference to Good Manufacturing Practice (GMP)").
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации и межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомления и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (gost.ru)
Введение
0.1 Общие положения
Настоящий стандарт идентифицирует принципы GMP и устанавливает требования к системе менеджмента качества, применимой к первичным упаковочным материалам для лекарственных средств. Реализация принципов GMP при производстве и контролировании первичных упаковочных материалов в рамках организации имеет чрезвычайно важное значение для обеспечения безопасности пациентов, пользующихся лекарственными средствами из-за непосредственного контакта с продукцией. Применение GMP в отношении упаковочных материалов для фармацевтической продукции помогает обеспечить выполнение этими материалами нужд и потребностей фармацевтической промышленности.
Настоящий стандарт является стандартом по применению первичных упаковочных материалов и содержит нормативный текст стандарта ИСО 9001:2008.
В отношении построения настоящего стандарта действуют следующие правила:
- разделы или подразделы, цитируемые полностью и без изменений из ИСО 9001:2008, заключены в рамку;
- текст, выделенный курсивом*, содержит дополнительную информацию по GMP, касающуюся первичных упаковочных материалов.
________________
* В бумажном оригинале обозначения и номера стандартов и нормативных документов в разделе "Предисловие" приводятся обычным шрифтом, остальные по тексту документа выделены курсивом. - .
Термины и определения по GMP содержатся в разделе 3. При включении в перечень источник приводится в скобках.
Для создания системы менеджмента качества необходимо стратегическое решение организации. На разработку и внедрение системы менеджмента качества организации влияют: a) ее внешняя среда, изменения или риски, связанные с этой средой; b) изменяющиеся потребности; c) конкретные цели; d) выпускаемая продукция; e) применяемые процессы; f) размер и структура организации. |
Настоящий стандарт не предполагает единообразия в структуре систем менеджмента качества или их документации. Требования к системе менеджмента качества, установленные настоящим стандартом, являются дополняющими по отношению к требованиям к продукции. Информация, обозначенная как "Примечание", носит характер методических указаний для понимания или разъяснения соответствующего требования. Настоящий стандарт может использоваться внутренними и внешними сторонами, включая органы по сертификации, в целях оценки способности организации выполнять требования потребителей, требования к продукции, являющиеся обязательными к исполнению в соответствии с действующим законодательством (далее - обязательные требования), и собственные требования. При разработке настоящего стандарта были учтены принципы менеджмента качества, установленные ИСО 9000 и ИСО 9004. [ИСО 9001:2008] |
Основной задачей настоящего стандарта является определение гармонизированных требований к первичным упаковочным материалам. Он содержит некоторые частные требования к первичным упаковочным материалам, заимствованные из правил GMP и касающиеся управления производством и т.п. лекарственных препаратов.
0.2 Процессный подход
Настоящий стандарт направлен на применение "процессного подхода" при разработке, внедрении и улучшении результативности системы менеджмента качества в целях повышения удовлетворенности потребителей путем выполнения их требований. Для успешного функционирования организация должна определить и осуществлять менеджмент многочисленных взаимосвязанных видов деятельности. Деятельность, использующая ресурсы и управляемая в целях преобразования входов в выходы, может рассматриваться как процесс. Часто выход одного процесса образует непосредственно вход следующего. Применение в организации системы процессов наряду с их идентификацией и взаимодействием, а также менеджмент процессов, направленный на получение желаемого результата, могут быть определены как "процессный подход". Преимущество процессного подхода состоит в непрерывности управления, которое он обеспечивает на стыке отдельных процессов в рамках их системы, а также при их комбинации и взаимодействии. |
При применении в системе менеджмента качества такой подход подчеркивает важность: a) понимания и выполнения требований; b) необходимости рассмотрения процессов с точки зрения добавляемой ими ценности; c) достижения запланированных результатов выполнения процессов и обеспечения их результативности; d) постоянного улучшения процессов, основанного на объективном измерении. Приведенная на рисунке 1 модель системы менеджмента качества, основанной на процессном подходе, иллюстрирует связи между процессами, представленными в разделах 4-8. Эта модель показывает, что потребители играют существенную роль в установлении требований, рассматриваемых в качестве входов. Мониторинг удовлетворенности потребителей требует оценки информации о восприятии потребителями выполнения их требований. Приведенная на рисунке 1 модель охватывает все основные требования настоящего стандарта, но не показывает процессы на детальном уровне. |
Примечание - Кроме того, ко всем процессам может быть применен цикл "Plan - Do - Check - Act" (PDCA). Цикл PDCA можно кратко описать так: - планирование (plan) - разработка целей и процессов, необходимых для достижения результатов в соответствии с требованиями потребителей и политикой организации; - осуществление (do) - внедрение процессов; - проверка (check) - постоянные контроль и измерение процессов и продукции в сравнении с политикой, целями и требованиями на продукцию и сообщение о результатах; - действие (act) - принятие действий по постоянному улучшению показателей процессов. |
[ИСО 9001:2008] |

0.3 Связь с ИСО 9004
ИСО 9001 и ИСО 9004 являются стандартами на системы менеджмента качества, которые дополняют друг друга, но их можно применять также независимо. ИСО 9001 устанавливает требования к системе менеджмента качества, которые могут быть использованы для внутреннего применения организациями, а также в целях сертификации или заключения контрактов. Стандарт направлен на результативность системы менеджмента качества при выполнении требований потребителей и соответствующих законодательных и других обязательных требований. Ко времени публикации стандарта ИСО 9001:2008 стандарт ИСО 9004 находился на стадии пересмотра. Новая версия ИСО 9004 будет содержать рекомендации для менеджмента по достижению устойчивого успеха любой организации в сложной, требовательной и постоянно изменяющейся среде. ИСО 9004 представляет более широкий взгляд на менеджмент качества, чем ИСО 9001; он нацеливает на удовлетворение потребностей и ожиданий всех заинтересованных сторон на основе систематического и постоянного улучшения деятельности организации. Однако этот стандарт не предназначен для целей сертификации, заключения контрактов и выполнения обязательных требований. [ИСО 9001:2008] |
0.4 Совместимость с другими системами менеджмента
При разработке настоящего стандарта должное внимание было уделено положениям ИСО 14001:2004 для улучшения совместимости этих двух стандартов в интересах сообщества пользователей. Приложение А показывает соответствие между ИСО 9001:2008 и ИСО 14001:2004. Настоящий стандарт не содержит конкретных требований к другим системам менеджмента, таким как менеджмент охраны окружающей среды, менеджмент профессионального здоровья и безопасности, финансовый менеджмент или менеджмент рисков. Однако стандарт позволяет организации согласовать или интегрировать свою собственную систему менеджмента качества с другими системами менеджмента с соответствующими требованиями. Организация может адаптировать действующую(ие) систему(ы) менеджмента для создания системы менеджмента качества, соответствующей требованиям настоящего стандарта. [ИСО 9001:2008] |
Настоящий стандарт содержит требования ИСО 9001:2008 и дополнительно частные требования к первичным упаковочным материалам, которые позаимствованы из правил GMP и адаптированы, если это целесообразно, к производству и контролю лекарственных средств.
1 Область применения
1.1 Общие положения
Настоящий стандарт устанавливает требования к системе менеджмента качества, когда организации требуется продемонстрировать свою способность поставлять первичные упаковочные материалы для лекарственных средств, систематически отвечающие требованиям потребителей, в том числе нормативным требованиям и требованиям международных стандартов, касающихся первичных упаковочных материалов.
В настоящем стандарте термин "если это целесообразно" используется неоднократно. Когда требование квалифицируется этой фразой, оно считается "целесообразным", если организация не предоставит документированного доказательства противного.
Настоящий стандарт устанавливает требования к системе менеджмента качества в тех случаях, когда организация: a) нуждается в демонстрации своей способности всегда поставлять продукцию, отвечающую требованиям потребителей и соответствующим обязательным требованиям; b) ставит своей целью повышение удовлетворенности потребителей посредством эффективного применения системы менеджмента качества, включая процессы постоянного ее улучшения, и обеспечение соответствия требованиям потребителей и соответствующим обязательным требованиям. |
Примечания 1 В настоящем стандарте термин "продукция" применим только: a) к предназначаемой для потребителя или затребованной им продукции; b) к любым заданным результатам процессов жизненного цикла. 2 Законодательные и другие обязательные требования могут быть выражены как правовые требования. [ИСО 9001:2008] |
Настоящий стандарт применяется при проектировании, изготовлении и поставке первичных упаковочных материалов для лекарственных средств. Он также применим в целях сертификации.
1.2 Применение
Требования настоящего стандарта являются общими и предназначены для применения всеми организациями независимо от их вида, размера и поставляемой продукции. Если какое-либо требование(я) настоящего стандарта нельзя применить вследствие специфики организации и ее продукции, допускается его исключение. При допущенных исключениях заявления о соответствии настоящему стандарту приемлемы, если эти исключения подпадают под требования раздела 7 и не влияют на способность или ответственность организации обеспечивать продукцией, соответствующей требованиям потребителей и соответствующим обязательным требованиям. [ИСО 9001:2008] |
2 Нормативные ссылки
Указанные в данном разделе документы* являются необходимыми для применения настоящего стандарта. Для датированных ссылок применяют только указанные версии издания. Для недатированных ссылок применяют самые последние издания (включая любые изменения к стандартам).
_______________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
Указанный ниже ссылочный документ необходим для использования настоящего стандарта. Для датированных ссылок применяют только ту версию, которая была упомянута в тексте. Для недатированных ссылок необходимо использовать самое последнее издание документа (включая любые поправки). ИСО 9000:2005 Системы менеджмента качества. Основные положения и словарь. [ИСО 9001:2008] |
ИСО 14644-1 Чистые помещения и связанные с ними контролируемые среды. Часть 1: Классификация чистоты воздуха по концентрации взвешенных частиц
ИСО 14644-2 Чистые помещения и связанные с ними контролируемые среды. Часть 2: Требования к мониторингу и периодическим испытаниям для подтверждения постоянного соответствия ИСО 14644-1
ИСО 14644-3 Чистые помещения и связанные с ними контролируемые среды. Часть 3: Методы испытаний
ИСО 14644-5 Чистые помещения и связанные с ними контролируемые среды. Часть 5: Эксплуатация
3 Термины и определения
В настоящем стандарте применены термины и определения, данные в ИСО 9000.
В настоящем стандарте применены термины и определения, данные в ИСО 9000. В тексте настоящего стандарта термин "продукция" может означать также "услугу". [ИСО 9001:2008] |
Дополнительные термины и определения, используемые в настоящем стандарте, относятся непосредственно к правилам GMP, применимым к производству первичных упаковочных материалов для лекарственных средств.
3.1 воздушный шлюз: Закрытое пространство для контролирования воздушного потока.
Примечание - Данное пространство, как правило, снабжено, по крайней мере, двумя взаимосвязанными дверями между двумя и более комнатами, и используется для перехода персонала или перемещения материалов при условии соблюдения различных условий, например, чистоты, потока воздуха при входе.
3.2 утвержденный: Имеющий статус соответствия.
Примечание - Соответствие может подтверждаться на любом этапе процесса (для исходных материалов, для вспомогательных средств, для упаковочных материалов или готовой продукции).
3.3 сборка: Соединение вместе первичных упаковочных материалов (3.35.1) и (или) их компонентов.
Примечание - Примерами могут служить сборка частей пипеток для заполнения аппаратов для инъекций или установка защитных колпачков для игл на предварительно наполненных шприцах.
3.4 автоматический контроль: Оценка соответствия, осуществляемая контрольным оборудованием без вмешательства человека.
Примечание - Контрольное оборудование может включать в себя оптоэлектронные компоненты (камеры), лазерные системы, ультразвуковые системы и связанные с ними системы обработки данных, а также другое оборудование.
3.5 серия, партия: Определенное количество первичных упаковочных материалов (3.35.1), изготовленных в ходе одного или нескольких процессов и имеющих стандартизированные характеристики и стабильное качество.
Примечания
1 Для удовлетворения производственных требований или требований потребителя партия может быть разделена на ряд подпартий, объединяемых позднее в единую однородную партию.
2 При непрерывном производстве серией считается доля продукции, определяемая либо заданным количеством, либо количеством, произведенным за установленный промежуток времени.
3.6 протокол на серию: Документы и записи, дающие представление о предыстории серии (3.5), включая информацию, относящуюся к производству и контролю, и облегчающие прослеживаемость (3.63).
3.7 номер серии, номер партии: Уникальный идентификатор серии или партии (3.5).
Примечание - Номер серии может представлять собой сочетание цифр, букв и (или) символов, идентифицирующих серию (или партию) и помогающих определить предысторию производства и распределения.
3.8 выпуск серии: Решение о выпуске серии (3.5) для продажи или поставки после официального рассмотрения протокола на серию (3.6), осуществляемого службой качества (3.41) или лицом, уполномоченным службой качества.
3.9 калибровка (поверка): Процесс проверки или регулирования (путем сопоставления с эталонным образцом) точности средства измерения.
Примечание - Калибровку (поверку) можно также описать как ряд операций, устанавливающих при заданных условиях зависимость между значениями, полученными с помощью средства измерения, или значениями, представляемыми вещественной мерой, и соответствующими известными значениями эталонного образца.
3.10 управление изменениями: Документированное управление изменениями.
Примечание - К изменениям могут, в частности, относиться изменения в сырье, технических условиях, производственной базе, оборудовании, производственных процессах и методах испытаний.
3.11 чистое помещение: Помещение, в котором контролируется концентрация взвешенных в воздухе частиц и которое сооружено и используется таким образом, чтобы минимизировать попадание, образование и сохранение частиц внутри помещения, и в котором другие соответствующие параметры, такие как температура, влажность и давление, контролируются по мере необходимости.
[ИСО 14644-1-99, 3.1.1]
3.12 чистая зона: Специальное пространство, в котором контролируется концентрация взвешенных в воздухе частиц и которое построено и используется так, чтобы минимизировать попадание, образование и сохранение частиц внутри зоны, и в котором другие соответствующие параметры, такие как температура, влажность и давление, контролируются по мере необходимости.
[ИСО 14644-1-99, 3.1.2]
Примечание - Зона может быть открытой или закрытой и находиться как внутри, так и вне чистого помещения.
3.13 загрязнение: Попадание любого нежелательного вещества в первичный упаковочный материал (3.35.1).
Примечания
1 Готовая продукция может быть загрязнена посредством физического (твердые частицы), химического или биологического (наличие био- и эндотоксинов) воздействия.
2 Загрязнение может возникать во время производства, упаковки, хранения и (или) транспортировки по вине загрязненных воздушных систем, персонала, оборудования для отбора проб, материалов, помещений или контейнеров.
3.14 контролируемая среда: Среда, созданная и эксплуатируемая таким образом, чтобы контролировать возможное попадание потенциальных загрязняющих веществ.
Примечания
1 Среда, как правило, создается и эксплуатируется таким образом, чтобы контролировать потенциальное загрязнение и последствия случайного выброса живых организмов.
2 Соответствующий перепад давления создает возможность эффективного удаления аэрозольных загрязнителей, предотвращения потенциального загрязнения и последствий случайного выброса.
3.15 перекрестное загрязнение: Загрязнение (3.13) материала или продукции другим материалом или продукцией.
Примечания
1 Перекрестное загрязнение может также называться смешиванием.
2 См. [24].
3.16 претензия потребителя: Информация, предоставленная потребителем относительно недостатков или несоответствий.
Примечания
1 Информация может передаваться в устной или письменной форме.
2 Предметом претензии может быть качество, количество или условия поставки первичного упаковочного материала.
3.17 дата изготовления: Дата наступления одной из первых стадий процесса изготовления первичного упаковочного материала, или процесса упаковки, или процесса окончательного выпуска, которая может быть предметом согласования с потребителем.
3.18 отклонение: Отступление от стандартной рабочей процедуры (3.58) или действующего стандарта.
3.19 документированная процедура: Процедура, которая установлена, документально оформлена, санкционирована, внедрена и сохраняется в рабочем состоянии.
3.20 повторная проверка: Документально оформленное подтверждение (3.65) деятельности, результатов или записей вторым лицом или второй системой.
Примечание - Частью такого процесса подтверждения может быть вторая подтверждающая подпись в ходе технологического контроля, производственная документация и записи о качестве для серии, подписанные вторым лицом, или контроль с помощью электронных приборов. Как правило, результаты повторной проверки подписываются вторым лицом.
3.21 срок годности: Предполагаемый предельный срок использования.
Примечания
1 См. также определение срока хранения (3.56).
2 Это, как правило, период, в течение которого ожидается, что первичный упаковочный материал остается пригодным для использования в случае хранения при определенных условиях, после чего он не должен использоваться.
3.22 выходной контроль: Испытания, проводимые на готовой продукции (3.23) для определения соответствия требованиям технических условий.
3.23 готовая продукция: Первичный упаковочный материал (3.35.1), прошедший все стадии производства (3.37).
3.24 надлежащая производственная практика (GMP): Процедуры управления качеством и обеспечения качества, используемые в процессе производства (3.29).
Примечания
1 См. определения понятий "управление качеством" и "обеспечение качества" в ИСО 9000:2005 (3.2.10 и 3.2.11).
2 Требования к надлежащей производственной практике в фармацевтической промышленности установлены в стандарте по обеспечению качества (см. [24]).
3 Требования надлежащей производственной практики (GMP) для материалов первичной упаковки включают, помимо наличия подходящего персонала, помещений и оборудования, наличие системы менеджмента качества, которая включает контролирование поступающего исходного материала, изготовление, наличие соответствующей документации, выполнение санитарно-гигиенических требований на производстве, контроль готовой продукции, записи о реализации, рассмотрение жалоб и самоконтроль.
4 Понятия GMP и текущая надлежащая производственная практика (cGMP) равнозначны. Методические указания GMP постоянно актуализируются в соответствии с меняющимися требованиями современного уровня развития. Это и привело к появлению термина cGMP, который иногда используется. Фармацевтическая промышленность предполагает, что организации принимают во внимание текущие GMP в своих постоянно совершенствующихся программах.
3.25 однородность, гомогенность: Единообразие значений характеристик материала для его определенного количества.
Примечание - Однородность может включать единообразие материалов или определенных характеристик материалов особой значимости.
3.26 контроль в процессе производства: Действия, предпринимаемые в ходе производственного процесса, для проверки соответствия продукции предъявляемым к ней техническим требованиям.
Примечания
1 Для выполнения требований к продукции могут потребоваться процессы мониторинга и наладка средств производства.
2 Контроль окружающей среды или оборудования может также рассматриваться как часть контроля в процессе производства.
3.27 полуфабрикат: Материал для первичной упаковки (3.35.1), прошедший некоторые, но не все стадии производства.
Примечание - Полуфабрикат требует дальнейшей обработки, прежде чем стать готовой продукцией.
3.28 очистка линии: Удаление всего, что связано с предыдущим производственным циклом (промывка линии).
Примечание - Как правило, очистка линии производится до очередного производственного цикла для предотвращения любой ошибки или перекрестного загрязнения. Как правило, требуется, чтобы производственное оборудование (линия) и соответствующий рабочий участок были полностью очищены от всех материалов, отходов, продукции, образцов, документов и т.п., которые использовались в ходе предыдущего производственного цикла, до завоза материалов, образцов продукции, документов и т.п., необходимых для начала следующего производственного цикла.
3.29 изготовление: Все операции по закупке материалов, включая материалы для первичной упаковки (3.35.1), производству (3.37), управлению качеством (3.39), выпуску, хранению, реализации продукции и связанные с ними средства управления.
3.30 лекарственное средство: Любое вещество или сочетание веществ, предназначенное для лечения или профилактики у людей или животных.
Примечания
1 Любое вещество или сочетание веществ, вводимое человеку или животным с диагностическими целями или для восстановления, корректировки или изменения физиологических функций человека или животных, также рассматривается как лекарственное средство.
2 См. [24].
3 Лекарственные средства могут также называться фармацевтическими или лекарственными препаратами, включая продукцию, подвергающуюся клиническим исследованиям.
3.31 организация: Группа лиц и средств с установленными обязанностями, полномочиями и взаимосвязями.
[ИСО 9000:2005, 3.3.1]
Примечание - В настоящем стандарте организация трактуется как компания, изготавливающая материалы для первичной упаковки.
3.32 подготовка печатного оригинала: Вся подготовительная работа до печати.
Примечание - Сюда относится концептуальное решение, дизайн, графическое исполнение, репрография, пленка, изготовление печатной формы, шелкотрафаретная печать, цифровые файлы и шрифтоносители.
3.33 за пределами технических требований: Результаты испытаний, не отвечающие требованиям технических условий (3.57).
3.34 аутсорсинг: Выполнение всего или части процесса другой организацией (3.3.1).
Примечание - Аутсорсинг часто называют субподрядом (см. определение термина "субподрядчик (6.61)").
3.35 упаковочные материалы
3.35.1 материалы для первичной упаковки: Упаковочные материалы, используемые в фармацевтической упаковке, которая содержит, герметизирует или используется для дозированного применения медикамента или находится в непосредственном контакте с лекарственным средством.
Примечания
1 Примерами материалов для первичной упаковки могут служить стекло, резина, пластмасса, алюминий, пленка, фольга, слоистый пластик. Может использоваться сочетание различных материалов/компонентов (например, шприцы, аэрозольные баллоны).
2 Настоящий стандарт распространяется также на материалы, находящиеся в непродолжительном контакте с лекарственными средствами (например, пипетки и шприцы).
3 На первичные упаковочные материалы могут наноситься печатная информация или декоративные элементы.
3.35.2 материалы для вторичной упаковки: Упаковочные материалы, не вступающие в контакт с лекарственным средством, например картонные коробки с текстом и без текста, этикетки, брошюрки или вкладыши (или рекламные листки), обертки и контейнеры для транзитных перевозок, такие как складные картонные коробки.
3.36 вспомогательные средства: Материалы, способствующие реализации процесса.
Примечание - Данные материалы не включены в технические требования к продукции и могут быть удалены при или до окончательной обработки.
Пример - Средства, способствующие разъему пресс-формы, сжатый воздух, технологическая прокатная смазка.
3.37 производство: Процессы, приводящие к получению материалов для первичной упаковки (3.35.1).
Примечание - Эти процессы образуют полный производственный цикл, начиная от приемки исходных материалов, включая переработку, упаковку и заканчивая получением готовой продукции.
3.38 процесс квалификации: Процесс, подтверждающий способность выполнять установленные требования.
[ИСО 9000:2005, 3.8.6]
Примечание - Квалификация и валидация (3.64) охватывают проектную квалификацию, установочную квалификацию, квалификацию функционирования, приемо-сдаточные испытания на предприятии и квалификацию эксплуатации, а также повторную квалификацию и повторную валидацию по мере необходимости. Эта деятельность может также проводиться одновременно путем группирования (матричная валидация) и (или) ретроспективно.
3.39 управление качеством: Часть менеджмента качества, направленная на выполнение требований к качеству.
[ИСО 9000:2005, 3.2.10]
Примечание - Управление качеством включает проверку или тестирование выполнения технических условий.
3.40 критическое состояние качества: Показатель, влияющий на качество для первичной упаковки материалов (3.35.1).
Примечание - Материал, операция или условия процесса, требования к испытаниям или любые другие соответствующие параметры могут рассматриваться как критические в случае, если невыполнение требований к ним приводит к существенному ущербу.
3.41 служба качества: Организационное подразделение, которое несет ответственность за обеспечение качества и управление качеством.
Примечание - Служба качества может состоять из отдельных подразделений по обеспечению качества и управлению качеством или из одного лица (группы лиц) в зависимости от размеров и структуры организации.
3.42 карантин: Статус материалов или продукции, изолированных до вынесения решения об их последующем одобрении или последующей выбраковки.
Примечание - Подкарантинный материал, как правило, изолируется физическим или иным действенным способом.
3.43 реализация: Общий термин, охватывающий все процессы, требуемые для достижения необходимого результата от проектирования до поставки продукции.
3.44 сопоставление, сверка: Сравнение между объемом готовой продукции, теоретически и фактически выпущенной или используемой, с учетом нормальной вариации.
Примечание - Сравнение проводится с учетом отходов, образцов и других потерь, присущих процессу.
3.45 восстановление: Обработка или повторная обработка материалов первичной упаковки для выполнения требований технических условий.
3.46 забракованный: Статус исходных материалов (3.59), вспомогательных средств (3.36), полуфабрикатов (3.27) или готовой продукции (3.23), результаты испытания которых не удовлетворяют одному или нескольким требованиям технических условий (3.57) и которые установлены (обычно службой качества (3.41)) непригодными для использования.
3.47 выбраковка: Процесс установления, выполняемый обычно службой качества (3.41), того, что исходные материалы (3.59), вспомогательные средства (3.36), полуфабрикаты (3.27) или готовая продукция (3.23) непригодны для использования.
3.48 повторная обработка: Повторение части технологического процесса.
Примечание - Продолжение части технологического процесса после того, как контроль в процессе производства показал, что данная часть процесса не завершена, является нормальной практикой и не относится к повторной обработке.
3.49 архивные образцы, арбитражные образцы: Материалы или готовая продукция (3.23), хранимые для будущего сравнения.
Примечание - Эти образцы обычно отбираются в достаточном количестве и хранятся в определенных условиях в течение заданного времени для последующего сравнения.
3.50 возврат: Процесс отправки материалов для первичной упаковки (3.35.1) обратно в организацию (3.31).
3.51 переделка: Действие, предпринятое в отношении несоответствующей продукции для того, чтобы она соответствовала требованиям.
[ИСО 9000:2005, 3.6.7]
Примечание - Сортировка может рассматриваться как переделка.
3.52 анализ риска: Систематическое использование информации для выявления опасности и количественной оценки риска.
[Руководство ИСО/МЭК 51:1999, 3.10]
3.53 оценка риска: Общий процесс анализа риска (3.52) и оценивания риска (3.54).
[Руководство ИСО/МЭК 51:1999, 3.12]
3.54 оценивание риска: Процедура, основанная на анализе риска (3.52) для определения того, достигнут ли допустимый уровень риска.
[Руководство ИСО/МЭК 51:1999, 3.11]
3.55 менеджмент риска: Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания, управления и мониторинга риска.
[ИСО 14971:2007, 2.22]
3.56 срок хранения: Период времени, в течение которого материалы для первичной упаковки (3.35.1) должны соответствовать требованиям (техническим условиям) при хранении при определенных условиях и по истечение которого материалы не должны использоваться.
Примечание - См. также срок годности (3.21).
3.57 технические условия: Документ, устанавливающий требования.
[ИСО 9000:2005, 3.7.3]
3.58 стандартная рабочая процедура: Санкционированная документально оформленная процедура или ряд процедур, инструкций по выполнению работы и испытаний, используемых в процессе производства (3.37) и управления.
3.59 исходный материал: Сырье, компоненты, вещества, используемые в производстве материалов для первичной упаковки (3.35.1).
3.60 стерильность: Отсутствие жизнеспособных микроорганизмов.
[ИСО 14937:2009, 3.26]
3.61 субподрядчик: Третья сторона, которая выполняет часть или всю работу по изготовлению материалов для первичной упаковки (3.35.1).
3.62 обработка поверхности: Процесс, направленный на улучшение поверхности материалов для первичной упаковки.
Пример - Покрытие силиконом или другая обработка внутренних стеклянных поверхностей, покрытие внутренних или внешних поверхностей стеклянных контейнеров или резиновых деталей.
3.63 прослеживаемость: Возможность проследить предысторию, применение или местонахождение рассматриваемого объекта.
Примечания
1 Прослеживаемость применительно к продукции может относиться к:
- происхождению материалов и комплектующих;
- предыстории обработки;
- распределению и местонахождению продукции после поставки.
2 В области метрологии принято определение, приведенное в VIM - 1993, подраздел 6.10.
[ИСО 9000:2005, 3.5.4]
3.64 валидация: Подтверждение посредством представления объективных свидетельств того, что требования, предназначенные для конкретного предполагаемого использования или применения, выполнены.
[ИСО 9000:2005, 3.8.5]
Примечание - См. примечание к 3.38.
3.65 верификация: Подтверждение посредством представления объективных свидетельств того что установленные требования были выполнены.
Примечания
1 Термин "верифицировано" используется для обозначения соответствующего статуса.
2 При разработке и проектировании верификация представляет собой процесс изучения результатов рассматриваемой деятельности с целью установления соответствия упомянутой деятельности заданным требованиям.
4 Система менеджмента качества
4.1 Общие требования Организация должна разработать, документировать, внедрить и поддерживать в рабочем состоянии систему менеджмента качества, постоянно улучшать ее результативность в соответствии с требованиями настоящего стандарта. Организация должна: a) определять процессы, необходимые для системы менеджмента качества, и их применение во всей организации (1.2); b) определять последовательность и взаимодействие этих процессов; c) определять критерии и методы, необходимые для обеспечения результативности как при осуществлении этих процессов, так и при управлении ими; |
d) обеспечивать наличие ресурсов и информации, необходимых для поддержания этих процессов и их мониторинга; e) осуществлять мониторинг, измерение, там, где это возможно, и анализ этих процессов; f) принимать меры, необходимые для достижения запланированных результатов и постоянного улучшения этих процессов. g) охарактеризовать свою общую политику, намерения и подход к обеспечению качества продукции. Организация должна осуществлять менеджмент процессов, необходимых для системы менеджмента качества, в соответствии с требованиями настоящего стандарта. Если организация решает передать сторонней организации выполнение какого-либо процесса, влияющего на соответствие продукции требованиям, она должна обеспечить со своей стороны управление таким процессом. Вид и степень управления процессами, переданными сторонним организациям, должны быть определены в системе менеджмента качества. |
Примечания 1 Упомянутые выше процессы, необходимые для системы менеджмента качества, включают в себя процессы управленческой деятельности руководства, обеспечения ресурсами, процессы жизненного цикла продукции, измерения, анализа и улучшения. 2 Процесс, переданный другой организации, является процессом, необходимым для системы менеджмента организации, но по выбору организации выполняемым внешней для нее стороной. 3 Обеспечение управления процессами, переданными сторонним организациям, не освобождает организацию от ответственности за соответствие всем требованиям потребителей и обязательным требованиям. Выбор вида и степени управления процессом, переданным сторонней организации, зависит от таких факторов, как: a) возможное влияние переданного сторонним организациям процесса на способность организации поставлять продукцию, соответствующую требованиям; b) степень участия в управлении процессом, переданным сторонней организации; c) возможность обеспечения необходимого управления посредством применения требований 7.4. [ИСО 9001:2008] |
4.2 Требования к документации
4.2.1 Общие положения
Документация системы менеджмента качества должна включать в себя: a) документально оформленные заявления о политике и целях в области качества; b) руководство по качеству; c) документированные процедуры и записи, требуемые настоящим стандартом; d) документы, включая записи, определенные организацией как необходимые ей для обеспечения эффективного планирования, осуществления процессов и управления ими. Примечания 1 Там, где в настоящем стандарте встречается термин "документированная процедура", это означает, что процедура разработана, документально оформлена, внедрена и поддерживается в рабочем состоянии. Один документ может содержать требования одной или более процедуры. Требование о наличии документированной процедуры может быть реализовано более чем одним документом. |
2 Степень документированности системы менеджмента качества одной организации может отличаться от степени документированности другой в зависимости: a) от размера организации и вида деятельности; b) от сложности и взаимодействия процессов; c) от компетентности персонала. 3 Документация может быть в любой форме и на любом носителе. [ИСО 9001:2008] |
Примечание 4 - Документированные процедуры, инструкции по проведению работы, испытаний в целях производства и управления, требуемые настоящим стандартом, можно назвать стандартными рабочими процедурами.
Необходимо документально оформить общую политику, намерения и подход к валидации.
4.2.2 Руководство по качеству
Организация должна разработать и поддерживать в рабочем состоянии руководство по качеству, содержащее: a) область применения системы менеджмента качества, включая подробности и обоснование любых исключений (1.2); b) документированные процедуры, разработанные для системы менеджмента качества, или ссылки на них; c) описание взаимодействия процессов системы менеджмента качества. [ИСО 9001:2008] |
4.2.2.1 Организация должна четко определить, в какой степени настоящий стандарт применим к ее процессам.
Примечание - Организация может определить, относится ли настоящий стандарт ко всей выпускаемой продукции (для фармацевтических и иных целей) или исключительно для фармацевтических целей.
4.2.2.2 Руководство по качеству описывает структуру документации, используемой в системе менеджмента качества.
4.2.3 Управление документами
Документы системы менеджмента качества должны быть управляемыми. Записи, представляющие собой специальный вид документов, должны быть управляемыми согласно требованиям 4.2.4. Для определения необходимых средств управления должна быть разработана документированная процедура, предусматривающая: a) официальное одобрение документов с точки зрения их достаточности до выпуска; b) анализ и актуализацию по мере необходимости и повторное официальное одобрение документов; c) обеспечение идентификации изменений и статуса пересмотра документов; |
d) обеспечение наличия соответствующих версий документов в местах их применения; e) обеспечение сохранения документов четкими и легко идентифицируемыми; f) обеспечение идентификации и управление рассылкой документов внешнего происхождения, определенных организацией как необходимые для планирования и функционирования системы менеджмента качества; g) предотвращение непреднамеренного использования устаревших документов и применение соответствующей идентификации таких документов, оставленных для каких-либо целей. [ИСО 9001:2008] |
4.2.3.1 Организация должна убедиться в том, что изменения в документах анализируются и утверждаются либо первичной согласительной инстанцией, либо другой назначаемой инстанцией, имеющей доступ к соответствующей исходной информации для обоснования своих решений.
4.2.3.2 Организация должна определить срок хранения устаревших контролируемых документов (см. также 4.2.4.8).
4.2.3.3 При использовании на документах электронных подписей они должны контролироваться для обеспечения безопасности, эквивалентной подписи от руки.
4.2.3.4 Управление документами должно включать однозначную идентификацию (например, название/номер документа, дата выпуска и номер страницы).
4.2.4 Управление записями
Записи, установленные для представления свидетельств соответствия требованиям и результативного функционирования системы менеджмента качества, должны находиться под управлением. Организация должна установить документированную процедуру для определения средств управления, необходимых для идентификации, хранения, защиты, восстановления, сохранения и изъятия записей. Записи должны оставаться четкими, легко идентифицируемыми и восстанавливаемыми. [ИСО 9001:2008] |
Примечание - К записям относятся данные о производстве серии продукции, а также другие записи о качестве, такие как отчеты об отклонениях и исследовании.
4.2.4.1 Электронные записи подлежат такому же управлению, которое требуется для других записей (см. 4.2.4 и 7.5.2.9).
4.2.4.2 Отдельные записи должны быть четкими, несмываемыми, выполненными сразу же после выполнения деятельности (в порядке выполнения работ), датированными и подписанными лицом, делающим запись. Исправления в записях должны быть датированными, подписанными и сопровождаемыми разъяснениями по мере необходимости, оставляя разборчивой первоначальную запись.
4.2.4.3 Организация должна определять процессы и показатели, важные для обеспечения качества, где для выпуска серии требуется двойная проверка. При проведении электронной проверки это должно быть четко определено.
4.2.4.4 Каждый этап и показатель в процессе производства и управления, важный для обеспечения качества, должен быть идентифицирован и подвергнут двойной проверке.
4.2.4.5 Для каждой серии первичных упаковочных материалов организация должна составлять и вести запись, которая обеспечивает прослеживаемость (см. 7.5.3) и определяет количество произведенной продукции и количество, утвержденное к реализации.
4.2.4.6 Организация должна определять те показатели документации по серии, которые подлежат верификации.
4.2.4.7 Документация по серии должна быть верифицирована и утверждена.
4.2.4.8 Все записи, касающиеся изготовления, управления, испытаний, распределения и расследования, должны сохраняться в течение, по крайней мере, пяти лет с даты изготовления или в течение срока, согласованного с потребителем или, по крайней мере, в течение одного года после истечения срока годности первичных упаковочных материалов, установленного организацией, если нет иной договоренности с потребителем.
Примечание - Может потребоваться сохранение записей о первичных упаковочных материалах до конца срока хранения лекарственного средства, как оговорено потребителем.
5 Ответственность руководства
5.1 Обязательства руководства
Высшее руководство должно обеспечивать наличие свидетельств принятия своих обязательств по разработке и внедрению системы менеджмента качества, а также постоянному улучшению ее результативности посредством: a) доведения до сведения персонала организации важности выполнения требований потребителей, а также законодательных и обязательных требований; b) разработки политики в области качества; c) обеспечения разработки целей в области качества; d) проведения анализа со стороны руководства; e) обеспечения необходимыми ресурсами. [ИСО 9001:2008] |
5.2 Ориентация на потребителя
Высшее руководство должно обеспечивать определение и выполнение требований потребителей для повышения их удовлетворенности (7.2.1 и 8.2.1). [ИСО 9001:2008] |
5.2.1 Проверки, проводимые потребителем
Организация должна разрешать потребителю или его представителям (по взаимному согласию) проводить проверки для анализа системы менеджмента качества.
Основные требования потребителя к организации связаны с приемлемыми производственными возможностями, компетентным и подготовленным персоналом, процессами, предназначенными для обеспечения безопасности продукции и отсутствия перекрестного загрязнения, а также со способностью постоянно выпускать продукцию, отвечающую техническим требованиям потребителя.
5.3 Политика в области качества
Высшее руководство должно обеспечивать, чтобы политика в области качества: a) соответствовала целям организации; b) включала в себя обязательство соответствовать требованиям и постоянно повышать результативность системы менеджмента качества; c) создавала основы для постановки и анализа целей в области качества; d) была доведена до сведения персонала организации и понятна ему; e) анализировалась на постоянную пригодность. [ИСО 9001:2008] |
5.4 Планирование
5.4.1 Цели в области качества
Высшее руководство организации должно обеспечивать, чтобы цели в области качества, включая необходимые для выполнения требований к продукции [7.1, перечисление а)], были установлены в соответствующих подразделениях и на соответствующих уровнях организации. Цели в области качества должны быть измеримыми и согласуемыми с политикой в области качества. [ИСО 9001:2008] |
5.4.2 Планирование создания и развития системы менеджмента качества
Высшее руководство должно обеспечивать: a) планирование создания, поддержания и улучшения системы менеджмента качества для выполнения требований 4.1, а также для достижения целей в области качества; b) сохранение целостности системы менеджмента качества при планировании и внедрении в нее изменений. [ИСО 9001:2008] |
5.5 Ответственность, полномочия и обмен информацией
5.5.1 Ответственность и полномочия
Высшее руководство должно обеспечивать определение и доведение до сведения персонала организации ответственности и полномочий. [ИСО 9001:2008] |
5.5.1.1 Организация должна вести текущие образцы (см. 4.2.4.) подписей ответственных лиц. Рекомендуется иметь перечни подписей и (или) идентификационные перечни всего персонала, проводящего проверки или повторные проверки этапов процесса, средств управления в ходе процесса и т.п.
5.5.1.2 Службы качества, отвечающие за принятие решений, важных для обеспечения качества, должны обладать полномочиями по принятию таких решений независимо от производственных процессов.
5.5.2 Представитель руководства
Высшее руководство должно назначить представителя из состава руководства организации, который независимо от других обязанностей должен нести ответственность и иметь полномочия, распространяющиеся: a) на обеспечение разработки, внедрения и поддержания в рабочем состоянии процессов, требуемых системой менеджмента качества; b) на представление отчетов высшему руководству о функционировании системы менеджмента качества и необходимости ее улучшения; c) на содействие распространению понимания требований потребителей по всей организации. Примечание - В ответственность представителя руководства может быть включено поддержание связи с внешними сторонами по вопросам, касающимся системы менеджмента качества. [ИСО 9001:2008] |
5.5.3 Внутренний обмен информацией
Высшее руководство должно обеспечивать установление в организации соответствующих процессов обмена информацией, включая информацию, относящуюся к результативности системы менеджмента качества. [ИСО 9001:2008] |
5.5.3.1 Правила GMP, описанные в настоящем стандарте, и нормативные требования должны по мере необходимости доводиться до сведения каждого уровня организации.
5.5.3.2 Высшее руководство должно быть своевременно уведомлено о ситуациях, критичных для обеспечения качества.
Примечание - К примерам процессов обмена информацией относятся те, которые касаются обмена информацией о политике в области качества, анализов со стороны руководства, результатов внутренних проверок качества и корректирующих и предупреждающих действий.
5.6 Анализ со стороны руководства
5.6.1 Общие положения
Высшее руководство должно анализировать через запланированные интервалы времени систему менеджмента качества организации в целях обеспечения ее постоянной пригодности, достаточности и результативности. Этот анализ должен включать в себя оценку возможностей улучшений и потребности в изменениях в системе менеджмента качества организации, в том числе в политике и целях в области качества. Записи об анализе со стороны руководства должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
5.6.2 Входные данные для анализа
Входные данные для анализа со стороны руководства должны включать в себя следующую информацию: a) результаты аудитов (проверок); b) обратную связь от потребителей; c) функционирование процессов и соответствие продукции; d) статус предупреждающих и корректирующих действий; e) последующие действия, вытекающие из предыдущих анализов со стороны руководства; f) изменения, которые могли бы повлиять на систему менеджмента качества; g) рекомендации по улучшению. [ИСО 9001:2008] |
h) результативность подготовки.
5.6.3 Выходные данные анализа
Выходные данные анализа со стороны руководства должны включать в себя все решения и действия, относящиеся: a) к повышению результативности системы менеджмента качества и ее процессов; b) к улучшению продукции по отношению к требованиям потребителей; c) к потребности в ресурсах. [ИСО 9001:2008] |
6 Менеджмент ресурсов
6.1 Обеспечение ресурсами
Организация должна определить и обеспечивать ресурсы, требуемые: a) для внедрения и поддержания в рабочем состоянии системы менеджмента качества, а также постоянного повышения ее результативности; b) для повышения удовлетворенности потребителей путем выполнения их требований. [ИСО 9001:2008] |
6.2 Человеческие ресурсы
6.2.1 Общие положения
Персонал, выполняющий работу, влияющую на соответствие продукции требованиям, должен быть компетентным на основе полученного образования, подготовки, навыков и опыта. Примечание - На соответствие продукции требованиям прямо или косвенно может влиять персонал, выполняющий любую работу в рамках системы менеджмента качества. [ИСО 9001:2008] |
6.2.2 Компетентность, осведомленность и подготовка
Организация должна: a) определять необходимую компетентность персонала, выполняющего работу, которая влияет на соответствие требованиям к качеству продукции; b) где это возможно, обеспечивать подготовку или предпринимать другие действия в целях достижения необходимой компетентности; c) оценивать результативность принятых мер; d) обеспечивать осведомленность своего персонала об актуальности и важности его деятельности и вкладе в достижение целей в области качества; e) поддерживать в рабочем состоянии соответствующие записи об образовании, подготовке, навыках и опыте (4.2.4). [ИСО 9001:2008] |
6.2.2.1 Обучение GMP
6.2.2.1.1 Должно регулярно проводиться дополнительное обучение, которое включает осведомленность о действующих правилах GMP и всех процедурах и методах, которые оказывают влияние на качество продукции и систему менеджмента качества. Такое обучение должно охватывать:
a) риск заражения и перекрестного заражения;
b) потенциальную опасность для конечного пользователя/пациента в случае заражения продукции;
c) влияние любых отклонений от установленных процедур, процессов или технических условий на качество продукции потребителя или конечного пользователя.
6.2.2.1.2 Следует уделять особое внимание подготовке персонала, занятого изготовлением стерильных компонентов или компонентов, подвергающихся последующей стерилизации.
6.2.2.1.3 Должно быть организовано специальное обучение микробиологическому загрязнению и загрязнению частицами и потенциальному риску такого загрязнения для пациента.
6.2.2.1.4 С определенной периодичностью должна проводиться дополнительная переподготовка.
6.2.2.1.5 Должна быть организована подготовка временного персонала или он должен находиться под наблюдением подготовленного лица.
6.2.2.1.6 При найме консультантов для консультирования по вопросам качества должен вестись учет их квалификации и видов оказываемых услуг.
6.3 Инфраструктура
Организация должна определять, обеспечивать и поддерживать в рабочем состоянии инфраструктуру, необходимую для достижения соответствия требованиям к продукции. Инфраструктура может включать в себя, если применимо: a) здания, рабочее пространство и связанные с ним средства труда; b) оборудование для процессов (как технические, так и программные средства); c) службы обеспечения (такие как транспорт, связь или информационные системы). [ИСО 9001:2008] |
6.4 Производственная среда
Организация должна создавать производственную среду, необходимую для достижения соответствия требованиям к продукции, и управлять ею. Примечание - Термин "производственная среда" относится к условиям, в которых выполняют работу, включая физические, экологические и другие факторы (такие как шум, температура, влажность, освещенность или погодные условия). [ИСО 9001:2008] |
6.4.1 Требования к производственной среде
6.4.1.1 Организация должна установить документированные требования по охране здоровья, чистоте, одежде и контролю доступа персонала, если контакт между таким персоналом и первичными упаковочными материалами или производственной средой может негативно отразиться на качестве первичных упаковочных материалов.
6.4.1.2 Если условия производственной среды могут отрицательно сказаться на качестве первичных упаковочных материалов, организация должна определить соответствующие условия производственной среды и разработать систему по их результативному мониторингу и контролю.
6.4.1.3 В случае необходимости должны быть созданы и документированы специальные условия по контролированию загрязненных или потенциально загрязненных первичных упаковочных материалов, чтобы препятствовать загрязнению других первичных упаковочных материалов, производственной среды или персонала.
6.4.2 Классификация чистых зон/чистых помещений
Чистые зоны/чистые помещения должны классифицироваться в соответствии с ИСО 14644-1 и должны контролироваться/использоваться в соответствии с ИСО 14644-2, ИСО 14644-3 и ИСО 14644-5 или их эквивалентом.
Относительно проектирования, строительства и ввода в действие чистых помещений см. ИСО 14644-2 и ИСО 14644-4.
Мониторинг может осуществляться в соответствии с ИСО 14698-1 и ИСО 14698-2.
6.4.3 Управление риском загрязнения
Организация должна определить и управлять рисками возможного загрязнения первичных упаковочных материалов, например:
a) личная гигиена и состояние здоровья;
b) личная одежда, ювелирные изделия, включая пирсинги, и косметика;
c) курение, еда, жевание, питье и личные медикаменты;
d) переработка и утилизация отходов;
e) микробиологическое загрязнение.
6.4.4 Борьба с вредителями
Должна быть реализована результативная программа по борьбе с вредителями.
6.4.5 Вспомогательные службы
6.4.5.1 Все инженерные сети (например, воздухо-, газо-, паро-, водоснабжение) должны быть оценены на предмет возможного влияния на качество первичных упаковочных материалов и любых связанных с ними рисков. Должны вестись записи проводимой оценки (см. 4.2.4).
При оценке должны использоваться другие жидкости (например, смазочные жидкости, охлаждающие жидкости, гидравлические масла и т.п.), которые могут вступать в контакт с первичными упаковочными материалами.
В зависимости от рисков должна быть рассмотрена возможность использования пищевых жидкостей.
6.4.5.2 Должны быть предусмотрены соответствующие вентиляционные и вытяжные системы в случае необходимости для сведения к минимуму риска загрязнения. Особое внимание следует уделять системам рециркуляции воздуха.
6.4.5.3 Если вода вступает в непосредственный контакт с первичными упаковочными материалами или исходным материалом или используется для мытья оборудования, контактирующего с продукцией, необходимо определять и контролировать ее качество.
6.5 Работы по техническому обслуживанию
6.5.1 Организация должна выработать документированные требования к техническому обслуживанию (например, в отношении технологических процессов, систем и оборудования), когда проводимые работы или их отсутствие могут отразиться на качестве продукции.
6.5.2 Следует вести учет работ по техническому обслуживанию.
6.5.3 Организация должна обеспечивать использование, эксплуатацию и поддержание в должном состоянии инфраструктуры в соответствии с правилами GMP и во избежание загрязнения продукции (включая контроль взвешенных частиц и микробиологический контроль в случае необходимости).
6.5.4 Компьютеризированные системы, которые могут оказывать влияние на качество первичных упаковочных материалов, должны быть оснащены достаточными средствами контроля для обеспечения монтажа, эксплуатации, технического обслуживания, внесения изменений и безопасности.
6.5.5 Необходимо вести комплект технической документации по оборудованию и установкам, имеющим важное значение для обеспечения качества.
7 Процессы жизненного цикла продукции
7.1 Планирование процессов жизненного цикла продукции
Организация должна планировать и разрабатывать процессы, необходимые для обеспечения жизненного цикла продукции. Планирование процессов жизненного цикла продукции должно быть согласовано с требованиями к другим процессам системы менеджмента качества (4.1). При планировании процессов жизненного цикла продукции организация должна установить подходящим для нее образом: a) цели в области качества и требования к продукции; b) потребность в разработке процессов и документов, а также в обеспечении ресурсами для конкретной продукции; c) необходимую деятельность по верификации и валидации, мониторингу, измерению, контролю и испытаниям для конкретной продукции, а также критерии приемки продукции; d) записи, необходимые для обеспечения свидетельства того, что процессы жизненного цикла продукции и продукция соответствуют требованиям (4.2.4). |
Результат этого планирования должен быть представлен в форме, соответствующей практике организации. Примечания 1 Документ, определяющий процессы системы менеджмента качества (включая процессы жизненного цикла продукции) и ресурсы, которые предстоит применять к конкретной продукции, проекту или контракту, может рассматриваться как план качества. 2 При разработке процессов жизненного цикла продукции организация может также применять требования 7.3. [ИСО 9001:2008] |
7.1.1 При планировании процессов жизненного цикла продукции следует учитывать требование к последовательной обработке первичных упаковочных материалов. При планировании следует также учитывать необходимость отбора и сохранения образцов в должном состоянии.
7.1.2 Организация должна предусматривать включение в планирование и реализацию на протяжении всего жизненного цикла продукции процессов менеджмента рисков; следует вести соответствующие записи (см. 4.2.4).
7.2 Процессы, связанные с потребителями
7.2.1 Определение требований, относящихся к продукции
Организация должна определить: a) требования, установленные потребителями, включая требования к поставке и деятельности после поставки; b) требования, не определенные потребителем, но необходимые для конкретного или предполагаемого использования, когда оно известно; c) законодательные и другие обязательные требования, применимые к продукции; d) любые дополнительные требования, рассматриваемые организацией как необходимые. Примечание - Деятельность после поставки может включать в себя действия по гарантийному обеспечению, контрактным обязательствам, таким как услуги по техническому обслуживанию, и дополнительные услуги, такие как утилизация или полное уничтожение. [ИСО 9001:2008] |
7.2.1.1 Должны быть определены и документированы требования, относящиеся к продукции, включая изменения, требующие уведомления.
7.2.1.2 Должны быть определены и документированы требования потребителей, касающиеся предотвращения несанкционированного использования отходов производства первичных упаковочных материалов (включая образцы, печатные материалы, ярлыки).
7.2.2 Анализ требований, относящихся к продукции
Организация должна анализировать требования, относящиеся к продукции. Этот анализ должен проводиться до принятия организацией обязательства поставлять продукцию потребителю (например, участие в тендерах, принятие контрактов или заказов, принятие изменений к контрактам или заказам) и должен обеспечивать: a) определение требований к продукции; b) согласование требований контракта или заказа, отличающихся от ранее сформулированных; c) способность организации выполнять определенные требования. Записи результатов анализа и последующих действий, вытекающих из анализа, должны поддерживаться в рабочем состоянии (4.2.4). |
Если потребители не выдвигают документированных требований, организация должна подтвердить их у потребителя до принятия к исполнению. Если требования к продукции изменены, организация должна обеспечить, чтобы соответствующие документы были исправлены, а заинтересованный персонал был поставлен в известность об изменившихся требованиях. Примечание - В некоторых ситуациях, таких как продажи, осуществляемые через Интернет, практически нецелесообразно проводить официальный анализ каждого заказа. Вместо этого анализ может распространяться на соответствующую информацию о продукции, такую как каталоги или другие рекламные материалы. [ИСО 9001:2008] |
7.2.3 Связь с потребителями
Организация должна определять и осуществлять эффективные меры по поддержанию связи с потребителями, касающиеся: a) информации о продукции; b) прохождения запросов, контракта или заказа, включая поправки; c) обратной связи от потребителей, включая жалобы потребителей. [ИСО 9001:2008] |
7.2.3.1 Организация должна разработать и поддерживать в рабочем состоянии систему обратной связи для заблаговременного оповещения о потенциальных и реальных проблемах качества и содействия вкладу потребителей в систему корректирующих и предупреждающих действий.
7.2.3.2 Если этого требует потребитель, организация должна согласовывать с потребителем, какие изменения требуют письменного подтверждения до утверждения, а какие только уведомления. О предлагаемых изменениях необходимо своевременно информировать, и следует согласовывать процесс внесения изменений (см. 7.2.1).
Между организацией и потребителем рекомендуется иметь документированное техническое соглашение об обеспечении качества, в котором рассматриваются меры, принимаемые по несоответствиям (см. 8.3).
7.3 Проектирование и разработка
7.3.1 Планирование проектирования и разработки
Организация должна планировать проектирование и разработку и управлять этими процессами. В ходе планирования проектирования и разработки организация должна устанавливать: a) стадии проектирования и разработки; b) проведение анализа, верификации и валидации, соответствующих каждой стадии проектирования и разработки; c) ответственность и полномочия в области проектирования и разработки. Организация должна управлять взаимодействием различных групп, занятых проектированием и разработкой, в целях обеспечения эффективной связи и четкого распределения ответственности. |
Результаты планирования должны актуализироваться, если это необходимо, в процессе проектирования и разработки. Примечание - Анализ, верификация и валидация проектирования и разработки имеют разные цели, поэтому их можно проводить и записи по ним вести как отдельно, так и в любых сочетаниях, подходящих для продукции и организации. [ИСО 9001:2008] |
7.3.1.1 Организация должна внедрять документированные процедуры проектирования и разработки. Эти процедуры включают оценку риска, определение соответствующих аспектов GMP и любого потенциального влияния на потребителя и, в конечном счете, на пациента.
7.3.1.2 Ответственность за проектирование и оценку риска должна быть согласована между потребителем и организацией.
7.3.1.3 В ходе процесса проектирования и разработки следует предусмотреть проверку пригодности выходных данных проектирования и разработки до окончательного согласования технических требований к производству.
7.3.2 Входные данные для проектирования и разработки
Входные данные, относящиеся к требованиям к продукции, должны быть определены, а записи должны поддерживаться в рабочем состоянии (4.2.4). Входные данные должны включать в себя: a) функциональные и эксплуатационные требования; b) соответствующие законодательные и другие обязательные требования; c) там, где это возможно, информацию, взятую из предыдущих аналогичных проектов; d) другие требования, важные для проектирования и разработки. Входные данные должны анализироваться на достаточность. Требования должны быть полными, недвусмысленными и непротиворечивыми. [ИСО 9001:2008] |
7.3.3 Выходные данные проектирования и разработки
Выходные данные проектирования и разработки должны быть представлены в форме, подходящей для проведения верификации относительно входных требований к проектированию и разработке, а также должны быть официально одобрены до их последующего использования. Выходные данные проектирования и разработки должны: a) соответствовать входным требованиям к проектированию и разработке; b) обеспечивать соответствующей информацией по закупкам, производству и обслуживанию; c) содержать критерии приемки продукции или ссылки на них; d) определять характеристики продукции, существенные для ее безопасного и правильного использования. Примечание - Информация по производству и обслуживанию может включать в себя подробные данные о сохранении продукции. [ИСО 9001:2008] |
7.3.4 Анализ проекта и разработки
На соответствующих стадиях должен проводиться систематический анализ проекта и разработки в соответствии с запланированными мероприятиями (7.3.1) в целях: a) оценивания способности результатов проектирования и разработки удовлетворять требованиям; b) выявления любых проблем и внесения предложений по необходимым действиям. В состав участников такого анализа должны включаться представители подразделений, имеющих отношение к анализируемой(ым) стадии(ям) проектирования и разработки. Записи результатов анализа и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
7.3.5 Верификация проекта и разработки
Верификация должна осуществляться в соответствии с запланированными мероприятиями (7.3.1) с целью удостовериться, что выходные данные проектирования и разработки соответствуют входным требованиям. Записи результатов верификации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
7.3.6 Валидация проекта и разработки
Валидация проекта и разработки должна осуществляться в соответствии с запланированными мероприятиями (7.3.1) с целью удостовериться, что полученная в результате продукция соответствует требованиям к установленному или предполагаемому использованию, если оно известно. Где это практически возможно, валидация должна быть завершена до поставки или применения продукции. Записи результатов валидации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
7.3.7 Управление изменениями проекта и разработки
Изменения проекта и разработки должны быть идентифицированы, а записи должны поддерживаться в рабочем состоянии. Изменения должны быть проанализированы, верифицированы и валидированы соответствующим образом, а также одобрены до внесения. Анализ изменений проекта и разработки должен включать в себя оценку влияния изменений на составные части и уже поставленную продукцию. Записи результатов анализа изменений и любых необходимых действий должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
7.3.7.1 Уведомление
Об изменениях, которые влияют на какие-либо предоставляемые данные, следует сообщать потребителю, а в случае предоставления организацией технического досье/основного файла - непосредственно регулирующим органам.
7.3.7.2 Изменение проекта
При осуществлении изменения действующая валидация и документы, на которые повлияло изменение, должны быть проанализированы и пересмотрены, а персонал должен пройти соответствующую переподготовку.
Примечания
1 К выходным данным проектирования и разработки относятся записи (технические условия, технология производства, технические чертежи, технические или исследовательские протоколы) и образцы.
2 Конфиденциальная научно-техническая информация (организации) может быть представлена в виде досье непосредственно в регулирующие органы (например, техническое досье и (или) основной файл).
7.4 Закупки
7.4.1 Процесс закупок
Организация должна обеспечивать соответствие закупленной продукции установленным требованиям к закупкам. Тип и степень управления, применяемые по отношению к поставщику и закупленной продукции, должны зависеть от ее воздействия на последующие стадии жизненного цикла продукции или готовую продукцию. Организация должна оценивать и выбирать поставщиков на основе их способности поставлять продукцию в соответствии с требованиями организации. Должны быть разработаны критерии отбора, оценки и повторной оценки. Записи результатов оценивания и любых необходимых действий, вытекающих из оценки, должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
7.4.1.1 Организация должна утвердить поставщиков:
a) исходных материалов;
b) вспомогательных средств, важных для обеспечения качества;
c) упаковочных материалов для использования в чистых помещениях.
7.4.1.2 Организация должна уведомить потребителя до передачи сторонней организации любой части производственного процесса.
7.4.1.3 Все услуги, предоставляемые сторонними организациями, которые могут повлиять на качество продукции, должны контролироваться, включая подготовку печатного оригинала, услуги лабораторий, стерилизацию, калибровочные услуги и квалификационные услуги, техническое обслуживание, очистку, перевозку, борьбу с вредителями и подрядчиков по переработке отходов в зависимости от рисков.
7.4.1.4 Консультанты, дающие рекомендации по производству и контролированию первичных упаковочных материалов, считаются поставщиками.
7.4.1.5 Поставщики материалов и услуг, важных для обеспечения качества, утверждаются службой качества или лицом, назначенным службой качества.
7.4.1.6 Организация должна оценивать и вести записи по компетентности лабораторий, выполняющих работы, важные для обеспечения качества. Для выполнения работ, важных для обеспечения качества, организация должна использовать только лаборатории, одобренные с точки зрения их компетентности.
7.4.1.7 Если выполнение процесса стерилизации поручено сторонней организации, организация должна убедиться в том, что процесс отвечает требованиям 7.5.1 и 7.5.2.
7.4.1.8 Изменение источника получения сырья, важного для обеспечения качества, подлежит процессу управления изменениями.
7.4.2 Информация по закупкам
Информация по закупкам должна описывать заказанную продукцию, включая, где это необходимо, требования: a) к официальному одобрению продукции, процедур, процессов и оборудования; b) к квалификации персонала; c) к системе менеджмента качества. Организация должна обеспечивать достаточность установленных требований к закупкам до их сообщения поставщику. [ИСО 9001:2008] |
7.4.2.1 Организация должна сохранять соответствующую информацию по закупкам, т.е. документы (см. 4.2.3) и записи (см. 4.2.4), в объеме, требуемом для обеспечения прослеживаемости согласно 7.5.3.
7.4.3 Верификация закупленной продукции
Организация должна разработать и осуществлять контроль или другую деятельность, необходимую для обеспечения соответствия закупленной продукции установленным требованиям к закупкам. Если организация или ее потребитель предполагает осуществить верификацию у поставщика, то организация должна установить меры по верификации и порядок выпуска продукции в информации по закупкам. [ИСО 9001:2008] |
7.4.3.1 Поступающие материалы подлежат физическому или административному карантину до их одобрения и выпуска в эксплуатацию.
Примечание - В исключительных случаях может использоваться испытываемый материал при условии наличия гарантированно надежных процедур, не допускающих выпуска первичных упаковочных материалов до подтверждения статуса таких материалов.
7.4.3.2 Следует вести записи верификации (см. 4.2.4).
7.4.3.3 Выборочный контроль должен осуществляться в соответствии с методикой выборочного контроля с использованием процедур, средств и оборудования, не допускающих загрязнения.
7.5 Производство и обслуживание
7.5.1 Управление производством и обслуживанием
Организация должна планировать и осуществлять производство и обслуживание в управляемых условиях. Управляемые условия должны включать в себя, там, где это применимо: a) наличие информации, описывающей характеристики продукции; b) наличие рабочих инструкций в случае необходимости; c) применение подходящего оборудования; d) наличие и применение оборудования для мониторинга и измерений; e) проведение мониторинга и измерений; f) осуществление выпуска, поставки и действий после поставки продукции. [ИСО 9001:2008] |
g) определение даты изготовления с учетом задействованных процессов;
h) уделение особого внимания маркировке, этикетированию и упаковке для обеспечения результативного контроля и предотвращения ошибок;
i) документированную процедуру с определением управления отклонениями в процессе. Должны быть исследованы отклонения, важные для обеспечения качества, и зарегистрированы полученные результаты (см. 4.2.4).
7.5.1.1 Управление чистотой продукции и загрязнением
7.5.1.1.1 Организация должна сформулировать и соблюдать документированные требования к чистоте первичных упаковочных материалов и процедуры по предотвращению загрязнения продукции и оборудования.
Должны оцениваться потенциальные риски, связанные с любыми материалами или вспомогательными средствами, которые могут нести угрозу безопасности пациента, например, трансмиссивная губчатая энцефалопатия.
7.5.1.1.2 Все технологические процессы, осуществляемые в чистых или контролируемых зонах, включая меры по охране окружающей среды, производство, контроль в процессе производства и упаковку первичных упаковочных материалов, должны отвечать установленным местным условиям и функциональным критериям. Чистые помещения должны быть оборудованы воздушными шлюзами.
7.5.1.1.3 Технологические процессы в контролируемых окружающих условиях должны быть согласованы между потребителем и организацией.
7.5.1.1.4 Организация должна также формулировать документированные требования к чистоте первичных упаковочных материалов, когда:
a) первичные упаковочные материалы очищаются организацией до проведения стерилизации и (или) их использования;
b) первичные упаковочные материалы должны поставляться нестерилизованными и их чистота важна для использования;
c) технологические реагенты должны быть удалены с продукции в период производства.
7.5.1.1.5 Должны быть определены контейнеры для хранения, сборники тары и линии загрузки и разгрузки.
7.5.1.1.6 Основное внимание (например, идентификация, безопасность, чистота) следует уделять в первую очередь загрузке и разгрузке контейнеров для насыпных грузов/бункеров.
7.5.1.1.7 Для очистки оборудования, используемого при производстве первичных упаковочных материалов, должны быть разработаны письменные процедуры. Должны вестись записи очистки оборудования, имеющие важное значение для обеспечения качества первичных упаковочных материалов (см. 4.2.4).
7.5.1.1.8 Следует определять содержание и уровень очистки производственного оборудования/производственных участков.
7.5.1.1.9 Изготовлению некоторых материалов свойственно применение переработанных материалов (например, изготовлению стекла, алюминия, бумаги, термопластиков). Параметры переработки должны быть определены и согласованы с потребителем.
7.5.1.1.10 Если это не согласовано с потребителем, термопластичные материалы не должны подвергаться повторному измельчению или не подлежат повторному использованию в первичных упаковочных материалах.
7.5.1.1.11 Должен быть организован контроль очистки линий между различными сериями для удаления всех материалов и документов, не требуемых для последующей операции. Должны вестись записи работ по очистке линий (см. 4.2.4).
7.5.1.2 Управление изменениями
7.5.1.2.1 Организация должна реализовать процесс результативного и эффективного управления изменениями, чтобы изменения отрицательно не влияли на качество продукции и чтобы они отвечали потребностям и ожиданиям заинтересованных сторон.
7.5.1.2.2 Изменения должны быть идентифицированными, зарегистрированными, оцененными, проанализированными и управляемыми для понимания их влияния на другие процессы.
7.5.1.2.3 Для обеспечения управляемости необходимо определить полномочия, связанные с инициацией, анализом и утверждением изменений.
7.5.1.3 Конкретные требования к стерильности первичных упаковочных материалов
Организация должна вести записи (см. 4.2.4) параметров процесса стерилизации, который использовался для стерилизации каждой серии. Записи о стерилизации должны быть прослеживаемыми для каждой серии первичных упаковочных материалов.
7.5.2 Валидация процессов производства и обслуживания
Организация должна валидировать все процессы производства и обслуживания, результаты которых не могут быть верифицированы последующим мониторингом или измерениями, из-за чего недостатки становятся очевидными только после начала использования продукции или после предоставления услуги. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов. Организация должна разработать меры по этим процессам, в том числе там, где это применимо: a) определенные критерии для анализа и утверждения процессов; b) утверждение соответствующего оборудования и квалификации персонала; c) применение конкретных методов и процедур; d) требования к записям (4.2.4); e) повторную валидацию. [ИСО 9001:2008] |
7.5.2.1 Организация должна идентифицировать процессы, важные для обеспечения качества, в рамках своей деятельности, а именно те, которые влияют на качество первичных упаковочных материалов. Контроль за любыми из этих процессов, выход которых не может быть верифицирован посредством последующего мониторинга или измерения, должен демонстрироваться путем валидации и документироваться.
7.5.2.2 Для определения того, какие процессы важны для обеспечения качества, и степени валидации, необходимой для демонстрации управления этими процессами, используется оценка риска. Анализ риска должен быть связан со свойствами продукции, определяющими ее качество.
7.5.2.3 Технические системы (например, машины, линии, структурные компоненты производства и т.п.), используемые для изготовления первичных упаковочных материалов, должны проходить верификацию и (или) аттестацию/валидацию в соответствии с документированной оценкой риска.
7.5.2.4 Верификация и (или) аттестация/валидация должны проводиться при возникновении существенных изменений в средствах производства, оборудовании и процессах, которые могут повлиять на качество продукции.
Примечание - Управление изменениями в процессе валидации является частью политики управления изменениями организации.
7.5.2.5 Насколько это целесообразно, валидация той или иной продукции должна проводиться по согласованию с потребителем.
7.5.2.6 Результаты валидации должны регистрироваться (см. 4.2.4). Записи о валидации должны сохраняться на протяжении всего срока службы оборудования и процесса и еще в течение двух лет после снятия с эксплуатации или по договоренности с потребителем.
7.5.2.7 В отношении программного обеспечения, используемого в процессах, важных для обеспечения качества, должны проводиться функциональные испытания в остаточном количестве и в соответствующих условиях для верификации прослеживаемости, точности передачи и сохранения данных. Система должна проверяться, например, путем ввода правильных и неправильных данных для определения прослеживаемости, точности передачи и сохранения данных или записей.
7.5.2.8 Результаты этих испытаний и проверок должны регистрироваться (4.2.4).
7.5.2.9 Электронные записи должны сохраняться и защищаться от потери и случайного повреждения и должны храниться в форме, допускающей восстановление; если это невозможно, должны сохраняться бумажные экземпляры в течение двух лет после списания оборудования или по договоренности с потребителем (см. 4.2.4.1).
Примечание - Дополнительные данные, касающиеся защищенности данных, менеджмента и валидации программного обеспечения содержатся в МЭК 60601-1-4, Руководстве по надлежащей практике автоматизированного производства и Кодексе Федеральных регламентов Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США 21, часть 11.
7.5.2.10 При передаче на сторону любого процесса, важного для обеспечения качества, организация должна убедиться в том, что процесс отвечает требованиям настоящего стандарта.
7.5.2.11 Если требуется стерилизация, организация должна установить документированные процедуры по валидации процессов стерилизации. Процессы стерилизации должны пройти валидацию до первоначального использования. Должны сохраняться записи результатов валидации процессов стерилизации (см. 4.2.4).
См. ИСО 11135, ИСО 11137-1 или ИСО 11137-2.
7.5.2.12 Если одним из требований является стерилизация, организация должна подвергнуть первичные упаковочные материалы валидированному процессу стерилизации и записать все контрольные параметры процесса стерилизации. При передаче на сторону процесса стерилизации организация должна убедиться в том, что процесс отвечает требованиям настоящего стандарта.
См. ИСО 14937.
7.5.3 Идентификация и прослеживаемость
Если это возможно и целесообразно, организация должна идентифицировать продукцию с помощью соответствующих средств на всех стадиях ее жизненного цикла. Организация должна идентифицировать статус продукции по отношению к требованиям мониторинга и измерений на всех стадиях ее жизненного цикла. Если прослеживаемость является требованием, то организация должна управлять специальной идентификацией продукции и поддерживать записи в рабочем состоянии (4.2.4). Примечание - В ряде отраслей промышленности менеджмент конфигурации является средством поддержания идентификации и прослеживаемости. [ИСО 9001:2008] |
7.5.3.1 Организация должна разработать и поддерживать в работоспособном состоянии систему прослеживания всех производственных материалов от источника получения до реализации продукции с определением масштаба и требуемых записей на основе оценки риска (см. 4.2.4, 8.3 и 8.5).
7.5.3.2 Записи о серийном производстве должны быть обозначены отличительным номером серии или идентификационным номером.
7.5.3.3 Должны сохраняться записи об использовании оборудования, важного для обеспечения качества (см. 4.2.4). Эти записи должны также включать очистку и техническое обслуживание наряду с производственными операциями. Работы по техническому обслуживанию должны документироваться и прослеживаться до конкретной производственной операции или единицы оборудования.
7.5.3.4 Организация должна разработать и вести документированные процедуры с тем, чтобы возвращаемые организации первичные упаковочные материалы, например, для повторной обработки по установленным требованиям, были идентифицированы и выделены из общего производства.
7.5.4 Собственность потребителей
Организация должна проявлять заботу о собственности потребителя, пока она находится под управлением организации или используется ею. Организация должна идентифицировать, верифицировать, защищать и сохранять собственность потребителя, предоставленную для использования или включения в продукцию. Если собственность потребителя утеряна, повреждена или признана непригодной для использования, организация должна известить об этом потребителя и поддерживать записи в рабочем состоянии (4.2.4). Примечание - Собственность потребителя может включать в себя интеллектуальную собственность и сведения личного характера. [ИСО 9001:2008] |
7.5.5 Сохранение соответствия продукции
Организация должна сохранять продукцию в ходе внутренней обработки и в процессе поставки к месту назначения в целях поддержания ее соответствия установленным требованиям. Если это применимо, сохранение соответствия продукции должно включать в себя идентификацию, погрузочно-разгрузочные работы, упаковку, хранение и защиту. Требование сохранения соответствия должно быть также применено и к составным частям продукции. [ИСО 9001:2008] |
7.5.5.1 Организация должна разработать и поддерживать в работоспособном состоянии систему контролирования продукции с ограниченным сроком хранения или требующую особых условий хранения. Такие особые условия хранения должны контролироваться и регистрироваться (см. 4.2.4). Сроки хранения должны быть оправданными.
7.5.5.2 Продукция должна быть четко обозначена, изолирована и должна подлежать ответственному хранению, а также должна быть защищена от попадания посторонних веществ или загрязнения. Упаковка, используемая для создания и вмещения продукции, должна быть чистой и подходящей. Поставки должны сопровождаться соответствующей документацией. Документация на поставку должна соответствовать конкретной серии.
7.5.5.3 В случае повторного использования контейнеров для упаковки предшествующие ярлыки должны быть удалены или надписи на них стерты. Контейнеры должны быть чистыми или проверенными на чистоту в соответствии с письменной процедурой.
7.5.5.4 В случае необходимости на ярлыке должны быть указаны и должны выполняться особые условия транспортировки или хранения первичных упаковочных материалов.
7.6 Управление оборудованием для мониторинга и измерений
Организация должна определить мониторинг и измерения, которые предстоит осуществлять, а также оборудование для мониторинга и измерений, необходимое для обеспечения свидетельства соответствия продукции установленным требованиям. Организация должна установить процессы для обеспечения того, чтобы мониторинг и измерения могли быть выполнены и в действительности были выполнены в соответствии с требованиями к ним. |
Там, где необходимо обеспечивать имеющие законную силу результаты, измерительное оборудование должно быть: a) откалибровано и/или поверено в установленные периоды или перед его применением по эталонам, передающим размеры единиц в сравнении с международными или национальными эталонами. При отсутствии таких эталонов база, использованная для калибровки или поверки, должна быть зарегистрирована (4.2.4); b) отрегулировано или повторно отрегулировано по мере необходимости; c) идентифицировано в целях установления статуса калибровки; d) защищено от регулировок, которые сделали бы недействительными результаты измерения; e) защищено от повреждения и ухудшения состояния в ходе обращения, технического обслуживания и хранения. |
Кроме того, организация должна оценить и зарегистрировать правомочность предыдущих результатов измерения, если обнаружено, что оборудование не соответствует требованиям. Организация должна предпринять соответствующее действие в отношении такого оборудования и любой измеренной продукции. Записи результатов калибровки и поверки должны поддерживаться в рабочем состоянии (4.2.4). Если при мониторинге и измерении установленных требований используют компьютерные программные средства, их способность удовлетворять предполагаемому применению предварительно должна быть подтверждена и повторно подтверждена по мере необходимости. Примечание - Подтверждение соответствия компьютерного программного обеспечения предполагаемому применению обычно предусматривает его верификацию и менеджмент конфигурации в целях поддержания его пригодности для использования. [ИСО 9001:2008] |
7.6.1 Должны проводиться регулярные регистрируемые провокационные испытания автоматического контрольного оборудования (например, фотокамерные инспекционные системы и считыватели штрих-кодов) для проверки постоянной функциональной пригодности.
7.6.2 Производственные и контрольные данные, относящиеся к продукции потребителя (за исключением секретной интеллектуальной собственности организации), должны предоставляться по требованию потребителя или представителя потребителя для проверки функциональной пригодности производственного процесса, контрольного и испытательного оборудования, используемого в ходе производства и на готовой продукции.
7.6.3 Испытательное оборудование, используемое для определения приемлемости исходных материалов, важных для обеспечения качества, полуфабрикатов или готовой продукции, должно калиброваться или должны проводиться дополнительные квалификационные испытания по мере необходимости.
8 Измерение, анализ и улучшение
8.1 Общие положения
Организация должна планировать и применять процессы мониторинга, измерения, анализа и улучшения, необходимые для: a) демонстрации соответствия требованиям к продукции; Это требование относится как к полуфабрикатам, так и к первичным упаковочным материалам. b) обеспечения соответствия системы менеджмента качества; c) постоянного повышения результативности системы менеджмента качества. Указанная деятельность должна включать в себя определение применимых методов, в том числе статистических, и область их использования. [ИСО 9001:2008] |
8.2 Мониторинг и измерение
8.2.1 Удовлетворенность потребителей
Организация должна проводить мониторинг информации, касающийся восприятия потребителем выполнения организацией его требований, как одного из способов измерения работы системы менеджмента качества. Должны быть установлены методы получения и использования этой информации. Примечание - Мониторинг восприятия потребителями может включать в себя получение информации из таких источников, как исследования удовлетворенности потребителей, данные от потребителей о качестве поставленной продукции, исследования мнений пользователей, анализ оттока клиентов, благодарности, претензии по гарантийным обязательствам и отчеты распространителей. [ИСО 9001:2008] |
8.2.2 Внутренние аудиты (проверки)
Организация должна проводить внутренние аудиты (проверки) через запланированные интервалы времени в целях установления того, что система менеджмента качества: a) соответствует запланированным мероприятиям (7.1), требованиям настоящего стандарта и требованиям к системе менеджмента качества, разработанным организацией; b) внедрена результативно и поддерживается в рабочем состоянии. Программа аудитов (проверок) должна планироваться с учетом статуса и важности процессов и участков, подлежащих аудиту, а также результатов предыдущих аудитов. Критерии, область применения, частота и методы аудитов должны быть определены. Выбор аудиторов и проведение аудитов должны обеспечивать объективность и беспристрастность процесса аудита. Аудиторы не должны проверять свою собственную работу. |
Должна быть установлена документированная процедура для определения ответственности и требований, связанных с планированием и проведением аудитов, ведением записей и составлением отчетов о результатах. Записи об аудитах и их результатах должны поддерживаться в рабочем состоянии (4.2.4). Руководство, ответственное за проверяемые области деятельности, должно обеспечить, чтобы все необходимые коррекции и корректирующие действия предпринимались без излишней отсрочки для устранения обнаруженных несоответствий и вызвавших их причин. Последующие действия должны включать в себя верификацию принятых мер и отчет о результатах верификации (8.5.2). Примечание - См. ИСО 19011 для руководства. [ИСО 9001:2008] |
8.2.3 Мониторинг и измерение процессов
Организация должна использовать подходящие методы мониторинга и, где это применимо, измерения процессов системы менеджмента качества. Эти методы должны демонстрировать способность процессов достигать запланированных результатов. Если запланированные результаты не достигаются, то должны предприниматься необходимые коррекции и корректирующие действия. Примечание - При определении подходящих методов организация должна учитывать тип и объем мониторинга или измерений, подходящих для каждого из таких процессов, в отношении их влияния на соответствие требованиям к продукции и на результативность системы менеджмента качества. [ИСО 9001:2008] |
Служба качества должна обеспечивать проведение исследования, устранения и документирования отклонений, важных для обеспечения качества.
8.2.4 Мониторинг и измерение продукции
Организация должна осуществлять мониторинг и измерять характеристики продукции в целях верификации соблюдения требований к продукции. Это должно осуществляться на соответствующих стадиях процесса жизненного цикла продукции согласно запланированным мероприятиям (7.1). Свидетельства соответствия критериям приемки должны поддерживаться в рабочем состоянии. Записи должны указывать лицо(а), санкционировавшее(ие) выпуск продукции (4.2.4). Выпуск продукции и предоставление услуги потребителю не должны осуществляться до тех пор, пока все запланированные действия (7.1) не будут удовлетворительно завершены, если не утверждено иное соответствующим полномочным лицом или органом и, где это применимо, потребителем. [ИСО 9001:2008] |
8.2.4.1 Расследование результатов за пределами заданных технических требований
Любой результат за пределами заданных технических требований должен расследоваться в соответствии с документированной процедурой, а полученные данные должны регистрироваться (см. 4.2.4).
8.2.4.2 Входной контроль и входные испытания
Должны быть установлены и должны выполняться требования для всех используемых материалов. Поступающие материалы должны контролироваться или иным образом проверяться на соответствие установленным требованиям.
8.2.4.3 Контроль в процессе производства
8.2.4.3.1 Организация должна, как того требуют документированные процедуры, контролировать и испытывать продукцию в процессе обработки.
8.2.4.3.2 Должны быть определены процедуры выборочного контроля, обеспечивающие репрезентативность образцов для оцениваемого процесса. Образцы не следует возвращать в производственную зону, если они были направлены на отдельную испытательную площадку.
8.2.4.3.3 После поломки оборудования или незапланированной остановки процесса должен быть проведен дополнительный контроль в процессе производства.
8.2.4.4 Контроль готовой продукции
Если требуется контроль готовой продукции, он должен быть проведен до выпуска серии.
Примечание - Контроль готовой продукции может проводиться не по всем установленным параметрам.
8.2.4.5 Сохраняемые образцы
Сохраняемые образцы отбираются в соответствии с требованиями организации и (или) потребителя.
8.2.4.6 Выпуск серии
Для выпуска серии требуется проведение анализа документации на серию.
8.3 Управление несоответствующей продукцией
Организация должна обеспечивать идентификацию продукции, не соответствующей требованиям, и управление ею в целях предотвращения непреднамеренного использования или поставки такой продукции. Должна быть установлена документированная процедура для определения средств управления и соответствующей ответственности и полномочий для действий с несоответствующей продукцией. Если применимо, организация должна предпринимать в отношении несоответствующей продукции следующие действия (одно или несколько): a) устранение обнаруженного несоответствия; b) санкционирование использования, выпуска или приемки продукции, если получено разрешение на отклонение от соответствующего полномочного лица или органа и, где это применимо, потребителя; |
c) предотвращение ее первоначального предполагаемого использования или применения; После того как несоответствующая продукция исправлена, она должна быть подвергнута повторной верификации для подтверждения соответствия требованиям. Записи о характере несоответствий и любых последующих предпринятых действиях, включая полученные разрешения на отклонения, должны поддерживаться в рабочем состоянии (4.2.4). [ИСО 9001:2008] |
8.3.1 Несоответствующая продукция или несоответствующие материалы должны подвергаться карантину в ожидании принятия решения о корректирующих или иных действиях. Когда идет речь об исправлении путем переделки или реконструкции, следует провести и зарегистрировать оценку риска любого отрицательного влияния переделки на продукцию (см. 4.2.4 и 7.5.1).
8.3.2 Переделка и (или) реконструкция должна проводиться в соответствии с документированной процедурой, одобренной службой качества. Порядок переделки должен быть согласован с потребителем, если это является оговоренным требованием.
8.3.3 Если первичные упаковочные материалы производились в условиях чистого помещения, любая переделка должна выполняться в тех же условиях.
8.3.4 Любое предложение о выпуске несоответствующей продукции должно быть сделано на основе документально оформленного разрешенного отступления, санкционированного потребителем.
8.3.5 В случае выбраковки первичные упаковочные материалы должны утилизироваться или уничтожаться в соответствии с документированной процедурой.
8.4 Анализ данных
Организация должна определять, собирать и анализировать соответствующие данные для демонстрации пригодности и результативности системы менеджмента качества, а также оценивания, в какой области возможно постоянное повышение результативности системы менеджмента качества. Данные должны включать в себя информацию, полученную в результате мониторинга и измерения и из других соответствующих источников. Анализ данных должен представлять информацию, относящуюся: a) к удовлетворенности потребителей (8.2.1); b) к соответствию требованиям к продукции (8.2.4); c) к характеристикам и тенденциям процессов и продукции, включая возможности проведения предупреждающих действий (8.2.3 и 8.2.4); d) к поставщикам (7.4). [ИСО 9001:2008] |
Организация должна разработать и поддерживать документированные процедуры, включая требования к анализу данных, для выявления существующих или потенциальных причин выпуска несоответствующей продукции или других проблем качества.
8.5 Улучшение
8.5.1 Постоянное улучшение
Организация должна постоянно повышать результативность системы менеджмента качества посредством использования политики и целей в области качества, результатов аудитов, анализа данных, корректирующих и предупреждающих действий, а также анализа со стороны руководства. [ИСО 9001:2008] |
Изменения, предлагаемые в качестве части постоянного улучшения, подлежат менеджменту риска.
8.5.2 Корректирующие действия
Организация должна предпринимать корректирующие действия в целях устранения причин несоответствий для предупреждения повторного их возникновения. Корректирующие действия должны быть адекватными последствиям выявленных несоответствий. Должна быть разработана документированная процедура для определения требований: a) к анализу несоответствий (включая жалобы потребителей); b) к установлению причин несоответствий; c) к оцениванию необходимости действий, чтобы избежать повторения несоответствий; d) к определению и осуществлению необходимых действий; e) к записям результатов предпринятых действий (4.2.4); f) к анализу результативности предпринятых корректирующих действий. [ИСО 9001:2008] |
8.5.2.1 Организация должна своевременно расследовать все жалобы потребителей и сообщить о проводимых корректирующих действиях на все производственные участки и участки, связанные с производством. Действия должны осуществляться как можно быстрее и в соответствии с согласованным графиком, следует вести записи расследования (см. 4.2.4).
8.5.2.2 Должно быть обосновано и зарегистрировано отсутствие корректирующих и (или) предупреждающих действий по результатам расследования жалоб потребителей (см. 4.2.4).
8.5.3 Предупреждающие действия
Организация должна определять действия в целях устранения причин потенциальных несоответствий для предупреждения их появления. Предупреждающие действия должны соответствовать возможным последствиям потенциальных проблем. Должна быть разработана документированная процедура для определения требований: a) к установлению потенциальных несоответствий и их причин; b) к оцениванию необходимости действий в целях предупреждения появления несоответствий; c) к определению и осуществлению необходимых действий; d) к записям результатов предпринятых действий (4.2.4); e) к анализу результативности предпринятых предупреждающих действий. [ИСО 9001:2008] |
Приложение А
(обязательное)
Требования GMP к первичным печатным упаковочным материалам
А.1 Средства получения печатных оттисков
А.1.1 Общие положения
Все средства получения печатных оттисков должны:
a) иметь четкое и однозначное обозначение, чтобы обеспечить прослеживаемость до исходного материала;
b) быть изготовлены из основного исходного материала, находящегося у потребителя, и обеспечивать прослеживаемость до этого материала;
c) проверяться на соответствие бумажному носителю или электронным данным, одобренным потребителем, и регистрироваться (см. 4.2.4);
d) храниться в надежном месте при наличии четкой системы санкционированного выпуска и возвращения в хранилище.
А.1.2 Сочетаемые печатные формы
Когда требуется больше одной печатной формы, должна существовать документированная система, обеспечивающая использование всех форм в комплекте. В том случае, когда комплект форм содержит типовой проект проведения нескольких работ, каждая отдельная форма в комплекте должна иметь четкое, однозначное обозначение и быть документирована.
А.1.3 Изменение текста/проекта
Когда проект требует использования нескольких форм и некоторые из них должны быть заменены из-за изменения в тексте/проекте, должна существовать документированная процедура, обеспечивающая замену непригодных форм и сохранение других средств в комплекте. Исходные формы подлежат повторной идентификации.
А.1.4 Верификация
А.1.4.1 Общие положения
Верификация проекта по средствам получения печатных оттисков должна проводиться в период подготовки к работе печатного станка и до получения разрешения на выпуск продукции.
А.1.4.2 Карантин и уничтожение
Организация должна иметь:
a) документированную процедуру, обеспечивающую надлежаще оформленную изоляцию средств подготовки печатного оригинала и печатания проекта, подвергающегося пересмотру;
b) документированную систему, детализирующую методику утилизации ненужных средств подготовки печатного оригинала и получения печатных оттисков; они должны приводиться в непригодное состояние и утилизироваться контролируемым и надежным способом.
А.2 Процессы печатания и преобразования
А.2.1 Наладка (подготовка к работе) печатного станка
А.2.1.1 Первоначальную подготовку к печатанию следует проводить с использованием чистых компонентов или материалов.
А.2.1.2 При подготовке к последующим процессам можно использовать материал из изначального процесса печатания той же серии.
А.2.1.3 Изначальный приправочный материал может повторно использоваться в ходе подготовительного процесса для получения правильного цвета.
А.2.1.4 Материал, используемый для приправки, должен быть изолирован, а затем утилизирован как производственные отходы.
А.2.2 Переходные системы
Переходные системы, предназначенные для сокращения времени, затрачиваемого на подготовку (например, автоматизированная смена формы) и не позволяющие проводить полную очистку пиний, подлежат документированной оценке рисков и должны использовать средства управления для обеспечения защиты продукции. Все печатные издания, полученные в ходе предыдущей операции, должны удаляться с линии до получения официального разрешения на выпуск тиража. Все средства управления должны регистрироваться (см. 4.2.4).
А.2.3 Сохраняемые образцы
А.2.3.1 Все сохраняемые печатные образцы должны быть четко обозначены и должны подлежать надежному хранению.
А.2.3.2 Образцы, используемые для иных целей (например, администрирование/продажа), должны уничтожаться, если они больше не контролируются организацией.
А.2.4 Запасные печатные средства
А.2.4.1 В ходе производства, если запасные формы изготавливают из фиксированных разрешенных исходных материалов (например, негативов или пошаговых снимков с использованием технологии компьютер-форма), операция может быть продолжена после проведения новой проверки первой партии.
А.2.4.2 Заменяемые печатные средства
А.2.4.1* В ходе производства, если заменяемые формы изготавливают из фиксированных одобренных исходных материалов (например, негативов или пошаговых снимков с использованием технологии компьютер-форма), операция может быть продолжена после проведения новой проверки первой партии.
_______________
* Нумерация соответствует оригиналу. - .
А.2.4.2* В ходе производства, если формы создают из новых материалов, операция должна быть отменена; заменяемые формы необходимо рассматривать как новые оригиналы. Последующую продукцию необходимо рассматривать как новую серию. Введение всех заменяемых печатных средств необходимо регистрировать (см. 4.2.4).
_______________
* Нумерация соответствует оригиналу. - .
А.2.5 Печать комплектов
Печать комплектов (процесс печатания более одного макета на подложке в ходе одного производственного цикла) не допускается из-за риска перекрестного загрязнения.
А.2.6 Серийное производство и хранение запасов
А.2.6.1 Серийное производство и хранение продукции на складе осуществляют, если это оговорено контрактом.
А.2.6.2 Организация должна управлять хранением для обеспечения безопасности и целостности продукции и сохранять ее прослеживаемость до производителя и используемых материалов.
А.2.7 Цифровая печать
А.2.7.1 Гибкие возможности цифровой печати внедряют новые виды деятельности, которыми необходимо управлять и документировать для обеспечения точности и безопасности печатной продукции.
А.2.7.2 Применение цифровой печати и других специальных требований к продукции должно быть согласовано с потребителем.
А.2.7.3 Организация должна разработать надежную систему доступа к файлам, предназначенную для предотвращения непреднамеренного использования несоответствующих исходных файлов.
А.2.7.4 Пока альтернативная система безопасности не разработана, контролирующий компьютер, связанный с цифровой печатной машиной, должен содержать только один исходный файл в своей памяти для текущего производственного цикла, и удаление этого файла должно быть частью документированной очистки линии.
А.2.7.5 Рабочие параметры для достижения приемлемого цвета необходимо устанавливать в виде официального процесса и регистрировать (см. 4.2.4).
А.3 Системы безопасности с применением штрих-кода
А.3.1 Основные положения
Для обеспечения безопасности продукции и предотвращения перекрестного загрязнения штрих-коды безопасности могут быть включены в макет печатных упаковочных материалов с целью верификации организацией в процессе производства и/или потребителем в процессе операции упаковывания.
Организация может добавить собственные идентификационные коды в макет продукции, если это предусмотрено контрактом.
Там, где организация ответственна за определение штрих-кода безопасности, каждый цвет макета должен быть включен в штрих-код. Для всех штрих-кодов, если цвет несовместим с требованиями сканирующего оборудования, необходимо уведомить потребителя.
А.3.2 Методы верификации и оборудование для верификации
А.3.2.1 Если это практически возможно, каждую единицу продукции со штрих-кодом безопасности необходимо подвергать верификации посредством использования онлайн сканирующего оборудования, для обеспечения того что коды читаемы и произведена необходимая продукция. Сканирование штрих-кода безопасности должно выполняться в течение последнего допустимого производственного процесса.
А.3.2.2 Конфигурация программного обеспечения/контроля сканирующего оборудования должна загружаться из независимого источника (например, технические условия или официальное подтверждение). После того как код будет введен и проверен, сканирующее оборудование должно быть заблокировано (например, применение механического замка или защита паролем) для предотвращения несанкционированного доступа.
А.3.2.3 Должна быть разработана эффективная система выбраковывания любого продукта, не прошедшего процесс сканирования. Любой продукт, забракованный онлайн системой сканирования, должен быть проверен для определения причины выбраковки, а непригодные компоненты впоследствии должны быть утилизированы. Выводы должны быть зарегистрированы и проанализированы до выпуска продукции.
А.3.2.4 Онлайн сканирующее оборудование и связанный с ним механизм выбраковки должны подвергаться испытаниям в ходе производства для проверки того, эффективны ли они в обнаружении и удалении материалов с несоответствующими штрих-кодами. Проведение такого мониторинга должно иметь место в начале процесса через запланированные интервалы времени и быть зарегистрировано (см. 4.2.4).
А.3.2.5 Любая продукция, при производстве которой определено применение электронного сканирования, но фактически оно не выполняется, должна быть одобрена и отражена в записях о качестве. Потребитель должен быть уведомлен об этом, и получено документальное одобрение до выпуска продукции.
А.3.2.6 Для рулонного многополосного производства все линии должны подлежать верификации при помощи штрих-кода. Там, где это выполнить невозможно, при согласовании с потребителем только одна линия может подвергаться верификации.
А.3.2.7 Должно быть предусмотрено выполнение измерений вне технологического процесса и/или верификации образцов штрих-кодов на всех линиях.
А.3.3 Товарный штрих-код
В том случае, когда товарный штрих-код включен в макет (EAN, Code 39, PZN и т.д.), в течение всего процесса производства необходимо выполнять документированную проверку образца.
А.3.4 Рулонные материалы и продукция
А.3.4.1 Если иное не указано потребителем:
a) склейка должна быть произведена с использованием склеивающей ленты ярких цветов на обеих сторонах полотна, и должны быть проверены обе стороны склейки, для обеспечения того что идентичные материалы соединены и зарегистрированы.
b) Может быть определен предел максимального количества склеек.
А.3.4.2 Количество материала (длина, вес, численность) для производства каждого рулона должно быть определено в точных пределах, согласованных с потребителем и указанных на рулоне.
А.3.4.3 Номер серии, номер рулона и дата изготовления должны быть указаны на внутренней поверхности для каждого рулона.
А.3.4.4 Для предотвращения перекрестного загрязнения полотно должно быть выработано до появления чистого материала в конце производственного цикла, чтобы гарантировать отсутствие материала в печатной машине.
А.3.4.5 Там, где необходимо оставить печатный материал в оборудовании по переработке в связи с трудностью восстановления целостности рулонного материала (например, продольно-фрезерный станок), должна быть разработана документированная процедура для удаления и утилизации материала, используемого для проведения нового макета через машину.
А.3.4.6 Если существует вероятность производства материала с пропусками печати по причине макета или работы печатного оборудования, организация должна иметь безопасную систему для обнаружения, удаления и отделения продукция, произведенной с пропущенными цветом или текстом.
Приложение В
(справочное)
Методические указания по требованиям к верификации и валидации первичных упаковочных материалов
В.1 Общие положения
В настоящем справочном приложении описан подход при необходимости проведения валидации, исходя из допущения, что та или иная форма верификации всегда может потребоваться.
Данные методические указания относятся к требованиям по валидации, приведенным в 7.5.2 и 7.5.2.1, а также к требованиям по проектированию и разработке, приведенным в 7.3.
К предметам, которые могут потребовать квалификацию/валидацию (анализ риска показал, что одной верификации недостаточно), относятся:
- технические системы, такие как станки и линии для производства первичных упаковочных материалов;
- испытательное оборудование, используемое для определения приемлемости исходных материалов, полуфабрикатов или готовой продукции, важных для обеспечения качества;
- валидация отдельного изделия, если это необходимо или согласовано с потребителем.
Целью валидации является подтверждение стабильного изготовления и упаковки продукции в соответствии с требованиями к продукции. Это средство получения документированного свидетельства того, что оборудование и процессы, используемые для производства и испытания первичных упаковочных материалов, обеспечивают стабильный выпуск продукции, отвечающей заранее установленным техническим требованиям.
Валидация требуется, в тех случаях, когда организация установила, либо потребитель решил, что одной только верификации недостаточно. При использовании одной только верификации должен быть комплект документации с указанием критериев приемки и результатов приемочных испытаний на месте изготовления (см. В.16), который устанавливает требования к мониторингу и измерению в процессе повседневного производства.
В.2 Факторы, которые необходимо учитывать
В.2.1 Общие положения
В соответствии с 7.5.2 официальная квалификация/валидации требуется исключительно для оценки определенных параметров, важных для обеспечения качества, которые не могут быть верифицированы с помощью обычного мониторинга или измерения. В настоящем разделе содержатся методические указания относительно того, когда может потребоваться валидация.
Калибровка проводится отдельно от валидации.
В.2.2 Факторы, которые необходимо учитывать до проведения верификации/квалификации/валидации
До проведения верификации, квалификации или валидации процесса, требуемого для изготовления первичных упаковочных материалов, необходимо учитывать следующие факторы:
- состав и технические характеристики материалов;
- оборудование, на котором они производятся и упаковываются;
- определение функций и обязанностей (организации и потребителя);
- потенциальное воздействие первичных упаковочных материалов на безопасность лекарственных средств и пациентов;
- определение критических параметров процесса;
- требования к уровню подготовки (операторы, специалисты по качеству, инженеры и т.п.);
- стандартные рабочие процедуры для получения сведений о том, как изготавливать/эксплуатировать и содержать оборудование.
В.3 Верификация/валидация программного обеспечения
Для верификации программного обеспечения могут использоваться так называемые функциональные испытания. Валидация программного обеспечения не является обязательным требованием для изготовления первичных упаковочных материалов, если отсутствуют иные указания.
В.4 Функциональные испытания
Программное обеспечение, используемое в процессах, критичных для обеспечения качества, должно подвергаться функциональным испытаниям в достаточном количестве и в соответствующих условиях для верификации прослеживаемости, точности передачи размера единицы физической величины, обработки и сохранения данных. Система должна проверяться, например, путем ввода верных и неверных данных, для определения прослеживаемости, точности передачи размера единицы физической величины и сохранения данных и записей. Следует регистрировать результаты этих испытаний и проверок.
В ходе функциональных испытаний следует проводить повторные проверки критических этапов и параметров.
В.5 Управление изменениями
Официальное документированное управление изменениями должно быть частью процесса валидации, от окончания квалификации проекта (см. В.13) и на протяжении всего срока службы оборудования. Управление изменениями на этапах верификации или в ходе процессов валидации должно быть частью политики управления изменениями организации.
В.6 Последующая деятельность
Должны быть приняты меры по выполнению незавершенных действий, например, составление дефектных ведомостей по результатам квалификации функционирования (см. В.15) и квалификации эксплуатации (см. В.17).
Все принимаемые меры должны документироваться.
В.7 Поддержание статуса валидации
Следует поддерживать статус валидации оборудования и процесса. Любые изменения, даже незначительные, должны оцениваться на предмет их влияния на статус валидации оборудования или продукции, и в случае необходимости должна проводиться подтверждающая проверка, эквивалентная квалификации эксплуатации (см. В.17).
Изменения/улучшения процедур валидации должны также проходить оценку для определения необходимости повторной валидации.
В.8 Подход к валидации
В.8.1 Перспективный подход к валидации
Валидация должна проводиться до начала производства в промышленных масштабах и следовать перспективному подходу. Альтернативные подходы к валидации приведены также в В.21.
На рисунке В.1 приведена блок-схема, демонстрирующая типичные этапы валидации.
В.8.2 Роли и обязанности
На рисунке В.1 показаны потенциальные сферы ответственности. В рамках этих функций до начала работ должны быть определены исполнители, ответственные за проведение процессов верификации или валидации и санкционирование результатов.
Служба качества несет ответственность за решения, связанные с выпуском продукции.
В.8.3 Планирование
В.8.3.1 Общие положения
Все стадии процесса верификации и (или) валидации должны планироваться и документироваться. Решение о виде требуемой документации должно приниматься в начале процесса, например, когда верификация является согласованным подходом, могут использоваться пользовательские технические требования для детализации процессов, требуемых для обеспечения верификации.
Целью валидации является установление выполнения требований пользователя, поэтому они должны быть четко определены.
В.8.3.2 Мастер-план валидации
Общий подход к валидации и план ее проведения должны содержаться в мастер-плане валидации.
Это документ, в котором описывается, как будет осуществляться валидация оборудования и (или) процесса, перечисляются основные обязанности и порядок выполнения деятельности. В плане указан подход к валидации, уделяя основное внимание аспектам, критичным для обеспечения качества оборудования и (или) процесса.
Он, как правило, включает следующие элементы:
- план проведения валидации;
- организационная структура работ по валидации - роли и обязанности;
- описание средств, систем, оборудования и процесса;
- формат документации - требуемый уровень документирования;
- планирование и составление графика;
- ссылки на существующие документы, если это необходимо;
- управление изменениями.
Когда требуется валидация разной продукции или различного оборудования, которое по результатам анализа риска считается равноценным, может понадобиться брэкетинг и (или) матричный подход к валидации. Обоснование такого решения должно быть документировано в плане валидации. Должен быть достаточный уровень знаний в отношении процесса и продукции для обоснования решения об использовании матричного подхода и (или) брэкетинга. При брэкетинге испытываются только крайние значения выделенных параметров. При матричном подходе может использоваться "план экспериментов" для сокращения количества требуемых валидационных серий.
В.9 Анализ риска
Анализ риска является неотъемлемой частью процесса принятия решений и может использоваться для определения необходимости проведения верификации и (или) валидации. Он может также использоваться в ходе квалификации проекта для определения любых потенциальных угроз для операторов или окружающей среды. Сюда относится доступ к станку (ограждение), движение материалов, воздействие лазеров и т.п. На рисунке В.1 показаны две стадии, когда требуется проведение анализа риска.
В.10 Технические требования пользователя
Стадия планирования включает разработку технических требований пользователя. Технические требования пользователя - это утверждаемый документ, в котором указаны все функциональные, рабочие и (или) технические свойства оборудования или процесса, необходимые для производства требуемого материала. Сюда может относиться схема компоновки оборудования, предусматривающая достаточное пространство для движения материалов и операторов, наличие запасных частей и легкость доступа для уборки и очистки линии. Как вариант эта информация может предоставляться в другой документации.
В.11 Условия контракта/заводские приемо-сдаточные испытания
Это юридически оформленное соглашение, заключаемое между организацией и поставщиком, подписываемое обеими сторонами и оговаривающее условия поставки. При необходимости проведения заводских приемо-сдаточных испытаний в документе должны быть оговорены требуемые испытания, где и кем они проводятся.
В.12 Этапы валидации
Существуют различные возможности проведения квалификации проекта (см. В.13), установочной квалификации (см. В.14), квалификации функционирования (см. В.15), приемо-сдаточных испытаний на площадке (см. В.16), квалификации эксплуатации (см. В.17) и анализа риска поставщиком и (или) организацией. Эта работа должна быть документально оформлена, например, организация может воспользоваться опытом поставщика при проведении исследований/оптимизации/приемо-сдаточных испытаний на площадке.
После составления плана проведения валидации может быть начат процесс квалификации. Квалификация доказывает, что продукция может систематически выпускаться в соответствии с требуемым стандартом (см. В.16).
В.13 Квалификация проекта
Это процесс верификации пригодности проекта. Он может включать соответствующий анализ риска, разработку пользовательских технических требований, условий контракта (возможно, в одном документе), заводские приемо-сдаточные испытания для определения готовности отправки оборудования от поставщика в организацию и испытаний на этапе разработки/оптимизационных испытаний до приемо-сдаточных испытаний на площадке (см. В.16).
Квалификация проекта не требуется для стандартизированного оборудования, изготовленного не на заказ.
Заводские приемо-сдаточные испытания могут проводиться с учетом внешних воздействий, т.е. нагрева, шума, света, пыли.
В.14 Установочная квалификация
Это утвержденное документально оформленное свидетельство того, что оборудование смонтировано в соответствии с письменными и одобренными техническими требованиями и чертежами и что после монтажа оно сохраняет все свойства согласно проектному заданию, калибровано в случае необходимости и снабжено всеми инструкциями по эксплуатации.
В.15 Квалификация функционирования
Это проведение испытаний на основе знаний процесса и систем, чтобы убедиться в том, что они функционируют, как и положено, на верхних и нижних эксплуатационных пределах с учетом колебаний в сырье и условиях, в которых работает оборудование.
Процесс и результаты испытаний должны быть официально документированы и одобрены, и могут быть отдельные документы, такие как план проведения испытаний/протокол испытаний (см. В.19.3.2), отчет/запись о проведении испытаний (см. В.19.3.4) и заключение (см. В.19.3.5).
Квалификация функционирования должна окончательно определять процедуры калибровки, оперирования (включая характер и периодичность проверок в процессе производства), очистки, подготовки операторов и профилактического технического обслуживания.
Для простого оборудования план проведения испытаний/протокол испытаний (см. В.19.3.2) и отчет/запись о проведении испытаний (см. В.19.3.4) могут быть одним документом, в каковом случае приемочные критерии устанавливаются этим документом.
В.16 Приемо-сдаточные испытания на площадке
Приемо-сдаточные испытания на площадке могут подтвердить успешное выполнение установочной квалификации и квалификации функционирования с помощью имитированного производства с использованием реальных производственных материалов. Выход подлежит мониторингу и измерению, а результаты должны использоваться для определения потребности в квалификации эксплуатации. Приемо-сдаточные испытания на площадке могут быть эквивалентны одной квалификации эксплуатации.
Приемо-сдаточные испытания на площадке могут также потребоваться для испытательного оборудования, включая верификацию калибровки и получения стабильных результатов.
В.17 Квалификация эксплуатации
Для квалификации эксплуатации используются проверочные испытания общей работы линии для подтверждения того, что она способна стабильно производить продукцию с одним и тем же уровнем качества. При испытаниях для квалификации эксплуатации используются производственные материалы для подтверждения стабильности процесса и возможности стабильного выпуска первичных упаковочных материалов в обычных производственных условиях. Процесс и результаты испытаний как минимум трех последовательных серий официально документируются и утверждаются.
Исключение из использования трех последовательных серий возможно в случае длительного производственного процесса, например, на выпуск одной серии материала может потребоваться несколько недель непрерывного производства. В этом случае квалификация эксплуатации может быть проведена на трех подпартиях, на выпуск каждой из которых требуется не менее одного дня.
В.18 Валидация продукции
Валидация продукции является необязательной либо в результате внутреннего решения организации, либо по требованию потребителя (см. рисунок В.1).
В случае особых требований рекомендуется проводить валидацию этапов процесса, критичных для обеспечения качества, для отдельного первичного упаковочного материала. Критичные этапы процесса должны испытываться на критичные параметры, выбранные в результате оценки риска. Валидация продукции включает определение возможностей оборудования и процесса и квалификацию средств управления полностью в онлайновом режиме.
В целом считается приемлемым, чтобы количество последовательных партий/серий с окончательно согласованными параметрами составляло валидацию процесса (см. руководство ЕС по GMP, приложение 15).
В.19 Документация по процессам верификации и валидации
В.19.1 Общие положения
Рекомендуется регистрировать подход к процессу верификации/валидации и каждую стадию процесса, а также полученные результаты (см. 4.2.4).
В.19.2 Верификация
Для документирования заводских приемо-сдаточных испытаний и приемо-сдаточных испытаний на площадке может использоваться единый документ. Он может содержать все или что-либо из следующего:
- историю развития или предыдущие испытания/верификацию/валидацию;
- обоснование используемого подхода;
- приемочные критерии;
- требуемые испытания;
- оценку результатов и заключений испытаний;
- дальнейшие действия;
- управление изменениями;
- получение разрешения.
В.19.3 Валидация
В.19.3.1 Объем документации
Основная документация по валидации должна включать:
- анализ риска;
- план проведения испытаний/протокол испытаний (план/протокол квалификации);
- отчет об испытаниях (запись испытаний);
- заключение и одобрение.
В.19.3.2 План проведения испытаний/протокол испытаний (план/протокол квалификации)
Сюда входят один или несколько документов с описанием испытаний на стадиях разработки и эксплуатации, включая квалификационные испытания. Эти документы должны содержать:
- основные этапы, которые считаются критичными для обеспечения качества;
- проводимые испытания и используемую методику;
- приемочные критерии;
- официальную визу для анализа и утверждения.
В.19.3.3 Контрольные файлы (в случае необходимости)
Речь идет об одном или нескольких документах с описанием и протоколированием испытаний на стадиях разработки и эксплуатации, включая квалификационные испытания. Эти документы должны содержать:
- результаты проводимых испытаний и описание используемой методики и используемого оборудования;
- приемочные критерии;
- официальную визу для анализа и утверждения.
В.19.3.4 Отчет/запись о проведении испытаний
Речь идет об одном или нескольких документах с описанием и протоколированием испытаний на стадиях разработки и эксплуатации, включая квалификационные испытания. Эти отчеты должны содержать:
- результаты проводимых испытаний;
- приемочные критерии;
- официальную визу для анализа и утверждения.
В зависимости от сложности выполняемой работы существует возможность объединения протокола испытаний, результатов испытаний и отчета о проведении испытаний в один документ по каждой стадии квалификации.
В.19.3.5 Заключение
Заключение представляет собой краткое описание результата квалификации. Заключение относится к задаче и требованиям, как они изложены в протоколе квалификации/валидации. Необходимо доказать соответствие задачи/требований полученным результатам. Отклонения должны быть обоснованы.
В.19.3.6 Итоговый отчет по валидации
Итоговый отчет по завершении всех стадий квалификации (квалификация проекта, установочная квалификация, квалификация функционирования, квалификация эксплуатации), который завершает процесс валидации. Он содержит:
- перекрестную ссылку на протоколы, результаты и рекомендации;
- изменения а планах и обоснование любого изменения;
- официальное одобрение и завершение.
В.20 Архивация
Записи о верификации и валидации должны сохраняться в течение всего срока службы оборудования и (или) процесса и в течение двух лет после списания.
В.21 Альтернативные подходы к валидации
В.21.1 Общие положения
Методические указания в В.1-В.20 касаются, прежде всего, перспективного подхода к валидации, т.е. валидации до начала коммерческого производства, поскольку этому в настоящее время отдается предпочтение в фармацевтической промышленности.
Организации могут решать, какой подход к валидации больше всего удовлетворяет требованиям с учетом его преимуществ. Альтернативными подходами являются следующие:
- совместная валидация;
- ретроспективная валидация;
- брэкетинг (матричная валидация).
В.21.2 Совместная валидация
Она включает квалификацию и (или) валидацию, проводимые параллельно с выпуском промышленной продукции, когда промышленная продукция выпускается до завершения валидации. Совместная валидация должна использовать принципы и процедуры, относимые к перспективной валидации. Совместная валидация относится исключительно к квалификации эксплуатации процесса.
В.21.3 Ретроспективная валидация
Ретроспективная валидация подразумевает, что промышленная продукция была выпущена до завершения процесса верификации/валидации.
Этот подход связан с получением документированного свидетельства того, что установленное и эксплуатируемое оборудование выпускает продукцию стабильного качества, путем проведения ретроспективного анализа данных, полученных по целому ряду функциональных параметров и сырьевых материалов. Такой анализ может распространяться на записи технического обслуживания и инженерные записи, записи о качестве и претензии потребителей. По завершении ретроспективной валидации заключения, относящиеся ко всем основным функциональным параметрам, должны быть документально оформлены в виде отчета о валидации.
В.21.4 Брэкетинг, или матричный подход
После подтверждения эквивалентности продукции или процесса существует возможность сокращения количества валидационных серий или количества технически идентичных станков/линий, подлежащих валидации, с использованием брэкетинга и (или) матричного подхода.
В.22 Дополнительные указания
Квалификация процессов, происходящих в ходе кампаний, является специфической особенностью производства первичных упаковочных материалов. В отдельных случаях станки/линии используются для планово-периодического производства. В этих случаях совместная валидация может быть прервана (на довольно длительное время) и возобновлена с началом новой производственной кампании.
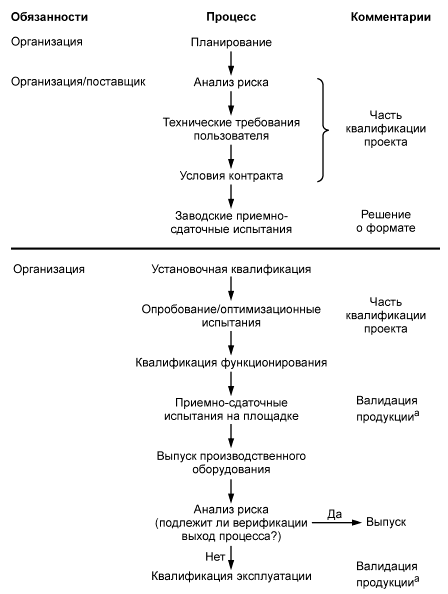
_______________
Как вариант: проведение валидации продукции:
- после внутреннего решения организации или
- по требованию потребителя.
Рисунок В.1 - Схема последовательности операций по верификации/валидации нового оборудования
Приложение С
(справочное)
Методические указания по менеджменту риска в связи с использованием первичных упаковочных материалов
С.1 Введение
В настоящем приложении содержатся методические указания, позволяющие организации определить опасности, связанные с проектированием/разработкой, изготовлением и поставкой первичных упаковочных материалов, дать количественную и качественную оценку рисков, управлять этими рисками и контролировать результативность такого управления. Оно не содержит рекомендаций по приемлемым уровням рисков в связи с большим ассортиментом первичных упаковочных материалов и широкими возможностями их применения.
Настоящее приложение следует рассматривать как основу для эффективного менеджмента организацией рисков, связанных с проектированием/разработкой и процессами изготовления и поставки первичных упаковочных материалов. Содержащиеся в нем методические указания создают структуру, в рамках которой систематически применяются опыт, понимание и оценочное суждение для менеджмента этих рисков и информирования о них.
Термины и определения содержатся в ИСО 14971 и Руководстве ИСО/МЭК 51.
С.2 Общие аспекты менеджмента рисков
Концепция риска обладает двумя основными элементами:
- вероятность возникновения опасности, т.е. как часто может возникать опасность;
- последствия опасности, т.е. насколько серьезными они могут быть.
На приемлемость риска влияют эти элементы и восприятие риска.
Менеджмент риска - это концепция, создающая основу для научных решений, будучи интегрированной в систему менеджмента качества или другие бизнес-процессы.
Хотя предпочтителен системный подход к менеджменту риска, не всегда целесообразно или необходимо использовать официальный процесс менеджмента риска, и формальность используемого подхода должна быть увязана со сложностью и (или) критичностью рассматриваемой проблемы.
Риск менеджмента, как он представлен на рисунке С.1, состоит из анализа риска, оценивания риска, управления риском, снижения риска и критического анализа. По завершении каждого этапа процесса менеджмента риска должны документироваться принятый подход, принятые решения и конечный результат. Комплект документов должен состоять, как минимум, из следующего:
a) описания продукции или процесса;
b) списка ответственных исполнителей или владельцев процесса;
c) даты завершения этапа процесса.
Деятельность по менеджменту рисков, как правило, осуществляется межфункциональными группами, ориентированными на выполнение определенной задачи. В группу могут входить технические специалисты помимо специалистов по менеджменту рисков.
Руководство должно нести ответственность за правильное применение процесса менеджмента рисков.
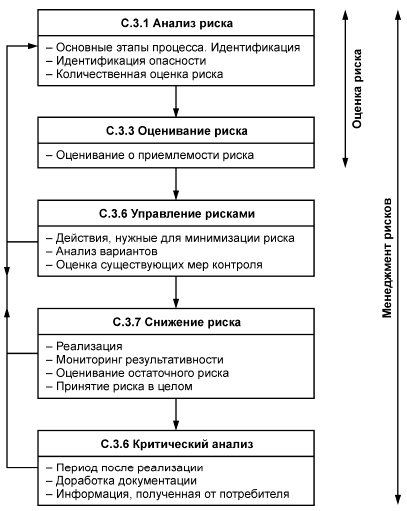
Рисунок С.1 - Схематическое представление процесса менеджмента рисков
С.3 Процесс оценки рисков
С.3.1 Общие положения
Оценка риска представляет собой систематический процесс организации информации в поддержку решения, которое принимается в ходе процесса менеджмента рисков (см. 7.5.2.2).
Процесс оценки рисков состоит из анализа риска и оценивания риска.
Оценка рисков должна использоваться для установления того, какие процессы являются критичными для обеспечения качества, и определения объема работ по валидации, необходимых для демонстрации управления этими процессами. Анализ риска должен быть связан с показателями качества продукции (см. 7.5.2).
С.3.2 Анализ риска
С.3.2.1 Общие положения
Анализ риска может быть количественным или качественным.
С.3.2.2 Выявление основных этапов процесса
Процесс оценки рисков должен охватывать все этапы цепочки поставок, которые контролируются организацией (плюс связи с поставщиками и потребителями), включая проектирование, изготовление и поставку (см. 7.1 и 7.5.2). Следует также принимать во внимание наличие совместной ответственности с другим предприятием или другой организацией, например, технические условия, подготовка печатного оригинала или аутсорсинг.
Выявление основных этапов должно охватывать используемые материалы, полуфабрикаты и готовую продукцию и производимые материалы, а также конкретные технические условия и (или) требования потребителя и должно также обеспечивать результативный обмен информацией.
С.3.3 Выявление опасностей, которые могут отразиться на качестве продукции
Следует убедиться в том, что известные зоны повышенного риска, жалобы потребителей и уязвимые места продукции включены в идентификацию опасностей. Подход может быть разным в зависимости от приоритетов организации и индивидуальных требований к продукции или процессу.
Для идентификации опасностей необходимо принимать во внимание следующие факторы.
С.3.3.1 Окружающая среда
Физические воздействия условий обработки и хранения (температура, время, влажность, дождь, давление воздуха, свет, вибрация и т.п.), зараженность вредителями, перекрестное загрязнение и ущерб от пожара или наводнения.
С.3.3.2 Оборудование
Тип, конструкция, состояние, возможности, местонахождение, установка, техническое обслуживание и калибровка.
С.3.3.3 Производственные мощности
Компоновка, системы инженерного обеспечения, техническое обслуживание, специализация и санитарно-гигиенические условия.
С.3.3.4 Управление использованием знаний
Интеллектуальная собственность, сохранение основных навыков.
С.3.3.5 Законодательство
Потенциальное нарушение действующего законодательства.
С.3.3.6 Материалы
Тождественность, контроль состояния, количество, обращение, технические требования, меры безопасности, контроль контрафакции и состояние материалов.
С.3.3.7 Персонал
Качества, уровень подготовки, образование, компетентность, подчиненность, обмен информацией, манипуляции, воровство и подмена.
С.3.3.8 Процессы и процедуры
Проверки, содержание, изменения, распределение, использование, состояние, управление изменениями, хранение, тенденции, транспортирование, плановые перерывы и аномальные события (включая переделки).
С.3.3.9 Технология
Системы информационной технологии, обработка электронных данных, информационная безопасность, защита данных.
С.3.4 Оценивание риска
Должны быть определены любые меры, принимаемые для контролирования каждой опасности, и должна быть оценена их результативность, например:
- анализ контракта;
- контроль в процессе производства;
- управление изменениями;
- анализ результатов проверок и последствий корректирующих действий;
- урегулирование претензий потребителей;
- показатели качества;
- обратная связь с персоналом.
С.3.5 Оценка риска
В отношении каждой опасности, по которой отсутствуют меры контроля, должны оцениваться последствия и вероятность возникновения (получая информацию от потребителей по мере необходимости).

Рисунок С.2 - Примеры рисков и потенциальных последствий
Следует оценить серьезность последствий и вероятность возникновения негативного события с учетом любых принимаемых мер контроля.
Простой подход к оцениванию серьезности последствий может быть следующим.
Необходимо классифицировать последствия каждой опасности как "Незначительные", "Средние" и "Значительные" и определить вероятность возникновения опасности в баллах. Баллы следует перемножить для получения оценки риска в баллах с указанием уровня риска.
Результаты оценки риска могут быть нанесены на карту и (или) занесены в таблицу, как показано на рисунке С.3.
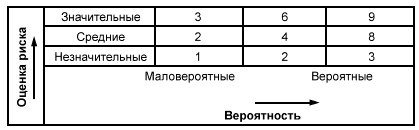
Рисунок С.3 - Рекомендации по нанесению на карту и (или) занесению в таблицу результатов оценки риска
Матрица может быть любого размера, насколько это требуется организации. Важнее всего способность классифицировать риск по трем категориям: тот, который требует немедленного воздействия, тот, который требует воздействия на определенном этапе, и тот, с которым можно ужиться и который не требует никакого воздействия.
Дополнительный пример таблицы оценки риска приведен на рисунке С.4.
С.3.6 Другие методы анализа риска
С.3.6.1 Анализ видов и последствий потенциальных отказов (FMEA)
FMEA представляет собой качественную методику планирования рисков и неблагоприятных факторов; данный процесс стремится к определению мест вероятных отказов, приоритизации их значимости/вероятности, а затем к составлению планов смягчения отрицательных последствий при возникновении этих отказов.
С.3.6.2 Анализ дерева отказов (FTA)
FTA - это в первую очередь средство анализа опасностей, выявленных с помощью других методов. Результаты анализа представляются наглядно в виде дерева режимов отказов. FTA главным образом используется при анализе риска как средство оценки вероятностей отказов. Наглядное представление помогает в упрощении понимания поведения системы и присутствующих факторов.
С.3.6.3 Анализ рисков и эксплуатационных возможностей (HAZOP)
HAZOP аналогичен FMEA. HAZOP основан на теории, которая исходит из допущения, что происшествия вызываются отклонениями от проекта или оперативных замыслов. Это системная методика выявления опасностей и проблем работоспособности. HAZOP применим, например к процессу или обработке (например, стерилизации), используемой при изготовлении первичных упаковочных материалов, которая может оказывать существенное влияние на функционирование первичных упаковочных материалов.
Целями данной методики являются получение полного описания первичных упаковочных материалов и их предположительного использования, проведение системного анализа каждой части предполагаемого использования/предполагаемого назначения для определения того, как могут возникать отклонения от нормальных эксплуатационных условий и планового проекта, для выявления последствий таких отклонений и для определения того, могут ли эти последствия привести к возникновению опасностей или проблем работоспособности.
С.3.6.4 Анализ рисков и критических контрольных точек (ХАССП)
ХАССП представляет собой систематический подход к выявлению, оценке рисков и их серьезности, контролю биологических, химических и физических рисков, связанных с конкретными процессами и практиками. После выявления стадий процесса, где возможно возникновение значительных рисков, определяются критические контрольные точки (ККТ). Должны быть установлены критические границы (пределы) для каждой ККТ и осуществляться мониторинг с целью выявления любых отклонений. Каждое отклонение предусматривает применение корректирующих действий, должно быть документировано, а также должен осуществляться мониторинг результатов.
С.3.7 Управление рисками
Управление рисками описывает действия по внедрению решений менеджмента рисков.
Предыдущие стадии должны выявить наличие любых слабых сторон. В отношении этих слабых сторон должны быть разработаны предупреждающие и корректирующие действия.
При управлении рисками должен применяться комплексный подход, учитывающий одно или несколько из следующих действий в порядке приоритетности:
a) безопасность конструкции;
b) проектирование процесса производства;
c) контроль в процессе производства;
d) промежуточная или законченная экспертиза товаров.
Для снижения риска должны быть определены возможности для дополнительного, альтернативного или улучшенного контроля. Если это возможно, проведение того или иного вида контроля должно исключить риск возникновения опасности. Там, где этого достигнуть невозможно, для исключения риска возникновения опасности применяются меры контроля или обеспечивается выявление любого возникновения опасности для того, чтобы можно было предпринять соответствующее действие.
Возможности для дополнительного, альтернативного или улучшенного контроля должны анализироваться, а действия должны быть официально одобрены с учетом:
- насколько сложно или легко внедрить меры контроля;
- вероятности реализации мер контроля;
- серьезность последствий, если опасность не контролируется;
- вероятность возникновения опасности при применяемых мерах контроля.
Подтверждение того, что действия являются подходящими/соответствующими, может быть определено посредством создания нового источника риска, принимая/допуская, что проверочный контроль был осуществлен/введен/внедрен.
Приоритет должен быть отдан снижению опасности тяжелых последствий или контролированию опасностей, вероятность возникновения которых очень высока.
Там, где оценка риска невысока или невозможно определить действие для снижения риска, ни одно действие не может быть целесообразным. В таких случаях должно быть принято решение не предпринимать никаких действий, и оно должно быть обосновано, документировано и, там где это необходимо, одобрено потребителем.
С.3.8 Снижение риска
Планы по внедрению новых видов контроля должны быть формализованы, содержать согласованные даты завершения/окончания и распределение ответственности. Должен осуществляться мониторинг прогресса по отношению к данному плану до его завершения/окончания. Должен также осуществляться мониторинг результативности улучшенных видов контроля для подтверждения их результативности.
После того, как наиболее значимые вопросы были исключены, процесс должен быть повторен для следующего по приоритетности уровня риска. Данный процесс должен быть повторен пока для всех опасностей не будут определены соответствующие действия.
С.3.9 Анализ
Результативность менеджмента рисков должна периодически оцениваться и представляться для анализа со стороны руководства.
Ста- | Номер риска | Риск | Потен- | Теку- | Пос- | Вероят- | Баллы риска (1, 2, 3, 4, 6, 9) | Допол- | Ответст- | Дата дости- |
Рисунок С.4 - Оценка рисков
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов ссылочным национальным стандартам Российской Федерации и действующим в этом качестве межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального, межгосударственного стандарта |
ИСО 9000:2008 | IDT | ГОСТ ISO 9000-2011 "Системы менеджмента качества. Основные положения и словарь" |
ИСО 9001:2008 | IDT | ГОСТ ISO 9001-2011 "Системы менеджмента качества. Требования" |
ИСО 9004:2009 | IDT | ГОСТ Р ИСО 9004-2010 "Менеджмент для достижения устойчивого успеха организации. Подход на основе менеджмента качества" |
ИСО 14644-1-99 | IDT | ГОСТ ИСО 14644-1-2002 "Чистые помещения и связанные с ними контролируемые среды. Часть 1. Классификация чистоты воздуха" |
ИСО 14644-2-2000 | IDT | ГОСТ Р ИСО 14644-2-2001 "Чистые помещения и связанные с ними контролируемые среды. Часть 2. Требования к контролю и мониторингу для подтверждения постоянного соответствия ГОСТ Р ИСО 14644-1" |
ИСО 14644-3:2005 | IDT | ГОСТ Р ИСО 14644-3-2007 "Чистые помещения и связанные с ними контролируемые среды. Часть 3. Методы испытаний" |
ИСО 14644-4:2001 | IDT | ГОСТ Р ИСО 14644-4-2002 "Чистые помещения и связанные с ними контролируемые среды. Часть 4. Проектирование, строительство и ввод в эксплуатацию" |
ИСО 14644-5:2004 | IDT | ГОСТ Р ИСО 14644-5-2005 "Чистые помещения и связанные с ними контролируемые среды. Часть 5. Эксплуатация" |
ИСО 14971:2007 | IDT | ГОСТ Р ИСО 14971-2011 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
ИСО 14937:2009 | IDT | ГОСТ Р ИСО 14937-2012 "Стерилизация медицинской продукции. Общие требования к определению характеристик стерилизующего агента и к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" |
ИСО 14698-1:2003 | IDT | ГОСТ Р ИСО 14698-1-2005 "Чистые помещения и связанные с ними контролируемые среды. Контроль биозагрязнений. Часть 1. Общие принципы и методы" |
ИСО 14698-2:2003 | IDT | ГОСТ Р ИСО 14698-2-2005 "Чистые помещения и связанные с ними контролируемые среды. Часть 2. Анализ данных о биозагрязнениях" |
ИСО 11135-1:2007 | - | * |
ИСО 11137-1:2006 | IDT | ГОСТ ISO 11137-1-2011 "Стерилизация медицинской продукции. Радиационная стерилизация. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" |
ИСО 11137-2:2006 | IDT | ГОСТ ISO 11137-2-2011 "Стерилизация медицинской продукции. Радиационная стерилизация. Часть 2. Установление стерилизующей дозы" |
* Соответствующий национальный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного международного стандарта. Перевод данного международного стандарта находится в Федеральном информационном фонде технических регламентов и стандартов. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - IDT - идентичные стандарты. | ||
Библиография
[1] | ISO 9001:2008 Quality management systems - Requirements | |
[2] | ISO 9004 Managing for the sustained success of an organization - A quality management approach | |
[3] | ISO 10007 Quality management systems - Guidelines for configuration management | |
[4] | ISO 10012 Measurement management systems - Requirements for measurement processes and measuring equipment | |
[5] | ISO 11135 Sterilization of health care products - Ethylene oxide - Requirements for the development, validation and routine control of a sterilization process for medical devices | |
[6] | ISO 11137-1 Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices | |
[7] | ISO 11137-2 Sterilization of health care products - Radiation - Part 2: Establishing the sterilization dose | |
[8] | ISO 13485:2003 Medical devices - Quality management systems - Requirements for regulatory purposes | |
[9] | ISO 14001:2004 Environmental management systems - Requirements with guidance for use | |
[10] | ISO 14644-4 Cleanrooms and associated controlled environments - Part 4: Design, construction and start-up | |
[11] | ISO 14644-7 Cleanrooms and associated controlled environments - Part 7: Separative devices (clean air hoods, gloveboxes, isolators and mini-environments) | |
[12] | ISO 14644-8 Cleanrooms and associated controlled environments - Part 8: Classification of airborne molecular contamination | |
[13] | ISO 14698-1 Cleanrooms and associated controlled environments - Biocontamination control - Part 1: General principles and methods | |
[14] | ISO 14698-2 Cleanrooms and associated controlled environments - Biocontamination control - Part 2: Evaluation and interpretation of biocontamination data | |
[15] | ISO 14937:2009 Sterilization of health care products - General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process for medical devices | |
[16] | ISO 14971:2007 Medical devices - Application of risk management to medical devices | |
[17] | ISO 19011 Guidelines for quality and/or environmental management systems auditing | |
[18] | ISO/IEC 90003:2004 Software engineering - Guidelines for the application of ISO 9001:2000 to computer software | |
[19] | ISO/IEC Guide 2 Standardization and related activities - General vocabulary | |
[20] | ISO/IEC Guide 51:1999 Safety aspects - Guidelines for their inclusion in standards | |
[21] | IEC 60601-1-4 Medical electrical equipment - Part 1-4: General requirements for safety - Collateral Standard: Programmable electrical medical systems | |
[22] | IEC 60812 Analysis techniques for system reliability - Procedure for failure mode and effects analysis (FMEA) | |
[23] | IEC 61025 Fault tree analysis (FTA) | |
[24] | IEC 61882 Hazard and operability studies (HAZOP studies) - Application guide | |
[25] | International vocabulary of basic and general terms in metrology (VIM). BIPM, IEC, IFCC, ISO, IUPAC, IUPAP, OIML, 1993 | |
[26] | EU Guidelines to Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, http://ec.europa.eu/health/documents | |
[27] | US/FDA Code of Federal Regulations | |
[28] | GAMP 5 A Risk-Based Approach to Compliant GxP Computerized Systems, http://www.ispe.org | |
[29] | PIC/S Guide PE 008-3, Explanatory Notes for Industry on the Preparation of a Site Master File, http://www.picscheme.org/pics.php | |
[30] | ICH Q7, Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, http://www.ich.org | |
[31] | EU Guidelines to Good Manufacturing Practice for Medical Products for Human and Veterinary Use, Annex 18 Good Manufacturing Practice for Active Pharmaceutical Ingredients Requirements, http://ec.europa.eu/health/documents/ | |
[32] | Global Harmonization Task Force (GHTF) - Study Group 1 (SG1), Document No. N029R15, 2005, http://ghtf.org/ | |
| ||
УДК 658.562.014:006.354 | ОКС 03.120.10 | Т59 | ОКСТУ 0025 |
11.040.01 | |||
Ключевые слова: первичные упаковочные материалы, лекарственные средства, система менеджмента качества, ответственность руководства, менеджмент рисков, процессы жизненного цикла продукции | |||
Электронный текст документа
и сверен по:
, 2015

{kind=link}