ГОСТ Р 56894-2016/GHTF/SG1/N063:2011
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Сводный комплект технической документации для демонстрации соответствия общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий для диагностики in vitro
Summary technical documentation for demonstrating conformity to the essential principles of safety and performance of in vitro diagnostic medical devices
ОКС 03.120.10
11.040.01
Дата введения 2017-03-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "МЕДИТЕСТ" на основе собственного перевода на русский язык англоязычной версии документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 17 марта 2016 г. N 158-ст
4 Настоящий стандарт идентичен международному документу Целевой группы по глобальной гармонизации (Global Harmonization Task Force, GHTF) GHTF/SG1/N063:2011* "Сводный комплект технической документации для демонстрации соответствия общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий для диагностики in vitro" (GHTF/SG1/N063:2011 "Summary Technical Documentation (STED) for demonstrating conformity to the essential principles of safety and performance of in vitro diagnostic medical devices", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных документов соответствующие им национальные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
6 ПЕРЕИЗДАНИЕ. Февраль 2020 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт разработан для целей сближения различных регулирующих систем в разных странах. Он предназначен для регулирующих органов и органов по оценке соответствия и призван обеспечить экономически целесообразный и результативный подход к регулированию медицинскими изделиями для диагностики in vitro. Настоящий стандарт содержит рекомендации по составу и содержанию сводного комплекта технической документации, помогающие изготовителю предоставить в регулирующий орган или орган по оценке соответствия документированные свидетельства того, что его медицинское изделие для диагностики in vitro соответствует общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий для диагностики in vitro. Использование сводного комплекта технической документации позволяет изготовителю и регулирующему органу сокращать финансовые затраты, устранять торговые барьеры и облегчать доступ к медицинским изделиям в международном масштабе.
Каждый изготовитель медицинских изделий должен подготовить техническую документацию на свои изделия и поддерживать ее в рабочем состоянии, а также обеспечивать своевременный доступ к данной технической документации, демонстрирующей, как было разработано, спроектировано и изготовлено медицинское изделие для диагностики in vitro. Данная техническая документация, обычно управляемая системой менеджмента качества (СМК) изготовителя, часто является обширной, и может содержать ссылки на части, которые могут находиться в разных местах. Документация подлежит пересмотру для включения каких-либо изменений, вносимых в течение жизненного цикла медицинских изделий для диагностики in vitro в рамках стандартного применения СМК изготовителя.
Это выгодно для обеих сторон: как для регулирующего органа или органа по оценке соответствия, так и регулируемой отрасли, если для выполнения установленных процедур по оценке соответствия используется подмножество полной технической документации - сводный комплект технической документации (далее - сводный комплект) на рассматриваемое изделие. Этот сводный комплект должен быть подготовлен в установленной регулирующими требованиями суммарной или сокращенной форме, со степенью детализации, достаточной для регулирующего органа или органа по оценке соответствия. Документы, содержащиеся в сводном комплекте, должны быть частью полной технической документации, хранящейся у изготовителя и позволяющей ему демонстрировать, что медицинское изделие, к которому относится данная техническая документация, соответствует общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий для диагностики in vitro (см. GHTF/SG1/N41:2005).
Наличие сводного комплекта технической документации помогает уменьшить различия в требованиях к документации, имеющиеся в разных юрисдикциях, сокращая таким образом финансовые затраты на достижение соответствия регулирующим требованиям и помогая пациентам получить доступ к новым технологиям и методикам лечения.
1 Область применения
Настоящий стандарт представляет собой руководство для информационного наполнения сводного комплекта технической документации, который необходимо собрать и предоставить в регулирующий орган или орган по оценке соответствия для предпродажного анализа и использования на послепродажной стадии, с тем, чтобы оценить соответствие рассматриваемого изделия для диагностики in vitro общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий для диагностики in vitro.
Настоящий стандарт применим к медицинским изделиям для диагностики in vitro.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие документы:
GHTF/SG1/N044:2008, Role of standards in the assessment of medical devices (Роль стандартов в оценке соответствия медицинских изделий)
GHTF/SG1/N45:2007, Principles of in vitro diagnostic medical devices classification (Принципы классификации медицинских изделий для диагностики in vitro)
GHTF/SG1/N29:2005, Information document concerning the definition of the term "medical device" (Информационный документ, содержащий определение "медицинское изделие")
GHTF/SG1/N46:2007, Principles of conformity assessment for in vitro diagnostic medical devices (Принципы оценки соответствия медицинских изделий для диагностики in vitro)
GHTF/SG1/N41:2005, Essential principles of safety and performance of medical devices (Общие принципы обеспечения безопасности и основных функциональных характеристик медицинских изделий)
GHTF/SG1/N43:2005, Labelling for medical devices (Маркировка медицинских изделий)
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 признанный стандарт (recognised standard): Стандарт, обеспечивающий презумпцию соответствия конкретным общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий.
3.2 техническая документация (technical documentation): Документированные свидетельства, обычно являющиеся выходом системы менеджмента качества, демонстрирующие соответствие изделия общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий.
4 Подготовка и использование сводного комплекта технической документации
4.1 Подготовка
Изготовители медицинских изделий для диагностики in vitro должны демонстрировать соответствие данных изделий общим принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (далее - общим принципам) посредством подготовки и хранения технической документации, демонстрирующей, как каждое медицинское изделие для диагностики in vitro было разработано, спроектировано и изготовлено, и содержащей описания и объяснения, необходимые для понимания намерений изготовителя в отношении такого соответствия. Данную техническую документацию обновляют по мере необходимости, для того чтобы отразить текущий статус, спецификации и конфигурацию изделия для диагностики in vitro.
В целях оценки соответствия изготовитель формирует сводный комплект технической документации из имеющейся полной технической документации на изделие, обеспечивая тем самым для регулирующего органа или органа по оценке соответствия свидетельства того, что рассматриваемое медицинское изделие для диагностики in vitro соответствует общим принципам. Сводный комплект технической документации отражает статус медицинского изделия для диагностики in vitro в конкретный момент времени (например, на предпродажной стадии при рассмотрении документации или, по требованию регулирующего органа, для послепродажных целей), его подготавливают для оценивания соответствия регулирующим требованиям. На рисунках 1 и 2 показан информационный поток, от полной технической документации к сводному комплекту технической документации.
Сводный комплект рекомендуется подготавливать на языке, требуемом регулирующим органом/органом по оценке соответствия.
Глубина и степень детализации информации, содержащейся в сводном комплекте технической документации, зависят от:
- классификации рассматриваемого изделия;
- сложности рассматриваемого изделия,
а также от того:
- имеет ли данное изделие новую технологию (при этом под новой технологией понимают как отсутствие на рынке ранее изделий для диагностики in vitro для того же аналита, так и применение новой аналитической технологии для известного аналита);
- отличается ли применение готового изделия от первоначального применения, предусмотренного изготовителем;
- является ли изделие новым для изготовителя;
- ассоциируется ли данный тип изделия со значительным числом неблагоприятных (нежелательных) событий, включая ошибки применения;
- включает ли изделие новые или потенциально опасные материалы;
- затрагивает ли данный тип изделия конкретные проблемы в области здравоохранения.
Сводный комплект технической документации должен содержать суммарную информацию по выбранным темам, подробную информацию по некоторым специальным темам (приведенным ниже) и таблицу соответствия общим принципам. Данная информация может включать в себя, например, краткое описание документов, итоговую информацию, проанализированную высшим руководством, или, когда целесообразно, имеющиеся в наличии управляемые документы, достаточные для передачи основной информации и позволяющие проверяющему лицу понять суть вопроса. Таблица соответствия общим принципам разработана как часть технической документации изготовителя и должна являться документом, управляемым системой менеджмента качества изготовителя. Это позволяет обеспечить краткий обзор общих принципов и идентифицировать те из них, которые применимы к рассматриваемому изделию, выбрать метод демонстрации соответствия изделия каждому относящемуся к нему общему принципу и указать ссылку на документы, поддерживающие конкретный общий принцип. Несмотря на то, что на поддерживающие документы рекомендуется иметь ссылки в таблице соответствия общим принципам, только некоторые из этих документов следует включать в сводный комплект технической документации. Упомянутые ссылки на поддерживающие документы облегчают выполнение запросов от регулирующего органа/органа по оценке соответствия, обеспечивая получение дополнительной информации.
4.2 Использование сводного комплекта технической документации на предпродажной стадии (до выпуска в обращение)
На предпродажной стадии сводный комплект подготавливают и передают в регулирующий орган/орган по оценке соответствия для изделий для диагностики in vitro классов C и D. Для изделий для диагностики in vitro классов A и B сводный комплект подготавливают и передают только по требованию регулирующего органа/органа по оценке соответствия (см. рисунок 1).
Примечания
1 Для изделий классов A и B, когда сводный комплект подготавливают по требованию, изготовитель должен быть готов собрать и передать сводный комплект в период времени, указанный регулирующим органом/органом по оценке соответствия. Данный период может быть очень коротким.
2 Копии переданного сводного комплекта должны храниться изготовителем для будущих ссылок.
|
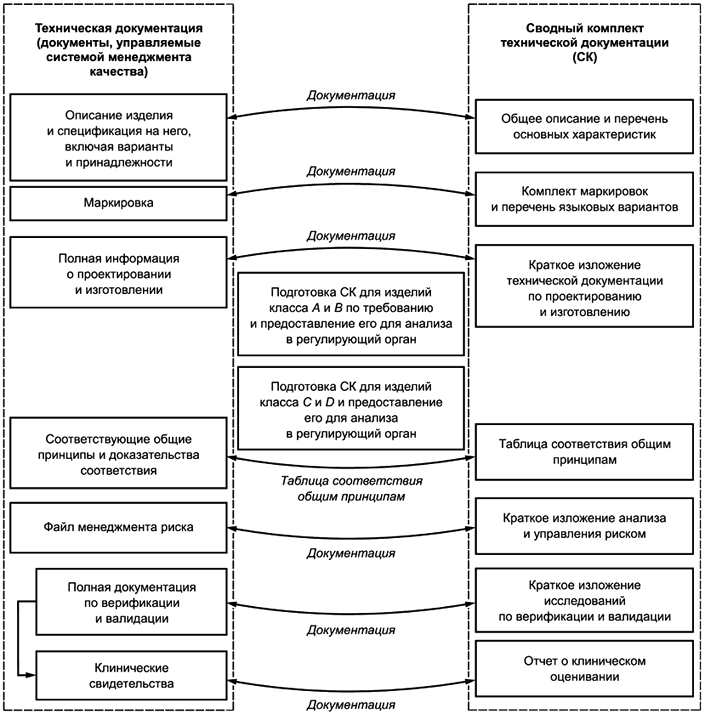
Рисунок 1 - Использование сводного комплекта на предпродажной стадии
4.3 Использование сводного комплекта технической документации на послепродажной стадии (после выпуска в обращение)
На послепродажной стадии регулирующий орган/орган по оценке соответствия может затребовать сводный комплект технической документации на рассматриваемое изделие либо для оценки соответствия медицинского изделия для диагностики in vitro класса A или B, либо для подтверждения оценки соответствия медицинского изделия для диагностики in vitro класса C или D (см. рисунок 2). Сводный комплект обычно не используют в целях послепродажного исследования неблагоприятных (нежелательных) событий, для сообщения информации, содержащейся в послепродажных регистрационных записях, либо для сообщения данных исследований, в которых использованы различные виды информации.
Примечания
1 Изготовитель должен быть готов собрать и передать сводный комплект в период времени, указанный регулирующим органом/органом по оценке соответствия. Данный период может быть очень коротким.
2 Копии переданного сводного комплекта должны храниться изготовителем для будущих ссылок.
|
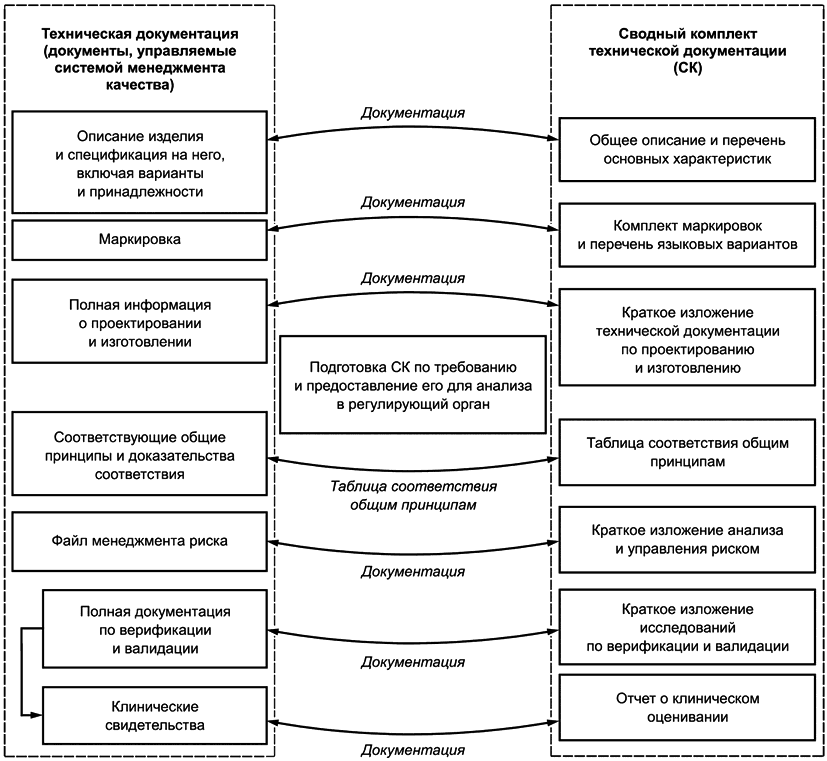
Рисунок 2 - Использование сводного комплекта на послепродажной стадии
4.4 Использование сводного комплекта технической документации для сообщения об изменениях в регулирующий орган/орган по оценке соответствия
Если для внесения изменения в медицинское изделие необходимо предварительное одобрение регулирующего органа, то для получения данного одобрения можно использовать сводный комплект технической документации. Для этих целей будет подготовлен отдельный руководящий документ.
5 Описание изделия и его технических характеристик, включая варианты (конфигурации) и принадлежности
5.1 Описание изделия
Сводный комплект технической документации должен содержать следующую информацию, описывающую изделие:
a) предусмотренное применение изделия, включая:
- что обнаруживается;
- основная функция (например, скрининг, мониторинг, диагностика или помощь в диагностике);
- специфическое заболевание, состояние или факторы риска, которые предполагается обнаружить, определить или дифференцировать;
- является ли изделие автоматизированным;
- является ли результат количественным или качественным;
- требуемый вид образца (например, сыворотка, плазма, цельная кровь, ткани после биопсии, моча);
- исследуемая популяция;
b) предусмотренный пользователь (неподготовленный пациент или профессионал);
c) принципы действия изделия или набора для диагностики in vitro;
d) класс потенциального риска и применяемое классификационное правило согласно установленным принципам классификации медицинских изделий для диагностики in vitro;
e) описание составных частей изделия (например, реагентов, контрольных проб и калибраторов) и, если применимо, описание активных составляющих соответствующих частей (таких как антитела, антигены, предшественники нуклеиновых кислот);
и, если применимо:
f) описание коллекции образцов и транспортных материалов, предусмотренных для изделия, или описание спецификаций, рекомендуемых к использованию;
g) для приборов с автоматическим отбором проб: описание соответствующих характеристик проб или типов проб;
h) для проб для автоматического отбора: описание соответствующих характеристик приборов или типов приборов;
i) описание какого-либо программного обеспечения, используемого с изделием для диагностики in vitro;
j) описание или исчерпывающий перечень конфигураций/вариантов изделия для диагностики in vitro, которые выпускаются на рынок;
k) описание принадлежностей, других медицинских изделий для диагностики in vitro и изделий, не являющихся медицинскими, но предусмотренных для использования в комбинации с рассматриваемым изделием для диагностики in vitro.
5.2 Ссылка на подобные и предыдущие варианты изделия, а также историю изделия
5.2.1 Изделие для диагностики in vitro еще не выпущено на рынок
В случае необходимости демонстрации соответствия общим принципам и представления общих сведений сводный комплект должен содержать краткое описание:
a) предыдущих поколений рассматриваемого изделия, если таковые существовали;
b) подобных изделий, имеющихся на региональных и международных рынках.
5.2.2 Изделие для диагностики in vitro выпущено на рынок в какой-либо стране
Данная информация может включать сводку отчетов по неблагоприятным событиям в отношении безопасности и функциональных характеристик данного изделия для диагностики in vitro в соотношении с количеством изделий, выпущенных на рынок.
Сертификаты, полученные от внешних организаций, а также документы, подтверждающие соответствие общим принципам, могут быть приложены к сводному комплекту.
6 Таблица соответствия общим принципам
Сводный комплект технической документации должен включать в себя таблицу соответствия общим принципам, определяющую:
a) общие принципы;
b) применим ли каждый общий принцип к рассматриваемому изделию для диагностики in vitro, и если нет, то почему;
c) метод(ы), используемый(ые) для демонстрации соответствия рассматриваемого изделия каждому применимому общему принципу;
d) ссылка на точное наименование документа(ов) из состава технической документации, обеспечивающего(их) свидетельство соответствия каждому применяемому методу.
Для демонстрации соответствия можно использовать один или несколько следующих методов:
a) соответствие признанным или иным стандартам;
b) соответствие общепринятым в промышленности методам испытаний;
c) соответствие методам испытаний, принятым в данной организации, которые были валидированы и верифицированы;
d) сравнение с медицинским изделием для диагностики in vitro, уже доступным на рынке.
Таблица соответствия общим принципам должна включать в себя перекрестные ссылки на местонахождение каждого свидетельства соответствия как в полной технической документации, находящейся у изготовителя, так и в сводном комплекте технической документации.
Образец таблицы соответствия общим принципам приведен в приложении А.
7 Основные результаты анализа и управления риском
Сводный комплект технической документации должен содержать краткий перечень рисков, идентифицированных в процессе анализа риска, и описание способов управления данными рисками в целях снижения их до допустимого уровня. Предпочтительно, чтобы данный анализ рисков был основан на признанных стандартах и являлся частью плана менеджмента риска изготовителя.
Аннотация по рискам должна включать возможные опасности, характерные для медицинского изделия для диагностики in vitro, такие как риск получения ложноположительных и ложноотрицательных результатов, косвенные риски, которые связаны с опасностями в контексте применения медицинского изделия для диагностики in vitro, например нестабильность его характеристик, которое может привести к ошибочным результатам, или опасности для пользователя, например при работе с реагентами, содержащими инфекционные агенты.
Результаты анализа риска должны представлять заключение, свидетельствующее о том, что все остаточные риски являются приемлемыми по сравнению с пользой.
Обычно для изделий для диагностики in vitro класса D требуется детальный отчет.
8 Информация по проектированию и производству
8.1 Проектирование изделия
Сводный комплект технической документации должен содержать информацию, позволяющую контролирующему лицу получить общее представление об основных стадиях проектирования рассматриваемого изделия для диагностики in vitro.
Информация должна включать описание важнейших компонентов анализа, таких как антитела, антигены, ферменты, и праймеров нуклеиновых кислот, предусмотренных и рекомендованных для использования с медицинским изделием для диагностики in vitro.
Для приборов информация должна включать описание основных подсистем, аналитической технологии (например, принципов работы, механизмов управления), специальных компьютерных аппаратных и программных частей.
Для приборов и программного обеспечения требуется обзор системы в целом, включая архитектуру, которая обычно представляет собой диаграмму с взаимосвязями между основными функциональными модулями в программном обеспечении, включая выводы на аппаратную часть и передачу данных, например по сети.
Для отдельного программного обеспечения требуется описание методологии интерпретации данных (т.е. алгоритмы).
Для изделий, предназначенных для самотестирования, проект должен включать описание аспектов конструкции, которые делают ее пригодной для неквалифицированного пользователя.
Обычно для изделий для диагностики in vitro класса D требуется представление подробной спецификации материалов.
При этом данную информацию нельзя использовать вместо более подробной информации, которая требуется для аудита системы менеджмента качества или для другой деятельности по оценке соответствия. Если проектирование проводилось несколькими подразделениями, то должно быть определено контролирующее подразделение.
8.2 Производственные процессы
Только для класса D сводный комплект технической документации должен содержать информацию, позволяющую инспектирующему лицу получить общее представление о производственных процессах. При этом данную информацию нельзя использовать вместо более подробной информации, которая требуется для аудита системы менеджмента качества или для другой деятельности по оценке соответствия. Упомянутая информация может быть представлена в виде блок-схемы процессов, дающей, например, общее представление о производстве, сборке, всех заключительных испытаниях изделия и об окончательной упаковке готового изделия для диагностики in vitro. Данная часть должна включать информацию по выходному и межоперационному контролю качества продукции (например, по программе контроля качества изготовителя).
8.3 Производственные площадки
Для видов деятельности, описанных в 8.2, сводный комплект технической документации должен идентифицировать производственные площадки, на которых эти виды деятельности были выполнены (это не включает площадки поставщиков исходных материалов, а только площадки, вовлеченные в критические производственные процессы). Если для данных площадок имеются сертификаты системы менеджмента качества или равноценные документы, то они должны быть приложены к сводному комплекту технической документации.
9 Деятельность по верификации и валидации
Сводный комплект технической документации должен содержать документы по верификации и валидации. При этом степень детализации может быть различной в зависимости от классификации изделия и прочих характеристик изделия (см. 4.1).
Как правило, в сводном комплекте должны быть кратко изложены результаты деятельности по верификации и валидации, которая предпринята для демонстрации соответствия применимым общим принципам. При необходимости такая информация может быть взята из литературных источников.
1 Краткое изложение (резюме) информации в сводном комплекте должно содержать в себе достаточно необходимой информации, чтобы позволить регулирующему органу/органу по оценке соответствия оценить ее достоверность. Представляемая информация должна содержать краткое описание:
a) протокола исследования;
b) результатов исследования;
c) заключение исследования.
Также такая информация может включать:
a) декларацию/свидетельство соответствия признанному(ым) стандарту(ам) и краткое изложение данных, если в упомянутом(ых) стандарте(ах) не установлены критерии приемки;
b) декларацию/свидетельство соответствия опубликованному(ым) стандарту(ам), который(е) не был(и) официально признан(ы), с обоснованием его (их) применения и краткое изложение данных, если в упомянутом(ых) стандарте(ах) не установлены критерии приемки;
c) декларацию/свидетельство соответствия профессиональному(ым) руководству(ам), производственному(ым) методу(ам) или методу(ам), используемым в организации, с обоснованием их применения, описанием данных методов и краткой информацией со степенью детализации, позволяющей оценить ее достаточность;
d) обзор опубликованных источников касательно рассматриваемого изделия или подобных изделий.
2 Подробная информация должна включать в себя:
a) полный протокол исследования;
b) метод анализа полученных данных;
c) полный отчет об исследовании;
d) заключение об исследовании.
В случае существования признанного стандарта, содержащего протокол и метод анализа данных, данную информацию допускается не включать, заменив ссылкой на соответствие признанному стандарту. Тем не менее следует предоставить краткое описание полученных результатов и выводов.
Для клинических характеристик (являющихся частью клинических доказательств) подробная информация обычно включает индивидуальные точки измерения ("сырые" данные) для класса D медицинских изделий для диагностики in vitro.
Если требуется, краткое изложение непосредственных результатов измерений с соответствующими критериями должны быть предоставлены, а не только заключения типа "соответствует/не соответствует".
9.1 Аналитические характеристики
Утверждения и описания нижеследующих частей относятся ко всем медицинским изделиям для диагностики in vitro. Применимость требований отличается между приборами и наборами реагентов, и сами реагенты могут быть предназначены для количественного, полуколичественного или качественного анализа. Поэтому применимость нижеследующих частей может быть ограничена для полуколичественных и качественных анализов. Ниже, где применимо, даны комментарии в отношении приборов или качественных анализов.
9.1.1 Вид образца
В этой части описывают различные виды образцов, которые могут быть использованы. Описание должно включать требования к стабильности и условиям хранения и, как правило, применимо ко всем системам и типам анализа. Стабильность включает хранение и, где применимо, условия транспортировки. Хранение включает такие параметры, как продолжительность, температурные ограничения и число циклов заморозки/размораживания.
Данная часть должна также включать краткую информацию по каждой матрице и антикоагулянту, если применимо, включая описание методики измерения для сравнения и оценки точности измерений. Это включает информацию о виде исследуемого образца, количестве образцов, диапазоне выборки образцов или анализируемые целевые концентрации, расчеты и статистические методы, результаты и заключения.
Как правило, для медицинских изделий для диагностики in vitro класса D предоставляется подробная информация.
9.1.2 Функциональные аналитические характеристики
9.1.2.1 Погрешность измерения
В этой части должны быть описаны достоверность и точность исследований.
Примечание - Общий термин погрешности используется в настоящее время и для достоверности, и для точности, в то время как ранее он использовался только для представления достоверности.
Достоверность измерений, на которую оказывают влияние систематические погрешности, обычно выражается в постоянном сдвиге, в то время как точность, на которую влияют случайные погрешности, обычно выражается в стандартном отклонении.
Погрешность определяется сочетанием систематических и случайных результатов, которые вместе вносят вклад в общую погрешность измерения.
a) Достоверность измерений
В этой части должна быть предоставлена информация по достоверности измерительной процедуры и сводка детализированных данных, позволяющих оценить корректность выбранных методов оценки достоверности. Характеристика достоверности применяется как к количественным, так и к качественным исследованиям при стандартном или референтном методе исследования.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется детальная информация.
b) Точность измерений
В этой части должны быть описаны повторяемость и воспроизводимость исследований.
1 Повторяемость
В этой части должны быть рассмотрены оценки и информация по повторяемости результатов при последовательных исследованиях образцов. Данные по повторяемости получают для конкретного оборудования и набора реагентов.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется детальная информация.
Примечание 1 - Такие исследования включают использование образцов, которые представляют весь диапазон возможных концентраций аналитов (измеряемых параметров), как заявлено изготовителем.
Примечание 2 - Если используется признанный стандарт, то вместе со сводкой данных и заключением должны быть предоставлены декларация/сертификат соответствия признанному стандарту.
2 Воспроизводимость
В эту часть должны быть включены оценка воспроизводимости и информация по исследованиям, используемым для проведения такой оценки в соответствующих случаях, вариабельность в результатах между днями исследований, режима работы, места исследования, серии (партии), операторов и приборов. Такая вариабельность также характеризуется как промежуточная (внутрилабораторная) прецизионность (Intermediate precision). Данные по воспроизводимости получают для приборов в сочетании с соответствующим анализом.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется подробная информация.
Примечание 1 - Такие исследования включают использование образцов, которые представляют весь диапазон возможных концентраций аналитов (измеряемых величин), как заявлено изготовителем.
Примечание 2 - Если используется признанный стандарт, то вместе со сводкой данных и заключением должны быть предоставлены декларация/сертификат соответствия признанному стандарту.
9.1.2.2 Аналитическая чувствительность
В этой части должны быть рассмотрены оценки и информация по методу исследований и результатам. Должно быть включено описание видов образцов и их подготовка, включая матрицы, уровни аналитов (измеряемых величин) и то, как эти уровни задаются. Число повторов, исследуемых для каждой концентрации, также должно быть указано наряду с описанием вычислений, используемых для определения чувствительности анализа. Например:
a) число стандартных отклонений свыше среднего значения для образца без аналита (измеряемой величиной), как правило, называют "пределом холостой пробы" (limit of blank, LoB);
b) самую низкую концентрацию, отличимую от нуля при использовании образцов, содержащих аналит (измеряемую величину), как правило, называют "пределом обнаружения" (limit of detection, LoD);
c) наименьшую концентрацию, при которой выполняются критерии точности и/или достоверности, как правило, называют "пределом количественного определения" (limit of quantification, LoQ).
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется детальная информация.
9.1.2.3 Аналитическая специфичность
В этой части должны быть описаны исследования в отношении интерференции (помех) и перекрестных реакций при установлении аналитической специфичности, определяемой как способность аналитической процедуры определить или измерить только заданный аналит (измеряемую величину, мюзерад) в присутствии других веществ/реагентов в образце.
Следует предоставить информацию по оценке возможной интерференции и перекрестных реакций между веществами/реагентами в анализе. Информация должна предусматривать тип субстанции/веществ и испытываемую концентрацию, вид образца, тестируемую концентрацию аналита (измеряемой величины) и результаты.
Интерферирующие и перекрестно реагирующие субстанции/вещества могут существенно различаться в зависимости от вида анализа и его исполнения, могут происходить от внешних и внутренних источников, таких как:
a) вещества, используемые при лечении пациента (например, лекарственные средства, антикоагулянты и т.п.);
b) вещества, употребленные пациентом (например, безрецептурные лекарственные средства, алкоголь, витамины, пищевые продукты и т.п.);
c) вещества, добавляемые во время подготовки образца (например, консерванты, стабилизаторы);
d) вещества, встречающиеся в конкретных видах образцов (например, гемоглобин, липиды, билирубин, белки);
e) аналиты со схожей структурой (например, прекурсоры, метаболиты) или клинические состояния, не относящиеся к исследованию, которые должны давать отрицательный результат, но дают положительный результат, так как имитируют условия исследования (например, при использовании набора для определения гепатита А, образец является отрицательным по гепатиту А, но положительным по гепатиту В).
Как правило, интерференционные исследования включают в себя добавление потенциального интерферирующего вещества в образец и определение любого отклонения от параметров испытания относительно контрольного образца, к которому интерферирующее вещество не было добавлено.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется подробная информация.
9.1.2.4 Метрологическая прослеживаемость калибраторов и контрольных материалов
Где применимо, следует обобщить информацию по метрологической прослеживаемости значений, заданных калибратором, и правильность контрольных материалов. Эта информация включает в себя, например методы и критерии приемки для метрологической прослеживаемости к референс-материалам и/или референс-методикам измерений и описание присвоенных значений и их валидации.
Точность контрольных материалов, используемых при установлении воспроизводимости методики измерения, не требует оценки метрологической прослеживаемости к референс-материалу и/или референс-методу.
Как правило, для медицинских изделий для диагностики in vitro класса D предоставляется подробная информация.
9.1.2.5 Анализ диапазона измерений
В эту часть должно быть включено краткое изложение исследований, которые определяют диапазон измерений (линейные и нелинейные системы измерений), включая предел обнаружения и информацию о том, как они были установлены. Эта информация должна включать описание видов образцов, количества образцов, количество повторов и их подготовку, включая информацию о материале, содержащем аналит, уровни аналита (измеряемой величины) и того, как эти уровни были установлены. Если применимо, следует добавить описание сверхдозового эффекта "сползания" (хук-эффект) и данные, подтверждающие уменьшение последствий данного эффекта (например, разведение).
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется подробная информация.
9.1.2.6 Определение анализа cut-off (пограничных значений, точки отсечки)
В этой части должны быть описаны сводные данные в отношении исследований аналитической процедуры, включая методы по определению ограничений по диагностике, в том числе:
a) исследованная популяция (демографические аспекты/принципы отбора/критерии включения и исключения/число исследуемых);
b) методы или способы идентификации образцов и
c) статистические методы, например параметрическая функция классификатора (ROC) для генерации результатов и, если применимо, определение серых и сомнительных зон.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется детальная информация.
9.2 Клинические характеристики
Если применимо, сводный комплект должен содержать данные по клиническим характеристикам изделия для диагностики in vitro. Эти данные по клиническим характеристикам являются частью клинических свидетельств, демонстрирующих соответствие изделия для диагностики in vitro применимым принципам обеспечения безопасности и основных функциональных характеристик.
Примечание - Аналитические характеристики и клинические характеристики являются частями клинических свидетельств. Более детальные рекомендации в отношении этих частей сводного комплекта будут представлены в отдельном руководстве, разработанном совместно с SG5 GHTF.
9.3 Стабильность (за исключением стабильности образцов)
В этой части должна быть представлена информация в отношении установленного срока годности, исследования по стабильности после вскрытия/разведения (продолжительности применения и исследования, особенности транспортирования).
9.3.1 Установленный срок годности
В этой части должна быть представлена информация по исследованиям в отношении стабильности изделия, подтверждающая установленный срок годности. Испытания должны быть выполнены в отношении по меньшей мере трех различных партий, изготовленных в условиях, эквивалентных серийному производству (данные партии не обязательно должны быть выпущены друг за другом). Ускоренные испытания или экстраполированные данные по результатам в реальном времени могут быть приняты для установления первоначального срока годности, но должны быть подкреплены исследованиями стабильности в реальном времени.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется детальная информация.
Такая детальная информация должна описывать:
a) отчет об исследовании (включая процедуру испытаний, число партий, критерии оценки и временные интервалы испытаний);
b) когда ускоренные испытания упреждают исследования в реальном времени, необходимо дать описание методов для ускоренных испытаний;
c) заключение и установленный срок годности.
Примечание - Окончательные данные по сроку годности допускается подтверждать в реальном времени для одной партии, в случае если предварительные данные по ускоренным испытаниям или экстраполяции результатов были сравнимы для всех испытанных партий.
9.3.2 Стабильность после вскрытия
В этой части должна быть представлена информация по исследованиям стабильности после вскрытия упаковки для одной партии, отражающим фактическое рутинное использование изделия (реальное или смоделированное). Данные исследования могут включать исследование стабильности открытых флаконов и/или для автоматизированных приборов стабильность на борту после установки флаконов в прибор.
В случае автоматизированных приборов, если стабильность калибровки заявлена, должны быть предоставлены соответствующие данные.
Такие данные могут включать:
a) отчет об исследовании (включая протокол, критерии приемки и временные интервалы испытаний);
b) заключение и заявленный срок стабильности после вскрытия.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется подробная информация.
9.3.3 Стабильность при транспортировании
В этой части должна быть представлена информация по исследованиям стабильности при транспортировании для одной партии, чтобы оценить устойчивость продукции к ожидаемым условиям транспортировки.
Исследования по транспортировке могут быть проведены в реальных и/или смоделированных условиях и должны включать вариабельные условия транспортировки, такие как максимальная и/или минимальная температура.
Данная информация должна описывать:
a) отчет об исследовании (включая протокол, критерии приемки);
b) метод, используемый для имитации условий;
c) заключение и рекомендуемые условия транспортировки.
Как правило, для медицинских изделий для диагностики in vitro классов С и D предоставляется подробная информация.
9.4 Верификация и валидация программного обеспечения
Сводный комплект технической документации должен содержать доказательства валидации программного обеспечения, используемого в готовом изделии. Данная информация обычно включает в себя краткое изложение результатов всей деятельности по верификации, валидации и результатов испытаний, выполненных как в организации-изготовителе, так и в моделируемой или реальной окружающей пользователя среде до окончательной версии программного обеспечения. Данная информация должна также включать в себя все имеющиеся конфигурации аппаратных средств и, когда применимо, операционные системы, идентифицированные в маркировке.
Как правило, для медицинских изделий для диагностики in vitro класса D предоставляется подробная информация.
10 Маркировка
Как правило, сводный комплект должен содержать набор всех маркировок, относящихся к рассматриваемому изделию для диагностики in vitro, и перечень языковых вариантов для стран, в которых будет реализовано данное изделие (см. GHTF/SG1/N43:2005). Информация по маркированию должна включать в себя:
- маркировки/этикетки на изделии для диагностики in vitro и на внутреннюю и внешнюю упаковки);
- инструкции по применению изделия.
Набор маркировок должен быть на языке, требуемом регулирующим органом или органом по оценке соответствия.
Для включения в сводный комплект маркировка должна содержать всю информацию, как установлено изготовителем, но при этом не обязательно должна быть представлена в окончательном (распечатанном) типографском формате.
11 Формат сводного комплекта технической документации
Несмотря на то, что настоящий стандарт не содержит конкретных рекомендаций по формату сводного комплекта, изготовителям и инспектирующим лицам желательно, чтобы сводный комплект включал в себя элементы, описанные в настоящем стандарте (описание изделия, перечень общих принципов и т.д).
12 Декларация о соответствии
Декларация о соответствии не является составной частью сводного комплекта технической документации. Однако она может быть приложена к сводному комплекту после завершения процесса оценки соответствия. Содержание декларации о соответствии приведено в GHTF/SG1/N46:2007.
Приложение А
(справочное)
Таблица соответствия общим принципам
Таблица соответствия общим принципам может быть использована регулирующими органами, органами по оценке соответствия и изготовителями; она позволяет быстро понять, как изготовитель демонстрирует соответствие конкретного изделия общим принципам. Таблица позволяет также легко идентифицировать документы и данные, необходимые для оценки соответствия.
Содержание таблицы соответствия меняется в зависимости от конкретного изделия. Для сложных изделий такая таблица, как правило, будет содержать большое количество ссылок на стандарты, протоколы испытаний и другие документы. В таких случаях таблица соответствия общим принципам может быть очень длинной. Очень простые изделия, вероятно, будут иметь более короткие таблицы, так как многие общие принципы не будут применимы к таким изделиям. В этих случаях в таблицы соответствия может быть включено минимальное количество поддерживающих ссылок.
Ниже приведен рекомендуемый формат. Подготовка таблицы с использованием данного формата полезна для анализа соответствия изделия общим принципам. Последовательное применение данного формата поддерживает гармонизацию между различными системами регулирования изделий для диагностики in vitro.
Как заполнять таблицу соответствия:
a) идентификация изделия для диагностики in vitro.
Изготовитель должен идентифицировать изделие для диагностики in vitro и, если применимо, различные конфигурации/варианты, охватываемые таблицей соответствия;
b) применимость таблицы соответствия к изделию.
Применим ли внесенный в таблицу общий принцип к рассматриваемому изделию для диагностики in vitro? Ответ может быть либо положительным, либо отрицательным. Если ответ отрицательный, то в графе, озаглавленной "Метод, применяемый для демонстрации соответствия", должно быть изложено краткое обоснование неприменения.
c) метод, применяемый для демонстрации соответствия.
В данной графе изготовитель должен установить метод(ы), выбранный(ые) для демонстрации соответствия, например признанный(ые) стандарт(ы), метод(ы) испытаний, применяемый(ые) в промышленности или в данной организации, сравнительное(ые) исследование(ия) или другой(ие) примененный(ые) метод(ы);
d) ссылка на метод.
Установив метод в предыдущей графе, в данной графе изготовитель должен привести наименование и ссылку на признанный(ые) стандарт(ы), метод(ы) испытаний, применяемый(ые) в промышленности или в данной организации, сравнительное(ые) исследование(ия) или другой метод, примененный для демонстрации соответствия. Для стандартов следует привести дату и при необходимости раздел(ы) и пункт(ы), демонстрирующие соответствие конкретному общему принципу;
e) ссылка на подтверждающие документы.
Данная графа должна содержать ссылку на фактическую техническую документацию, демонстрирующую соответствие общим принципам, сертификаты, протоколы испытаний, протоколы валидации, аналитические отчеты или другие документы, появившиеся в результате применения конкретного метода демонстрации соответствия, а также на местонахождение данных документов.
Примечание - Приведенная ниже таблица предназначена только для иллюстративных целей. Общие принципы, перечисленные в первой графе, взяты из GHTF/SG1/N41:2005.
Таблица А.1 - Таблица соответствия общим принципам
Изделие: | |||||
Общий принцип | Применим ли к изделию? | Метод, применяемый для демонстрации соответствия | Ссылка на метод | Ссылка на подтвер- | |
1 Общие требования | |||||
A.1 Медицинские изделия должны быть спроектированы и изготовлены так, чтобы при использовании их по назначению в надлежащих условиях и (при необходимости) с применением технических знаний, опыта, образования или подготовки потенциальных пользователей они не подвергали риску клинические условия или безопасность пациентов либо безопасность и здоровье пользователей или других лиц; любые риски, связанные с их применением, должны быть допустимыми и меньшими, чем приносимая польза пациенту, а также соответствовать высокому уровню защиты здоровья и обеспечения безопасности | |||||
А.2 Решения, принятые изготовителем при проектировании и разработке изделий, должны соответствовать принципам безопасности с учетом современного состояния науки и техники. | |||||
А.3 Изделия должны функционировать в соответствии со своим назначением и быть спроектированы, изготовлены и упакованы так, чтобы выполнять одну или несколько функций из официально утвержденной области применения конкретного медицинского изделия | |||||
А.4 Технические и эксплуатационные характеристики медицинских изделий, описанные в А.1-А.3 настоящей таблицы, не должны оказывать вредное воздействие настолько, чтобы подвергать риску клинические условия, а также безопасность пациентов и других лиц в течение срока службы изделия, указанного изготовителем, если изделие находится в нормальных условиях эксплуатации и поддерживается в рабочем состоянии надлежащим образом и в соответствии с инструкциями изготовителя | |||||
А.5 Изделия должны быть спроектированы, изготовлены и упакованы так, чтобы их технические и эксплуатационные характеристики при использовании по назначению не испытывали вредного воздействия при транспортировании и хранении, осуществляемыми в соответствии с инструкциями и с учетом информации изготовителя | |||||
А.6 При функционировании в соответствии с назначением польза от применения медицинского изделия должна превышать вред от нежелательных побочных эффектов | |||||
2 Требования к проектированию и разработке | |||||
А.7 Химические, физические и биологические свойства | |||||
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных документов национальным стандартам
Таблица ДА.1
Обозначение ссылочного международного документа | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
GHTF/SG1/N044:2008 | - | * |
GHTF/SG1/N45:2007 | - | * |
GHTF/SG1/N29:2005 | - | * |
GHTF/SG1/N46:2007 | - | * |
GHTF/SG1/N41:2005 | - | * |
GHTF/SG1/N43:2005 | - | * |
* Соответствующий национальный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного документа. | ||
УДК 006.83:006.354 |
|
| ОКС 03.120.10 |
| 11.040.01 | ||
Ключевые слова: сводный комплект технической документации, демонстрация соответствия, общие принципы, обеспечение безопасности, основные функциональные характеристики, медицинские изделия для диагностики in vitro | |||
Электронный текст документа
и сверен по:
, 2020

{kind=link}