ГОСТ 28178-89
Группа С19
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ДРОЖЖИ КОРМОВЫЕ
Методы испытаний
Fodder yeast. Test methods
ОКСТУ 9209
Дата введения 1990-07-01
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. РАЗРАБОТАН И ВНЕСЕН Министерством медицинской и микробиологической промышленности СССР
РАЗРАБОТЧИКИ В.Н.Балахонцева, канд. с.-х. наук (руководитель темы); P.M.Федорович, канд. техн. наук; М.Е.Поспелов, канд. биол. наук; Е.И.Коночкина
2. УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Государственного комитета СССР по стандартам от 29.06.89 N 2267
3. ВВЕДЕН ВПЕРВЫЕ
4. ССЫЛОЧНЫЕ НОРМАТИВНО-ТЕХНИЧЕСКИЕ ДОКУМЕНТЫ
Обозначение НТД, на который дана ссылка | Номер пункта |
ГОСТ 9-92 | 13.1 |
ГОСТ 61-75 | 8.1; 12.1; 22.1 |
ГОСТ 245-76 | 20.1 |
ГОСТ 334-73 | 14.1 |
ГОСТ 427-75 | 23.1 |
ГОСТ 435-77 | 22.1 |
ГОСТ 450-77 | 4.1; 5.1; 9.1; 10.1 |
ГОСТ 860-75 | 13.1 |
ГОСТ 975-88 | 19.1; 20.1 |
ГОСТ 1027-67 | 12.1 |
ГОСТ 1467-93 | 15.1 |
ГОСТ 1770-74 | 6.1; 8.1; 9.1; 11.1; 12.1; 13.1; 14.1; 15.1; 17.1; 19.1; 20.1; 22.1; 23.1 |
ГОСТ 1973-77 | 12.1 |
ГОСТ 2603-79 | 9.1; 23.1 |
ГОСТ 2768-84 | 9.1 |
ГОСТ 3022-80 | 23.1 |
ГОСТ 3118-77 | 8.1; 9.1; 11.1; 12.1; 13.1; 14.1; 15.1 |
ГОСТ 3778-98 | 11.1 |
ГОСТ 4108-72 | 7.1 |
ГОСТ 4145-74 | 6.1 |
ГОСТ 4146-74 | 6.1 |
ГОСТ 4160-74 | 12.1 |
ГОСТ 4165-78 | 6.1; 7.1 |
ГОСТ 4166-76 | 10.1 |
ГОСТ 4172-76 | 19.1; 20.1 |
ГОСТ 4174-77 | 22.1 |
ГОСТ 4197-74 | 22.1 |
ГОСТ 4198-75 | 19.1 |
ГОСТ 4204-77 | 6.1; 10.1; 11.1; 12.1; 13.1; 15.1 |
ГОСТ 4208-72 | 19.1 |
ГОСТ 4209-77 | 19.1 |
ГОСТ 4217-77 | 22.1 |
ГОСТ 4233-77 | 18.1; 19.1; 20.1 |
ГОСТ 4236-77 | 11.1 |
ГОСТ 4328-77 | 6.1; 7.1; 8.1; 10.1; 12.1; 18.1 |
ГОСТ 4386-89 | 14.4 |
ГОСТ 4456-75 | 15.1 |
ГОСТ 4461-77 | 11.1; 12.1; 13.1; 15.1 |
ГОСТ 4463-76 | 14.1 |
ГОСТ 5230-74 | 12.1; 13.1 |
ГОСТ 5456-79 | 13.1 |
ГОСТ 5457-75 | 11.1; 15.1 |
ГОСТ 5821-78 | 22.1 |
ГОСТ 5833-75 | 19.1 |
ГОСТ 5962-67 | 23.1 |
ГОСТ 6038-79 | 19.1; 20.1 |
ГОСТ 6217-74 | 22.1 |
ГОСТ 6259-75 | 8.1 |
ГОСТ 6672-75 | 17.1; 18.1; 19.1 |
ГОСТ 6691-77 | 19.1 |
ГОСТ 8677-76 | 8.1 |
ГОСТ 9147-80 | 2.3; 5.1; 6.1; 10.1; 12.1; 14.1; 23.1 |
ГОСТ 9284-75 | 17.1; 18.1; 19.1 |
ГОСТ 9293-74 | 10.1; 23.1 |
ГОСТ 9656-75 | 6.1 |
ГОСТ 10157-79 | 13.1 |
ГОСТ 10444.1-84 | 17.1; 18.1; 19.1; 20.1 |
ГОСТ 10444.12-88 | 17.1 |
ГОСТ 10485-75 | 12.1 |
ГОСТ 10929-76 | 6.1; 11.1; 12.1; 15.1 |
ГОСТ 10930-74 | 8.1 |
ГОСТ 11773-76 | 20.1 |
ГОСТ 12026-76 | 7.1; 8.1; 12.1; 19.1 |
ГОСТ 13496.0-80 | 1.1; 1.3 |
ГОСТ 13805-76 | 19.1; 20.1 |
ГОСТ 14261-77 | 9.1 |
ГОСТ 17206-96 | 18.1; 19.1; 20.1 |
ГОСТ 17299-78 | 6.1; 9.1; 10.1 |
ГОСТ 17626-81 | 20.1 |
ГОСТ 17792-72 | 14.1 |
ГОСТ 18300-87 | 6.1; 8.1; 9.1; 10.1; 12.1; 19.1; 23.1 |
ГОСТ 18963-73 | 19.3 |
ГОСТ 20015-88 | 13.1; 23.1 |
ГОСТ 20288-74 | 13.1 |
ГОСТ 20490-75 | 13.1 |
ГОСТ 20730-75 | 19.1 |
ГОСТ 22180-76 | 19.1 |
ГОСТ 22280-76 | 14.1 |
ГОСТ 24104-88 | 4.1; 5.1; 6.1; 8.1; 9.1; 10.1; 11.1; 12.1; 13.1; 14.1; 15.1; 17.1; 18.1; 19.1; 20.1; 21.1; 22.1; 23.1 |
ГОСТ 24363-80 | 10.1 |
ГОСТ 25336-82 | 4.1 5.1; 6.1; 7.1; 8.1; 9.1; 10.1; 11.1; 12.1; 13.1; 14.1; 15.1; 17.1; 18.1; 19.1; 20.1; 22.1; 23.1 |
ГОСТ 25706-83 | 23.1 |
ГОСТ 25794.1-83 | 6.1 |
ГОСТ 27068-86 | 19.1 |
ТУ 3-3.404-83 | 17.1; 18.1; 19.1; 20.1 |
ТУ 6-09-5467 | 12.1 |
ТУ 6-09-07-1684-89 | 13.1 |
ТУ 6-09-07-1703-90 | 2.2.1 |
5. Ограничение срока действия снято по протоколу N 4-93 Межгосударственного Совета по стандартизации, метрологии и сертификации (ИУС 4-94)
6. ИЗДАНИЕ с Изменением N 1, утвержденным в июне 1991 г. (ИУС 10-91)
1. ОТБОР ПРОБ
1.1. Для проведения испытаний от партии кормовых дрожжей отбирают точечные пробы по ГОСТ 13496.0.
1.2. Из точечных проб составляют объединенную пробу, помещают ее в чистую тару и перемешивают. В тару вкладывают этикетку с указанием наименования продукта, номера партии, даты отбора точечных проб, наименования предприятия-изготовителя.
1.3. Из объединенной пробы выделяют среднюю пробу по ГОСТ 13496.0, делят ее путем квартования на две равные части и помещают в чистые сухие банки с плотно закрывающимися крышками или пробками. Одну из них используют для анализов, а другую опечатывают или пломбируют и хранят не менее 2 мес на случай проведения контрольных испытаний при разногласиях в оценке качества кормовых дрожжей.
1.4. К банке со средней пробой прикрепляют этикетку, на которой должны быть обозначены:
наименование продукта;
наименование предприятия-изготовителя;
номер партии;
дата отбора пробы и подпись лица, отобравшего пробу.
1.5. Для испытаний по микробиологическим показателям пробы отбирают в стерильную посуду. Все оборудование, с помощью которого отбирают пробы, должно быть простерилизовано в лабораторных условиях фламбированием (протиранием ватой, смоченной спиртом, с последующим обжиганием на огне) или в сушильном шкафу в течение 1,5 ч при температуре 150-170 °С.
1.6. Масса объединенной пробы для испытаний по микробиологическим показателям должна быть не менее 1 кг.
1.7. Допускается для испытаний по всем показателям качества кормовых дрожжей отбирать по п.1.5 одну объединенную пробу массой не менее 5 кг, из которых 1 кг используют для испытания кормовых дрожжей по микробиологическим показателям.
2. ПОДГОТОВКА ПРОБ К ИСПЫТАНИЯМ
2.1. Часть средней пробы (около 400 г) просеивают через штампованное сито или решетное полотно с ячейками диаметром 0,2-0,3 мм. Остаток на сите измельчают на лабораторной мельнице и просеивают до тех пор, пока весь остаток не пройдет через сито. Просеянный продукт насыпают в тару вместимостью 0,5 дм с плотно закрывающейся крышкой или пробкой и используют для проведения испытания.
Гранулированный продукт перед просеиванием измельчают.
2.2. Для определения массовой доли сырого протеина и белка по Барнштейну продукт измельчают до пылеобразного состояния.
2.3. Для проведения испытаний по микробиологическим показателям гранулированный продукт растирают в фарфоровой ступке по ГОСТ 9147. Ступку и пестик перед использованием стерилизуют фламбированием.
3. ОПРЕДЕЛЕНИЕ ВНЕШНЕГО ВИДА, ЦВЕТА И ЗАПАХА
3.1. Для определения внешнего вида и цвета навеску продукта массой около 250 г рассыпают на белую чистую поверхность и рассматривают при естественном свете.
3.2. Запах определяют органолептически.
4. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ ВЛАГИ
Сущность метода заключается в высушивании навески продукта до постоянной массы при установленных температуре и времени; массовую долю влаги определяют как отношение потери в массе после высушивания к массе исходной навески продукта.
(Измененная редакция, Изм. N 1).
4.1. Аппаратура и реактивы*
________________
* Во всех методах при проведении испытаний допускается использовать импортные аппаратуру, материалы и реактивы с характеристиками не ниже отечественных аналогов.
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104*.
________________
* С 1 июля 2002 г. вводится в действие ГОСТ 24104-2001 (здесь и далее).
Шкаф сушильный любого типа, обеспечивающий постоянство температуры (130±5) °С.
Эксикатор по ГОСТ 25336.
Стаканчики для взвешивания (бюксы) СВ-24/10 или СВ-34/12 по ГОСТ 25336 или стаканчики металлические для высушивания, поставляемые с сушильными шкафами СЭШ-1 или СЭШ-3М.
Щипцы тигельные.
Кальций хлористый технический по ГОСТ 450 или кальций хлористый обезвоженный, прокаленный.
4.2. Проведение испытания
Во взвешенную бюксу помещают 2-4 г продукта, разравнивая его равномерным слоем по дну бюксы. Бюксу с продуктом закрывают крышкой и взвешивают.
Открытую бюксу и крышку от нее помещают в сушильный шкаф, нагретый до температуры (130±5) °С, и высушивают 40 мин. По истечении 40 мин бюксу с помощью тигельных щипцов закрывают крышкой, вынимают из сушильного шкафа и ставят в эксикатор для охлаждения до комнатной температуры. Затем бюксу с закрытой крышкой взвешивают. Все результаты взвешиваний записывают с точностью до четвертого десятичного знака.
Проводят два параллельных определения.
4.3. Обработка результатов
Массовую долю влаги () в процентах вычисляют по формуле
![]() , (1)
, (1)
где - масса бюксы с навеской до высушивания, г;
- масса бюксы с навеской после высушивания, г;
- масса пустой бюксы, г.
Результат округляют до второго десятичного знака. Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 5%, округленных до целого числа.
За окончательный результат испытаний принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
5. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ ЗОЛЫ
Сущность метода заключается в сожжении навески продукта и прокаливании полученного остатка до постоянной массы.
5.1. Аппаратура и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Печь муфельная, обеспечивающая нагрев до 800 °С, с регулятором температуры.
Тигли низкие 4 или 5 по ГОСТ 9147 или фарфоровые чашки диаметром 30-40 мм или тигли и чашки из кварцевого стекла, платины.
Эксикатор по ГОСТ 25336.
Щипцы тигельные.
Кальций хлористый технический по ГОСТ 450 или кальций хлористый обезвоженный, прокаленный.
5.2. Подготовка к испытанию
Чистый тигель прокаливают в муфельной печи при температуре (600±20) °С в течение 5 ч, затем тигель переносят в эксикатор и после охлаждения до комнатной температуры взвешивают.
Тигель вновь помещают на 1 ч в муфельную печь для повторного прокаливания при той же температуре и после охлаждения в эксикаторе до комнатной температуры взвешивают. Если его масса изменилась более чем на 0,0004 г, прокаливание повторяют. Прокаливание считают законченным, если разница между двумя последовательными взвешиваниями составит не более 0,0004 г.
Все результаты взвешиваний записывают с точностью до четвертого десятичного знака.
Подготовленные тигли хранят в эксикаторе над хлористым кальцием.
5.3. Проведение испытания
В предварительно доведенный до постоянной массы и взвешенный тигель помещают 0,8-0,1 г продукта. Тигель с продуктом взвешивают и ставят у открытой дверцы муфельной печи, нагретой до температуры (600±20) °С. По мере прогревания тигля с продуктом происходит интенсивное выделение паров и газов, которые не должны воспламеняться; если воспламенение произошло, испытание повторяют.
После обугливания продукта и прекращения выделения паров и газов тигель с навеской переставляют в зону более высокой температуры и дверцу муфельной печи закрывают. Прокаливание проводят в течение 4-5 ч, затем тигель вынимают, переносят в эксикатор и после охлаждения до комнатной температуры взвешивают.
Тигель вновь ставят в муфельную печь и проводят повторное прокаливание в течение 1 ч. После охлаждения в эксикаторе тигель с золой взвешивают. Прокаливание повторяют до достижения постоянной массы. Массу считают постоянной, если разница между двумя последовательными взвешиваниями составит не более 0,0004 г. Если при очередном прокаливании масса увеличивается, то за конечную величину принимают наименьшую массу предыдущего взвешивания. Все результаты взвешиваний записывают с точностью до четвертого десятичного знака.
Проводят два параллельных определения.
5.4. Обработка результатов
Массовую долю золы () в процентах вычисляют по формуле
![]() , (2)
, (2)
где - масса тигля с золой, г;
- масса пустого тигля, г;
- масса тигля с навеской до озоления, г;
- массовая доля влаги продукта, %.
Результат округляют до второго десятичного знака. Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 5%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
6. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ СЫРОГО ПРОТЕИНА
Сущность метода заключается в восстановлении азота органических соединений при минерализации продукта серной кислотой до аммиака, титрометрическом определении аммиака и пересчете его количества на содержание сырого протеина.
6.1. Аппаратура, материалы и реактивы
Весы лабораторные технические любого типа.
Весы лабораторные 1-го класса точности с наибольшим пределом взвешивания 1 кг по ГОСТ 24104.
Устройства нагревательные любого типа: электрические, инфракрасные, газовые, снабженные устройствами для регулирования степени нагрева.
Колба Кьельдаля 2-250-29 ТХС или 2-500-29 ТХС по ГОСТ 25336.
Втулка Кьельдаля или воронка лабораторная В 36-80 по ГОСТ 25336.
Пробирка П2-16-180, или П 2-19-180 или П 2-21-200 по ГОСТ 25336.
Цилиндры мерные 1-10; 1-25 или 3-25; 1-100 или 3-100 по ГОСТ 1770.
Палочка стеклянная диаметром 5-6 мм.
Прибор для отгонки аммиака (черт.1, 2).
Прибор для отгонки аммиака
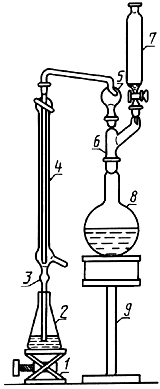
1 - подъемный столик; 2 - приемная колба; 3 - удлиненная муфта; 4 - холодильник; 5 - каплеуловитель;
6 - переход; 7 - делительная воронка; 8 - отгонная колба; 9 - нагревательное устройство
Черт.1
Прибор для отгонки аммиака с водяным паром
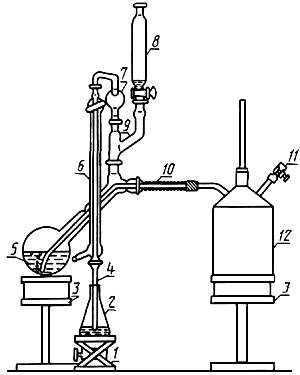
1 - подъемный столик; 2 - приемная колба; 3 - нагревательное устройство; 4 - удлиненная муфта;
5 - отгонная колба; 6 - холодильник; 7 - каплеуловитель; 8 - делительная воронка; 9 - переход;
10 - изоляция паропроводящей трубки; 11 - отвод с зажимом; 12 - парообразователь
Черт.2
Колба круглодонная К-1-500-29/32 ТС или К-1-1000-29/32 ТС, или колба плоскодонная П-1-500-29/32 ТС, или П-1-1000-29/32 по ГОСТ 25336.
Переход П2П-29/32-14/23-14/23 по ГОСТ 25336.
Каплеуловитель КО-14/23-100 ХС по ГОСТ 25336.
Холодильник ХПТ-1-300-14/23 ТС или ХПТ-1-400-14/23 ТС, или ХШ-1-200-14/23 ХС, или ХШ-1-300-14/23 ХС, или ХШ-1-400-14/23 ХС по ГОСТ 25336.
Муфта МПО-14/23 ТС по ГОСТ 25336 с удлиненным до 160-170 мм отводом.
Колба Кн-2-250-34 ТХС по ГОСТ 25336.
Воронка делительная ВД-2-100 ХС по ГОСТ 25336.
Столик подъемный типа СПД.
Кольца резиновые.
Пипетки 2-1-50 или 2-2-50 по НТД.
Бюретки 1-1-50 или 1-2-50 по НТД или в исполнениях 2, 3, 4, 5.
Ступка фарфоровая диаметром 90 или 110 мм с пестиком по ГОСТ 9147.
Промывалка для дистиллированной воды.
Капельница для индикатора.
Бумага индикаторная универсальная.
Асбест листовой.
Кислота серная по ГОСТ 4204, х.ч., концентрированная и раствор концентрации (1/2H
SO
)=0,1 моль/дм
(0,1 н.); готовят из стандарт-титра или по ГОСТ 25794.1 из концентрированной кислоты.
Натрия гидроокись по ГОСТ 4328, водный раствор с массовой долей 30-40% и раствор концентрации (NaOH)=0,1 моль/дм
(0,1 н.); готовят из стандарт-титра или по ГОСТ 25794.1 из натрия гидроокиси.
Калий сернокислый по ГОСТ 4145 любой квалификации или калий надсернокислый по ГОСТ 4146, х.ч. или ч.д.а.
Медь (II) сернокислая 5-водная по ГОСТ 4165 любой квалификации.
Селен металлический, ч.
Водорода перекись по ГОСТ 10929, раствор с массовой долей 30%.
Кислота борная по ГОСТ 9656, ч. д. а. или х. ч., раствор с массовой долей 4%.
Метиловый красный, спиртовой раствор с массовой долей 0,2 и 0,4%.
Метиленовый голубой, спиртовой раствор с массовой долей 0,2%.
Бромкрезоловый зеленый, спиртовой раствор с массовой долей 0,1%.
Спирт этиловый ректификованный технический по ГОСТ 18300 или спирт этиловый технический по ГОСТ 17299.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
6.2. Подготовка к испытанию
6.2.1. Приготовление твердых катализаторов
Катализатор 1. Смешивают 10 весовых частей сернокислой меди, 100 весовых частей сернокислого калия и 2 весовые части селена, смесь тщательно растирают в ступке до получения однородного мелкозернистого порошка.
Катализатор 2. Смешивают 1 весовую часть сернокислой меди и 3 весовые части сернокислого калия, смесь тщательно растирают в ступке до получения однородного мелкозернистого порошка.
Катализатор 3. Смешивают 10 весовых частей сернокислой меди, 100 весовых частей сернокислого калия, смесь тщательно растирают в ступке до получения однородного мелкозернистого порошка.
Допускается заменять сернокислый калий надсернокислым калием в том же количестве.
Катализаторы хранят в склянках (стеклянных или тефлоновых) с плотно закрывающимися крышками.
(Измененная редакция, Изм. N 1).
6.2.2. Приготовление серной кислоты, содержащей селен в качестве катализатора
Аморфный или растертый в порошок селен из расчета 0,5 г на 100 см концентрированной серной кислоты растворяют при нагревании в термостойкой колбе до обесцвечивания раствора.
Серную кислоту с растворенным селеном хранят в склянках с плотно закрывающимися пробками.
6.2.3. Приготовление смешанных индикаторов
Индикатор 1. Смешивают равные объемы 0,4%-ного спиртового раствора метилового красного и 0,2%-ного спиртового раствора метиленового голубого.
Индикатор 2. Смешивают 1 объем 0,2%-ного спиртового раствора метилового красного и 3 объема 0,1%-ного спиртового раствора бромкрезолового зеленого.
Индикаторы хранят в склянках из темного стекла или в защищенном от света месте.
6.2.4. Определение поправки к титру раствора гидроокиси натрия концентрации (NaOH)=0,1 моль/дм
(0,1 н.)
Поправка к титру представляет собой отношение эквивалентной концентрации приготовленного раствора к заданной концентрации.
Для определения этой поправки в три конические колбы вместимостью до 250 см пипеткой вносят по 20 см
раствора серной кислоты, приготовленного из стандарт-титра, концентрации 0,1 моль/дм
(0,1 н.), добавляют 5-6 капель индикатора 1 или 2 и титруют приготовленным раствором гидроокиси натрия до перехода окраски индикатора от розовой до светло-серой (одна капля избытка гидроокиси натрия приводит к образованию зеленой окраски).
Из результатов трех параллельных титрований вычисляют среднее арифметическое.
Эквивалентную концентрацию раствора гидроокиси натрия () в молях на кубический дециметр вычисляют по формуле
![]() , (3)
, (3)
где - концентрация раствора серной кислоты, приготовленного из стандарт-титра, 0,1 моль/дм
(0,1 н.);
- объем раствора серной кислоты, приготовленной из стандарт-титра, взятый для титрования, см
;
- объем раствора гидроокиси натрия, израсходованный на титрование взятого количества серной кислоты, см
.
Поправку () к титру раствора гидроокиси натрия вычисляют по формуле
![]() , (4)
, (4)
где - установленная эквивалентная концентрация приготовленного раствора гидроокиси натрия, моль/дм
;
- заданная эквивалентная концентрация раствора гидроокиси натрия 0,1 моль/дм
(0,1 н.).
6.3. Проведение испытания
6.3.1. Минерализация образца
В длинную сухую пробирку, свободно входящую в горло колбы Кьельдаля, насыпают (0,5±0,02) г продукта и взвешивают. Затем продукт осторожно высыпают в сухую колбу Кьельдаля, возможно глубже опуская пробирку в горло колбы. Пробирку вновь взвешивают. По разнице между первым и вторым взвешиваниями определяют массу навески продукта, взятую для анализа. Все результаты взвешиваний записывают с точностью до четвертого десятичного знака.
Для внесения в колбу Кьельдаля навески продукта можно пользоваться следующим приемом. Перед тем, как в пробирку насыпают продукт, на нее надевают два резиновых кольца на расстоянии 6-8 см. Затем в пробирку насыпают продукт, пробирку взвешивают, помещая ее на весах в стакан или подвешивая с помощью проволочного кольца. После того, как произведено взвешивание, пробирку держат открытым концом вверх и под кольца подсовывают стеклянную палочку длиной на 4-5 см больше, чем длина колбы Кьельдаля, в которой будет производиться сжигание. Затем одной рукой на пробирку надевают перевернутую вверх дном колбу Кьельдаля так, чтобы дно колбы оказалось на расстоянии 1-2 см от открытого конца пробирки. Далее колбу с находящейся в ней пробиркой перевертывают дном вниз, удерживая пробирку за стеклянную палочку. При этом продукт высыпается непосредственно на дно колбы и не попадает на стенки. После того, как продукт высыпался, пробирку вытягивают из колбы, переворачивают, палочку из-под резиновых колец вынимают и пробирку вместе с резиновыми кольцами вновь взвешивают.
Минерализацию проводят одним из следующих способов.
Способ 1. В колбу с навеской добавляют 2-3 г катализатора 1 или 2 и приливают 25 см концентрированной серной кислоты.
Способ 2. В колбу с навеской приливают 10 см раствора перекиси водорода с массовой долей перекиси водорода 30% и 25 см
концентрированной серной кислоты. Для ускорения минерализации рекомендуется использовать серную кислоту, содержащую селен, приготовленную по п.6.2.2.
Способ 3. Добавляют в колбу 8 г катализатора. После прибавления катализатора осторожно приливают 15 см концентрированной серной кислоты.
Серную кислоту приливают по стенкам колбы, одновременно смывая на дно колбы частицы катализатора и продукта, оставшиеся на стенках колбы.
После добавления всех реактивов содержимое колбы тщательно перемешивают легкими круговыми движениями колбы, обеспечивая полное смешение навески продукта с кислотой.
После этого колбу устанавливают на нагреватель; допускается перед установкой на нагреватель колбу оставить на 1,5-2 ч при комнатной температуре для предварительного окисления продукта.
На нагреватель колбу ставят так, чтобы ее ось была наклонена к вертикали под углом 30-45 °. В горло колбы вставляют втулку или маленькую воронку для уменьшения испарения кислоты. Минерализацию проводят под тягой, так как при этом процессе происходит выделение сернистого ангидрида.
В начале минерализации обычно происходит образование пены, поэтому нагрев сначала производят умеренно, чтобы предотвратить выброс пены из колбы, по мере прекращения пенообразования нагрев усиливают, пока жидкость не станет равномерно кипеть. Нагрев считают нормальным, если жидкость кипит, а пары конденсируются около середины - двух третей горла колбы.
Избегают перегрева стенок колбы выше уровня жидкости. Если используют нагрев в пламени горелки, то во избежание такого перегрева колбу помещают на лист асбеста с отверстием по диаметру несколько меньшим, чем диаметр колбы на уровне жидкости.
При минерализации по способу 1 допускается производить обработку продукта перекисью водорода, как и при минерализации по способу 2.
Кроме того, для ускорения минерализации допускается добавлять перекись водорода по ходу процесса. В этом случае колбу снимают с нагревателя, дают ей немного остыть и добавляют к содержимому колбы перекись водорода небольшими порциями, почти по каплям, давая жидкости стекать по стенке колбы. Как только стекающая порция перекиси водорода коснется жидкости, содержимое колбы перемешивают круговыми движениями колбы. Следующую порцию перекиси водорода добавляют только после того, как кончится бурная реакция окисления. Внесение перекиси водорода как вначале, до добавления кислоты, так и потом, по ходу минерализации, проводят под тягой, следя за тем, чтобы отверстие горла колбы было направлено в сторону от исполнителя и других работающих.
Во время минерализации содержимое колбы периодически помешивают круговыми движениями колбы. Если на стенках колбы выше уровня жидкости оказались частицы продукта или брызги кислоты, их смывают при помешивании в основной объем жидкости.
По ходу минерализации жидкость в колбе постепенно осветляется и, когда она станет прозрачной, обесцветится или будет иметь зеленоватый или голубоватый оттенок, минерализацию продолжают еще 30 мин, после чего колбу снимают с нагревателя и дают ей охладиться. Затем в колбу добавляют 100-150 см дистиллированной воды и содержимое колбы перемешивают.
6.3.2. Отгонка аммиака
Отгонку аммиака проводят в приборе, изображенном на черт.1. Допускается проводить отгонку аммиака с водяным паром в приборе, изображенном на черт.2. В этом случае в парообразователь наливают водопроводную воду, которую подкисляют серной кислотой, чтобы исключить выделение из нее имеющегося аммиака.
Допускается для отгонки аммиака применение приборов, изготовленных в соответствии с черт.1 из других аналогичных деталей с соединением этих деталей на резиновых пробках и с помощью полиэтиленовых трубок. Конец отгонки аммиака проверяют с помощью индикаторной бумаги.
6.3.2.1. Отгонка аммиака в серную кислоту
Смесь, содержащуюся в колбе Кьельдаля, количественно с помощью дистиллированной воды переносят в колбу прибора для отгонки аммиака. Допускается отгонку аммиака проводить из колбы Кьельдаля вместимостью 500 см. В приемную колбу пипеткой вносят 50 см
раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.) и добавляют 5-6 капель индикатора 1 или 2. Собирают прибор. В делительную воронку наливают 100 см
раствора гидроокиси натрия с массовой долей 30-40%. Приемную колбу устанавливают так, чтобы удлиненный конец трубки холодильника был погружен в кислоту на глубину 0,5-1 см.
В холодильник пускают воду, включают нагрев и из капельной воронки в колбу осторожно приливают раствор гидроокиси натрия, оставляя в воронке небольшое количество раствора. Капельную воронку промывают два-три раза порциями по 10-15 см дистиллированной воды, которую сливают в колбу, следя за тем, чтобы при каждом сливе, включая последний, в воронке оставалось небольшое количество воды в качестве гидрозатвора.
Допускается прибавлять раствор гидроокиси натрия до присоединения отгонной колбы к аппарату. В этом случае раствор гидроокиси натрия добавляют в отгонную колбу по стенке, стараясь перемешивать его с минерализатом. После добавления всего объема колбу сразу соединяют с аппаратом для отгонки аммиака.
В процессе отгонки аммиака следят за тем, чтобы конец трубки холодильника был погружен в раствор кислоты на 0,5-1 см, но не более. Для этого по мере увеличения объема жидкости в приемной колбе колбу постепенно опускают, что удобно производить с помощью подъемного столика.
Отгонку ведут до тех пор, пока объем раствора в приемной колбе увеличится примерно в три раза, что гарантирует полную отгонку аммиака. По окончании отгонки приемную колбу опускают так, чтобы конец трубки холодильника не касался раствора. Обогрев прекращают, холодильник отсоединяют от каплеуловителя и перегонной колбы и внутреннюю поверхность трубки холодильника, а также ее наружный конец, который был погружен в кислоту, промывают дистиллированной водой из промывалки, давая промывным водам стечь в приемную колбу.
Можно использовать другой прием. Отгонку ведут до тех пор, пока объем жидкости в приемной колбе не увеличится примерно в два с половиной раза, тогда приемную колбу опускают так, чтобы конец трубки холодильника оказался на 2-2,5 см выше уровня жидкости. Отгонку продолжают, отгоняя еще 50-70 см жидкости. Окончание отгонки аммиака проверяют с помощью индикаторной бумаги. Затем конец трубки холодильника, который раньше был погружен в кислоту, промывают снаружи дистиллированной водой из промывалки, давая промывной воде стечь в приемную колбу.
6.3.1, 6.3.2, 6.3.2.1. (Измененная редакция, Изм. N 1).
6.3.2.2. Отгонка аммиака в борную кислоту
Отгонку проводят так же, как при отгонке в серную кислоту, с той разницей, что в приемную колбу вносят 50 см раствора борной кислоты с массовой долей 4%. В начале отгонки, когда аммиак начнет поступать в приемную колбу, цвет индикатора принимает зеленую окраску.
6.3.3. Титрометрическое определение аммиака
6.3.3.1. Титрование при отгонке в серную кислоту
Содержимое приемной колбы титруют раствором гидроокиси натрия концентрации 0,1 моль/дм (0,1 н.) до перехода окраски индикатора от фиолетовой в зеленую при применении индикатора 1 и от розовой в зеленую при применении индикатора 2.
6.3.3.2. Титрование при отгонке в борную кислоту
Содержимое колбы титруют раствором серной кислоты концентрации 0,1 моль/дм (0,1 н.) до перехода окраски от зеленой в фиолетовую при применении индикатора 1 и от зеленой в розовую при применении индикатора 2.
6.3.4. Проведение контрольных опытов
Одновременно с рабочим опытом проводят контрольный опыт для определения степени загрязнения воды и реактивов аммиаком. Контрольный опыт повторяет все стадии, исключая взятие навески продукта; его проводят при смене хотя бы одного применяемого реактива, а если реактивы не заменяли, то не реже, чем каждые 5 сут.
Контрольный опыт считают удовлетворительным, если при отгонке аммиака в серную кислоту объем раствора гидроокиси натрия, израсходованный на титрование, окажется не менее 45,0 см, а при отгонке аммиака в борную кислоту объем раствора серной кислоты, израсходованный на титрование, окажется более 5,0 см
.
При превышении указанных норм выявляют источник загрязнения реактивов аммиаком и устраняют его.
Проводят два параллельных определения.
6.4. Обработка результатов
6.4.1. Массовую долю сырого протеина () в процентах при отгонке аммиака в серную кисло вычисляют по формуле
![]() , (5)
, (5)
где - объем раствора гидроокиси натрия концентрации 0,1 моль/дм
(0,1 н.), израсходованный на титрование серной кислоты в контрольном опыте, см
;
- объем раствора гидроокиси натрия концентрации 0,1 моль/дм
(0,1 н.), израсходованный на титрование серной кислоты в испытуемом растворе, см
;
- поправка к титру раствора гидроокиси натрия концентрации 0,1 моль/дм
(0,1 н.);
![]() - масса азота, эквивалентная 1 см
- масса азота, эквивалентная 1 см раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.);
- коэффициент пересчета массовой доли азота на массовую долю сырого протеина;
- масса навески продукта, г;
- массовая доля влаги в испытуемом прод
укте, %.
6.4.2. Массовую долю сырого протеина () в процентах при отгонке аммиака в борную кислоту вычисляют по формуле
![]() , (6)
, (6)
где - объем раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.), израсходованный на титрование испытуемого раствора, см
;
- объем раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.), израсходованный на титрование в контрольном опыте, см
;
- поправка к титру раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.), если он приготовлен не из стандарт-титра;
![]() - масса азота, эквивалентная 1 см
- масса азота, эквивалентная 1 см раствора серной кислоты концентрации 0,1 моль/дм
(0,1 н.), г;
- коэффициент пересчета массовой доли азота на массовую долю сырого протеина;
- масса навески продукта, г;
- массовая доля влаги в испытуемом продукте, %.
Результат округляют до первого десятичного знака. Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 1%, округленного до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до целого числа.
Допускаемое относительное расхождение между окончательными результатами, получеными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до целого числа. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 3%, округленных до целого числа.
7. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ БЕЛКА ПО БАРНШТЕЙНУ
Сущность метода заключается в удалении из продукта водорастворимых небелковых азотосодержащих соединений при обработке продукта горячей водой, восстановлении азота оставшихся органических соединений при минерализации продукта серной кислотой до аммиака, титрометрическом определении аммиака и пересчете его количества на содержание белка по Барнштейну.
7.1. Аппаратура, материалы и реактивы
Аппаратура, материалы и реактивы - по п.6.1 и дополнительно:
Воронка В-56-80 ХС или В-75-110 ХС или В-75-140 ХС по ГОСТ 25336.
Стакан В-1-250 ТХС или Н-1-250 ТХС по ГОСТ 25336.
Колба Кн-2-500-34 ТХС по ГОСТ 25336.
Фильтр бумажный обеззоленный (синяя лента).
Бумага фильтровальная лабораторная по ГОСТ 12026.
Натрия гидроокись по ГОСТ 4328, раствор с массовой долей 2,5%.
Медь сернокислая 5-водная по ГОСТ 4165, раствор с массовой долей 10%.
Барий хлористый по ГОСТ 4108, раствор с массовой долей 1-2%.
7.2. Проведение испытания
В химический стакан отвешивают (0,5±0,2) г продукта (результат взвешивания записывают с точностью до четвертого десятичного знака), приливают 100 см кипящей дистиллированной воды. Стакан ставят на нагреватель и кипятят содержимое 2-3 мин. Затем стакан снимают и, не охлаждая жидкости, приливают 20 см
раствора сернокислой меди, содержимое перемешивают, добавляют 20 см
раствора гидроокиси натрия с массовой долей 2,5%, снова перемешивают и оставляют при комнатной температуре на 1 ч. Отстоявшуюся жидкость декантируют через фильтр, осадок в стакане промывают горячей водой, декантацией сливая промывные воды через тот же фильтр, затем осадок переносят на фильтр и промывают его горячей водой до исчезновения в промывных водах реакции на сульфат-ион, для чего в пробирку отбирают несколько капель промывных вод и добавляют 2-3 капли раствора хлористого бария; отсутствие помутнения указывает на полноту промывки.
Промытый осадок вместе с фильтром подсушивают на воздухе или в сушильном шкафу при температуре не выше (105±2) °С и помещают в колбу Кьельдаля. Испытание проводят по п.6.3 одним из установленных способов минерализации.
Проводят два параллельных определения.
7.3. Обработка результатов
Обработка результатов - по п.6.4.
Массовую долю белка по Барнштейну () в процентах при отгонке аммиака в серную кислоту вычисляют по формуле (5); при отгонке аммиака в борную кислоту - по формуле (6).
8. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ ЛИЗИНА
Сущность метода заключается в кислотном гидролизе белковых веществ продукта до свободных аминокислот с последующим спектрофотометрическим определением окрашенных производных, полученных в результате специфической реакции лизина с фурфуролом в среде ледяной уксусной кислоты.
8.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Шкаф сушильный, обеспечивающий постоянство температуры (110±2) °С.
Баня водяная, обеспечивающая постоянство температуры (100±2) °С.
Колбонагреватель любого типа, обеспечивающий постоянство температуры (100±2) °С.
Спектрофотометр или фотоэлектроколориметр любого типа, обеспечивающий измерение в области 530-545 нм.
Фен или вентилятор комнатный с обогревом любого типа.
Цилиндры мерные 1-1000, 2-100 или 3-100 по ГОСТ 1770.
Холодильники ХПТ-2-400-29/32 ХС, ХПТ-1-200-14/23 ХС по ГОСТ 25336.
Колба круглодонная К-1-1000-29/32 ТС или К-1-2000-29/32 ТС, К-1-50-14/23 ТС по ГОСТ 25336.
Колбы плоскодонные П-1-1000-29/32 ТС и П-1-100-14/23 ТС или конические Кн-1-1000-29/32 ТС и Кн-1-100-14/23 ТС по ГОСТ 25336.
Колба грушевидная Гр-25-14/23 ТС или колба остродонная ОГ-2-50-14/23 ТС, или ОГ-3-25-14/23 ТС, или круглодонная К-1-25-14/23 ТС по ГОСТ 25336.
Трубки хлоркальциевые ТХ-П-2-19, ТХ-П-1-17 по ГОСТ 25336.
Насадки типа Н1-14/23-14/23-14/23 ТС или Н2-14/23-14/23 ТС по ГОСТ 25336.
Изгиб U < 75° 2К-29/32-14/23 ТС по ГОСТ 25336.
Алонжи АИО-29/32-29/32-75 ТС, АИО-14/23-14/23-60 ТС по ГОСТ 25336.
Стаканчики для взвешивания СВ-14/8 или СВ-19/9 или СВ-24/10 или СН-34/12 по ГОСТ 25336.
Пробирки мерные П-2-15-14/23, П-2-5-14/23 ХС по ГОСТ 1770.
Пипетки 4-1-2 или 4-2-2, 5-1-2 или 5-2-2, 6-1-5 или 6-2-5, 7-1-5 или 7-2-5, 6-1-10 или 6-2-10, 7-1-10 или 7-2-10 по НТД.
Воронка В-36-50 или В-36-80 ХС по ГОСТ 25336.
Склянка из темного стекла вместимостью 50-100 см с пришлифованной пробкой.
Микрошприц или автоматические пипетки любого типа вместимостью 10-50 мкл или автоматические пипетки вместимостью 10 или 20 мкл.
Бумага индикаторная универсальная.
Фильтры обеззоленные с синей полосой диаметром 8 см.
Бумага фильтровальная по ГОСТ 12026.
Кислота ледяная уксусная по ГОСТ 61.
Кислота соляная концентрированная по ГОСТ 3118, х.ч.
Спирт этиловый ректификованный технический по ГОСТ 18300 или спирт этиловый абсолютный.
Глицерин по ГОСТ 6259, ч.
Фурфурол по ГОСТ 10930 или технический.
Калий пиросернистокислый, ч.
Натрия гидроокись по ГОСТ 4328, х.ч.
Кальций хлористый обезвоженный, ч.
L - лизин моногидрохлорид, или дигидрохлорид, х.ч.
Кальций оксид (окись кальция) по ГОСТ 8677, ч.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
8.2. Подготовка к испытанию
8.2.1. Приготовление абсолютированного спирта
В круглодонную колбу вместимостью 2 дм наливают 1 дм
этилового спирта и помещают 250 г оксида кальция. Смесь кипятят в течение 10 ч с обратным холодильником, закрытым хлоркальциевой трубкой с хлористым кальцием. Кипячение допускается проводить в два приема с перерывом на ночь. На время перерыва прибор не разбирают, оставляя его закрытым хлоркальциевой трубкой. После окончания кипячения нагреватель оставляют, прибор не разбирают и дают ему охладиться. Далее колбу со спиртом через изгиб соединяют с установкой для перегонки, состоящей из прямого холодильника, алонжа и приемной колбы; к свободному концу алонжа присоединяют хлоркальциевую трубку с хлористым кальцием.
Отгонку проводят до тех пор, пока в приемную колбу не отгонится 0,7 дм спирта. Далее приемник заменяют и отгоняют оставшийся спирт.
Первый отгон - абсолютированный спирт - хранят в сосуде с закрытой пробкой.
Допускается абсолютирование спирта проводить другими способами, установленными в нормативно-технической документации.
8.2.2. Перегонка фурфурола в вакууме
Перегонку осуществляют в приборе, состоящем из грушевидной или остродонной, или круглодонной колбы холодильника, алонжа и приемной круглодонной колбы. Во избежание выбросов в прямое горло колбы на резиновой пробке вводится стеклянная трубка с оттянутым в капилляр концом, доходящим почти до конца дна колбы. На выходящий из пробки наружный конец трубки надевается кусок резиновой трубки, через которую вводится небольшой кусок нитки или тонкой проволоки. Резиновая трубка пережимается винтовым зажимом, которым во время отгонки регулируют поступление в колбу через капилляр пузырьков воздуха. Пузырьки воздуха во время отгонки должны выходить из капилляра и подниматься в виде цепочки отдельных мелких пузырьков. Второе горло отгонной колбы закрывают пришлифованной пробкой. Отгонку ведут под вакуумом водоструйного насоса на водяной бане при температуре 60-70 °С или на колбонагревателе при температуре не выше 95 °С.
При абсолютировании спирта, его разгонке и перегонке фурфурола в вакууме допускается сборка приборов из других элементов.
8.2.3. Приготовление фурфурольного реактива
В стаканчик для взвешивания (бюксу) отвешивают (0,065±0,005) г пиросернистокислого калия (результат взвешивания записывают с точностью до четвертого десятичного знака). Навеску возможно более полно пересыпают в коническую или плоскодонную колбу вместимостью 100 см с пришлифованной пробкой. Количество пиросернистокислого калия не должно быть точным, поэтому второе взвешивание стаканчика и определение точной массы соли по разности между взвешиваниями не производят.
В колбу добавляют 40 см ледяной уксусной кислоты, дают соли раствориться, добавляют 30 см
абсолютированного или абсолютного этилового спирта и 10 см
свежеперегнанного фурфурола или фурфурола перегнанного, хранящегося в запаянных вакуумированных ампулах.
Раствор перемешивают.
Приготовленный реактив хранят в холодильнике не более 2 недель.
8.2.1-8.2.3. (Измененная редакция, Изм. N 1).
8.2.4. Приготовление водного раствора глицерина
В стеклянной колбе или склянке с пришлифованной пробкой смешивают один объем глицерина с четырьмя объемами дистиллированной воды.
Раствор хранят в холодильнике 2 мес.
8.2.5. Приготовление раствора соляной кислоты концентрации (НСl)=6 моль/дм
(6 н.).
В стеклянной колбе или склянке с пришлифованной пробкой смешивают соответствующие в зависимости от плотности концентрированной соляной кислоты объемы этой кислоты и дистиллированной воды.
(Измененная редакция, Изм. N 1).
8.2.6. Приготовление раствора гидроокиси натрия концентрации (NaOH)=6 моль/дм
(6 н.)
В фарфоровый сосуд отвешивают гидроокись натрия и добавляют дистиллированную воду. После растворения гидроокиси натрия раствор охлаждают и переливают в мерную колбу. Затем к раствору порциями добавляют дистиллированную воду, постепенно доводя объем раствора до метки. После добавления каждой порции раствор перемешивают и, если произошло нагревание, раствору дают охладиться. Для получения 250 см раствора берут 60 г гидроокиси натрия и 200 см
дистиллированной воды. Для получения других объемов раствора гидроокиси натрия концентрации
(NaOH)=6 моль/дм
(6 н.) количество гидроокиси натрия и воды рассчитывают.
8.2.7. Приготовление стандартных растворов лизина
8.2.7.1. Раствор А, содержащий 2,5 мг/см лизина
В стаканчик для взвешивания отвешивают (0,078±0,001) г монохлорида лизина или соответствующее количество дигидрохлорида (результат записывают с точностью до четвертого десятичного знака). Навеску соли количественно переносят дистиллированной водой в мерную колбу вместимостью 25 см, монохлориду дают раствориться, раствор в колбе доводят до метки 25 см
дистиллированной водой и перемешивают.
(Измененная редакция, Изм. N 1).
8.2.7.2. Раствор Б, содержащий 1,0 мг/см лизина
От раствора А пипеткой вместимостью 10 см отбирают 10 см
раствора, переносят в мерную колбу вместимостью 25 см
, доводят дистиллированной водой до метки 25 см
и перемешивают.
Стандартные растворы лизина хранят в холодильнике не более месяца.
8.2.8. Построение градуировочного графика
Из листа фильтровальной бумаги вырезают шесть полосок длиной 5 и шириной 0,5 см, которые нумеруют карандашом.
На полоски, примерно на середине, микрошприцем или автоматической пипеткой наносят капли растворов: на первую полоску - 10 мкл раствора Б, на вторую - 20 мкл раствора Б, на третью - 10 мкл раствора А, на четвертую - по 10 мкл растворов А и Б, на пятую - 20 мкл раствора А и на шестую - 20 мкл дистиллированной воды. После нанесения растворов и воды полоски по очереди берут пинцетом и сушат в токе теплого воздуха.
Номер полоски | Количество лизина, мкг |
1 | 10 |
2 | 20 |
3 | 25 |
4 | 35 |
5 | 50 |
6 | 0 |
Количество лизина на полосках фильтровальной бумаги должно соответствовать указанному в таблице.
Для построения градуировочного графика допускается использование и других количеств лизина.
Участок бумаги, занятый пробой, вырезают и помещают в пробирки вместимостью 5 см с пришлифованными пробками. Затем в каждую пробирку добавляют пипеткой 1,2 см
фурфурольного реактива, пробирки плотно закрывают пробками, помещают в водяную баню, предварительно нагретую до 95 °С, и выдерживают при этой температуре в течение 40 мин. Затем пробирки вынимают из бани, быстро охлаждают, опустив пробирки в баню с холодной водой или помещая их под струю воды из водопровода. После охлаждения сразу в каждую пробирку пипеткой приливают по 2 см
раствора глицерина и содержимое пробирок тщательно перемешивают.
Далее раствор спектрофотометрируют, измеряя поглощение света на длине волны 540 нм в кюветах с длиной поглощающего слоя 5 мм. В кювету сравнения наливают раствор, приготовленный, как раствор 6, но без помещения в пробирку фильтровальной бумаги.
Для каждого количества лизина готовят две параллельные пробы.
За окончательный результат величины оптической плотности каждого раствора лизина принимают среднюю арифметическую величину двух параллельных определений, значения которых не должны отличаться от средней величины более чем на 5%, рассчитанных до целого числа.
По полученным данным строят градуировочный график, откладывая по оси ординат показатели оптической плотности, а по оси абсцисс - содержание лизина в микрограммах.
8.3. Проведение испытания
В грушевидную колбу отвешивают 0,2-0,3 г продукта (результат записывают с точностью до четвертого десятичного знака). Пипеткой приливают 5 см раствора соляной кислоты концентрации 6 моль/дм
(6 н.). Колбу плотно закрывают пришлифованной пробкой и помещают в сушильный шкаф, предварительно нагретый до температуры (110±2) °С, и выдерживают при этой температуре 18 ч. Через 1 ч колбу вынимают, просматривают и, если в реакционной смеси заметны комочки, их разбивают встряхиванием колбы и снова ставят в сушильный шкаф.
Через 18 ч колбу вынимают и охлаждают. Затем доводят рН раствора по универсальной индикаторной бумаге до 4-5, добавляя необходимое количество раствора гидроокиси натрия концентрации 6 моль/дм (6 н.).
Гидролизат фильтруют через бумажный фильтр в мерную пробирку вместимостью 15 см. Реакционную колбу обмывают 4 см
дистиллированной воды (двумя порциями по 2 см
) большим количеством дистиллированной воды (1-2 см
) и этой же водой промывают осадок на фильтре. Раствор тщательно перемешивают и измеряют объем полученного раствора гидролизата.
Микрошприцем или автоматической пипеткой на полоску фильтровальной бумаги размером 5х0,5 см наносят 20 мкл полученного раствора гидролизата. Полоску высушивают в токе теплого воздуха. Участок бумаги, занятый раствором гидролизата, вырезают, помещают в пробирки вместимостью 5 см с пришлифованной пробкой и проводят определение по п.8.2.8.
Содержание лизина в растворе гидролизата определяют по калибровочному графику.
Проводят два параллельных определени
я.
8.4. Обработка результатов
Массовую долю лизина () в процентах вычисляют по формуле
![]() , (7)
, (7)
где - количество лизина в растворе гидролизата, найденное по калибровочному графику, мкг;
- объем полученного раствора гидролизата, см
;
- масса навески продукта, г;
- объем раствора гидролизата, взятый на определение, мкл;
- массовая доля влаги в продукте, %.
Результат округляют до второго десятичного знака. Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после занятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 20%, округленных до целого числа.
8.2.8-8.4. (Измененная редакция, Изм. N 1).
9. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ ЛИПИДОВ
Сущность метода заключается в экстракции липидов смесью органических растворителей из продукта, обработанного раствором соляной кислоты при нагревании, гравиметрическом определении суммы экстрагированных веществ после удаления растворителя.
(Измененная редакция, Изм. N 1).
9.1. Аппаратура и реактивы
Весы лабораторные технические любого типа.
Весы лабораторные 1-го класса точности с наибольшим пределом взвешивания 1 кг по ГОСТ 24104.
Баня водяная, обеспечивающая постоянство температуры (75±2) °С.
Шкаф сушильный, обеспечивающий постоянство температуры (105±2) °С.
Испаритель ротационный ИР-1М или другой марки.
Насос водоструйный лабораторный.
Колба К-1-100-29/32 ТХС или К-1-250-29/32 ТХС, или П-1-100-29/32 ТХС, или Кн-1-100-29/32 ТХС по ГОСТ 25336.
Холодильник ХПТ-2-400-29/32 ХС или ХШ-1-300-29/32 ХС, или ХШ-1-400-29/32 ХС по ГОСТ 25336.
Воронка делительная ВД-1-100 ХС или ВД-3-100 ХС, или ВД-1-250 ХС, или ВД-3-250 ХС по ГОСТ 25336.
Цилиндр мерный 1-50 или 3-50 по ГОСТ 1770.
Пипетки 1-1-2 или 1-2-2, 1-1-10 или 1-2-10 или в исполнениях 2, 4, 5 по НТД.
Эксикатор по ГОСТ 25336.
Эфир этиловый очищенный.
Эфир петролейный (фракция 40-70 °С).
Спирт ректификованный технический по ГОСТ 18300 или спирт этиловый технический по ГОСТ 17299.
Ацетон по ГОСТ 2603 или ацетон технический по ГОСТ 2768.
Кислота соляная по ГОСТ 3118 или кислота соляная особой чистоты по ГОСТ 14261, х.ч. или ч., разбавленная в соотношении 2:1.
Кальций хлористый технический по ГОСТ 450 или кальций обезвоженный, прокаленный.
Вода дистиллированная.
9.2. Проведение испытания
В круглодонную или плоскодонную колбу вместимостью 100 см отвешивают 2,0 г продукта (результат записывают с точностью до второго десятичного знака). В колбу добавляют 2 см
этилового спирта и содержимое колбы перемешивают встряхиванием. Добавляют 10 см
разбавленной водой соляной кислоты (2:1) и содержимое колбы вновь тщательно перемешивают.
Колбу соединяют с обратным холодильником и реакционную смесь нагревают на водяной бане при температуре (75±2) °С в течение 1 ч. Затем в колбу через холодильник добавляют 10 см этилового спирта, колбу снимают с бани и оставляют для охлаждения. Допускается охлаждать колбу в бане с холодной водой или под струей холодной воды.
Содержимое колбы переливают в делительную воронку, остатки гидролизата в колбе вместе с твердой фазой смывают в ту же делительную воронку 25 см этилового эфира. Воронку закрывают пробкой и энергично встряхивают, придерживая пробку одной рукой, а кран на спускной трубке - другой. Затем делительную воронку переворачивают краном вверх и осторожно открывают кран, выпуская образовавшиеся пары эфира. Кран делительной воронки закрывают, воронку энергично встряхивают в течение 30 с, переворачивают пробкой вверх, пробку вынимают и в воронку добавляют 25 см
петролейного эфира. Воронку закрывают пробкой и содержимое воронки вновь энергично встряхивают в течение 30 с, используя описанный выше прием для удаления образовавшихся паров эфира. 3атем воронку укрепляют в штативе и оставляют для расслаивания жидкостей. В случае образования устойчивой эмульсии к смеси в делительной воронке добавляют 10-15 см
этилового спирта, содержимое воронки встряхивают и оставляют для расслаивания.
После расслаивания отделяют нижний кислотный слой, сливая его обратно в круглодонную или плоскодонную колбу, а верхний эфирный слой переливают в чистую делительную воронку.
Экстракцию липидов из кислотного слоя повторяют еще дважды, как описано выше, используя каждый раз смесь 15 см этилового эфира и 15 см
петролейного эфира; при плохом расслаивании добавляют 10-15 см
этилового спирта.
По окончании экстракции реакционную смесь вместе с твердой фазой отбрасывают, а объединенный экстракт в делительной воронке промывают дистиллированной водой дважды порциями по 20 см. Затем экстракт упаривают в 2-3 приема на ротационном испарителе при остаточном давлении не ниже 50 мм рт.ст. и температуре водяной бани не более 35 °С, перенося экстракт из делительной воронки порциями по 50-60 см
в предварительно взвешенную круглодонную колбу вместимостью 100 или 250 см
(результат взвешивания записывают с точностью до четвертого десятичного знака). Когда весь растворитель испарится, в колбу добавляют 20 см
ацетона и растворитель вновь упаривают. Допускается проводить отгонку растворителя обычным способом с дефлегматором.
После испарения растворителя колбу с липидами помещают в сушильный шкаф, нагретый до температуры (105±2) °С, и выдерживают при этой температуре в течение 30 мин. Затем колбу переносят в эксикатор и после охлаждения до комнатной температуры взвешивают (результат записывают с точностью до четвертого десятичного знака).
Колбы вновь помещают в сушильный шкаф для повторного высушивания в течение 15 мин и после охлаждения в эксикаторе взвешивают. Высушивание повторяют до достижения постоянной массы.
Массу считают постоянной, если разница между двумя последовательными взвешиваниями составит не более 0,004 г.
Проводят два параллельных опре
деления.
9.1; 9.2. (Измененная редакция, Изм. N 1).
9.3. Обработка результатов
Массовую долю липидов () в процентах вычисляют по формуле
![]() , (8)
, (8)
где - масса колбы с липидами после высушивания, г;
- масса пустой колбы, г;
- масса навески продукта, г;
- массовая доля влаги в продукте, %.
Результат округляют до второго десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 5%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до целых единиц.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
10. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ ОБЩЕГО КОЛИЧЕСТВА
УГЛЕВОДОРОДОВ И МАССОВОЙ ДОЛИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Сущность метода заключается в экстракции углеводородов гексаном из продукта, предварительно обработанного спиртовым раствором едкого кали, очистки углеводородов хроматографией на окиси алюминия от других экстрагированных веществ, количественном определении в элюате суммы углеводородов гравиметрически и спектрофотометрическом определении ароматических углеводородов как фракции суммы углеводородов.
(Измененная редакция, Изм. N 1).
10.1. Аппаратура и реактивы
Весы лабораторные технические любого типа.
Весы лабораторные 1-го класса точности с наибольшим пределом взвешивания 1 кг по ГОСТ 24104.
Спектрофотометр ультрафиолетовый, пригодный для измерения в интервале длин волн 200-250 нм.
Шкаф сушильный лабораторный типа СНОЛ, обеспечивающий постоянство температуры (400±8) °С.
Испаритель ротационный ИР-1М.
Баня водяная.
Колбы К-1-100-29/32 ТХС, К-1-250-29/32 ТХС, К-1-500-29/32 ТХС или П-1-500-29/32 ТХС по ГОСТ 25336.
Колбы Кн-1000-29/32 ТХС по ГОСТ 25336.
Колбы Кн-2-250-34 ТХС по ГОСТ 25336.
Холодильник ХПТ-2-400-29/32 ХС или ХШ-1-300-29/32 ХС, или ХШ-1-400-29/32 ХС по ГОСТ 25336.
Холодильник ХПТ-1-300-14-23 ХС или ХПТ-1-400-14/23 ХС по ГОСТ 25336.
Дефлегматор 250-19/26-29/32 ТС или дефлегматор 300-19/26-39/32 ТС по ГОСТ 25336.
Насадка Н 1-19/26-14/23-14/23 ТС или Н 2-19/26-14/23 ТС по ГОСТ 25336.
Алонж АИО-14/23-50 ТС или АИО-14/23-14/23-65 ТС по ГОСТ 25336.
Воронка делительная ВД-1-500 или ВД-3-500 по ГОСТ 25336.
Цилиндры мерные 1-100, 1-250 или 3-100, 3-250 по ГОСТ 25336.
Стакан химический В-1-150 по ГОСТ 25336.
Стаканчики стеклянные вместимостью 25 см или бюксы той же вместимости по ГОСТ 25336.
Палочка стеклянная диаметром 5-6 мм.
Пробка-насадка от склянки для промывания газов (Дрекселя) СН 2-100 по ГОСТ 25336 с укороченной до 60-70 мм трубкой.
Колонка стеклянная хроматографическая длиной 195-205 мм, внутренним диаметром 19-21 мм, с оттянутым внизу кольцом и резервуаром вместимостью 50-60 см.
Пипетка 1-1-10 или 1-2-10 по НТД.
Эксикатор по ГОСТ 25336.
Насос водоструйный лабораторный.
Банки стеклянные с плотно закрывающимися пробками вместимостью 1-2 дм.
Чашки выпарительные 4, 5, 6 по ГОСТ 9147.
Натрий сернокислый безводный по ГОСТ 4166.
Натрия гидроокись по ГОСТ 4328, водный раствор с массовой долей 10%.
Кислота серная по ГОСТ 4204, х.ч., концентрированная.
Бумага индикаторная универсальная.
Гексан, ч.
Спирт этиловый ректификованный технический по ГОСТ 18300 или спирт этиловый технический по ГОСТ 17299.
Эфир этиловый (серный).
Калия гидроокись по ГОСТ 24363.
Алюминия окись для хроматографии.
Кальций хлористый технический по ГОСТ 450 или кальций хлористый, обезвоженный, прокаленный.
Воздух сжатый с давлением в линии в пределах 0,25-0,70 мПа (2,5-7,0 кгс/см) или азот газообразный по ГОСТ 9293.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
10.2. Подготовка к испытанию
10.2.1. Приготовление раствора гидроокиси калия в этиловом спирте
В коническую колбу вместимостью 250 см отвешивают (10±0,2) г гидроокиси калия. В колбу приливают 10 см
дистиллированной воды, содержимое колбы перемешивают, добавляют 100 см
этилового спирта. Полученный раствор перемешивают до полного растворения гидроокиси калия.
Раствор хранят в банке с плотно закрывающейся крышкой или в колбе с пришлифованной пробкой.
10.2.2. Подготовка окиси алюминия
Окись алюминия насыпают в фарфоровые чашки. Чашки с окисью алюминия помещают в сушильный шкаф и прокаливают при температуре (400±10) °С в течение 6-8 ч, затем чашки с окисью алюминия переносят в эксикатор с хлористым кальцием для охлаждения.
Подготовленную окись алюминия хранят в банках или колбах с пришлифованной пробкой или в чашках над хлористым кальцием в эксикаторе.
10.2.3. Подготовка гексана
В сухую банку с плотно закрывающейся пробкой вместимостью 1 дм помещают 500 см
гексана и 100 см
концентрированной серной кислоты. Банку закрывают пробкой, устанавливают на качалку и реакционную смесь перемешивают в течение 2-3 ч. Обработку гексана серной кислотой повторяют еще 2-3 раза.
Гексан отделяют от серной кислоты в делительной воронке, промывают его дистиллированной водой, 10%-ным водным раствором едкого натра и вновь дистиллированной водой до нейтральной реакции по индикаторной бумаге. Гексан из делительной воронки переносят в сухую банку или коническую колбу, добавляют сернокислый натрий из расчета 100 г осушителя на 1 дм гексана и оставляют для высушивания на 1,5-2 ч. Гексан отделяют от осушителя декантацией и перегоняют, собирая фракцию при 68-69 °С.
Степень чистоты полученного гексана проверяют спектрофотометрически относительно воздуха в кварцевых кюветах с толщиной оптического слоя 10 мм при длине волны 210 нм. Оптическая плотность не должна превышать величину 0,5.
(Введен дополнительно, Изм. N 1).
10.3. Проведение испытания
В круглодонную, плоскодонную или коническую колбу вместимостью 500 см отвешивают 20 г продукта. (Результат записывают с точностью до второго десятичного знака). В колбу с навеской продукта добавляют 200 см
этилового спирта, 20 см
дистиллированной воды и 20 г гидроокиси калия или 250 см
спиртового раствора гидроокиси калия. Содержимое колбы перемешивают встряхиванием. Колбу соединяют с обратным холодильником и реакционную смесь нагревают на кипящей водяной бане в течение 3 ч. Затем в колбу через холодильник добавляют 100 см
дистиллированной воды, колбу снимают с бани и оставляют для охлаждения.
После охлаждения жидкую фазу реакционной смеси декантацией переносят в делительную воронку, оставляя остаток продукта в колбе. В колбу с остатком добавляют 100 см гексана, содержимое колбы энергично перемешивают и гексан декантируют в делительную воронку.
Воронку закрывают пробкой и энергично встряхивают в течение 30 с, придерживая пробку одной рукой, а кран на спускной трубке - другой. Затем делительную воронку переворачивают краном вверх и осторожно открывают кран, выпуская образовавшиеся пары растворителя. Кран делительной воронки закрывают, воронку переворачивают пробкой вверх, пробку вынимают, воронку укрепляют в штативе и оставляют для расслаивания жидкостей.
В случае образования устойчивой эмульсии к смеси в делительной воронке добавляют 20 см этилового спирта, содержимое воронки встряхивают и оставляют для расслаивания жидкостей. После расслаивания нижнюю водно-спиртовую фазу сливают обратно в реакционную колбу с осадком, а гексановый экстракт переливают в круглодонную колбу вместимостью 250 см
.
Такую обработку реакционной смеси повторяют еще два раза, используя для экстракции гексан порциями по 50 см и этиловый спирт для расслаивания эмульсии порциями по 20 см
.
По окончании экстракции остаток в колбе и гидролизат отбрасывают, а объединенный экстракт в круглодонной колбе упаривают до объема 2-5 см на ротационном испарителе при остаточном давлении не ниже 50 мм рт.ст. и температуре водяной бани не более 60 °С. Допускается проводить отгонку обычным способом с дефлегматором.
В стакан вместимостью 150 см отвешивают 50 г окиси алюминия, добавляют 70-100 см
гексана. Содержимое стакана перемешивают стеклянной палочкой и полученную суспензию переносят в хроматографическую колонку. После того как избыток гексана вытечет из колонки и слой гексана над окисью алюминия составит 1-2 мм, в колбу количественно небольшой порцией гексана переносят остаток экстракта из круглодонной колбы. Углеводороды с колонки элюируют 150 см
смеси гексана и этилового эфира (95:5), собирая элюат в коническую колбу вместимостью 250 см
.
По окончании хроматографирования элюат упаривают, для этого элюат небольшими порциями переносят в предварительно взвешенную круглодонную колбу вместимостью 100 см и упаривают до объема 2-3 см
на ротационном испарителе при остаточном давлении не ниже 50 мм рт.ст. и температуре водяной бани не более 60 °С или обычным способом с дефлегматором на водяной бане.
Колбу с остатком элюата закрывают пробкой-насадкой от склянки Дрекселя и удаляют остатки растворителя, пропуская через насадку слабый ток газа (азот, воздух). Интенсивность пропускания газа устанавливают следующим образом: в резиновую трубку, через которую газ будет подаваться в колбу через насадку Дрекселя, вставляют стеклянную трубку с оттянутым концом диаметром 0,8-1,0 мм. Этот конец опускают в сосуд с водой и регулируют подачу газа по количеству выходящих из трубки пузырьков, их количество не должно превышать 120 пузырьков в минуту.
Колбу с углеводородами последовательно взвешивают первоначально после пропускания газа в течение 20 мин, а затем через каждые 5 мин до достижения постоянной массы.
Допускается упаривать элюат после хроматографической колонки в круглодонной колбе вместимостью 250 см до объема 2-3 см
, далее остаток элюата переносят в предварительно взвешенный стакан или бюксу и гексан удаляют испарением на воздухе, создавая ток воздуха над стаканчиком или бюксой с помощью комнатного вентилятора. Содержимое бюксы или стаканчика доводят до постоянной массы.
Массу считают постоянной, если разница между двумя последовательными взвешиваниями составит не более 0,0004 г. Все результаты взвешиваний записывают с точностью до четвертого десятичного знака. Для определения содержания ароматических углеводородов в колбу с общей суммой углеводородов пипеткой добавляют 10 см гексана для спектральных работ и содержимое колбы тщательно перемешивают. Полученным раствором заполняют кювету спектрофотометра с толщиной поглощающего свет слоя 10 мм и измеряют оптическую плотность в максимумах длин волн 210, 225 и 250 нм, используя в качестве раствора сравнения гексан.
Если величины оптических плотностей полученного раствора при указанных значениях длин волн превышают 0,8, проводят ряд последовательных разведении в 20, 50, 100 и более раз с тем, чтобы значения величин оптических плотностей в максимумах длин волн находились в диапазоне 0,2-0,8.
Проводят два параллельных определения и одновременно контрольный опыт, который проводит через все стадии анализа с использованием всех реактивов согласно прописи методики, но без навески продукта.
(Измененная редакц
ия, Изм. N 1).
10.4. Обработка результатов
10.4.1. Массовую долю общего количества углеводородов () в процентах вычисляют по формуле
![]() , (9)
, (9)
где - масса колбы с углеводородами, г;
- масса пустой круглодонной колбы, г;
- масса навески продукта, г;
- массовая доля влаги продукта, %.
Результат округляют до второго десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 6%, округленных до целого числа.
За окончательный результат определения принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
10.4.2. Массовую долю ароматических углеводородов () в процентах вычисляют по формуле
![]() , (10)
, (10)
где - количество ароматических углеводородов, содержащееся в выделенных из продукта суммарных углеводородов, мг;
- количество ароматических углеводородов, найденное в контрольном опыте, мг;
- масса навески продукта, г;
- перевод миллиграмм в граммы;
- массовая доля влаги продукта, %.
Величины и
определяют расчетным путем по найденным значениям оптической плотной по формуле
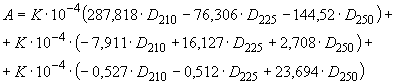 (11)
(11)
или ![]() ,
,
где ,
и
- величины оптической плотности раствора углеводородов в гексане при длинах волн 210, 225 и 250 нм;
- коэффициент разведения пробы (в данном случае
=10).
Числовые значения при величинах оптических плотностей - эмпирические коэффициенты, полученные расчетным путем.
Результат округляют до третьего десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 20%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до второго десятичного знака.
Допускаемые относительные расхождения между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака для результатов определения общего количества углеводородов и до второго десятичного знака для результатов определения ароматических углеводородов. Далее определяют расхождения между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10% для результатов определения общего количества углеводородов и 30% для результатов определения ароматических углеводородов, округленных до целого числа.
(Измененная редакция, Изм. N 1).
11. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ СВИНЦА
Сущность метода заключается в минерализации навески продукта при нагревании смесью концентрированных серной, азотной и хлорной кислот, с последующим растворением минерализата в концентрированной соляной кислоте и количественном определении свинца в полученном растворе методом атомно-абсорбционной спектроскопии.
11.1. Аппаратура и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Спектрофотометр атомно-абсорбционный, обеспечивающий чувствительность определения свинца 0,5 мкг/см, соответствующую 1% поглощения.
Лампа свинцовая с полым катодом типа ЛСП-1 или аналогичного типа.
Устройство нагревательное любого типа: электрическое, инфракрасное, газовое, снабженное устройством для регулирования температур.
Колба Кьельдаля 2-250-29 ТХС или 2-500-29 ТХС по ГОСТ 25336.
Колба Кн-2-250-34 ТХС по ГОСТ 25336.
Колбы мерные 2-25-1 или 2-25-2, 2-100-1 или 2-100-2, 2-1000-1 или 2-1000-2 по ГОСТ 1770.
Пипетки 1-1-1, 1-1-2 или 2-го класса точности и в исполнениях 2, 4, 5; 1-1-5, 1-1-10 или 2-го класса точности и в исполнениях 2, 6, 7 или 2-2-20 по ГОСТ 25336.
Цилиндры мерные 1-50 или 3-50, 1-500 по ГОСТ 1770 или мензурка 500 по ГОСТ 1770.
Пробирка П 2-16-180 ХС или П 2-19-180 ХС, или П 2-21-200 ХС по ГОСТ 25336.
Стакан В-1-100 ТХС или Н-1-100 ТХС по ГОСТ 25336.
Флаконы полиэтиленовые цилиндрические типа ФЦ вместимостью 100 и 1000 см.
Ацетилен растворенный технический по ГОСТ 5457 или пропан бытовой.
Воздух сжатый с давлением в линии в пределах 0,25-0,70 мПа (2,5-7,0 кгс/см).
Кислота серная по ГОСТ 4204, х.ч., концентрированная.
Кислота азотная по ГОСТ 4461, х.ч., концентрированная и разбавленная в соотношении 1:3.
Кислота хлорная, ч., концентрированная.
Кислота соляная по ГОСТ 3118, х.ч., концентрированная и раствор концентрации (НСl)=0,1 моль/дм
(0,1 н.).
Свинец по ГОСТ 3778, х.ч. или свинец (II) азотнокислый, х. ч., по ГОСТ 4236.
Водорода перекись по ГОСТ 10929, раствор с массовой долей 30%.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
11.2. Подготовка к испытанию
11.2.1. Приготовление стандартных растворов свинца
11.2.1.1. Раствор А, содержащий 1 мг/см свинца
Приготовление раствора из металлического свинца
В колбу Кьельдаля вместимостью 500 см отвешивают 1,0000 г свинца (результат записывают с точностью до четвертого десятичного знака), приливают цилиндром 50 см
раствора азотной кислоты (1:3), колбу устанавливают на нагреватель, раствор упаривают до 2-3 см
, слегка охлаждают, добавляют 5 см
концентрированной соляной кислоты и выпаривают содержимое почти досуха, охлаждают, снова добавляют 5 см
соляной кислоты и вторично упаривают почти досуха. После охлаждения в колбу приливают 300 см
раствора соляной кислоты концентрации 0,1 моль/дм
(0,1 н.), остаток растворяют при слабом нагревании и после охлаждения количественно переносят раствором соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) в мерную колбу вместимостью 1000 см
, раствор в колбе доводят до метки раствором соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) и перемешивают. Раствор переливают в полиэтиленовую посуду и хранят не более года.
Приготовление раствора из азотнокислого свинца
В стакан вместимостью 100 см отвешивают 1,6 г азотнокислого свинца (результат записывают с точностью до четвертого десятичного знака), предварительно высушенного до постоянной массы при температуре 100-105 °С, приливают цилиндром 50 см
дистиллированной воды, добавляют 1 см
концентрированной азотной кислоты. Содержимое стакана перемешивают и после полного растворения соли раствор количественно переносят с помощью раствора соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) в мерную колбу вместимостью 1000 см
. Раствор в колбе доводят до метки раствором соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) и перемешивают.
Раствор переливают в полиэтиленовую посуду и хранят не
более года.
11.2.1.2. Раствор Б, содержащий 0,1 мг/см свинца
Из раствора А пипеткой отбирают 10 см, переносят в мерную колбу вместимостью 100 см, раствор в колбе доводят до метки раствором соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) и перемешивают.
Раствор переливают в полиэтиленовую посуду и хранят не более 3 мес.
11.2.2. Построение градуировочного графика
В четыре мерные колбы вместимостью по 100 см пипетками соответствующей вместимости вносят 1, 2, 5 и 10 см
раствора Б, раствор в колбах доводят до метки раствором соляной кислоты концентрации 0,1 моль/дм
(0,1 н.) и перемешивают. Концентрация полученных растворов свинца в колбах соответственно составляет: 1,0; 2,0; 5,0; 10 мкг/см
. Растворы переливают в полиэтиленовую посуду и хранят не более 3 мес.
Производят градуировку атомно-абсорбционного спектрофотометра. Для этого устанавливают нуль прибора по дистиллированной воде и полученные растворы в порядке возрастания концентрации свинца последовательно распыляют в воздушно-ацетиленовое или воздушно-пропановое пламя горелки и измеряют величину абсорбции при аналитической линии свинца 283,3 нм или при 217 нм, если позволяет чувствительность прибора. Условия спектрофотометрирования подбирают по инструкции к прибору.
После каждого замера в пламя горелки распыляют дистиллированную воду до полного возврата стрелки прибора в нулевое положение.
По полученным данным строят градуировочный график, откладывая по оси ординат значение величины абсорбции, и по оси абсцисс - концентрацию свинца в мкг/см.
Проверку градуировочного графика проводят ежедневно после каждого зажигания горелки по двум стандартным растворам.
11.3. Проведение испытания
В сухую пробирку, свободно входящую в горло конической колбы или колбы Кьельдаля вместимостью 250 см, насыпают 2,5 г продукта и пробирку взвешивают (результат записывают с точностью до четвертого десятичного знака). Затем продукт осторожно высыпают в колбу (коническую или Кьельдаля) и пробирку взвешивают вновь. По разности между первым и вторым взвешиваниями определяют массу продукта.
В колбу с продуктом вносят пипеткой 3 см концентрированной серной кислоты, колбу устанавливают на нагреватель и проводят минерализацию продукта до его обугливания и получения однородной черной массы. После обугливания колбу снимают с нагревателя, слегка охлаждают, приливают пипетками 5 см
концентрированной азотной кислоты и 1 см
хлорной кислоты. Колбу снова помещают на нагреватель и проводят дальнейшую минерализацию продукта. Следят за тем, чтобы в колбе все время находилась азотная кислота и по мере ее испарения добавляют порциями по 5 см
. Наличие азотной кислоты определяют по выделяющимся из жидкости бурым парам окислов азота. Когда раствор приобретет равномерную устойчивую светло-желтую окраску, его упаривают почти досуха. Если при этом не наблюдается почернение осадка, минерализацию считают законченной. Если осадок почернеет, добавляют очередную порцию азотной кислоты и минерализацию продолжают.
Допускается в конце минерализации использовать раствор перекиси водорода взамен концентрированной азотной кислоты.
После полного разложения продукта азотную кислоту удаляют упариванием до исчезновения бурых паров окислов азота и появления белых паров окислов серы. Полное удаление азотной кислоты осуществляют двукратным добавлением по 10 см дистиллированной воды и испарением ее почти досуха.
Почти сухой остаток растворяют при нагревании в 5 см концентрированной соляной кислоты, полученный раствор количественно переносят дистиллированной водой в мерную колбу вместимостью 25 см
, в колбу добавляют дистиллированную воду до метки, раствор перемешивают.
Проводят два параллельных определения.
Допускается прерывать анализ после стадии минерализации и продолжать его на следующий день.
В тех же условиях проводят контрольный опыт без продукта, начиная с внесения в коническую колбу 3 см концентрированной серной кислоты.
В растворах, полученных после разложения продукта, проводят спектрофотометрическое определение содержания свинца в тех же условиях, при которых анализировали стандартные растворы свинца при построении градуировочного графика.
После подготовки прибора в пламя горелки распыляют последовательно в порядке возрастания концентрации свинца (по концентрациям, близким к определяемой) два стандартных раствора. В случае сохранения пропорциональности изменения величины абсорбции с увеличением концентрации свинца в растворе в пламя горелки распыляют анализируемые растворы и раствор контрольного опыта, строго соблюдая постоянство условий спектрофотометрирования.
Для повышения точности измерения величины абсорбции шкалу измерительного прибора атомно-абсорбционного спектрофотометра расширяют в 2,5 или 10 раз. При этом условия спектрофотометрирования подбирают так, чтобы отношение сигнал - шум было минимальным.
(Измененная редакция, Из
м. N 1).
11.4. Обработка результатов
Содержание свинца () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (12)
, (12)
где - содержание свинца в 1 см
анализируемого раствора, найденное по градуировочному графику, мкг/см
;
- содержание свинца в 1 см
контрольной пробы, найденное по градуировочному графику, мкг/см
;
- объем мерной колбы, см
;
- масса навески продукта, г.
Результат округляют до второго десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до целого числа.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до целого числа. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 50%, округленных до целого числа.
12. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МЫШЬЯКА
Сущность метода заключается в минерализации навески продукта смесью концентрированных серной, азотной и хлорной кислот, с последующим растворением минерализата в концентрированной соляной кислоте, восстановлением мышьяковистых соединений до мышьяковистого водорода и поглощением образующегося газа бромно-ртутной бумагой с образованием окрашенного пятна арсенида ртути. Сравнением интенсивности окраски полученного пятна по шкале окрасок стандартных пятен определяют содержание мышьяка.
12.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Прибор для определения содержания мышьяка по ГОСТ 10485 или прибор, изготовленный по черт.3 и состоящий из:
плоскодонной склянки, на которой нанесена метка, соответствующая объему 60 см. Метку наносят следующим образом: мерным цилиндром вместимостью 100 см
наливают в склянку 60 см
воды и на уровне мениска восковым карандашом или краской наносят штрих длиной 20-25 мм. К склянке через резиновую пробку присоединена стеклянная трубка с оттянутым концом, к верхнему концу которой пришлифована вторая короткая трубка того же диаметра; на верхнем конце длинной трубки и на нижнем конце короткой трубки припаяны стеклянные крючки так, чтобы обе трубки можно было стянуть резиновыми кольцами.
Прибор для определения содержания мышьяка
1 - метка, соответствующая объему 60 см; 2 - склянка; 3 - ватный тампон; 4 - длинная стеклянная трубка
(перетяжка в трубке не обязательна); 5 - бромно-ртутная бумага; 6 - короткая стеклянная трубка
(перетяжка в трубке не обязательна)
Черт.3
Устройства нагревательные электрические, инфракрасные, газовые, снабженные устройствами для регулирования степени нагрева.
Шкаф электрический, обеспечивающий поддержание температуры 60-70 °С с колебаниями не более ±2 °С.
Колбы Кьельдаля 2-250-29 ТХС или 2-500-29 ТХС по ГОСТ 25336.
Колбы Кн-2-250-34 ТХС по ГОСТ 25336.
Колбы мерные 2-200-1 или 2-200-2, 2-1000-2 по ГОСТ 1770.
Пипетки 1-1-0,5; 1-1-2 или 2-го класса точности и в исполнениях 2, 4, 5; 1-1-5, 1-1-10 или 2-го класса точности и в исполнениях 2, 6, 7 по НТД.
Цилиндры мерные 1-100 или 3-100, 1-500 или мензурка 500 по ГОСТ 1770.
Микробюретка 1-2-5-0,02 по НТД.
Пробирки П 2-16-180 или П 2-19-180, или П 2-21-200 по ГОСТ 25336.
Стакан В-1-100 ТХС, В-1-1000 или Н-1-1000 ТХС по ГОСТ 25336.
Воронка Бюхнера диаметром 100 мм по ГОСТ 9147.
Чашки Петри по ГОСТ 25336.
Чашки выпарительные 3, 4, 5 по ГОСТ 9147.
Флаконы полиэтиленовые цилиндрические типа ФЦ вместимостью 100 см.
Бумага фильтровальная лабораторная по ГОСТ 12026.
Фильтры обеззоленные диаметром 90 мм.
Пинцет.
Вата гигроскопическая.
Пергамент растительный.
Парафин твердый.
Ангидрид мышьяковистый по ГОСТ 1973, 1-й сорт или мышьяковистокислый ортодвузамещенный, ч. или ангидрид мышьяковистый, медицинский препарат ГФХ ст.5.
Кислота серная по ГОСТ 4204, х.ч., концентрированная и раствор серной кислоты с массовой долей 10%.
Кислота азотная по ГОСТ 4461, х.ч., концентрированная и раствор в соотношении 1:8.
Кислота хлорная, ч., концентрированная.
Кислота соляная по ГОСТ 3118, х.ч., концентрированная.
Кислота уксусная по ГОСТ 61, х.ч., раствор с массовой долей 30%.
Натрия гидроокись по ГОСТ 4328, х.ч., раствор с массовой долей 25%.
Цинк гранулированный.
Свинец уксуснокислый по ГОСТ 1027, ч.д.а., раствор с массовой долей 0,1%.
Метиловый красный, спиртовой раствор с массовой долей 0,1%.
Олово гранулированное, ч.д.а.
Олово двухлористое, ч.д.а., раствор с массовой долей 10%.
Натрий бромид, ч.д.а., или калий бромистый по ГОСТ 4160, ч.д.а.
Ртути окись желтая по ГОСТ 5230, ч.д.а.
Дифениламин по ТУ 6-09-5467, ч.д.а., раствор с массовой долей 1% в серной кислоте.
Спирт этиловый ректификованный технический по ГОСТ 18300.
Водорода перекись по ГОСТ 10929, раствор с массовой долей 30%.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
12.2. Подготовка к испытанию
12.2.1. Приготовление раствора двухлористого олова
В стакан вместимостью 100 см отвешивают (20±1) г двухлористого олова (результат записывают с точностью до второго десятичного знака), добавляют 80 см
концентрированной соляной кислоты и небольшое количество (одну гранулу) металлического олова. Реакционную смесь нагревают на кипящей водяной бане до полного растворения соли. После охлаждения смесь количественно переносят дистиллированной водой в мерную колбу вместимостью 200 см
, добавляют в колбу до метки дистиллированную воду и смесь перемешивают. Раствор хранят в темном месте.
12.2.2. Приготовление стандартных растворов мышьяка
12.2.2.1. Раствор А, содержащий 0,1 мг/см мышьяка, приготовленный из мышьяковистого ангидрида
В стакан вместимостью 100 см отвешивают (0,1320±0,0005) г мышьяковистого ангидрида (результат взвешивания записывают с точностью до четвертого десятичного знака), добавляют 25 см
раствора гидроокиси натрия с массовой долей 25%, содержимое стакана перемешивают до полного растворения соли, в стакан добавляют 3-5 капель раствора индикатора метилового красного и раствор нейтрализуют раствором серной кислоты с массовой долей 10%. Содержимое стакана количественно переносят дистиллированной водой в мерную колбу вместимостью 1000 см
, добавляют в колбу до метки дистиллированную воду и смесь перемешивают. Раствор переливают в полиэтиленовую посуду и хранят не более года.
12.2.2.2. Раствор А, содержащий 0,1 мг/см мышьяка, приготовленный из мышьяковистого натрия
В стакан вместимостью 100 см отвешивают (0,4165±0,0005) г соли (результат записывают с точностью до четвертого десятичного знака), растворяют в дистиллированной воде и количественно переносят с помощью дистиллированной воды в мерную колбу вместимостью 1000 см
, доводят объем до метки и перемешивают.
12.2.2.3. Раствор Б, содержащий 0,001 мг/см мышьяка
Из раствора А пипеткой отбирают 10 см, переносят в мерную колбу вместимостью 1000 см
, добавляют в колбу до метки дистиллированную воду и раствор перемешивают. Раствор переливают в полиэтиленовую посуду и хранят не более 3 мес.
12.2.3. Синтез бромной ртути
В стакан вместимостью 1000 см наливают 450 см
азотной кислоты, разбавленной дистиллированной водой в соотношении 1:8. К кислоте постепенно прибавляют 120 г желтой окиси ртути. После растворения окиси ртути образовавшейся мути дают отстояться, раствор фильтруют через бумажный складчатый фильтр в другой стакан той же вместимости.
В полученный фильтрат приливают раствор бромида натрия, содержащего 130 г соли в 200 см дистиллированной воды или бромистого калия, содержащего 140 г соли в 200 см
дистиллированной воды. Образовавшийся белый осадок бромной ртути отделяют на воронке Бюхнера под вакуумом. Осадок на воронке промывают дистиллированной водой (2-3 раза по 200 см
) до полного удаления нитрат-иона по пробам с дифениламином. Отмытый осадок сушат на пергаментной бумаге в сушильном шкафу при температуре 60-70 °С до получения сухого рассыпающегося порошка. Бромную ртуть хранят в банке с пришлифованной пробкой в темном месте.
12.2.4. Приготовление бромно-ртутной бумаги
В стакан вместимостью 100 см отвешивают 2,5 г бромной ртути (результат взвешивания записывают с точностью до второго десятичного знака), добавляют 50 см
этилового спирта и содержимое стакана перемешивают стеклянной палочкой до полного растворения соли. В полученный раствор на 1 ч погружают 15-20 бумажных фильтров или 15-20 кусков фильтровальной бумаги, нарезанной в виде квадратов. Затем фильтры вынимают из раствора пинцетом, дают избытку раствора стечь, раскладывают на стекле, покрывают листом фильтровальной бумаги и высушивают при комнатной температуре. Бромно-ртутную бумагу хранят в банке из темного стекла с притертой пробкой в темном месте не более месяца.
12.2.5. Приготовление ваты, пропитанной раствором уксуснокислого свинца
В стакан вместимостью 100 см отвешивают 1,2 г уксуснокислого свинца (результат записывают с точностью до второго десятичного знака), добавляют 20 см
дистиллированной воды и 2,5 см
концентрированной уксусной кислоты для предотвращения образования мути. После охлаждения раствор количественно переносят в мерную колбу вместимостью 100 см
, добавляют в колбу до метки дистиллированную воду и перемешивают. Вату помещают в выпарительную чашку, выливают в нее раствор уксуснокислого свинца так, чтобы раствор полностью покрыл вату, оставляют в вытяжном шкафу на 1-2 ч. После чего раствор из чашки сливают, вату раскладывают на фильтровальную бумагу и сушат на воздухе. Вату хранят в темных склянках с притертыми пробками.
12.2.6. Приготовление стандартной шкалы окрасок мышьяка
Для приготовления стандартной шкалы используют 6 приборов для определения мышьяка (см. черт.3). Трубку каждого прибора заполняют следующим образом: в длинную часть трубки помещают тампон из ваты, пропитанной раствором уксуснокислого свинца и высушенной на воздухе. Тампон должен быть рыхлым, заполнять все сечение трубки и занимать по высоте 4,5-5,5 см. На верхний шлифованный торец трубки укладывают квадрат бромно-ртутной бумаги размером 10х10 мм и прижимают его короткой частью трубки, закрепляя ее резинками, надеваемыми на крючки обеих трубок.
В склянки приборов из микробюретки или пипетками соответствующей вместимости приливают 0,50; 1,00; 1,50; 2,00; 2,50; 3,00 см раствора Б мышьяка, что соответствует количествам мышьяка 0,0005; 0,0010; 0,0015; 0,0020; 0,0025; 0,0030 мг. Допускается 0,50 см
приливать градуированной пипеткой вместимостью 5 см
.
В каждую склянку добавляют пипетками 5 см концентрированной серной кислоты, 5 см
концентрированной соляной кислоты и 1 см
раствора двухлористого олова, объем реакционной смеси доводят до метки дистиллированной водой, добавляют 5-6 г (10-11 гранул) цинка, склянки быстро закрывают пробками с приготовленными трубками. Через 1,5 ч приборы разбирают, снимают квадраты бромно-ртутной бумаги. Проявившуюся окраску закрепляют, опуская бумагу пинцетом в парафин, доведенный в фарфоровой чашке до расплавленного, но не перегретого состояния. Затем бумагу вынимают, давая избытку парафина стечь и бумажки в порядке возрастания концентрации мышьяка укладывают на дно стеклянной чашки Петри, подписывая под каждым квадратом концентрацию мышьяка. После охлаждения парафина чашку закрывают крышкой. Полученные бумажки с определенной концентрацией мышьяка хранят в защищенном от света месте не более 3 мес.
Допускается в конце минерализации использовать раствор перекиси водорода взамен концентрированной азотной кислоты.
(Измененная редакция, Изм. N 1).
12.3. Проведение испытания
В сухую пробирку, входящую свободно в горло конической колбы вместимостью 250 см или горло колбы Кьельдаля вместимостью 250 см
, насыпают около 2,5 г продукта и пробирку взвешивают (результат записывают с точностью до четвертого десятичного знака). Продукт осторожно пересыпают в колбу и пробирку взвешивают вновь. По разнице между первым и вторым взвешиваниями определяют массу навески продукта. В колбу с продуктом пипеткой вносят 3 см
концентрированной серной кислоты.
Колбу устанавливают на нагреватель и проводят обугливание продукта в течение 2-3 мин до получения однородной черной вязкой массы. После обугливания колбу снимают с нагревателя, слегка охлаждают и приливают пипетками 5 см концентрированной азотной кислоты и 1 см
концентрированной хлорной кислоты. Колбу помещают на нагреватель и проводят минерализацию продукта, следя за тем, чтобы в колбе все время находилась азотная кислота, и по мере ее испарения добавляют порциями по 5 см
. Наличие азотной кислоты определяют по выделяющимся из жидкости бурым парам окислов азота. Полноту сожжения продукта определяют следующим образом. Во время сожжения раствор постепенно становится прозрачным и приобретает желтую окраску. Когда окраска станет светло-желтой и устойчивой, раствор выпаривают досуха и, если при этом не наблюдается почернение осадка, сожжение считают законченным. Если осадок почернеет, то добавляют азотную кислоту и сожжение продолжают.
Допускается в конце минерализации использовать раствор перекиси водорода взамен концентрированной азотной кислоты.
После окончания сожжения остаток азотной кислоты удаляют двукратным добавлением по 10 см дистиллированной водой и испарением ее почти досуха.
К полученному таким образом почти сухому остатку пипеткой приливают 5 см концентрированной соляной кислоты, слегка нагревают на нагревателе до полного растворения остатка и содержимое колбы количественно переносят дистиллированной водой в склянку прибора, добавляя пипетками 5 см
концентрированной серной кислоты и 1 см
раствора двухлористого олова, объем реакционной смеси доводят до метки дистиллированной водой, добавляют 5-6 г (10-11 гранул) цинка и далее поступают так, как описано в п.12.2.6, вплоть до парафинирования бумаги.
Концентрацию мышьяка определяют сравнением интенсивности окраски полученного пятна бромно-ртутной бумаги с интенсивностью окраски пятен стандартной шкалы мышьяка.
Проводят два параллельных определения, при этом навески продукта не должны отличаться более чем на 0,1 г.
В тех же условиях проводят контрольный опыт без продукта, начиная со стадии внесения в коническую колбу 3 см концентрированной серной кислоты.
(Измененная редакция, Изм.
N 1).
12.4. Обработка результатов
Содержание мышьяка () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (13)
, (13)
где - количество мышьяка во взятой навеске, определенное по стандартной шкале, мг;
- количество мышьяка, определенное по стандартной шкале в контрольном опыте, мг;
- коэффициент пересчета содержания мышьяка на килограмм продукта;
- масса навески продукта, г.
Результат округляют до третьего десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до целого числа. Если среднее арифметическое значение составит менее 0,5 мг/кг, окончательный результат записывают - "менее 1 мг/кг".
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до целого числа. Далее определяют расхождение между окончательными результатами испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 50%, округленных до целого числа.
13. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ РТУТИ
Сущность метода заключается в минерализации навески продукта при умеренном нагреве последовательно концентрированной кислотой и раствором марганцовокислого калия, с последующим восстановлением двухлористым оловом до паров металлической ртути, отгонки ее в виде паров инертным газом и поглощением раствором серной кислоты. В полученном растворе количественно определяют содержание ртути колориметрическим методом в виде дитизоната.
13.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Спектрофотометр видимого спектра.
Баня водяная, обеспечивающая постоянство температуры (60±2) °С.
Прибор для выделения ртути (черт.4).
Прибор для определения содержания ртути
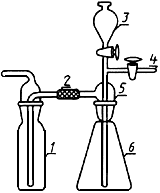
1 - барбатер-приемник ртути; 2 - резиновая трубка; 3 - воронка; 4 - ввод инертного газа;
5 - насадка; 6 - коническая колба вместимостью 250 см
Черт.4
Колбы Кн-1-250-24-29 ТХС по ГОСТ 25336.
Колбы мерные 2-100-1 или 2-100-2, 2-1000-1 или 2-1000-2 по ГОСТ 1770.
Пипетки 1-1-0,5, 1-1-1, 1-1-2 или 2-го класса точности и в исполнениях 2, 4, 5; 1-1-5, 1-1-10 или 2-го класса точности в исполнениях 2, 6, 7 по НТД.
Цилиндры мерные 1-10, 1-25, 1-100, 1-1000 по ГОСТ 1770 или в исполнении 3.
Пробирки П 2-16-180 ХС или П 2-19-180 ХС или П 2-21-200 ХС по ГОСТ 25336.
Стакан В-1-100 ТХС или Н-1-100 ТХС по ГОСТ 25336.
Воронки ВД-1-250 ХС, ВД-1-500 ХС или в исполнениях 2, 3 по ГОСТ 25336.
Трубка резиновая.
Бумага индикаторная универсальная.
Шарики стеклянные.
Кислота соляная по ГОСТ 3118, х.ч., концентрированная.
Кислота серная по ГОСТ 4204, х.ч., концентрированная и раствор с массовой долей 15%.
Кислота азотная по ГОСТ 4461, х.ч., концентрированная.
Хлороформ технический по ГОСТ 20015, х.ч., или углерод четыреххлористый по ГОСТ 20288, х.ч., свежеперегнанные.
Олово по ГОСТ 860.
Олово двухлористое, ч., раствор с массовой долей 10%.
Ртути окись желтая по ГОСТ 5230, ч.д.а.
Гелий газообразный или аргон газообразный по ГОСТ 10157.
Калий марганцовокислый по ГОСТ 20490, х.ч., раствор с массовой долей 6%.
Гидроксиламина гидрохлорид по ГОСТ 5456, х.ч., раствор с массовой долей 10% в растворе серной кислоты с массовой долей 5%.
Дитизон по ТУ 6-09-07-1684, ч.д.а., дополнительно очищенный.
Аммиак водный технический по ГОСТ 9.
Лед (водяной).
Вода дистиллированная.
13.2. Подготовка к испытанию
13.2.1. Приготовление раствора двухлористого олова с массовой долей 10%.
В стакан вместимостью 100 см отвешивают (20±0,1) г двухлористого олова (результат записывают с точностью до второго десятичного знака), добавляют 80 см
концентрированной соляной кислоты и небольшое количество (одну гранулу) металлического олова. Реакционную смесь нагревают на кипящей водяной бане до полного растворения соли. После охлаждения смесь количественно переносят дистиллированной водой в мерную колбу вместимостью 200 см
, добавляют в колбу до метки дистиллированную воду и смесь перемешивают. Раствор хранят в темном месте.
13.2.2. Очистка и приготовление растворов дитизона
13.2.2.1. Раствор А
В стакан вместимостью 100 см отвешивают 0,1 г дитизона (результат записывают с точностью до третьего десятичного знака), добавляют мерным цилиндром 50 см
свежеперегнанного хлороформа. После растворения дитизона раствор переносят в делительную воронку вместимостью 250 см
и добавляют мерным цилиндром 50 см
разбавленного дистиллированной водой в отношении 1:100 раствора аммиака. Содержимое воронки энергично встряхивают в течение 2 мин. При этом дитизон переходит в водно-аммиачную фазу и его окраска из синей переходит в оранжевую. После разделения нижний органический слой отбрасывают, а верхний водно-аммиачный раствор дитизона подкисляют до рН 3-4 по универсальной индикаторной бумаге, добавляя по каплям разбавленную дистиллированной водой в отношении 1:1 соляную кислоту. Окраска дитизона в кислой среде переходит в синюю. Дитизон из раствора экстрагируют хлороформом три раза порциями по 20 см
, собирая экстракт во вторую делительную воронку. Объединенный экстракт промывают три раза дистиллированной водой порциями по 20 см
. Промывные воды отбрасывают, а промытый раствор дитизона переносят в мерную колбу вместимостью 100 см
. Объем раствора доводят хлороформом до 100 см
и раствор перемешивают. Полученный раствор хранят в холодильнике не более 1 год
а.
13.2.2.2. Раствор Б
Из раствора А пипеткой отбирают 1 см, переносят в мерную колбу вместимостью 100 см
, добавляют в колбе до метки хлороформ и перемешивают. Оптическая плотность раствора Б не должна превышать величину 0,2 при длине волны 490 нм и толщине слоя 10 мм относительно хлороформа, в противном случае повторно проводят очистку раствора А. Раствор Б готовят непосредственно перед определением.
Очистку дитизона при приготовлении раствора А допускается не проводить, если оптическая плотность раствора Б, приготовленного из неочищенного дитизона, не превышает величину 0,2 при длине волны 490 нм и толщине слоя 10 мм относительно хлороформа.
(Измененная редакция, Изм. N 1).
13.2.3. Приготовление стандартных растворов ртути
13.2.3.1. Раствор А, содержащий 1 мг/см ртути
В стакан вместимостью 100 см отвешивают 0,1080 г окиси ртути (результат взвешивания записывают с точностью до четвертого десятичного знака), добавляют пипеткой 5 см
концентрированной азотной кислоты. Реакционную смесь нагревают на водяной бане до полного растворения окиси ртути. После охлаждения полученный раствор количественно переносят дистиллированной водой в мерную колбу вместимостью 100 см
и добавляют в колбу до метки дистиллированную воду.
13.2.3.2. Раствор Б, содержащий 0,001 мг/см ртути
Из раствора А отбирают пипеткой 1 см, переносят в мерную колбу вместимостью 1000 см
, добавляют до метки дистиллированную воду и перемешивают. Раствор Б используют свежеприготовленным.
13.2.4. Построение градуировочного графика
В четыре делительные воронки вместимостью по 250 см наливают по 50 см
дистиллированной воды и 0,0; 0,5; 1,0; 1,5 и 2,0 см
раствора Б ртути пипетками соответствующей вместимости, что соответствует количеству ртути 0,0; 0,0005; 0,0010; 0,0015 и 0,0020 мг. Затем в каждую воронку приливают по 20 см
раствора серной кислоты с массовой долей 15%, по 5 см
раствора Б дитизона и встряхивают смесь в течение 2 мин. После разделения фаз хлороформный слой отделяют и сразу измеряют величину его оптической плотности относительно хлороформа. Измерения проводят при длине волны 490 нм и толщине оптического слоя 10 мм.
По полученным данным строят градуировочный график, откладывая по оси ординат величину оптической плотности () растворов, а по оси абсцисс - количество ртути (
) в миллиграммах.
В случае если значение оптической плотности контрольной пробы (без добавки раствора ртути) превышает величину 0,25, необходимо провести очистку раствора серной кислоты с массовой долей 15% и заменить дистиллированную воду на бидистиллированную. Для очистки берут 250 см раствора серной кислоты с массовой долей 15%, переносят в делительную воронку вместимостью 500 см
, приливают 5 см
исходного раствора А дитизона и встряхивают 2-3 мин. После разделения дитизоновый слой отбрасывают, промывают трижды порциями по 20 см
свежеперегнанного хлороформа для удаления остатков дитизона и отфильтровывают кислоту через беззольный фильтр.
(Измененная редакция, Изм.
N 1).
13.3. Проведение испытания
В коническую колбу вместимостью 250 см приливают пипеткой 20 см
концентрированной серной кислоты и вносят 3-4 стеклянных шарика диаметром 0,5-0,7 см.
В сухую пробирку, свободно входящую в горло этой конической колбы, насыпают около 2,5 г продукта и пробирку взвешивают, продукт осторожно высыпают в колбу с кислотой и пробирку вновь взвешивают. По разнице между первым и вторым взвешиваниями определяют массу навески продукта. Все результаты взвешиваний записывают с точностью до четвертого десятичного знака.
Колбу закрывают стеклянной пробкой и помещают на водяную баню с температурой 60-70 °С на 3 ч. Затем колбу снимают с бани, охлаждают до комнатной температуры, помещают в баню со льдом и по каплям приливают 25 см раствора марганцовокислого калия с массовой долей 6%, постоянно перемешивая реакционную смесь. Колбу выдерживают еще 5-10 мин в ледяной бане, а затем снова помещают на водяную баню с температурой 60-70 °С до полного разложения продукта, сопровождающегося ослаблением окраски получаемого раствора. Далее реакционную смесь охлаждают до комнатной температуры, колбу присоединяют к насадке прибора (см. черт.4), через воронку насадки приливают цилиндром 7 см
раствора гидроксиламина для разложения избыточного количества марганцовокислого калия, 25 см
раствора серной кислоты с массовой долей 15%. Колбу с насадкой соединяют резиновой трубкой с приемником ртути, в который предварительно наливают 20 см
раствора серной кислоты с массовой долей 15%. Через газовый ввод насадки в колбу подают гелий (или аргон) для вытеснения из колбы в приемник получаемых паров ртути.
После продувки системы инертным газом в колбу через воронку насадки приливают цилиндром или пипеткой 10 см раствора двухлористого олова с массовой долей 10%. Восстановление в токе инертного газа ведут 2 ч, затем приемник отделяют от насадки с колбой.
Полученный раствор из приемника количественно переносят 50 см дистиллированной воды в делительную воронку вместимостью 250 см
, приливают 5 см
раствора Б дитизона, делительную воронку закрывают и встряхивают в течение 2 мин. После разделения фаз хлороформный слой отделяют и сразу измеряют значение оптической плотности раствора при длине волны 490 нм и толщине оптического слоя 10 мм относительно хлороформа. Аналогичное испытание проводят без навески продукта (контрольный опыт).
Проводят два параллельных определе
ния.
13.4. Обработка результатов
Содержание ртути () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (14)
, (14)
где - количество ртути во взятой навеске продукта, найденное по градуировочному графику, мг;
- количество ртути в контрольном опыте, мг;
- коэффициент пересчета содержания ртути на килограмм продукта;
- масса навески, г.
Результат округляют до третьего десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до первого десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 50%, округленных до целого числа.
14. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФТОРА
Сущность метода заключается в экстракции хлорной кислотой при 50 °С фтористых соединений из навески продукта с последующим определением содержания фтора в полученной суспензии потенциометрическим методом со фтор-селективным электродом.
14.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
рН-метр-милливольтметр лабораторный типа рН-121 или иономер универсальный типа ЭВ-74.
Электрод фторидный селективный типа ЭФ-VI.
Электрод сравнения хлорсеребряный насыщенный образцовый 2-го разряда, типа ЭВЛ-1МЗ по ГОСТ 17792.
Микротермостат МТ-0,3 или любая магнитная мешалка с подогревом.
Шкаф сушильный электрический, обеспечивающий поддержание температуры (105±2) °С.
Секундомер или часы песочные на 3 мин.
Колбы мерные 2-100-1 или 2-100-2, 2-500-1 или 2-500-2, 2-1000-1 или 2-1000-2 по ГОСТ 1770.
Пипетки 1-1-1, 1-1-2 или 2-го класса точности и в исполнениях 2, 6, 7; 2-1-20 или 2-2-20 по НТД.
Пробирки П 2-16-180 ХС или П 2-19-180 ХС или П 2-21-200 ХС по ГОСТ 25336.
Стаканы В-1-100 ТС или Н-1-100 ТС и В-1-1000 ТС или Н-1-1000 ТС по ГОСТ 25336.
Цилиндры мерные 1-25 или 3-25 по ГОСТ 1770.
Мензурки полиэтиленовые вместимостью 30 см.
Флаконы полиэтиленовые цилиндрические типа ФС вместимостью 100, 500 и 1000 см.
Чашки Петри с крышкой пластмассовые лабораторные, диаметром 400 мм.
Ступка фарфоровая с пестиком по ГОСТ 9147.
Бумага фильтровальная.
Бумага масштабно-координатная марки ПЛН, формат 200х300 мм по ГОСТ 334.
Кислота соляная по ГОСТ 3118, х.ч., концентрированная и разбавленная в соотношении 1:1.
Кислота хлорная, ч., раствор рН 1,0.
Натрий лимоннокислый трехзамещенный по ГОСТ 22280, ч., раствор с массовой долей 30%, рН 6,5.
Натрий фтористый по ГОСТ 4463, х.ч.
Вода дистиллированная.
14.2. Подготовка к испытанию
14.2.1. Приготовление раствора хлорной кислоты
14 см концентрированной хлорной кислоты цилиндром вместимостью 25 см
переносят в мерную колбу вместимостью 1000 см
, приливают до метки дистиллированную воду и с помощью рН-метра устанавливают рН раствора 1,0, добавляя по каплям хлорную кислоту.
14.2.2. Приготовление раствора лимоннокислого натрия
В стакан вместимостью 1000 см отвешивают 300 г соли (результат записывают с точностью до второго десятичного знака), добавляют 10 см
раствора соляной кислоты в соотношении 1:1 и 600 см
дистиллированной воды. После растворения соли раствор переносят в мерную колбу вместимостью 1000 см
, доводят до метки дистиллированной водой и с помощью рН-метра устанавливают рН раствора 6,5±0,3, добавляя по каплям раствор соляной кислоты, разбавленной в соотношении 1:1.
14.2.3. Приготовление стандартных растворов фтористого натрия
14.2.3.1. Приготовление раствора фтористого натрия концентрации 0,1 моль/дм (0,1 н.)
В стакан вместимостью 100 см отвешивают 2,100 г соли (результат записывают с точностью до четвертого десятичного знака), предварительно высушенной до постоянной массы при температуре (105±3) °С, приливают 100 см
раствора лимоннокислого натрия и полученный раствор количественно переносят раствором хлорной кислоты с рН 1 в мерную колбу вместимостью 500 см
, доводят объем до метки раствором хлорной кислоты и смесь перемешивают. Этот раствор имеет величину pF=1. Раствор хранят в полиэтиленовой посуде с плотно закрытой пробкой до 6 мес.
14.2.3.2. Приготовление раствора фтористого натрия концентрации 0,01 моль/дм (0,01 н.)
10 см раствора фтористого натрия концентрации 0,1 моль/дм
(0,1 н.) пипеткой переносят в мерную колбу вместимостью 100 см
, приливают 20 см
раствора лимоннокислого натрия, доводят объем колбы до метки раствором хлорной кислоты с рН 1, смесь перемешивают. Раствор имеет величину pF=2.
14.2.3.3. Приготовление раствора фтористого натрия концентрации 0,001 моль/дм (0,001 н.)
10 см раствора фтористого натрия концентрации 0,01 моль/дм
(0,01 н.) пипеткой переносят в мерную колбу вместимостью 100 см
, приливают 20 см
раствора лимоннокислого натрия, доводят объем колбы до метки раствором хлорной кислоты с рН 1 и перемешивают. Раствор имеет величину pF=3.
14.2.3.4. Приготовление раствора фтористого натрия концентрации 0,0001 моль/дм (0,0001 н.)
10 см раствора фтористого натрия концентрации 0,001 моль/дм
(0,001 н.) пипеткой переносят в мерную колбу вместимостью 100 см
, приливают 20 см
лимоннокислого натрия, доводят объем колбы до метки раствором хлорной кислоты с рН 1 и перемешивают. Раствор имеет величину pF=4.
12.4.3.5. Приготовление раствора фтористого натрия концентрации 0,00001 моль/дм (0,00001 н.)
10 см раствора фтористого натрия концентрации 0,0001 моль/дм
(0,0001 н.) пипеткой перевесят в мерную колбу вместимостью 100 см
, приливают 20 см
раствора лимоннокислого натрия, доводят объем колбы до метки раствором хлорной кислоты с рН 1 и перемешивают. Раствор имеет величину pF=5.
Растворы фтористого натрия концентраций 0,01; 0,001; 0,0001 и 0,00001 моль/дм (соответственно 0,01; 0,001; 0,0001 и 0,00001 н.) готовят в день проведения анализа.
14.2.4. Подготовка фторидного селективного электрода
Новый фторидный электрод предварительно выдерживают в растворе фтористого натрия концентрации 0,001 моль/дм (0,001 н.). При ежедневной работе с электродом его хранят, погрузив в раствор фтористого натрия концентрации 0,0001 моль/дм
(0,0001 н.). При длительных перерывах в работе электрод хранят в сухом состоянии.
14.2.5. Построение градуировочного графика
В полиэтиленовые мензурки вместимостью 30 см наливают по 20 см
каждого стандартного раствора, погружают на дно мензурок магнит и в порядке возрастания концентраций фтористого натрия в растворе устанавливают мензурки на магнитную мешалку, погружают в раствор электроды, включают перемешивание и через 3 мин замеряют потенциал в милливольтах.
После каждого замера потенциала электроды тщательно промывают большим количеством дистиллированной воды до установления исходного значения водного потенциала, а затем осушают фильтровальной бумагой.
При выполнении измерений следят за тем, чтобы на поверхности мембраны фторидного электрода не налипали пузырьки воздуха.
По полученным данным строят градуировочный график на бумаге с полулогарифмической сеткой, откладывая по оси абсцисс концентрацию фтора (г·моль/дм), а по оси ординат - значение потенциала (
) в милливольтах.
Градуировочный график проверяют ежедневно по трем стандартным растворам фтористого натрия. При построении градуировочного графика проверяют правильность работы фторидного электрода - крутизну характеристики электрода.
При измерении потенциалов рабочих стандартных растворов потенциал должен изменяться oт раствора к раствору на величину =(56±3) мв при температуре анализируемой среды (20±2) °С. Если такая зависимость значения потенциала не соблюдается, то фторидный электрод следует регенерировать вымачиванием в растворе фтористого натрия концентрации 0,001 моль/дм
(0,001 н.) в течение суток, а затем тщательно отмыть дистиллированной водой.
(Измененная редакция, Изм. N 1).
14.3. Проведение испытания
Перед испытанием продукт тщательно растирают в фарфоровой ступке до пудрообразного состояния и хранят в пластмассовых чашках Петри.
В полиэтиленовую мензурку вместимостью 30 см отвешивают от 0,05 до 0,3 г продукта (результат записывают с точностью до четвертого десятичного знака), с тем чтобы количество фтора составляло в навеске от 1 до 50 мкг. Пипеткой приливают по 15 см
раствора хлорной кислоты рН 1, предварительно нагретого до температуры 50 °С, погружают на дно мензурки магнит, ставят на магнитную мешалку с подогревом до 50 °С, включают перемешивание и выдерживают 15 мин. Полученную суспензию охлаждают до комнатной температуры, приливают по 3 см
раствора лимоннокислого натрия. В полученную суспензию погружают электроды, помещают на магнитную мешалку и через 3 мин замеряют потенциал.
Проводят два параллельных определения.
Одновременно проводят контрольный опыт без продукта. Для этого в полиэтиленовую мензурку пипеткой приливают 15 см раствора хлорной кислоты и выдерживают при температуре нагрева до 50 °С на магнитной мешалке в течение 15 мин. Охлаждают до комнатной температуры, приливают 3 см
раствора лимоннокислого натрия, погружают электроды в мензурку с полученным раствором контрольного опыта и через 3 мин замеряют потенциал. После замера потенциала в контрольном опыте электроды только осушают фильтровальной бумагой и сразу погружают в анализируемую суспензию, перемешивают 3 мин и замеряют потенциал. После измерения потенциала в каждой анализируемой суспензии электроды промывают большим объемом дистиллированной воды при интенсивном помешивании до установления значения потенциала 280-300 мв, осушают фильтровальной бумагой и погружают в раствор с контрольным опытом, выдерживая их до установления первоначального значения потенциала контрольного опыта.
Если после многоразового анализа исходное значение потенциала контрольного опыта не достигается, необходимо приготовить свежий раствор контрольного опыта.
(Измененная редакция, Изм. N 1).
14.4. Обработка результатов
Содержание фтора () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (15)
, (15)
где - концентрация фтора в суспензии продукта, найденная по градуировочному графику, г·моль/дм
;
- грамм-эквивалент фтора;
- объем анализируемой суспензии, равный объему раствора хлорной кислоты и раствора лимоннокислого натрия, см
;
- коэффициент пересчета граммов в миллиграммы;
- масса навески продукта, г.
Результат округляют до первого десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, которое округляют до целого числа.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до целого числа. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 20%, округленных до целого числа.
В случае отсутствия масштабно-координатной бумаги с полулогарифмической сеткой производят расчет содержания фтора по ГОСТ 4386.
15. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ КАДМИЯ
Сущность метода заключается в минерализации навески продукта при нагревании смесью концентрированных серной, азотной и хлорной кислот, с последующим растворением минерализата в концентрированной соляной кислоте и количественном определении кадмия атомно-абсорбционной спектроскопией.
15.1. Аппаратура и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Спектрофотометр атомно-абсорбционный, обеспечивающий чувствительность определения кадмия 0,3 мкг/см, соответствующую 1% поглощения.
Лампа кадмиевая с полым катодом типа ЛСП-1 или аналогичного типа.
Устройства нагревательные электрические, инфракрасные, газовые, снабженные устройством для регулирования степени нагрева.
Шкаф сушильный с терморегулятором типа СЭШ-1 или СЭШ-3М, обеспечивающий поддержание температуры (105±2) °С.
Колбы Кьельдаля 1-250-29/32 ТС или 1-500-29/32 ТС по ГОСТ 25336.
Колбы Кн-2-250-34 ТС по ГОСТ 25336.
Колбы мерные 2-25-1, 2-100-1, 2-1000-1 или 2-25-2, 2-100-2, 2-1000-2 по ГОСТ 1770.
Пипетки 1-1-1, 1-1-2 или 2-го класса точности и в исполнениях 2, 4, 5; 1-1-5, 1-1-10 или 2-го класса точности и в исполнениях 2, 6, 7; 2-1-20 или 2-2-20 по НТД.
Пробирки П 2-16-180 ХС или П 2-19-180 ХС, или Р 2-21-200 ХС по ГОСТ 25336.
Стакан В-1-100 ТС или Н-1-100 ТС по ГОСТ 25336.
Флаконы полиэтиленовые цилиндрические типа ФЦ вместимостью 100 и 1000 см.
Ацетилен растворенный технический по ГОСТ 5457 или пропан бытовой.
Воздух сжатый с давлением в линии в пределах 0,25-0,70 МПа (2,5-7,0 кгс/см).
Кадмий по ГОСТ 1467, х.ч., или кадмий сернокислый по ГОСТ 4456, х.ч.
Кислота серная по ГОСТ 4204, х.ч., концентрированная.
Кислота азотная по ГОСТ 4461, х.ч., концентрированная.
Кислота хлорная, ч., концентрированная.
Кислота соляная по ГОСТ 3118, х.ч., концентрированная.
Водорода перекись по ГОСТ 10929, раствор с массовой долей 30%.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
15.2. Подготовка к испытанию
15.2.1. Приготовление стандартных растворов кадмия
15.2.1.1. Раствор А, содержащий 100 мг/дм кадмия
В стакан вместимостью 100 см отвешивают 0,100 г металлического кадмия (результат записывают с точностью до четвертого десятичного знака), добавляют пипеткой 10 см
концентрированной соляной кислоты и 4-5 капель концентрированной азотной кислоты, стакан помещают на кипящую водяную баню до полного растворения кадмия. Полученный раствор количественно переносят дистиллированной водой в мерную колбу вместимостью 1000 см
, в колбу добавляют до метки дистиллированную воду и перемешивают.
Раствор переливают в полиэтиленовую посуду и хранят не более 1 года.
Допускается приготовлять раствор А из сернокислого кадмия, предварительно подсушенного до постоянной массы при температуре 100-105 °С. Для этого в стакан вместимостью 100 см отвешивают 0,1855 г сернокислого кадмия (результат записывают с точностью до четвертого десятичного знака), приливают пипеткой 10 см
концентрированной соляной кислоты и цилиндром 50 см
дистиллированной воды. Содержимое стакана перемешивают и после полного растворения соли полученный раствор количественно переносят в мерную колбу вместимостью 1000 см
. Раствор в колбе доводят до метки и перемешивают.
Раствор переливают в полиэтиленовую посуду и хранят не более 1 год
а.
15.2.1.2. Раствор Б, содержащий 20 мг/дм кадмия
Из раствора А пипеткой отбирают 20 см, переносят в мерную колбу вместимостью 100 см
, в колбу добавляют до метки дистиллированную воду и перемешивают.
Раствор переливают в полиэтиленовую посуду и хранят не более 3 мес.
15.2.2. Построение градуировочного графика
В четыре мерные колбы вместимостью по 100 см пипетками соответствующей вместимости вносят 1, 2, 5 и 10 см
раствора Б, добавляют в колбу до метки дистиллированную воду, смесь перемешивают. Концентрация полученных растворов соответственно составляет 0,2; 0,4; 1,0 и 2,0 мкг/см
.
Растворы переливают в полиэтиленовую посуду и хранят не более 3 мес.
После установки нуля прибора по дистиллированной воде полученные растворы в порядке возрастания концентрации кадмия последовательно распыляют в воздушно-ацетиленовое (или воздушно-пропановое) пламя горелки и измеряют величину абсорбции при аналитической линии кадмия 228,9 нм. Условия спектрофотометрирования подбирают по инструкции к прибору. После каждого замера в пламя горелки распыляют дистиллированную воду до полного возврата стрелки в нулевое положение.
По полученным данным строят градуировочный график, откладывая по оси ординат значение величины абсорбции, а по оси абсцисс - концентрацию кадмия в мкг/см. Проверку градуировочного графика проводят ежедневно после каждого зажигания горелки по двум стандартным растворам кадмия.
15.3. Проведение испытания
В сухую пробирку, свободно входящую в горло конической колбы вместимостью 250 см или колбы Кьельдаля, насыпают около 2,5 г продукта и пробирку взвешивают (результат записывают с точностью до четвертого десятичного знака). Продукт осторожно высыпают в колбу и пробирку взвешивают вновь. По разнице между первым и вторым взвешиваниями определяют массу навески продукта.
В колбу с продуктом пипеткой вносят 3 см концентрированной серной кислоты, колбу устанавливают на нагреватель и проводят минерализацию продукта до получения однородной черной массы. После обугливания продукта колбу снимают с нагревателя, слегка охлаждают, приливают пипетками 5 см
концентрированной азотной кислоты и 1 см
хлорной кислоты. Колбу снова помещают на нагреватель и проводят дальнейшую минерализацию продукта. Следят за тем, чтобы в колбе все время находилась азотная кислота и по мере ее испытания добавляют порциями по 5 см
. Наличие азотной кислоты определяют по выделяющимся из жидкости бурым парам окислов азота.
Когда раствор приобретет устойчивую светло-желтую окраску, его упаривают почти досуха. Если при этом не наблюдается почернение минерального осадка, минерализацию считают законченной. Если осадок почернеет, добавляют очередную порцию азотной кислоты и минерализацию продолжают.
Допускается в конце минерализации использовать раствор перекиси водорода взамен концентрированной азотной кислоты.
После полного разложения продукта азотную кислоту удаляют упариванием до исчезновения бурых паров окислов азота и появления белых паров окислов серы. Полное удаление азотной кислоты осуществляют двукратным добавлением по 10 см дистиллированной воды и испарением ее почти досуха. Почти сухой остаток растворяют при нагревании в 5 см
концентрированной соляной кислоты, полученный раствор количественно переносят дистиллированной водой в мерную колбу вместимостью 25 см
, в колбу до метки добавляют дистиллированную воду, смесь перемешивают.
Проводят два параллельных определения.
В тех же условиях проводят контрольный опыт без продукта, начиная с внесения в коническую колбу 3 см концентрированной серной кислоты.
В полученных после разложения продукта растворах проводят спектрофотометрическое определение содержания кадмия в тех же условиях, при которых анализировали стандартные растворы кадмия при построении градуировочного графика.
После подготовки прибора в пламя горелки распыляют последовательно в порядке возрастания концентрации кадмия два стандартных раствора. В случае сохранения пропорциональности изменения величин абсорбции с увеличением концентрации кадмия в растворе в пламя горелки распыляют анализируемые растворы и раствор контрольного опыта, строго соблюдая постоянство условий спектрофотометрирования.
Для повышения точности измерения величины абсорбции шкалу измерительного прибора растягивают в 2, 5 или 10 раз. При этом условия спектрофотометрирования подбирают так, чтобы отношение сигнал - шум было минимальн
ым.
15.4. Обработка результатов
Содержание кадмия () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (16)
, (16)
где - содержание кадмия в 1 см
анализируемого раствора, найденное по градуировочному графику, мкг/см
;
- содержание кадмия в 1 см
контрольной пробы, найденное по градуировочному графику, мкг/см
;
- объем мерной колбы, см
;
- масса навески продукта, г.
Результат округляют до второго десятичного знака.
Из результатов двух параллельных определений вычисляют среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, округленных до целого числа.
За окончательный результат испытания принимают среднее арифметическое двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разным оборудованием, с разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое округляют до целого числа. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 50%, округленных до целого чис
ла.
15.3; 15.4. (Измененная редакция, Изм. N 1).
16. МЕТОД ОПРЕДЕЛЕНИЯ КРУПНОСТИ ГРАНУЛ
16.1. Аппаратура
Весы лабораторные технические любого типа.
Сито штампованное с отверстиями диаметром 2, 3, 0,15 мм; диаметр сит должен быть не менее 150 мм.
Анализатор ситовой механический марки АЛГ-М.
Штангенциркуль или масштабная линейка.
(Измененная редакция, Изм. N 1).
16.2. Проведение испытания
Навеску продукта массой 100 г просеивают через сито прибора в течение 10 мин при 190-210 мин. Допускается просеивать вручную при 110-210 мин
и размахе сита около 10 см. Продукт, прошедший через сито, или остаток на сите взвешивают (результат записывают с точностью до одного десятичного знака). Затем с погрешностью не более 1 мм измеряют длину и диаметр 10 случайно отобранных гранул продукта.
16.3. Обработка результатов
Проход через сито или остаток на сите выражают в процентном отношении массы продукта, прошедшего через сито или оставшегося на сите, к массе навески.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений.
Допускаемое относительное расхождение между результатами параллельных определений не должно превышать 0,1%, а между результатами, полученными в разных лабораториях - 0,2%.
За размер гранул (диаметр и длину) принимают среднее арифметическое значение десяти измерений, которое округляют до целого числа.
17. МЕТОД ОПРЕДЕЛЕНИЯ ДРОЖЖЕВЫХ КЛЕТОК
17.1. Аппаратура, растворы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Микроскоп световой биологический типов I, III по ТУ 3-3.404.
Термостат электрический с автоматическим терморегулятором.
Колбы вместимостью 250 или 500 см по ГОСТ 25336.
Цилиндры мерные вместимостью 100 см по ГОСТ 1770.
Пипетки вместимостью 1 см по НТД; для анализа конец пипетки расширяют.
Стекла покровные по ГОСТ 6672.
Стекла предметные по ГОСТ 9284.
Чашки Петри по ГОСТ 25336.
Баня водяная.
Стерилизатор.
Шпатель металлический.
Раствор физиологический рН 5,6; готовят по ГОСТ 10444.1.
Сусло-агар; готовят по инструкции, утвержденной в установленном порядке.
Окситетрациклина дегидрат в таблетках по 0,25 (250000 ед).
Стрептомицина сульфат во флаконах по 0,25 (250000 ед); 0,5 (500000 ед) или 1 г (1000000 ед).
Раствор окситетрациклина дегидрата; готовят по ГОСТ 10444.12.
Раствор стрептомицина; готовят по ГОСТ 10444.12.
Сусло-агар с окситетрациклином; готовят по ГОСТ 10444.12.
Вода дистиллированная.
17.2. Проведение испытания
В стерильную коническую колбу или плоскодонную вместимостью 250 или 500 см берут навеску продукта массой 10 г, заливают 90 см
физиологического раствора рН 5,6 и тщательно перемешивают до получения однородной суспензии.
Из первого разведения 1:10 готовят ряд десятикратных разведений до такой степени, чтобы можно было определить предполагаемое количество дрожжей в 1 г исследуемого продукта.
Высевают по 1 см каждого разведения в две стерильные чашки Петри. В каждую чашку, содержащую инокулум, наливают не позднее чем через 15 мин 12-15 см
сусло-агара, охлажденного до (45±1) °С. При необходимости в сусло-агар добавляют антибиотик окситетрациклин или стрептомицин. Среду немедленно тщательно перемешивают и оставляют до застывания. После застывания среды посевы инкубируют крышкой вниз при температуре 30-32 °С в течение 5 сут. Через 3 сут инкубирования проводят предварительный учет типичных колоний, а через 5 сут - окончательный, наблюдая за ростом дрожжей визуально, при необходимости и микроскопически.
17.1; 17.2. (Измененная редакция, Изм. N 1).
17.3. Обработка результатов
Для подсчета используют посев того разведения, где на чашках выросло от 15 до 150 колоний дрожжей. По результатам подсчета вычисляют среднее арифметическое значение числа колоний во всех посевах этого разведения. Если же не в одном, а в двух следующих друг за другом разведениях количество колоний на чашках находится в пределах 15-150, то подсчитывают число колоний в каждом из разведений раздельно и вычисляют среднее арифметическое, если полученные результаты не отличаются друг от друга более чем в два раза. В противном случае результаты оценивают по результатам высева наибольшего разведения.
Количество дрожжей в 1 г продукта вычисляют умножением среднего арифметического значения числа колоний на соответствующее разведение навески.
18. МЕТОД ОПРЕДЕЛЕНИЯ ОБЩЕЙ БАКТЕРИАЛЬНОЙ ОБСЕМЕНЕННОСТИ
18.1. Аппаратура, растворы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Прибор для счета колоний бактерий.
Термостат электрический с автоматическим терморегулятором.
Баня водяная.
рН-метр.
Электроплитка с регулятором температуры или электронагреватель.
Автоклав или стерилизатор.
Микроскоп световой биологический типов I, III по ТУ 3-3.404.
Шпатель металлический.
Пробирки вместимостью 25 или 50 см по ГОСТ 25336.
Чашки Петри по ГОСТ 25336.
Пипетки вместимостью 1 см по НТД; конец пипетки для анализа предварительно расширяют.
Колбы конические вместимостью 250, 500 и 750 см по ГОСТ 25336.
Стекла предметные по ГОСТ 9284.
Стекла покровные по ГОСТ 6672.
Агар микробиологический по ГОСТ 17206.
Агар питательный сухой или агар мясо-пептонный (готовят по ГОСТ 10444.1), или агаризованная среда на основе ферментолизата микроорганизмов (готовят по п.18.2.1 настоящего стандарта).
Агар голодный; готовят по ГОСТ 10444.1.
Раствор физиологический рН 7,0; готовят по ГОСТ 10444.1.
Ферментолизат биомассы микроорганизмов осветленный.
Натрия гидроокись по ГОСТ 4328, раствор с массовой долей 20%.
Натрий хлористый по ГОСТ 4233.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
18.2. Подготовка к испытанию
18.2.1. Приготовление агаризованной среды на основе ферментолизата биомассы микроорганизмов
Среду готовят исходя из содержания аминного азота в порошке ферментолизата. Необходимое количество порошка разводят в 1 дм водопроводной воды до содержания аминного азота около 100 мг/100 см
; устанавливают рН среды 7,0-7,2 раствором гидроокиси натрия с массовой долей 20%; к жидкому ферментолизату добавляют 5 г хлористого натрия и нагревают на слабом огне при постоянном помешивании до полного растворения ингредиентов.
Приготовленный бульон фильтруют через ватно-марлевый фильтр, устанавливают слабощелочную реакцию (рН 7,0-7,2) и добавляют 20 г агара. После добавления агара жидкость кипятят на слабом огне при постоянном помешивании до полного растворения агара. Приготовленную среду фильтруют через ватно-марлевый фильтр, разливают в колбы и стерилизуют в автоклаве при температуре 120 °С в течение 20 мин.
18.3. Проведение испытания
В стерильную пробирку фламбированным шпателем берут навеску продукта массой 1 г.
Разведения для посевов готовят следующим образом: к навеске массой 1 г добавляют 9 см физиологического раствора рН 7,0 и тщательно перемешивают стерильной пипеткой до получения однородной суспензии. Из первого разведения 1:10 готовят последующие разведения 1:100 и т.д. Порядок разведения выбирают в зависимости от предполагаемой обсемененности продукта, с тем чтобы на чашках развивалось 30-300 колоний бактерий. Для приготовления первого разведения используют пипетку с расширенным концом.
Посев проводят из двух-трех (а в отдельных случаях более) последовательных 10-кратных разведений. Каждое разведение высевают на 2-3 чашки Петри. По 1 см суспензии вносят в чашку с заранее маркированным дном и заливают не позднее чем через 15 мин 12-15 см
расплавленного и охлажденного до температуры 45 °С мясо-пептонного агара или одной из сред, указанных в п.18.1. Сразу после заливания среды содержимое чашки тщательно перемешивают путем вращательного покачивания для равномерного распределения посевного материала.
Для того, чтобы помешать развитию на поверхности агара спорообразующих микробов и бактерий протея, допускается наслоение расплавленного и охлажденного до температуры 45-50 °С голодного агара толщиной 3-4 мм.
После застывания среды чашки Петри переворачивают крышками вниз и инкубируют в термостате при температуре 37 °С в течение 48 ч.
(Измененная редакция, Изм. N 1).
18.4. Обработка результатов
Количество выросших колоний подсчитывают через 24-48 ч в каждой чашке, поместив ее вверх дном в приборе для счета колоний бактерий.
Оценку результатов проводят по посеву того разведения, в котором количество выросших колоний на чашках составляет от 30 до 300. По результатам подсчета вычисляют среднее арифметическое значение числа колоний во всех посевах этого разведения. Если же не в одном, а в двух следующих друг за другом разведениях количество колоний на чашках находится в пределах 30-300, то подсчитывают количество бактерий в продукте по результатам подсчета колоний в каждом из разведений раздельно и вычисляют среднее арифметическое значение, если полученные результаты не отличаются друг от друга более чем в два раза. В противном случае результаты посева оценивают по результатам высева наибольшего разведения.
Количество бактерий в 1 г продукта вычисляют умножением среднего арифметического значения числа колоний на соответствующее разведение навески.
19. МЕТОД ВЫЯВЛЕНИЯ БАКТЕРИЙ РОДА САЛЬМОНЕЛЛА
19.1. Аппаратура, материалы, реактивы и среды
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Автоклав или стерилизатор.
Баня водяная с терморегулятором.
Микроскоп световой биологический типов I, III по ТУ 3-3.404.
Термостат электрический с автоматическим терморегулятором.
Шкаф сушильный лабораторный.
Колбы конические плоскодонные вместимостью 50, 100, 500 см по ГОСТ 25336.
Пипетки вместимостью 1, 2, 5, 10 см по НТД.
Цилиндры и колбы мерные вместимостью 100, 250, 500 см по ГОСТ 1770.
Чашки Петри по ГОСТ 25336.
Бумага фильтровальная.
Петля бактериологическая.
Стекла предметные по ГОСТ 9284.
Стекла покровные по ГОСТ 6672.
Глюкоза кристаллическая гидратная по ГОСТ 975 или -глюкоза по ГОСТ 6038.
Калий фосфорнокислый однозамещенный по ГОСТ 4198, х.ч.
Кислота щавелевая по ГОСТ 22180.
Магний хлористый 6-водный по ГОСТ 4209, х.ч.
Натрий хлористый по ГОСТ 4233, х.ч.
Натрий серноватистокислый (натрия тиосульфат) 5-водный по ГОСТ 27068, х.ч.
Натрий фосфорнокислый двузамещенный 12-водный по ГОСТ 4172.
Ферментолизат биомассы микроорганизмов осветленный.
Спирт ректификованный технический по ГОСТ 18300.
Соль закиси железа и аммония двойная сернокислая (соль Мора) по ГОСТ 4208.
Мочевина по ГОСТ 6691, х.ч.
Лактоза, х.ч.
Агар микробиологический по ГОСТ 17206.
Агар питательный сухой.
Агар с эозин-метиленовым синим, сухой (среда Левина).
Бактоагар Плоскирева, сухой (среда Плоскирева).
Бульон мясо-пептонный по ГОСТ 20730.
Висмут-сульфит-агар, сухой.
Набор адсорбированных поливалентных сальмонеллезных О-сывороток основных пяти групп (A, В, С, D, Е) и редких групп.
Пептон сухой ферментативный для бактериологических целей по ГОСТ 13805.
Среда питательная для первичной дифференциации энтеробактерий (типа Клиглера), сухая.
Среда питательная с индикатором ВР и лактозой, сухая (Гисса).
Раствор физиологический рН 7,0; готовят по ГОСТ 10444.1.
Сахароза по ГОСТ 5833.
Феноловый красный.
Экстракт дрожжевой жидкий; готовят по ГОСТ 10444.1 или экстракт дрожжевой.
Цитрат агар Симмонса (среда Симмонса).
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
19.2. Подготовка к испытанию
19.2.1. Приготовление магниевой среды
Пептон сухой | 4,2 г | |||
Натрий хлористый | 7,0 г | |||
Калий фосфорнокислый однозамещенный | 1,5 г | |||
Экстракт дрожжевой | 2,0 г или | |||
Экстракт дрожжевой жидкий | 20,0 см | |||
Вода дистиллированная | 890,0 см | |||
Растворяют при кипении, затем добавляют растворы:
а) магний хлористый - 36,0 г,
вода дистиллированная - 90,0 см;
б) водный раствор бриллиантового зеленого с массовой долей 0,1% - 5,0 см.
Среду фильтруют через бумажный фильтр, разливают в колбу и стерилизуют при температуре 112 °С в течение 20 мин.
(Измененная редакция, Изм. N 1).
19.2.1a. Приготовление магниевой среды на основе ферментолизата биомассы микроорганизмов
Ферментолизат биомассы микроорганизмов осветленный | 10,0 г | |||
Натрий хлористый | 7,0 г | |||
Калий фосфорнокислый однозамещенный | 1,5 г | |||
Вода дистиллированная | 890 см | |||
Приготовление среды аналогично приготовлению магниевой среды по п.19.2.1. Готовая среда должна иметь рН 7,0.
19.2.16. Приготовление среды предобогащения на основе пептона
Пептон сухой | 10,0 г | |||
Натрий хлористый | 5,0 г | |||
Натрий фосфорнокислый двузамещенный 12-водный | 9,0 г | |||
Калий фосфорнокислый однозамещенный | 1,5 г | |||
Вода дистиллированная | 1000 см | |||
Компоненты растворяют в воде при подогревании до кипения. Фильтруют через бумажный фильтр. Стерилизуют при температуре 120 °С в течение 20 мин. Готовая среда должна иметь рН 7,0.
19.2.1в. Приготовление среды предобогащения на основе ферментолизата биомассы микроорганизмов
Ферментолизат биомассы микроорганизмов осветленный | 15,0 г | |||
Натрий хлористый | 5,0 г | |||
Натрий фосфорнокислый двузамещенный 12-водный | 9,0 г | |||
Калий фосфорнокислый однозамещенный | 1,5 г | |||
Вода дистиллированная | 1000 см | |||
Приготовление аналогично приготовлению среды предобогащения на основе пептона по 19.2.16. Готовая среда должна иметь рН 7,0.
19.2.1a-19.2.1в. (Введены дополнительно, Изм. N 1).
19.2.2. Приготовление пептонной воды
Пептон сухой | 20,0 г | |||
Натрий хлористый | 5,0 г | |||
Вода дистиллированная | 1000 см | |||
К дистиллированной воде добавляют пептон и хлористый натрий, устанавливают значение рН 7,2-7,4, кипятят 30 мин, проверяют рН, фильтруют через бумажный фильтр до полной прозрачности и стерилизуют при температуре 120 °С в течение 30 мин.
(Измененная редакция, Изм. N 1).
19.2.3. Приготовление комбинированной двууглеводной среды с мочевиной (КДСМ)
Среда питательная с индикатором ВР и лактозой, сухая (Гисса) | 40,0 г | |||
Глюкоза кристаллическая гидратная | 1,0 г | |||
Агар питательный, сухой | 5,0 г | |||
Мочевина | 10,0 г | |||
Соль Мора | 0,2 г | |||
Натрия тиосульфат | 0,3 г | |||
Вода дистиллированная | 1000 см | |||
Компоненты среды, кроме мочевины, растворяют в воде при нагревании до кипения. Затем добавляют мочевину и растворяют (не нагревая среду).
Среду разливают в пробирки по 6-8 см и стерилизуют текучим паром 20 мин. Скашивают среду в пробирках, оставив столбик высотой 2-3 см. Цвет готовой среды бледно-сиреневый. Посев на КДСМ производят штрихом на поверхность косяка, а затем уколом в глубину столбика.
19.2.4. Приготовление комбинированной среды Олькеницкого
Вода дистиллированная | 100 см | |||
Агар-агар | 2,5 г | |||
Лактоза | 1,0 г | |||
Сахароза | 1,0 г | |||
Глюкоза | 0,1 г | |||
Мочевина | 1,0 г | |||
Соль Мора | 0,02 г | |||
Натрия тиосульфат | 0,03 г | |||
Водный раствор фенолового красного с массовой долей 0,4% | 1 см | |||
Все тщательно смешивают, устанавливают рН 7,2-7,4. Среду разливают в пробирки и стерилизуют текучим паром по 20 мин 3 дня подряд. После стерилизации среду скашивают. Готовая среда имеет бледно-розовый цвет.
19.2.4а. Приготовление среды Клиглера с мочевиной
5,5 г сухого порошка среды Клиглера засыпают в колбу с 100 см холодной дистиллированной воды, тщательно перемешивают и на слабом огне доводят до кипения. Кипятят 3-5 мин, закрыв колбу ватной пробкой. Среду фильтруют через предварительно смоченный водой и отжатый ватный фильтр. Добавляют 1 см
20%-ного раствора мочевины, тщательно перемешивают. Разливают в пробирки по 7 см
и стерилизуют при температуре 112 °С 20 мин или текучим паром 30 мин.
Допускается приготавливать среду без добавления мочевины, но в этом случае в расплавленную после стерилизации среду в пробирке вносят в асептических условиях 0,1 см стерильного раствора мочевины.
(Введен дополнительно, Изм. N 1).
19.2.5. Приготовление полужидкого агара
Агар питательный сухой | 3,0 г | |||
Вода дистиллированная | 1000 см | |||
Среду разливают в пробирки по 5 см и стерилизуют при температуре 120 °С в течение 30 мин.
19.2.6. Приготовление индикаторной бумаги для обнаружения индола
Листы фильтровальной бумаги размером 20х30 см обильно смачивают горячим 60-70 °С насыщенным раствором щавелевой кислоты с массовой долей 12%, сушат при температуре (23±2) °С, нарезают полосками шириной 0,2-0,4 см, длиной 5-6 см и хранят в банках из темного стекла с пробками.
19.3. Проведение испытания
Навеску продукта массой 50 г, взятую с соблюдением правил асептики, переносят в асептических условиях в колбу с 350 см среды предобогащения на основе пептона (п.19.2.1б) или ферментолизата (п.19.2.1в). Посев инкубируют при температуре 37 °С в течение 16-20 ч. После инкубирования стерильно переносят 20 см
культуральной жидкости в колбу с 200 см
магниевой среды на основе пептона или ферментолизата. Посев инкубируют при температуре 37 °С в течение 48 ч.
Через 18-24 ч термостатирования из колбы с магниевой средой производят посевы бактериологической петлей на чашки с твердыми дифференциально-диагностическими средами: висмут-сульфит агаром и по выбору со средой Плоскирева или Левина (по две чашки) для получения изолированных колоний.
На висмут-сульфит агаре S. typhi и S. paratyphi А. растут в виде мелких, нежных, серовато-зеленых колоний с черным центром, S. cholerae suis - в виде зеленых колоний. Колонии почти всех других сальмонелл значительно крупнее, темно-коричневого цвета с металлическим блеском, окруженные металлическим или светлым ореолом, цвет участка среды под колонией - черный.
На среде Плоскирева сальмонеллы растут в виде прозрачных или нежно-розовых колоний, на среде Левина - в виде прозрачных, бледных нежно-розовых и розовато-фиолетовых колоний.
В случае обнаружения колоний, подозрительных на сальмонеллы, 3-6 из них отвивают по 1/3 колонии на одну из сред: КДСМ, Олькеницкого или Клиглера. Для определения подвижности проводят посев уколом исследуемой колонии в полужидкий агар. Оставшуюся часть колонии отвивают на косяк Симмонса для проверки ассимиляции цитрата и в МПБ или пептонную воду для проверки индолообразования, вкладывая под пробку индикаторную бумагу на индол. Бумагу помещают таким образом, чтобы она удерживалась пробкой, но не прикасалась к среде. При наличии индола через 1-3 дня термостатирования при 37 °С нижняя часть бумажки окрашивается в розовый цвет, хорошо заметный в проходящем свете.
Посевы на комбинированные среды КДСМ, Олькеницкого или Клиглера делают сначала штрихом на скошенную поверхность, а затем уколом в глубину столбика, посев на среду Симмонса - штрихом. Посевы инкубируют при температуре 37 °С 18-24 ч.
Культуры сальмонелл ассимилируют и сбраживают глюкозу, не ассимилируют лактозу, образуют сероводород, не разлагают мочевину, утилизируют цитраты, не образуют индол.
При росте сальмонелл цвет скошенной поверхности всех трех комбинированных сред не меняется (лактозу не ассимилируют) или слегка розовеет. При ассимиляции глюкозы растущей культурой столбик среды КДСМ приобретает голубовато-зеленый цвет, столбик среды Олькеницкого окрашивается в оранжево-розовый цвет, среды Клиглера - в желтый. Вследствие образования газа при ферментации глюкозы происходит разрыв столбика агара со скоплением в нем пузырьков газа у большинства культур сальмонелл. При росте штаммов, образующих сероводород, на границе скошенной поверхности среды и столбика или на дне пробирки происходит почернение среды. Некоторые культуры продуцируют сероводород замедленно, либо не продуцируют его (S. paratyphi A.,S. cholerae suis, S. bareilly, S. typhi suis, S. pullorum).
При разложении мочевины среда КДСМ меняет цвет скошенной части и столбика на ярко-оранжевый. При этом учет ферментации глюкозы и лактозы становится невозможным. Среда Олькеницкого при разложении мочевины остается розовой.
Сальмонеллы растут на среде Симмонса, ассимилируя цитрат, и изменяют ее цвет из оливково-зеленого в синий. Большинство культур сальмонелл подвижны (кроме S. pullorum, S. gallinarum), что выражается равномерным ростом культуры по всему столбику полужидкого агара после инкубации в термостате.
Культуры, ферментирующие на комбинированных средах лактозу или расщепляющие мочевину, не относятся к роду сальмонелл.
Культуры, подозрительные на сальмонеллы, окрашивают по Грамму по ГОСТ 18963.
Культуры, представляющие грамотрицательные подвижные палочки, ферментирующие глюкозу, не ферментирующие лактозу, не разлагающие мочевину, не образующие индол, утилизирующие цитрат, подвергают серологическому исследованию - испытанию в реакции агглютинации с поливалентной адсорбированной сальмонеллезной О-сывороткой основных пяти групп (А, В, С, D и Е) и редких групп.
Для проведения реакции агглютинации на предметное стекло наносят каплю физиологического раствора и рядом каплю агглютинирующей поливалентной сальмонеллезной сыворотки ABCDE и каплю агглютинирующей поливалентной сыворотки к сальмонеллам редких групп. Затем в каждую из капель, начиная с физиологического раствора, вносят петлей часть анализируемой колонии и равномерно растирают.
При положительной реакции через 0,5-2,0 мин в капле сыворотки образуются хлопья, жидкость просветляется. В капле с физиологическим раствором заметно равномерное помутнение.
Реакция агглютинации с поливалентными сальмонеллезными сыворотками считается положительной при наличии феномена хлопьеобразования с поливалентной сальмонеллезной О-сывороткой ABCDE групп, а также феномен агглютинации указывает на наличие бактерий рода сальмонелла.
19.4. Обработка результатов
Обнаружение подвижных грамотрицательных палочек, дающих характерный рост на плотных питательных средах и не ферментирующих лактозу, ферментирующих глюкозу, утилизирующих цитраты, не образующих индол, образующих сероводород (большинство), дающих положительную реакцию агглютинации с поливалентной адсорбированной О-сывороткой или поливалентной О-сывороткой редких групп, а также феномен агглютинации указывает на наличие бактерий рода сальмонелла.
19.3; 19.4. (Измененная редакция, Изм. N 1).
20. МЕТОД ОПРЕДЕЛЕНИЯ ТОКСИЧНОСТИ С ПРИМЕНЕНИЕМ ТЕСТ-КУЛЬТУРЫ
ИНФУЗОРИЙ ТЕТРАХИМЕНА ПИРИФОРМИС
20.1. Аппаратура, материалы, реактивы и среды
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104 или весы торзионные.
Термостат электрический с автоматическим терморегулятором.
Микроскоп световой биологический типов I, III по ТУ 3-3.404.
Автоклав.
рН-метр.
Колбы конические вместимостью 100 см по ГОСТ 25336.
Пипетки вместимостью 0,2; 1; 2; 10 см по НТД.
Пипетки пастеровские.
Палочки стеклянные.
Мензурки мерные вместимостью 250 см по ГОСТ 1770.
Казеин по Гаммерстену, ч. или казеин технический по ГОСТ 17626*.
________________
* В Российской Федерации см. также ГОСТ Р 51463-99.
Натрий фосфорнокислый однозамещенный 2-водный по ГОСТ 245.
Натрий фосфорнокислый двузамещенный 12-водный по ГОСТ 4172 или натрий фосфорнокислый двузамещенный по ГОСТ 11773.
Натрий хлористый по ГОСТ 4233.
Буфер фосфатный рН 7,4.
Д-глюкоза по ГОСТ 6038 или глюкоза кристаллическая гидратная по ГОСТ 975.
Агар мясо-пептонный; готовят по ГОСТ 10444.1, или агар микробиологический по ГОСТ 17206, или агар питательный, сухой.
Соль морская для ванн.
Экстракт дрожжевой.
Инокулят инфузории (3-4-суточная культура инфузории).
Триптон бактериологический фирм "Ферак", "Дифко" или пептон сухой ферментативный для бактериологических целей по ГОСТ 13805.
Среда УСД для определения токсичности (углеводно-солевая дрожжевая); готовят следующим в образом: в мерную мензурку вносят 1,5 г Д-глюкозы или кристаллической гидратной глюкозы, 0,1 г дрожжевого экстракта, 0,1 г морской соли и доливают дистиллированной водой до 100 см, устанавливают рН 7,1-7,2, среду разливают в конические колбы по 10 см
, закрывают ватно-марлевыми пробками и стерилизуют в автоклаве 15-20 мин при 760 мм рт.ст.
Среда триптонная или пептонная (для поддержания инокулята инфузории); готовят следующим образом: в мерную мензурку вносят 2,0 г сухого ферментативного пептона для бактериологических целей или бактериологического триптона, 0,5 г Д-глюкозы или гидратной глюкозы, 0,1 г дрожжевого экстракта, 0,1 г морской соли и доводят объем дистиллированной водой до 100 см, устанавливают рН 7,1-7,2, среду разливают в конические или плоскодонные колбы вместимостью 100 см
по 10 см
, закрывают ватно-марлевыми пробками и стерилизуют в автоклаве 15-20 мин при 760 мм рт.ст.; среду хранят в течение месяца при температуре 4 °С.
(Измененная редакция, Изм. N 1
).
20.2. Подготовка к испытанию
20.2.1. Культивирование инфузорий
Пересевают 0,2 см среды с культурой на свежую триптонную или пептонную среду. Пересев проводят каждые 7 дней. Раз в месяц проводят бактериологический контроль среды на стерильность путем посева на мясо-пептонный агар.
20.2.2. Приготовление фосфатного буфера рН 7,4
В мерную колбу вместимостью 200 см берут 81 см
0,2 моль/дм
(0,2 н.) раствора фосфорнокислого натрия двузамещенного и 19 см
0,1 моль/дм
фосфорнокислого натрия однозамещенного, объем доводят дистиллированной водой до 200 см
.
20.2.3. Расчет массы навески продукта
Массу навесок продукта () в граммах, соответствующую заданным количествам (концентрациям) азота: 3, 6, 10, 15 и 20 мг вычисляют по формуле
![]() , (17)
, (17)
где - заданные количества (концентрации) азота в навеске продукта, мг;
- коэффициент пересчета содержания азота в 100 г продукта;
- массовая доля азота в продукте, %.
20.3. Проведение испытания
Соответствующие навески переносят в конические колбы со средой УСД. Проверяют рН среды, рН не должно резко меняться. Если рН снижается до 5 и ниже, среду готовят, используя вместо дистиллированной воды фосфатный буфер рН 7,4. Затем засевают 0,2 см инокулята 3-4-суточной инфузории и полученные пробы инкубируют в термостате при температуре 24-26 °С в течение 72 ч. Для лучшей аэрации колбы 2-3 раза в сутки встряхивают.
В качестве контроля за состоянием культуры используют казеин в заданных концентрациях азота: 3, 6, 10, 15 и 20 мг.
20.4. Обработка результатов
Культуру инфузории в исследуемых пробах просматривают в капле на предметном стекле (каплю берут стеклянной палочкой или пастеровской пипеткой) под микроскопом (объектив 10, окуляр 7) в 5-6 полях зрения через 72 ч.
В случае гибели инфузории в течение учетного времени хотя бы в одной из концентраций или наличия единичных (до 5) клеток в поле зрения при одновременном снижении активности тест-культуры в двух последних концентрациях и получение аналогичного результата в повторном эксперименте с предварительным кипячением среды УСД с навесками в колбах в течение 10 мин на водяной бане продукт считается токсичным. Если в опыте без кипячения токсичность выявлена, но в повторном опыте с предварительным кипячением токсичность не обнаруживается, то следует ввести контроль токсичности исследуемой партии продукта на белых мышах в соответствии с требованиями п.21.
При большом количестве анализируемых проб допускается использовать микрометод при 10-кратном уменьшении питательной среды, навески и количества засеваемой культуры. Для исследований используют флакончики из-под антибиотиков с резиновыми пробками, имеющими боковой срез для доступа воздуха, или доски для постановки реакции гемоагглютинации.
(Измененная редакция, Изм. N 1).
21. ОПРЕДЕЛЕНИЕ ТОКСИЧНОСТИ НА БЕЛЫХ МЫШАХ
Метод применяют при разногласиях в оценке качества кормовых дрожжей.
21.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г или весы 3-го класса точности с наибольшим пределом взвешивания 1 кг по ГОСТ 24104.
Баня водяная.
Центрифуга лабораторная.
Гомогенизатор лабораторный.
Аппарат для встряхивания жидкости в сосудах (шуттель-аппарат).
Колбы вместимостью 100 см с притертой пробкой.
Стакан химический мерный вместимостью 50 см.
Шприц вместимостью 1 или 2 см с тупыми иглами, слегка загнутыми, длиной 2,5-3,0 см
, диаметром внутреннего отверстия не менее 1,5 мм.
Фильтры бумажные (белая лента).
Мыши белые, беспородные самцы, массой 18-20 г.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
21.2. Подготовка к испытанию
21.2.1. Приготовление суспензии
Навеску продукта в количестве 10 г измельчают в фарфоровой ступке до пылевидного состояния, помещают в колбу вместимостью 100 см с притертой пробкой, приливают 40 см
горячей дистиллированной воды (температура 65-70 °С) и встряхивают на шуттель-аппарате в течение 30-40 мин, затем суспензию центрифугируют при 3-5 тыс. об/мин в течение 15 мин или фильтруют через бумажный фильтр. Полученный экстракт (центрифугат) используют для приготовления суспензии. Для этого взвешивают 2,5 г измельченного испытуемого продукта, переносят в мерный стакан вместимостью 50 см
и доводят до объема 10 см
экстрактом (центрифугатом). Суспензию тщательно перемешивают или гомогенизируют в течение 2-3 мин.
21.3. Проведение испытания
Для опыта берут 5 белых мышей массой 18-20 г каждая, выдерживают без корма 4-5 ч, после чего с помощью шприца с зондом (тупая слегка загнутая игла) вводят однократно через рот в желудок 1 см суспензии.
Наблюдают за мышами в течение 5 сут, не ограничивая их в корме и питье. Оставшихся в живых мышей убивают и вскрывают.
В качестве контроля белым мышам вводят по 1 см дистиллированной воды.
21.4. Обработка результатов
Продукт не токсичный - мыши живы. На вскрытии у убитых мышей патологоанатомических изменений не обнаруживают.
Продукт слабо токсичный - мыши живы. На вскрытии у всех или у большинства (не менее трех) убитых мышей выявляют геморрагическое воспаление желудочно-кишечного тракта, часто сопровождающееся дегенерацией печени, почек, кровоизлияниями в паренхиматозных органах.
Продукт токсичный - гибнут все или хотя бы одна мышь и на вскрытии павших и убитых мышей выявляют геморрагическое воспаление желудочно-кишечного тракта, часто сопровождающееся дегенерацией печени, почек или кровоизлияниями в паренхиматозных органах.
В случае падежа одной мыши, если остальные живы и патологоанатомических изменений у них не обнаруживают, опыт повторяют.
(Измененная редакция, Изм. N 1).
22. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ НИТРАТОВ
Сущность метода заключается в извлечении нитратов из проб дистиллированной водой, осаждении белков, обесцвечивании водной вытяжки и колориметрическом определении нитратов по реакции Грисса-Илосвая с образованием азосоединения розово-красного цвета.
22.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104 или торзионные.
Весы лабораторные 3-го класса точности с наибольшим пределом взвешивания 500 г или 1 кг по ГОСТ 24104.
Фотоэлектроколориметр.
Аппарат для встряхивания жидкости в сосудах.
Центрифуга лабораторная.
Холодильник.
Штатив лабораторный.
Пробирки стеклянные по ГОСТ 25336.
Цилиндры мерные вместимостью 100 см по ГОСТ 1770.
Колбы мерные вместимостью 100, 500 и 1000 см по ГОСТ 1770.
Колбы конические вместимостью 250 см по ГОСТ 25336.
Воронки стеклянные по ГОСТ 25336.
Пипетки вместимостью 1, 2, 5, 10, 20, 50 и 100 см по НТД.
Палочки стеклянные.
Шкаф сушильный лабораторный.
Фильтры обеззоленные, среднепористые.
Калий азотнокислый по ГОСТ 4217, х.ч.
Калий железистосинеродистый (желтая кровяная соль) 3-водный.
Цинк сернокислый по ГОСТ 4174, ч.д.а.
Кислота уксусная по ГОСТ 61, х.ч., растворы с массовой долей 10 и 12%.
Кислота сульфаниловая безводная по ГОСТ 5821, х.ч.
-нафгиламин по ТУ 6-09-07-1703, х.ч.
Пыль цинковая.
Уголь активный древесный дробленый марки БАУ по ГОСТ 6217.
Марганец сернокислый по ГОСТ 435.
Вода дистиллированная.
(Измененная редакция, Изм. N 1).
22.2. Подготовка к испытанию
22.2.1. Приготовление реактива Грисса-Илосвая
а - 0,6 г сульфаниловой кислоты растворяют в 180 см раствора уксусной кислоты с массовой долей 12%;
б - 0,2 г -нафтиламина растворяют при нагревании (не доводя до кипения) в 20 см
дистиллированной воды в течение 30 мин, фильтруют и смешивают с 180 см
раствора уксусной кислоты с массовой долей 12%.
Растворы хранят в холодильнике 2 мес. При использовании реактивы "а" и "б" смешивают в соотношении 1:1.
(Измененная редакция, Изм. N 1).
22.2.2. (Исключен, Изм. N 1).
22.2.3. Приготовление растворов уксусной кислоты с массовой долей 10 и 12%
Соответственно 100 см и 120 см
уксусной кислоты смешивают в колбах вместимостью 1000 см
с 750 см
дистиллированной воды и доводят объем дистиллированной водой до метки.
22.2.4. (Исключен, Изм. N 1).
22.2.5. Приготовление раствора Карреза 1
150 г желтой кровяной соли количественно переносят в мерную колбу вместимостью 1000 см, растворяют в дистиллированной воде и доводят объем дистиллированной водой до метки. Раствор хранят в склянке из темного стекла.
22.2.6. Приготовление раствора Карреза 2
300 г сульфата цинка количественно переносят в мерную колбу вместимостью 1000 см, растворяют в дистиллированной воде и доводят объем дистиллированной водой до метки.
22.2.7. Приготовление смеси цинковой пыли с сернокислым марганцем
0,5 г цинковой пыли и 50 г сернокислого марганца смешивают и растирают в ступке.
22.2.8. Приготовление основного стандартного раствора азотнокислого калия
1,6322 г азотнокислого калия, высушенного до постоянной массы при температуре 105 °С, взвешивают (результат записывают с точностью до четвертого десятичного знака), количественно переносят в мерную колбу вместимостью 1000 см, растворяют в дистиллированной воде и доводят объем дистиллированной водой до метки. 1 см
раствора содержит 1 мг NO
. Раствор хранят 2 мес.
22.2.9. Построение градуировочного графика
В восемь колб вместимостью 100 см пипетками соответствующей вместимости вносят 0,5; 1; 1,5; 2; 3; 4; 5; 6 см
стандартного раствора азотнокислого калия, доводят до метки дистиллированной водой и перемешивают.
В девять сухих пробирок вносят по 0,1 г смеси цинковой пыли с сернокислым марганцем. В восемь пробирок из каждой мерной колбы приливают 1 см стандартного раствора, затем добавляют 5 см
дистиллированной воды, а в девятую пробирку (контрольный опыт) - 6 см
дистиллированной воды. В девять пробирок добавляют 2 см
раствора уксусной кислоты с массовой долей 10% и закрывают пробками. Содержимое пробирок встряхивают в течение 2 мин до полного растворения цинковой пыли с сернокислым марганцем. Затем в пробирки добавляют 1 см
свежеприготовленного реактива Грисса-Илосвая, содержимое перемешивают и оставляют на 30 мин до проявления окраски. Содержимое NO
в пробирках соответственно составляет 0,005; 0,01; 0,015; 1,02; 0,03; 0,04; 0,05; 0,06 мг/см
.
Колориметрирование проводят при зеленом светофильтре N 5 с длиной волны 490 нм в кювете с расстояниями между рабочими гранями 10 мм. Раствором сравнения служит дистиллированная вода.
Колориметрирование начинают в порядке возрастания содержания NO, начиная с девятой пробирки (контрольный опыт), затем 1, 2, 3, 4, 5, 6, 7, 8 пробирки.
По полученным данным строят градуировочный график, откладывая по оси абсцисс количества NО в мг/см
, а по оси ординат - соответствующие оптические плотности растворов.
Градуировочный график строят по мере приготовления новых растворов сульфаниловой кислоты и -нафти
ламина.
22.3. Проведение испытания
5,0 г продукта взвешивают (результат записывают с точностью до третьего десятичного знака) в колбе вместимостью 250 см. Приливают 70 см
дистиллированной воды и встряхивают в течение 10 мин, затем центрифугируют при 6000 мин
. Центрифугат переносят в мерную колбу вместимостью 100 см
. Осадок промывают 15 см
дистиллированной воды и снова центрифугируют. Надосадочную жидкость приливают в мерную колбу и доводят объем до метки дистиллированной водой.
Затем 40 см водной вытяжки переносят в колбу вместимостью 250 см
, приливают по 20 см
растворов Карреза 1 и Карреза 2, взбалтывают и фильтруют через плотный фильтр. Если вытяжку не удается обесцветить полностью растворами Карреза, тогда для удаления оставшейся окраски к раствору, полученному после осаждения белков растворами Карреза, прибавляют около 0,4 г активированного угля марки Б, взбалтывают 2-3 мин и фильтруют через плотный фильтр.
6 см фильтрата вносят в пробирку для анализа, добавляют 0,1 г смеси цинковой пыли с сернокислым марганцем, приливают 2 см
раствора уксусной кислоты с массовой долей 10% и закрывают пробкой. Содержимое пробирки встряхивают в течение 2 мин до полного растворения цинковой пыли с сернокислым марганцем. Затем в пробирку приливают 1 см
свежеприготовленного реактива Грисса-Илосвая, содержимое перемешивают и через 30 мин колориметрируют при зеленом светофильтре N 5 с длиной волны 490 нм в кювете с расстояниями между рабочими гранями 10 мм. Раствором сравнения служит дистиллированная вода. Проводят два параллельных определ
ения.
22.2.9; 22.3. (Измененная редакция, Изм. N 1).
22.4. Обработка результатов
Содержание нитратов () в миллиграммах на килограмм продукта вычисляют по формуле
![]() , (18)
, (18)
где - объем водной вытяжки, равный 100 см
;
- количество нитратов, найденное по градуировочному графику, мг;
- пересчет на 1 кг продукта;
- разбавление раствора при обесцвечивании;
- объем фильтрата, взятый для анализа, см
;
- масса навески продукта, г.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, допускаемые относительные расхождения между которыми не должны превышать 20% от средней величины результатов испытаний, округленных до целого числа.
(Измененная редакция, Изм. N 1).
23. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ НЕФРАСА
Сущность метода заключается в экстракции нефраса из продукта при обработке водой и этиловым спиртом и количественном определении содержания нефраса в полученном экстракте газохроматографически.
23.1. Аппаратура, материалы и реактивы
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Хроматограф аналитический газовый с пламенно-ионизационным детектором, обеспечивающий чувствительность определения по нефрасу 0,001% в смеси этиловый спирт-вода.
Колонка из нержавеющей стали длиной 2 м и диаметром 3-5 мм.
Микрошприц хроматографический типа "Газохром 101" или аналогичный.
Линейка измерительная с ценой деления 1 мм по ГОСТ 427 или любая другая с делениями, расположенными по скошенной грани.
Лупа измерительная с ценой деления 0,1 мм по ГОСТ 25706.
Шприц медицинский вместимостью 1-2 см с иглой.
Испаритель ротационный лабораторный любого типа.
Чашка фарфоровая диаметром не менее 130 мм по ГОСТ 9147.
Аппарат для встряхивания жидкости в сосудах.
Ступка фарфоровая с пестиком диаметром 75-110 мм по ГОСТ 9147.
Склянка вместимостью 50 см с завинчивающейся пластмассовой крышкой; перед приготовлением растворов под крышку подкладывают резиновую прокладку с диаметром, равным диаметру горла склянки, и толщиной 2-2,5 мм; для прокладок используют силиконовую резину или любую другую, не набухающую от паров органических растворителей; в центре крышки просверливают отверстие диаметром 3-4 мм.
Колбы конические -1-50-14/19,
-1-500-29/32 по ГОСТ 25336.
Пипетки 1-1-2 или 1-2-2; 2-1-5; или 2-2-5; 2-1-25, или 2-2-25 по НТД.
Колбы мерные 2-100-1 или 2-100-2; 2-50-1, или 2-50-2 по ГОСТ 1770.
Цилиндры мерные 1-100 и 1-500 по ГОСТ 1770.
Стаканчики для взвешивания СВ-14/8 по ГОСТ 25336.
Гелий газообразный или азот газообразный по ГОСТ 9293.
Водород технический, высший сорт по ГОСТ 3022.
Воздух сжатый с давлением в линиях в пределах 0,25-0,70 МПа (2,5-7,0 кгс/см).
Ацетон по ГОСТ 2603.
Хлороформ технический по ГОСТ 20015.
Спирт этиловый ректификованный технический по ГОСТ 18300 или спирт пищевой по ГОСТ 5962*.
________________
* В Российской Федерации действует ГОСТ Р 51652-2001.**
** Вероятно ошибка оригинала. Следует читать ГОСТ Р 51652-2000. - Примечание "КОДЕКС".
Нефрас, используемый для экстракции на промышленной установке.
Гексан, ч.
Триэтаноламин.
Хроматон N-AW или хроматон N-AW-HMДS, или хроматон N-AW-ДMCS, фракция 0,16-0,20 мм производства ЧССР или другой диатомитовый носитель для газовой хроматографии.
Вода дистиллированная.
23.2. Подготовка к испытанию
23.2.1. Подготовка газохроматографической колонки
В стаканчике взвешивают 1,5 г триэтаноламина (результат записывают с точностью до второго десятичного знака). С помощью этилового спирта навеску количественно переносят в круглодонную колбу вместимостью 500 см и добавляют этиловый спирт для растворения триэтаноламина. Общее количество спирта должно составлять около 150 см
. Мерным цилиндром вместимостью 100 см
отмеривают 60 см
хроматона или другого диатомитового носителя и вносят в ту же колбу. Смесь тщательно перемешивают в течение 15-20 мин. Затем спирт отгоняют на ротационном испарителе насколько возможно полно. Носитель вместе с триэтаноламином переносят в фарфоровую чашку и оставляют при комнатной температуре на 10-12 ч для удаления остатков этилового спирта.
Колонку последовательно промывают хлороформом, ацетоном, дистиллированной водой, этиловым спиртом, гексаном, высушивают током воздуха при комнатной температуре и заполняют носителем с триэтаноламином. Затем колонку помещают в термостат хроматографа, один конец ее присоединяют к испарителю, другой - оставляют свободным.
Колонку подвергают стабилизации. Для этого через нее пропускают поток газа-носителя со скоростью 30 см/мин, затем нагревают до 80 °С, выдерживают 5 ч, далее поднимают температуру до 100 °С и выдерживают еще 1,5-2 ч. Колонку охлаждают в токе газа-носителя и свободный конец ее присоединяют к детектору.
23.2.2. Приготовление смеси этиловый спирт-вода
В коническую колбу вместимостью 500 см вливают 200 см
этилового спирта и 100 см
дистиллированной воды, отмеряя их количество мерным цилиндром. Смесь тщательно перемешивают и используют для приготовления стандартных растворов.
23.2.3. Условия газохроматографического испытания
В зависимости от марки используемого хроматографа (конструкции пламенно-ионизационного детектора, формы и размера колонок и др.) условия газохроматографического испытания подбирают так, чтобы обеспечить достаточное разделение пиков нефраса, этилового спирта и примесей, присутствующих в водно-спиртовых экстрактах при малых концентрациях нефраса. Для хроматографа ЛХМ-80 рекомендуются следующие условия, которые могут быть уточнены в процессе работы:
колонка стальная длиной 2 м и внутренним диаметром 3 мм;
насадка - 10% триэтаноламина на хроматоне N-AW-HMДS (0,16-0,20 мм);
температура колонки - 80 °С;
температура испарителя - 140 °С;
скорость газа-носителя и водорода -30 см/мин;
скорость воздуха - 250-300 см/мин;
объем пробы - 1 мкл;
скорость движения бумага - 720 мм/ч.
Количество нефраса рассчитывают по любому из параметров пика: высоте или площади, полученной путем умножения высоты пика на его ширину, измеренную на середине высоты с помощью лупы.
Типичная хроматограмма нефраса в водно-спиртовом экстракте приведена на черт.5.
Хроматограмма нефраса в водно-спиртовом экстракте

1 - ввод пробы; 2 - нефрас; 3, 4, 5 - примеси, присутствующие в водно-спиртовом экстракте; 6 - этиловый спирт
Черт.5
23.2.4. Проверка чистоты смеси этиловый спирт-вода
Смесь этиловый спирт-вода, приготовленную по п.23.2.2, подвергают хроматографическому анализу в условиях, выбранных по п.23.2.3, при чувствительности детектора, которая необходима для анализа стандартного раствора с минимальной концентрацией нефраса. Если на хроматограмме появляется пик, время удерживания которого совпадает с временем удерживания нефраса, то анализ смеси проводят 3 раза и рассчитывают среднее арифметическое значение высоты или площади этого пика, которое учитывают при проведении градуировки прибора и выполнении анализа.
23.2.5. Приготовление стандартных растворов
В предварительно взвешенную склянку (результат записывают с точностью до четвертого десятичного знака) с завинчивающейся крышкой приливают мерным цилиндром 40 см смеси этиловый спирт-вода, приготовленной по п.23.2.2. Склянку плотно закрывают завинчивающейся крышкой с резиновой прокладкой, взвешивают и рассчитывают массу добавленной смеси. Затем медицинским шприцем через отверстие в крышке и прокладку в склянку вводят 0,5-0,55 см
нефраса. Склянку взвешивают и рассчитывают массу добавленного нефраса. Раствор тщательно перемешивают. Рассчитывают массовую долю нефраса в процентах в исходном стандартном растворе, исходя из определенных взвешиванием количеств нефраса и смеси этиловый спирт-вода.
Пипеткой отбирают 2 см этого исходного стандартного раствора, переносят их в мерную колбу вместимостью 100 см
, содержимое колбы доводят до метки смесью этиловый спирт-вода и раствор перемешивают. Рассчитывают точное значение массовой доли нефраса в полученном растворе (раствор А). Далее из раствора А с помощью мерных колб и пипеток соответствующей вместимости приготовляют стандартные растворы с точно рассчитанными массовыми долями, приблизительно равными 0,002; 0,005 и 0,01%, по следующей схеме:
Взятый объем раствора А, см | 25 | 25 | 5 | |||
Объем мерной колбы, см | 50 | 100 | 50 | |||
Примерная концентрация раствора, % | 0,01 | 0,005 | 0,002. | |||
23.2.6. Построение градуированного графика
Линейность зависимости площади пика нефраса от его массовой доли проверяют в пределах концентраций от 0,002 до 0,02% при налаживании метода, а затем периодически после вынужденной остановки прибора, связанной с заменой колонки, с ремонтом или другими причинами.
Для каждого стандартного раствора, приготовленного по п.23.2.5, записывают три хроматограммы, вводя микрошприцем по 1 мкл каждого из них в хроматограф при соответствующей чувствительности и измеряют высоты или площади пиков нефраса по п.23.2.3. Для каждого раствора рассчитывают среднее арифметическое значение выбранного параметра пика нефраса.
Если при проверке чистоты смеси этиловый спирт-вода было обнаружено наличие примеси, мешающей определению нефраса, то величина среднего арифметического значения параметра пика этой примеси вычитается из величин средних арифметических значений параметров пиков нефраса для стандартных растворов с учетом чувствительности детектора, при которой были записаны соответствующие хроматограммы.
Полученные значения высот или площадей пиков, определенных при различных чувствительностях детектора, пересчитывают умножением на соответствующие множители на одну чувствительность, наиболее удобную в работе. По полученным значениям строят градуировочный график, откладывая по оси абсцисс концентрации нефраса в стандартных растворах, а по оси ординат - соответствующие им средние арифметические значения высот или площадей пиков нефраса. Градуировочный график должен представлять собой прямую линию.
23.3. Проведение испытания
Продукт растирают в фарфоровой ступке.
В предварительно взвешенную склянку или коническую колбу вместимостью 50 см взвешивают (1,0±0,1) г измельченного продукта (результат записывают с точностью до четвертого десятичного знака) и затем пипеткой вносят 5 см
дистиллированной воды и плотно закрывают. Смесь тщательно перемешивают и оставляют стоять 10 мин. По истечении 10 мин туда же пипеткой вносят 10 см
этилового спирта, смесь перемешивают и склянку или колбу плотно закрывают и вновь взвешивают с той же точностью. Рассчитывают массу смеси этиловый спирт-вода. Затем содержимое колбы перемешивают на аппарате для встряхивания 30 мин.
Записывают две хроматограммы, вводя микрошприцем в хроматограф по 1 мкл раствора, образовавшегося после оседания продукта.
Проводят два параллельных определения.
Затем записывают две хроматограммы для любого стандартного раствора так же вводя в хроматограф по 1 мкл пробы. Высоты или площади пиков нефраса на всех хроматограммах измеряют как указано в п.23.2.3 и рассчитывают средние арифметические значения из параметров пиков для каждого из растворов. Расхождение между высотами или площадями пиков и их средним арифметическим значением не должно превышать 10%.
Если в смеси этиловый спирт-вода были обнаружены мешающие примеси, их содержание учитывают, как указано в п.23.2.6.
23.4. Обработка результатов
Массовую долю нефраса () в процентах вычисляют по формуле
![]() , (19)
, (19)
где и
- среднее значение высоты или площади пика нефраса в растворе, полученном после оседания продукта, и стандартном растворе, соответственно, мм или мм
;
- массовая доля нефраса в стандартном растворе, %;
- масса смеси этиловый спирт-вода, добавленной к продукту, г;
- масса навески продукта, г.
Результат округляют до третьего десятичного знака.
Из результатов двух параллельных определений рассчитывают среднее арифметическое значение с тем же числом знаков после запятой и определяют расхождение между каждым результатом и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 10%, рассчитанных до целого числа.
За окончательный результат испытания принимают среднее арифметическое двух параллельных определений, которое округляют до первого десятичного знака.
Допускаемое относительное расхождение между окончательными результатами, полученными в разных условиях (в разных лабораториях, в разное время, при работе с разной аппаратурой, разными материалами и реактивами), вычисляют следующим образом: из окончательных результатов испытаний, полученных в разных условиях, определяют среднее арифметическое значение, которое рассчитывают и округляют до третьего десятичного знака. Далее определяют расхождение между каждым окончательным результатом испытания и средним арифметическим значением. Допускаемое относительное расхождение не должно превышать 30%, рассчитанных и округленных до целого числа.
23, 23.1-23.4. (Измененная редакция, Изм. N 1).
Текст документа сверен по:
Комбикорма. Часть 6.
Жмыхи, шроты и горчичный порошок.
Дрожжи. Методы анализа: Сб. ГОСТов. -
М.: ИПК Издательство стандартов, 2002

{kind=link}