ГОСТ Р 56720-2015
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
НЕФТЕПРОДУКТЫ И КОНДЕНСАТ ГАЗОВЫЙ СТАБИЛЬНЫЙ
Определение фракционного состава методом газовой хроматографии
Petroleum products and stable gas condensate. Determination of boiling range distribution by gas chromatography method
ОКС 75.160.30
Дата введения 2016-07-01
Предисловие
1 РАЗРАБОТАН Открытым акционерным обществом "Газпром" и Обществом с ограниченной ответственностью "Научно-исследовательский институт природных газов и газовых технологий" - Газпром ВНИИГАЗ" (ООО "Газпром ВНИИГАЗ")
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 31 "Нефтяные топлива и смазочные материалы"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 18 ноября 2015 г. N 1848-ст
4 В настоящем стандарте учтены основные нормативные положения следующих стандартов:
ИСО 3924:2010* "Нефтепродукты. Определение распределения компонентов по температурам кипения. Метод газовой хроматографии" (ISO 3924:2010 "Petroleum products - Determination of boiling range distribution - Gas chromatography method", NEQ);
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
ASTM D 2887-15 "Стандартный метод определения распределения фракций нефти по температурам кипения методом газовой хроматографии" (ASTM D 2887-15 "Standard test method for boiling range distribution of petroleum fractions by gas chromatography", NEQ)
5 ВВЕДЕН ВПЕРВЫЕ
6 ПЕРЕИЗДАНИЕ. Сентябрь 2019 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
1 Область применения
1.1 Настоящий стандарт устанавливает два метода (А и Б) определения фракционного состава (распределения углеводородных компонентов по температурам кипения) нефтепродуктов и стабильного газового конденсата (СГК) с использованием газовой хроматографии.
1.2 Метод А предназначен для определения фракционного состава нефтепродуктов с температурой кипения в диапазоне от 55°С до 545°С.
1.3 Метод Б предназначен для определения индивидуальных углеводородов ![]() и фракционного состава углеводородов
и фракционного состава углеводородов ![]() (далее - компонентно-фракционного состава) СГК с учетом нелетучего остатка, выкипающего при температуре выше 545°С.
(далее - компонентно-фракционного состава) СГК с учетом нелетучего остатка, выкипающего при температуре выше 545°С.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ 2477 Нефть и нефтепродукты. Метод определения содержания воды
ГОСТ 2517 Нефть и нефтепродукты. Методы отбора проб
ГОСТ 3022 Водород технический. Технические условия
ГОСТ 9293 (ИСО 2435-73) Азот газообразный и жидкий. Технические условия
ГОСТ 17323 Топливо для двигателей. Метод определения меркаптановой и сероводородной серы потенциометрическим титрованием
ГОСТ 17433 Промышленная чистота. Сжатый воздух. Классы загрязненности
ГОСТ 17567 Хроматография газовая. Термины и определения
ГОСТ 25336 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 29169 (ИСО 648-77) Посуда лабораторная стеклянная. Пипетки с одной отметкой
ГОСТ 29227 (ИСО 835-1-81) Посуда лабораторная стеклянная. Пипетки градуированные. Часть 1. Общие требования
ГОСТ 31873 Нефть и нефтепродукты. Методы ручного отбора проб
ГОСТ ISO 7886-1 Шприцы инъекционные однократного применения стерильные. Часть 1. Шприцы для ручного использования
ГОСТ Р 50779.42 (ИСО 8258-91) Статистические методы. Контрольные карты Шухарта
_____________________
Заменен на ГОСТ Р ИСО 7870-2-2015 "Статистические методы. Контрольные карты. Часть 2. Контрольные карты Шухарта".
ГОСТ Р 50802 Нефть. Метод определения сероводорода, метил- и этилмеркаптанов
ГОСТ Р 52501 (ИСО 3696:1987) Вода для лабораторного анализа. Технические условия
ГОСТ Р 53228 Весы неавтоматического действия. Часть 1. Метрологические и технические требования. Испытания
ГОСТ Р ИСО 5725-6 Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности на практике
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя "Национальные стандарты" за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины по ГОСТ 17567, а также следующие термины с соответствующими определениями.
3.1
стабильный газовый конденсат: Газовый конденсат, получаемый путем очистки нестабильного газового конденсата от примесей и выделения из него углеводородов [ГОСТ Р 53521-2009, статья 9] |
3.2
дистиллят (газового конденсата): Жидкая углеводородная смесь, получаемая в результате конденсации паров при перегонке газового конденсата при атмосферном или пониженном давлении. [ГОСТ Р 53521-2009, статья 34] |
3.3 хроматограмма: Графическое представление сигнала газохроматографического детектора как функции времени.
3.4 пик: Участок хроматограммы, соответствующий выходу из колонки одного из компонентов пробы.
3.5 площадь пика: Площадь, ограниченная контуром пика и его основанием.
3.6 ширина пика: Длина отрезка базовой линии, ограниченного двумя касательными, которые проведены через точку перегиба каждой из сторон хроматографического пика.
3.7 скорректированная площадь хроматограммы пробы: Разность площадей хроматограмм пробы и холостого опыта.
3.8 имитированная дистилляция: Метод определения фракционного состава углеводородных проб с использованием газовой хроматографии; для разделения углеводородов в соответствии с их температурами кипения применяют колонку с неполярной неподвижной фазой.
3.9 температура начала кипения : Температура, соответствующая времени удерживания, при котором скорректированная площадь хроматограммы пробы составляет 0,5% ее суммарной площади (или теоретической общей площади на хроматограмме СГК).
3.10 температура конца кипения : Температура, соответствующая времени удерживания, при котором скорректированная площадь хроматограммы пробы составляет 99,5% ее суммарной площади (или теоретической общей площади на хроматограмме СГК).
3.11 метод внутреннего стандарта: Способ вычисления концентраций определяемых компонентов пробы, основанный на добавлении к ней определенного количества известного соединения (внутреннего стандарта), которое регистрируется на хроматограмме в виде хорошо разрешенного пика.
3.12 доля отгона: Массовая доля пробы, выкипающая к определенному значению температуры, в процентах.
3.13 нелетучий остаток: Часть анализируемой пробы, выкипающая при температуре выше 545°С.
3.14 теоретическая общая площадь хроматограммы Т: Площадь хроматограммы, которая была бы получена при элюировании из колонки всей пробы.
4 Сущность методов
4.1 При проведении анализа по методу А пробу нефтепродукта с температурой кипения до 545°С анализируют на газовом хроматографе с пламенно-ионизационным детектором в режиме программирования температуры термостата колонок. Используют газохроматографическую колонку, обеспечивающую разделение углеводородов в соответствии с их температурами кипения. Для установления зависимости между значениями времен удерживания и температурами кипения углеводородов на этой же колонке хроматографируют смесь углеводородов известного состава, охватывающую диапазон температур кипения компонентов пробы. Полученную зависимость используют для определения фракционного состава пробы с помощью метода внутренней нормализации.
4.2 При проведении анализа по методу Б компонентно-фракционный состав пробы СГК определяют по процедуре, аналогичной приведенной в 4.1, с учетом содержания индивидуальных углеводородов ![]() и нелетучего остатка - тяжелой части пробы, выкипающей при температуре выше 545°С. Долю нелетучего остатка определяют путем проведения дополнительного анализа пробы с добавлением внутреннего стандарта.
и нелетучего остатка - тяжелой части пробы, выкипающей при температуре выше 545°С. Долю нелетучего остатка определяют путем проведения дополнительного анализа пробы с добавлением внутреннего стандарта.
5 Условия выполнения измерений
Измерения проводят при:
- температуре окружающей среды (20±5)°С;
- относительной влажности окружающей среды от 30% до 80%;
- атмосферном давлении от 80,0 до 106,7 кПа (от 630 до 800 мм рт.ст.);
- напряжении переменного тока (220) В;
- частоте переменного тока (50±1) Гц;
- отсутствии механических воздействий, внешних электрических и магнитных полей, влияющих на работу аппаратуры;
- отсутствии агрессивных газов и паров.
6 Отбор проб
Отбор и хранение проб нефтепродуктов и СГК - по ГОСТ 2517 или ГОСТ 31873.
Метод А. Определение фракционного состава нефтепродуктов
7 Средства измерений, вспомогательные устройства, материалы и реактивы
7.1 Средства измерений и вспомогательные устройства
7.1.1 Газовый хроматограф, включающий:
- блок управления;
- пламенно-ионизационный детектор (ПИД) с пределом детектирования не более 2·10 г/с (по углероду), способный работать в непрерывном режиме при максимальной температуре используемой колонки (детектор должен быть подсоединен к колонке таким образом, чтобы между ними не было холодных зон);
- систему ввода пробы в изотермическом режиме при температуре, соответствующей максимальной температуре колонки, или в режиме температурно-программируемого нагрева, или обеспечивающую прямой ввод пробы в колонку;
- термостат, обеспечивающий программируемое регулирование скорости подъема температуры и поддержание заданной температуры с погрешностью не более 0,1°С во всем интервале рабочих температур [воспроизводимость скорости нагрева термостата должна быть достаточной для обеспечения повторяемости времени удерживания для каждого компонента градуировочной смеси (см. 7.2.18) в пределах 6 с];
- систему охлаждения термостата колонок (для анализа проб с начальной температурой кипения ниже 90°С);
- программное обеспечение для сбора, обработки и хранения хроматографической информации.
Примечание - Используемый программный комплекс должен обеспечивать:
- преобразование непрерывно интегрируемого сигнала детектора в площади узких участков хроматограммы шириной не более 0,1 с;
- вычитание площади узкого участка нулевой линии из площади соответствующего участка хроматограммы анализируемой пробы.
7.1.2 Капиллярная хроматографическая колонка с полидиметилсилоксаном в качестве неподвижной фазы (НФ):
- DB-1 или CP-Sil 5 СВ длиной 30 м, внутренним диаметром 0,53 мм (толщина пленки НФ - 1,5 мкм) или
- НР-1 длиной 10 м, внутренним диаметром 0,53 мм (толщина пленки НФ - 2,65 мкм), или
- НР-1 длиной 5 м, внутренним диаметром 0,53 мм (толщина пленки НФ - 0,88 мкм).
7.1.3 Весы лабораторные по ГОСТ Р 53228 высокого класса точности с наибольшим пределом взвешивания 0,2 кг.
7.1.4 Микрошприц фирмы SGE, Hamilton или аналогичного типа вместимостью 1 или 10 мм с ценой деления 0,01 мм
.
7.1.5 Газонепроницаемый шприц фирмы Agilent, Hamilton или аналогичного типа из стекла и политетрафторэтилена вместимостью 250 мм с ценой деления 5 мм
, вместимостью 500 и 1000 мм
с ценой деления 10 мм
.
7.1.6 Шприц медицинский по ГОСТ ISO 7886-1 вместимостью 1 и 2 см.
7.1.7 Пипетки вместимостью 10, 15, 20 см по ГОСТ 29227 и ГОСТ 29169.
7.1.8 Флаконы стеклянные медицинские вместимостью 40 см по стандарту [1] с навинчивающимися крышками с высверленным отверстием диаметром 2 мм, оснащенными прокладками из силиконовой резины.
7.1.9 Посуда лабораторная стеклянная по ГОСТ 25336.
Примечание - Допускается использовать другие средства измерений и вспомогательные устройства, технические характеристики которых не уступают указанным, если их применение не ухудшает метрологические характеристики метода.
7.2 Материалы и реактивы
7.2.1 Гелий газообразный очищенный марки А с содержанием гелия не менее 99,99% по [2].
7.2.2 Водород газообразный высокой чистоты по [3], водород марки А по ГОСТ 3022.
7.2.3 Воздух сжатый класса 0 (свободный от углеводородов) по ГОСТ 17433.
7.2.4 Азот технический по ГОСТ 9293.
7.2.5 Пентан квалификации х.ч. по [4].
7.2.6 Гексан квалификации х.ч. по [5].
7.2.7 н-Гексан квалификации х.ч. по [6].
7.2.8 Гептан квалификации х.ч. по [7].
7.2.9 Октан квалификации х.ч. по [8].
7.2.10 Нонан квалификации х.ч. по [9].
7.2.11 Декан квалификации х.ч. по [10].
7.2.12 Додекан квалификации х.ч. по [11].
7.2.13 Тетрадекан квалификации ч. по [12].
7.2.14 Пентадекан квалификации ч. по [13].
7.2.15 Гексадекан квалификации ч. по [14].
7.2.16 Гептадекан квалификации х.ч. по [15].
7.2.17 Индивидуальные н-алканы для приготовления градуировочных смесей - октадекан ![]() , эйкозан
, эйкозан ![]() , тетракозан
, тетракозан ![]() , октокозан
, октокозан ![]() , дотриаконтан
, дотриаконтан ![]() , гексатриаконтан
, гексатриаконтан ![]() , тетраконтан
, тетраконтан ![]() , тетратетраконтан
, тетратетраконтан ![]() с массовой долей основного вещества не менее 97%.
с массовой долей основного вещества не менее 97%.
7.2.18 Градуировочная смесь - смесь н-алканов от пентана ![]() до тетратетраконтана
до тетратетраконтана ![]() в соответствии со стандартом [16] с температурой кипения до 545°С.
в соответствии со стандартом [16] с температурой кипения до 545°С.
Примечание - В градуировочной смеси должны присутствовать одно или несколько соединений с температурой кипения, равной (или ниже) начальной температуре кипения пробы. При необходимости в герметично закрытый сосуд с градуировочной смесью можно с помощью газового шприца неколичественно добавить небольшие объемы пропана и бутана.
7.2.19 Стандартный образец фракционного состава нефтепродукта в соответствии со стандартом [16].
7.2.20 Вода дистиллированная по ГОСТ Р 52501.
7.2.21 Дисульфид углерода с содержанием основного вещества не менее 99%.
Примечание - Допускается использовать другие материалы и реактивы, характеристики которых не уступают указанным, если их применение не ухудшает метрологические характеристики метода. Можно использовать генераторы водорода и азота для получения чистых газов, соответствующих требованиям 7.2.
8 Подготовка к проведению измерений
8.1 Подключение хроматографа к электрической сети, проверку на герметичность и вывод на режим выполняют в соответствии с руководством по эксплуатации хроматографа.
8.2 Капиллярную колонку устанавливают в термостат хроматографа и, не присоединяя к детектору, кондиционируют в потоке газа-носителя (гелия) с расходом от 5 до 10 см/мин, повышая температуру со скоростью 5°С/мин-10°С/мин до 340°С-350°С. При этой температуре колонку выдерживают в течение 30 мин.
После кондиционирования колонку охлаждают до температуры окружающей среды, подсоединяют к ПИД, проверяют герметичность газовой линии и записывают нулевую линию в рабочем режиме (см. таблицу 1). При стабильной нулевой линии определяют разрешающую способность колонки в соответствии с 8.3.
Таблица 1 - Условия проведения измерений
Параметр | Значение | ||||
Капиллярная колонка | DB-1 | НР-1 | НР-1 | ||
Длина колонки, м | 30 | 5 | 10 | ||
Диаметр колонки (внутренний), мм | 0,53 | ||||
Неподвижная фаза капиллярной колонки | Полидиметилсилоксан | ||||
Толщина пленки неподвижной фазы, мкм | 1,5 | 0,88 | 2,65 | ||
Детектор | ПИД | ||||
Расход газа-носителя (азота/гелия), см | 10 | 12 | 10-20 | ||
Расход добавочного газа через детектор, см | 20 | ||||
Расход водорода, см | 30 | ||||
Расход воздуха, см | 300 | ||||
Начальная температура термостата колонки* (время выдерживания - 1-2 мин), °С | 0-35 | ||||
Скорость нагрева термостата колонки, °С/мин | 15-20 | 10 | 15-20 | ||
Конечная температура термостата колонки (время выдерживания 6 мин), °С | 340-350 | 350 | 350 | ||
Условия ввода пробы в хроматограф | Температура испарителя 320°С - 340°С | Прямой ввод пробы в колонку | Температурно- | ||
Температура детектора, °С | 340-350 | 380 | 350 | ||
Разбавление пробы (стандартного образца) дисульфидом углерода | 1:4 | 1:10 | Без разбавления | ||
Объем вводимой пробы, мм | 0,5-1,0 | 1 | 0,2 | ||
* Как правило, чем ниже температура начала кипения пробы, тем ниже значение начальной температуры термостата колонки. Примечание - Можно использовать другие колонки, в том числе насадочные, элюирование компонентов с которых происходит в порядке возрастания температур кипения компонентов, а значение разрешающей способности колонки R - не менее трех по 8.3. | |||||
8.3 Перед проведением анализа хроматографируют смесь, содержащую от 0,1% масс. до 0,5% масс. гексадекана и октадекана в дисульфиде углерода (см. рисунок 1). Вычисляют разрешающую способность колонки R по формуле
![]() , (1)
, (1)
где и
- время удерживания для пиков гексадекана и октадекана соответственно, с;
- ширина пика гексадекана на половине его высоты, с;
- ширина пика октадекана на половине его высоты, с.
При значении разрешающей способности R не менее трех колонка готова к работе.
8.4 Хроматографируют градуировочную смесь (см. 7.2.18), содержащую известные количества н-алканов при условиях, приведенных в таблице 1. Для пика самой большой высоты определяют коэффициент асимметрии как отношение А/В (см. рисунок 2). В соответствии со стандартом [17] значение коэффициента асимметрии пика должно быть от 0,5 до 2,0. Если значение коэффициента асимметрии выходит за указанный предел, уменьшают объем смеси н-алканов, вводимой в хроматограф, или повышают степень разбавления смеси дисульфидом углерода.
Примечание - Асимметрия пика(ов) часто указывает на перегрузку колонки пробой. Это может привести к искажению значений времен удерживания компонентов и, соответственно, ошибке в определении значений их температуры кипения.
|
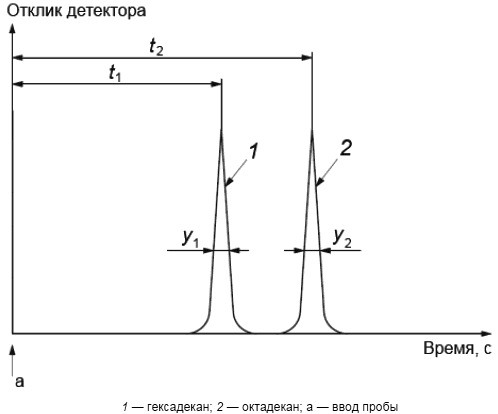
Рисунок 1 - Типовая хроматограмма смеси гексадекана и октадекана для определения разрешающей способности колонки
|
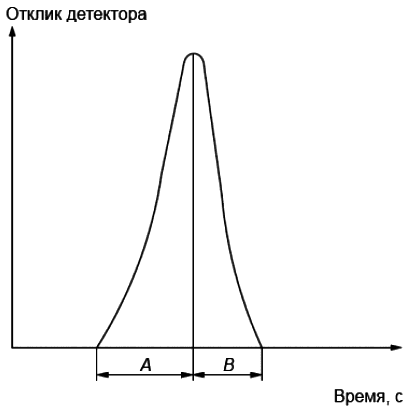
A - ширина передней части пика на уровне 5% его высоты; B - ширина задней части пика на уровне 5% его высоты
Рисунок 2 - Параметры, характеризующие асимметрию хроматографического пика
8.5 Для сведения к минимуму изменения чувствительности детектора (ПИД) следует регулярно удалять образующиеся на нем отложения.
Примечание - Отложения образуются при сгорании неподвижной фазы, частично уносимой потоком газа-носителя из колонки при высокой температуре. Удаление отложений проводят в соответствии с руководством по эксплуатации прибора.
8.6 Регенерация хроматографической колонки
Регенерацию колонки проводят:
- при превышении значения уровня шумов нулевой линии, приведенного в методике поверки или в руководстве по эксплуатации хроматографа;
- при разрешающей способности хроматографической колонки менее трех по 8.3.
Хроматографическую колонку регенерируют по 8.2, не снимая с хроматографа.
9 Проведение измерений
9.1 Процедура проведения измерений
9.1.1 После установки условий хроматографирования в соответствии с таблицей 1 повышают температуру колонки до максимального значения и выдерживают не менее чем 10 мин. Затем охлаждают колонку до температуры начала анализа и выдерживают при этой температуре не менее 3 мин для установления равновесия.
9.1.2 После ввода в хроматограф градуировочной смеси по 7.2.18, анализируемой пробы или чистого дисульфида углерода (холостой опыт) запускают температурную программу термостата колонки (см. таблицу 1). Условия выполнения измерений (см. таблицу 1) и их продолжительность при проведении холостого опыта, градуировке хроматографа и анализе проб должны быть одинаковыми.
9.2 Проведение холостого опыта
9.2.1 После выхода хроматографа на рабочий режим вводят в него от 0,1 до 1,0 мм дисульфида углерода и регистрируют нулевую линию при условиях, приведенных в таблице 1.
Примечания
1 Холостой опыт следует проводить для учета эффекта повышения нулевой линии вблизи максимума температуры колонки. Факторы, влияющие на стабильность нулевой линии, - фон колонки, точность контроля температуры детектора, степень стабильности потоков газа-носителя и газов, питающих детектор, утечки, инструментальный дрейф и т.д.
2 При анализе неразбавленных проб холостой опыт проводят без введения в хроматограф дисульфида углерода.
9.2.2 Перед проведением вычислений из хроматограммы пробы или градуировочной смеси вычитают хроматограмму холостого опыта.
Примечание - Холостой опыт позволяет получить дополнительную информацию о работе оборудования и проконтролировать остаточное содержание в системе компонентов предыдущей пробы. Холостой опыт выполняют перед анализом градуировочной смеси и проб, но также рекомендуется проводить его в конце серии анализов.
9.3 Градуировка хроматографа
9.3.1 Для построения градуировочного графика используют зависимость времени удерживания углеводородов от их температуры кипения, которую получают в день проведения анализа. Для этого используют смесь н-алканов от до
по 7.2.18 или смесь, приготовленную по 9.3.3.
9.3.2 При использовании смеси н-алканов по 7.2.18 содержимое ампулы разбавляют дисульфидом углерода (см. примечание), тщательно перемешивают и вводят аликвоту полученного раствора в испаритель хроматографа при условиях, приведенных в таблице 1. Объем раствора вводимой в хроматограф смеси н-алканов (от 0,2 до 1,0 мм) подбирают таким образом, чтобы получаемые пики имели правильную форму (см. 8.4).
Примечание - Рекомендуется на насадочных колонках анализировать растворы с объемным отношением смеси углеводородов к дисульфиду углерода 1:10, а на капиллярных - 1:100.
9.3.3 Смесь индивидуальных н-алканов от до
по 7.2.5-7.2.17 в дисульфиде углерода с массовой долей каждого н-алкана от 0,05% до 0,50% готовят следующим образом. В стеклянный сосуд вместимостью от 15 до 40 см
с завинчивающейся крышкой с отверстием, оснащенной прокладкой из силиконовой резины, поочередно вносят от 6,5 до 200 мг твердых н-алканов (
,
,
,
,
,
,
,
). Сосуд взвешивают до и после внесения каждого компонента, результаты взвешиваний в граммах записывают с точностью до четвертого десятичного знака. Затем в сосуд пипеткой добавляют дисульфид углерода (от 10 до 30 см
); сосуд герметично закрывают и взвешивают. Далее в сосуд вводят микрошприцем через прокладку в крышке от 6,5 до 200 мг жидких н-алканов. По разности результатов двух последовательных взвешиваний вычисляют массу каждого н-алкана и рассчитывают его массовую долю в смеси
, %, по формуле
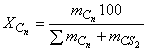 , (2)
, (2)
где - масса навески н-алкана с числом углеродных атомов n;
- масса дисульфида углерода.
9.3.4 По хроматограмме смеси н-алканов определяют значения их времен удерживания, соответствующие времени регистрации максимума пиков индивидуальных н-алканов. По результатам анализа составляют таблицу, в которую вносят время удерживания и температуру кипения для каждого компонента смеси. Температуры кипения н-алканов приведены в таблице 2.
9.3.5 Строят график зависимости времени удерживания углеводородов от их температуры кипения. Типовой градуировочный график приведен на рисунке 3. При проведении анализов полученный градуировочный график используют для перевода шкалы времени удерживания в шкалу температуры кипения.
Таблица 2 - Температуры кипения н-алканов
н-Алкан | Температура кипения, °С |
| -89 |
-42 | |
| 0 |
| 36 |
| 69 |
| 98 |
| 126 |
| 151 |
| 174 |
| 196 |
| 216 |
| 235 |
| 254 |
| 271 |
| 287 |
| 302 |
| 316 |
| 330 |
| 344 |
| 356 |
| 369 |
| 380 |
| 391 |
| 402 |
| 412 |
| 422 |
| 430 |
| 440 |
| 449 |
| 458 |
| 466 |
| 474 |
| 481 |
| 489 |
| 496 |
| 503 |
| 509 |
| 516 |
| 522 |
| 528 |
| 534 |
| 540 |
| 545 |
- | - |
9.3.6 Область градуировки должна охватывать нижнюю и верхнюю границы температуры кипения пробы. Градуировочный график имеет линейный характер, однако на практике возможно отклонение графика от линейности.
Примечания
1 Наибольшее отклонение от линейности наблюдают для низкокипящих н-алканов, для которых характерны небольшие значения времен удерживания и значительные различия в температурах кипения.
2 Если содержание н-алканов в анализируемой пробе достаточно для идентификации их пиков на хроматограмме, такую пробу можно использовать для построения графика зависимости температуры кипения от времени удерживания н-алканов (см. рисунок 3).
|
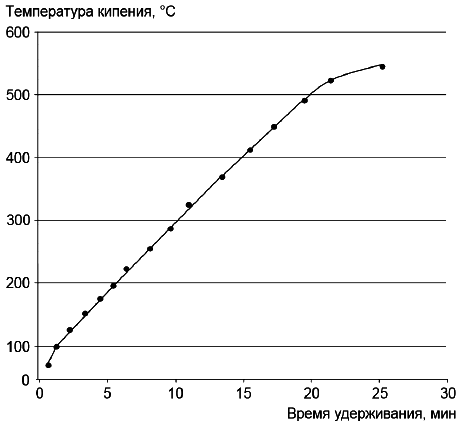
Рисунок 3 - Типовой график зависимости времени удерживания от температуры кипения н-алканов
9.4 Анализ стандартного образца
9.4.1 Правильность проведения имитированной дистилляции проверяют путем анализа стандартного образца (см. 7.2.19), разбавленного дисульфидом углерода (9.3.2) в соответствии с таблицей 1. После тщательного перемешивания раствор стандартного образца анализируют при условиях, приведенных в таблице 1. Отклонение полученных значений температуры, соответствующих фиксированным значениям доли отгона стандартного образца, от паспортных данных (таблица 3) не должно превышать значений воспроизводимости, приведенных в таблице 5. При несоблюдении приведенных требований готовят новый раствор стандартного образца.
Примечание - Раствор стандартного образца в дисульфиде углерода хранят в холодильнике в стеклянном сосуде с притертой пробкой или с навинчивающейся крышкой, оснащенной прокладкой из силиконовой резины.
9.4.2 Правильность проведения имитированной дистилляции с использованием стандартного образца проверяют не реже одного раза в неделю.
9.5 Анализ пробы
9.5.1 Количество пробы, вводимой в хроматограф (см. таблицу 1), не должно приводить к перегрузке колонки.
Примечание - Пробы с узким диапазоном температур кипения требуют введения в хроматограф меньшего объема пробы по сравнению с пробами с широким диапазоном температур кипения.
9.5.2 Пробы с достаточно низкой вязкостью при комнатной температуре можно вводить в хроматограф без разбавления дисульфидом углерода. Пробы вязких или парафинистых нефтепродуктов следует разбавлять дисульфидом углерода; степень разбавления и объем вводимого в хроматограф раствора пробы - в соответствии с таблицей 1.
9.5.3 Пробу нефтепродукта (или раствор пробы в дисульфиде углерода по 9.5.2) вводят в хроматограф и анализируют при условиях, приведенных в таблице 1.
Таблица 3 - Доля отгона в зависимости от температуры для типовых стандартных образцов по стандарту [17]
Доля отгона, % | Температура, °С | |
Партия N 1 | Партия N 2 | |
Н. к. | 114 | 115 |
5 | 143 | 151 |
10 | 169 | 176 |
15 | 196 | 201 |
20 | 221 | 224 |
30 | 258 | 259 |
40 | 287 | 289 |
50 | 312 | 312 |
60 | 332 | 332 |
70 | 354 | 354 |
80 | 376 | 378 |
90 | 404 | 407 |
95 | 425 | 428 |
К. к. | 475 | 475 |
Примечание - Н. к. - температура начала кипения; К. к. - температура конца кипения. | ||
10 Обработка результатов
10.1 На полученной хроматограмме нефтепродукта проводят идентификацию пиков н-алканов сравнением значений их времени удерживания со значениями времени удерживания, полученными при анализе смеси н-алканов от до
(см. 9.3).
10.2 По разности площадей хроматограмм анализируемой пробы и холостого опыта вычисляют скорректированную площадь хроматограммы пробы (далее - суммарная скорректированная площадь). Для этого необходимо, чтобы времена регистрации вышеуказанных хроматограмм были одинаковыми.
10.3 Определяют значение времени, при котором площадь хроматограммы составляет 99,5% ее суммарной площади. Температура, соответствующая этому значению времени, является температурой конца кипения пробы .
Примечание - Определение точки конца кипения пробы является достаточно сложной проблемой. Хроматограммы некоторых проб имеют весьма длинный "хвост", обусловленный наличием в пробе малых содержаний высококипящих углеводородов. В сочетании с тенденцией повышения нулевой линии вблизи максимума температуры колонки, а также элюирования из колонки следов компонентов от предыдущих проб это приводит к невозможности снижения сигнала детектора до исходного уровня базовой линии. Поэтому ближе к концу анализа рекомендуется вычислять площадь каждого участка хроматограммы для установления точки, в которой скорость изменения хроматографического сигнала в секунду достигает постоянного значения, не превышающего 0,00001% общей площади хроматограммы. Это точку принимают за конечную точку хроматограммы.
10.4 Затем определяют значение времени (в начале хроматограммы), при котором площадь хроматограммы равна 0,5% ее суммарной площади. Температура, соответствующая данному времени, является температурой начала кипения пробы . При анализе пробы, растворенной в дисульфиде углерода, сигнал растворителя исключают из расчетов.
10.5 Обработку результатов имитированной дистилляции проводят в соответствии с рекомендациями разработчиков используемого программного обеспечения. Методом нормализации вычисляют значение доли отгона пробы, соответствующее каждой точке временной шкалы хроматограммы. Для этого умножают скорректированную площадь хроматограммы пробы, соответствующую каждой точке временной шкалы, на 100 и делят на суммарную скорректированную площадь.
10.6 Затем, используя линейную интерполяцию, в диапазоне между 1% отгона пробы и долей отгона, достигаемой к температуре кипения ![]() (545°С), определяют значения времени удерживания, соответствующие увеличению доли отгона пробы на 1%.
(545°С), определяют значения времени удерживания, соответствующие увеличению доли отгона пробы на 1%.
10.7 По графику зависимости времени удерживания углеводородов от их температуры кипения (см. рисунок 3) полученные данные пересчитывают в зависимость доли отгона пробы от температуры и представляют в виде таблицы, аналогичной таблице 3. Таблица должна содержать значения температуры (с точностью до 0,5°С), соответствующие доле отгона пробы от 1% до 99% с шагом 1%, а также значения температур начала и конца кипения пробы.
11 Прецизионность метода
11.1 Повторяемость
Расхождение результатов двух измерений температуры Т (°С) для фиксированного значения доли отгона пробы, полученных одним и тем же оператором на одном и том же приборе при постоянных рабочих условиях в течение длительного времени, может превышать значения, приведенные в таблице 4, только в одном случае из двадцати.
Таблица 4 - Значения повторяемости
Доля отгона, % | Показатель | Предел повторяемости r, °С, |
0,5 (начало кипения) | 0,004 | 0,011 |
5-40 | 0,29 | 0,8 |
50-90 | 0,36 | 1,0 |
95 | 0,43 | 1,2 |
99,5 (конец кипения) | 1,2 | 3,3 |
Примечание - | ||
11.2 Воспроизводимость
Расхождение результатов двух единичных независимых измерений температуры Т (°С) для фиксированной доли отгона одной и той же пробы, полученных разными операторами в разных лабораториях в течение длительного времени, может превышать значения, приведенные в таблице 5, только в одном случае из двадцати.
Таблица 5 - Значения воспроизводимости
Доля отгона, % | Показатель | Предел воспроизводимости R, °С, |
0,5 (начало кипения) | 0,024 | 0,066 |
5-30 | 0,0054 ( | 0,015 ( |
40-90 | 1,5 | 4,3 |
95 | 1,8 | 5 |
99,5 (конец кипения) | 4,2 | 12 |
Примечание - | ||
Метод Б. Определение компонентно-фракционного состава газового конденсата
12 Средства измерений, вспомогательные устройства, материалы и реактивы
Средства измерений, вспомогательные устройства, материалы, реактивы - в соответствии с разделом 7, а также внутренний стандарт - смесь четырех углеводородов с числом атомов углерода от до
, взятых приблизительно в равных количествах*.
________________
* Например, смесь углеводородов "Internal Standard Mixture 20" фирмы Agilent, США, состоящая из н-алканов (% масс.): 33,3% тетрадекана, 13,3% пентадекана, 26,7% гексадекана, 26,7% гептадекана.
13 Проведение измерений
13.1 Подготовку к проведению измерений проводят по 9.1.
13.2 Холостой опыт проводят по 9.2.
13.3 Градуировку хроматографа проводят по 9.3.
13.4 Подготовка пробы
13.4.1 Для обеспечения однородности пробу СГК перемешивают, при необходимости нагревают до комнатной температуры и отбирают в отдельный сосуд примерно 100 см пробы.
13.4.2 Подготовка пробы с внутренним стандартом
Взвешивают стеклянный сосуд вместимостью приблизительно 40 см с крышкой с отверстием, оснащенной прокладкой из силиконовой резины. Пипеткой вносят в сосуд примерно 10-15 см
дисульфида углерода, быстро его закрывают и снова взвешивают. Затем в сосуд вносят от 10 до 15 см
пробы, закрывают, взвешивают и добавляют внутренний стандарт. Для этого через прокладку из силиконовой резины медицинским шприцем в сосуд вводят последовательно 0,3-0,4 см
каждого из четырех н-алканов от
до
. После внесения последнего н-алкана сосуд взвешивают. При наличии готового внутреннего стандарта его вводят медицинским шприцем в сосуд с пробой в количестве от 1,0 до 1,5 см
, затем сосуд взвешивают. Результаты взвешиваний в граммах записывают с точностью до четвертого десятичного знака. Массы пробы СГК и внутреннего стандарта определяют по разности масс сосуда до и после их внесения. Содержимое сосуда энергично перемешивают в течение 2-3 мин до полного растворения пробы.
13.4.3 Подготовка пробы без внутреннего стандарта
Во втором сосуде такое же количество пробы анализируемого СГК разбавляют таким же объемом дисульфида углерода без добавления внутреннего стандарта и тщательно перемешивают до растворения пробы.
13.5 Анализ пробы
13.5.1 Микрошприцем вводят в испаритель хроматографа раствор СГК с внутренним стандартом (13.4.2) и хроматографируют при тех же условиях, при которых проводят холостой опыт и анализ градуировочной смеси н-алканов (см. таблицу 1). Регистрируют хроматограмму до конца анализа [до времени удерживания, соответствующего температуре кипения ![]() (545°С)].
(545°С)].
13.5.2 Затем микрошприцем вводят в испаритель хроматографа раствор СГК без внутреннего стандарта (13.4.3) и проводят анализ по 13.5.1.
13.5.3 Допускается проводить анализ пробы СГК с внутренним стандартом и без него в любой последовательности.
Типовые хроматограммы растворов СГК с внутренним стандартом и без него приведены на рисунке 4.
14 Обработка результатов
14.1 Корректировка площади
14.1.1 Хроматограммы проб СГК обрабатывают аналогично хроматограммам нефтепродуктов по разделу 10.
14.1.2 Для каждой полученной хроматограммы пробы СГК (см. рисунок 4) вычисляют скорректированные площади ее узких участков вычитанием площадей соответствующих узких участков хроматограммы холостого опыта (см. 13.2).
14.1.3 Суммированием скорректированных площадей узких участков хроматограммы определяют суммарную скорректированную площадь каждой хроматограммы до времени удерживания, соответствующего температуре кипения ![]() (545°С) (см. площади А и В на рисунке 4).
(545°С) (см. площади А и В на рисунке 4).
|
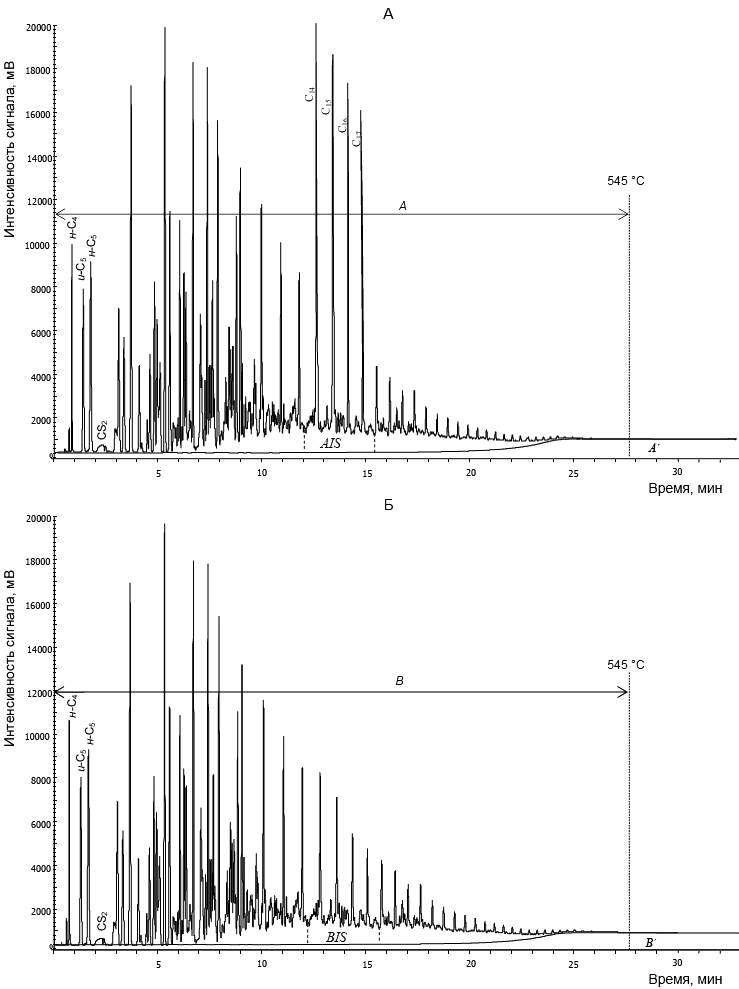
А - проба СГК с внутренним стандартом; Б - проба СГК без внутреннего стандарта
Рисунок 4 - Типовые хроматограммы стабильного газового конденсата (СГК)
14.2 Теоретическая общая площадь
14.2.1 Используя значения времени удерживания, полученные для смеси н-алканов от до
, выбирают время удерживания, на 5% меньшее, чем время удерживания компонента внутреннего стандарта, регистрируемого первым (
![]() ), и время удерживания, на 5% большее, чем для компонента внутреннего стандарта, регистрируемого последним (
), и время удерживания, на 5% большее, чем для компонента внутреннего стандарта, регистрируемого последним (![]() ).
).
Выбранные значения времени удерживания соответствуют участку хроматограммы, на котором регистрируются пики внутреннего стандарта. Далее определяют общую площадь этого участка на хроматограмме анализируемой пробы с внутренним стандартом (площадь AIS, см. рисунок 4, хроматограмма А). Также определяют общую площадь соответствующего участка на хроматограмме пробы СГК без внутреннего стандарта (площадь BIS, см. рисунок 4, хроматограмма Б).
14.2.2 Массовую долю внутреннего стандарта в его смеси с СГК, , вычисляют по формуле
![]() , (3)
, (3)
где - масса внутреннего стандарта, г;
- масса СГК, г.
14.2.3 Отношение суммы площадей пиков всех углеводородов на обеих хроматограммах до времени удерживания, соответствующего температуре кипения ![]() (545°С), полученных при анализе пробы СГК с добавлением и без добавления внутреннего стандарта, за вычетом площадей участков с пиками внутреннего стандарта z вычисляют по формуле
(545°С), полученных при анализе пробы СГК с добавлением и без добавления внутреннего стандарта, за вычетом площадей участков с пиками внутреннего стандарта z вычисляют по формуле
![]() , (4)
, (4)
где В - сумма площадей всех пиков на хроматограмме Б до температуры кипения ![]() (545°С), единицы счета;
(545°С), единицы счета;
BIS - площадь участка на хроматограмме Б, соответствующего положению пиков внутреннего стандарта, единицы счета;
А - сумма площадей всех пиков на хроматограмме А (см. рисунок 4) до температуры кипения ![]() (545°С), единицы счета;
(545°С), единицы счета;
AIS - площадь участка на хроматограмме А с пиками внутреннего стандарта, единицы счета.
14.2.4 Теоретическую общую площадь хроматограммы пробы СГК с учетом нерегистрируемых компонентов нелетучего остатка (площадь В+В'; см. рисунок 4, хроматограмма Б) вычисляют по формуле
![]() , (5)
, (5)
где AIS - площадь участка хроматограммы А с пиками внутреннего стандарта, единицы счета;
z - отношение суммы площадей пиков углеводородов на хроматограммах, полученных при анализе СГК с добавлением и без добавления внутреннего стандарта, вычисленное по формуле (4);
BIS - площадь участка хроматограммы Б, соответствующего положению пиков внутреннего стандарта, единицы счета;
- массовая доля внутреннего стандарта в смеси с СГК по формуле (3).
14.2.5 Массовую долю нелетучего остатка RES, выкипающего при температурах выше 545°С, вычисляют по формуле
![]() , (6)
, (6)
где В - сумма площадей пиков всех углеводородов до ![]() на хроматограмме Б (рисунок 4), единицы счета;
на хроматограмме Б (рисунок 4), единицы счета;
- теоретическая общая площадь хроматограммы СГК, единицы счета, по формуле (5).
14.3 Вычисление распределения углеводородов пробы по температурам кипения
14.3.1 На хроматограмме пробы СГК (рисунок 4, хроматограмма Б) определяют точку на оси абсцисс, соответствующую 0,5% теоретической общей площади по формуле (5). Температура, соответствующая данному времени, является температурой начала кипения пробы
.
14.3.2 Затем вычисляют зависимость доли отгона пробы от времени. Для этого умножают значение скорректированной площади хроматограммы пробы, соответствующее каждой точке временной шкалы, на 100 и делят на значение теоретической общей площади .
14.3.3 Полученные данные представляют в виде таблицы, отражающей зависимость доли отгона от времен удерживания.
14.3.4 Затем, используя линейную интерполяцию, в диапазоне между 1% отгона пробы и долей отгона, достигаемой к температуре кипения ![]() (545°С), определяют значения времени удерживания, соответствующие увеличению доли отгона пробы на 1%.
(545°С), определяют значения времени удерживания, соответствующие увеличению доли отгона пробы на 1%.
14.3.5 По графику зависимости времени удерживания углеводородов от их температуры кипения (см. рисунок 3) полученные данные пересчитывают в зависимость доли отгона пробы от температуры и представляют в виде таблицы. Таблица должна содержать значения температуры (с точностью до 0,5°С), соответствующие доле отгона пробы от 1% до 99% с шагом 1%, значения температур начала и конца кипения пробы, а также значение массовой доли нелетучего остатка, выкипающего при температуре выше 545°С.
14.4 Вычисление компонентно-фракционного состава стабильного газового конденсата
14.4.1 Суммарное содержание всех углеводородов, зарегистрированных на хроматограмме, принимают равным 100%. Для вычисления содержания индивидуальных легких углеводородов ![]() используют значения массовых коэффициентов чувствительности, приведенные в таблице 6. При этом значения массовых коэффициентов чувствительности углеводородов от гексанов и выше принимают равными 1,00.
используют значения массовых коэффициентов чувствительности, приведенные в таблице 6. При этом значения массовых коэффициентов чувствительности углеводородов от гексанов и выше принимают равными 1,00.
Таблица 6 - Массовые коэффициенты чувствительности К (газ-носитель - гелий) по стандарту [18])
Компонент | К |
Метан | 1,114 |
Этан | 1,047 |
Пропан | 1,023 |
Изобутан, н-бутан | 1,012 |
Изопентан, н-пентаны | 1,005 |
н-Гексан | 1,000 |
14.4.2 Массовую долю индивидуальных углеводородов ![]() и фракций углеводородов до
и фракций углеводородов до в пробе СГК
(%) вычисляют методом нормализации площадей пиков всех углеводородов на хроматограмме пробы СГК по формуле
![]() , (7)
, (7)
где - площадь пика i-го индивидуального углеводорода
![]() или фракций углеводородов до температуры кипения
или фракций углеводородов до температуры кипения ![]() (545°С), единицы счета;
(545°С), единицы счета;
- относительные массовые коэффициенты чувствительности (см. таблицу 6).
14.4.3 Полученные значения массовых долей индивидуальных углеводородов ![]() и фракций углеводородов до
и фракций углеводородов до корректируют для учета массовой доли нелетучего остатка по формуле
![]() , (8)
, (8)
где - массовая доля индивидуальных углеводородов
![]() и фракций углеводородов до
и фракций углеводородов до в пробе СГК с учетом содержания нелетучего остатка, %;
- массовая доля индивидуальных углеводородов
![]() и фракций углеводородов до
и фракций углеводородов до в пробе СГК, вычисленная по формуле (7), без учета содержания нелетучего остатка, %;
RES - массовая доля нелетучего остатка, выкипающего при температуре выше 545°С, вычисленная по формуле (6), %.
Примечание - Если проба СКГ содержит серосодержащие соединения и воду, то вычисленные по формуле (8) результаты хроматографического анализа корректируют для учета содержания компонентов, не регистрируемых с помощью ПИД, по формуле
![]() , (9)
, (9)
где - массовая доля индивидуальных углеводородов
![]() и фракций углеводородов до
и фракций углеводородов до в пробе СГК с учетом содержания нелетучего остатка, серосодержащих соединений и воды, %;
- массовая доля сероводорода по ГОСТ Р 50802 или ГОСТ 17323, %;
![]() - массовая доля меркаптановой серы по ГОСТ 17323, %;
- массовая доля меркаптановой серы по ГОСТ 17323, %;
![]() - массовая доля воды по ГОСТ 2477, %.
- массовая доля воды по ГОСТ 2477, %.
15 Метрологические характеристики
15.1 За результат определения температуры кипения Т, °С, для фиксированной доли отгона пробы принимают среднеарифметическое значение результатов двух параллельных определений, если выполняется условие приемлемости
![]() , (10)
, (10)
где - результаты параллельных определений температуры кипения для фиксированной доли отгона пробы, °С;
- значение предела повторяемости для фиксированной доли отгона пробы по таблице 7, °С.
Таблица 7 - Метрологические характеристики результатов определения температур кипения, соответствующих фиксированным долям отгона пробы
Доля отгона, % | Показатель точности (границы абсолютной погрешности) | Показатель повторяемости (среднеквадратическое отклонение повторяемости) | Показатель воспроизводимости (среднеквадратическое отклонение воспроизводимости) | Предел повторяемости r, °С, |
0,5 (начало кипения) | 7 | 1,8 | 3,6 | 5,0 |
5 | 8 | 2,0 | 4,0 | 5,5 |
10 | 10 | 2,4 | 4,8 | 7,0 |
20 | 11 | 2,7 | 5,4 | 7,5 |
30 | 12 | 3,0 | 6,0 | 8,0 |
40 | 13 | 3,3 | 6,6 | 9,0 |
50 | 15 | 3,8 | 7,6 | 11 |
60 | 17 | 4,2 | 8,4 | 12 |
70 | 20 | 5,0 | 10 | 14 |
80 | 22 | 5,5 | 11 | 15 |
90 | 24 | 6,0 | 12 | 17 |
95 | 26 | 6,5 | 13 | 18 |
99,5 (конец кипения) | 28 | 7,0 | 14 | 19 |
15.2 Если условие (10) не выполняется, то выясняют причины превышения предела повторяемости, устраняют их и повторяют измерение по разделу 13.
15.3 Результат анализа представляют в виде:
начало кипения |
| |
от 5% до 95% отгона (с шагом 5%) получено при температуре |
| (11) |
конец кипения |
|
где - среднеарифметическое значение результатов параллельных определений температуры кипения (признанных приемлемыми по 15.1), соответствующее фиксированной доле отгона пробы, °С;
- границы абсолютной погрешности по таблице 7, °С.
15.4 За результат определения массовой доли индивидуальных углеводородов ![]() и фракций углеводородов
и фракций углеводородов ![]() в СГК принимают среднеарифметическое значение результатов двух параллельных определений, если выполняется условие
в СГК принимают среднеарифметическое значение результатов двух параллельных определений, если выполняется условие
![]() , (12)
, (12)
где ![]() - результаты двух параллельных определений массовой доли i-го компонента (индивидуальных углеводородов
- результаты двух параллельных определений массовой доли i-го компонента (индивидуальных углеводородов ![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка) в СГК;
, нелетучего остатка) в СГК;
- предел повторяемости для соответствующего значения массовой доли i-го компонента (индивидуальных углеводородов
![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка) по таблицам 8, 9, %.
, нелетучего остатка) по таблицам 8, 9, %.
Таблица 8 - Метрологические характеристики результатов определения массовой доли индивидуальных углеводородов, фракций углеводородов в СГК
Диапазон измерений массовой доли углеводородов (температурных фракций) в нефтепродуктах и СГК, % | Показатель точности (границы относительной погрешности) | Показатель повторяемости (относительное среднеквадрати- | Показатель воспроизводимости (относительное среднеквадрати- | Предел повторяемости r, % отн., Р=0,95, n=2 |
От 0,005 до 0,10 включ. | 51-126 X | 10-31 Х | 20-63 X | 28-87 X |
Св. 0,10 до 1,0 включ. | 38 | 7 | 14 | 19 |
Св. 1,0 до 2,0 включ. | 32 | 6 | 12 | 17 |
Св. 2,0 до 5,0 включ. | 22 | 4 | 8 | 11 |
Св. 5,0 до 25 включ. | 17 | 3 | 6 | 8 |
Таблица 9 - Метрологические характеристики результатов определения массовой доли нелетучего остатка
Диапазон измерений массовой доли нелетучего остатка, % | Показатель точности (границы относительной погрешности) | Показатель повторяемости (относительное среднеквадрати- | Показатель воспроизводимости (относительное среднеквадрати- | Предел повторяемости r, % отн., Р=0,95, n=2 |
От 0,50 до 10 включ. | 17 | 3 | 6 | 8 |
Метрологические характеристики результатов измерений по настоящему стандарту не должны превышать значений, приведенных в таблицах 8 и 9, для соответствующих диапазонов измерений.
15.5 Если условие по формуле (12) не выполняется, то проводят еще одно измерение по разделу 13. За результат анализа принимают среднеарифметическое значение результатов трех измерений, если выполняется условие
![]() , (13)
, (13)
где ![]() - максимальное и минимальное значения результатов трех параллельных определений массовой доли i-го компонента (индивидуальных углеводородов
- максимальное и минимальное значения результатов трех параллельных определений массовой доли i-го компонента (индивидуальных углеводородов ![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка), %;
, нелетучего остатка), %;
![]() - значение критического диапазона для уровня вероятности Р=0,95 и n результатов определений, вычисляемое по формуле
- значение критического диапазона для уровня вероятности Р=0,95 и n результатов определений, вычисляемое по формуле
![]() , (14)
, (14)
где f(n) - коэффициент критического диапазона, f(n)=3,3 для n=3 по ГОСТ Р ИСО 5725-6;
- показатель повторяемости определения для соответствующего значения массовой доли i-го компонента (индивидуальных углеводородов
![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка) по таблицам 8, 9, %.
, нелетучего остатка) по таблицам 8, 9, %.
15.6 Если условие по формуле (13) не выполняется, то выясняют причины превышения критического диапазона, устраняют их и повторяют измерение по разделу 13.
15.7 Результат определения массовой доли i-го компонента (индивидуальных углеводородов ![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка), %, при Р=0,95 представляют в виде
, нелетучего остатка), %, при Р=0,95 представляют в виде ![]() , где
, где - среднеарифметическое значение результатов n определений, признанных приемлемыми в соответствии с 15.4 или 15.5, %; ±
- границы относительной погрешности, %, соответствующие значению X для i-го компонента, - см. таблицы 8, 9.
При массовой доле i-го компонента (индивидуальных углеводородов ![]() , фракции углеводородов
, фракции углеводородов ![]() , нелетучего остатка) ниже нижней или выше верхней границы концентраций, охватываемых настоящим методом, полученный результат представляют в виде: "массовая доля i-го компонента (индивидуальных углеводородов
, нелетучего остатка) ниже нижней или выше верхней границы концентраций, охватываемых настоящим методом, полученный результат представляют в виде: "массовая доля i-го компонента (индивидуальных углеводородов ![]() , фракции углеводородов, нелетучего остатка) менее (более) _____%" (приводят нижнюю или верхнюю границу определяемых концентраций по таблицам 8, 9).
, фракции углеводородов, нелетучего остатка) менее (более) _____%" (приводят нижнюю или верхнюю границу определяемых концентраций по таблицам 8, 9).
16 Контроль качества результатов измерений
16.1 Качество результатов измерений проверяют по ГОСТ Р ИСО 5725-6 с помощью контроля стабильности среднеквадратического (стандартного) отклонения промежуточной прецизионности и показателя правильности. Стабильность результатов измерений проверяют с помощью контрольных карт Шухарта по ГОСТ Р 50779.42 или в соответствии с рекомендациями [19].
16.2 Периодичность контроля стабильности результатов измерений устанавливают в Руководстве по качеству лаборатории. Контролируемый период рекомендуется устанавливать таким образом, чтобы количество результатов контрольных измерений находилось в интервале 20-30.
16.3 При неудовлетворительных результатах контроля, например при превышении предела действия или регулярном превышении предела предупреждения, выясняют причины этих отклонений, в том числе проверяют качество работы оператора.
Библиография
[1] | OCT 64-2-71-80 | Банки и флаконы из стекломассы с винтовой горловиной. Типы и размеры |
[2] | ТУ 0271-135-31323949-2005 | Гелий газообразный (сжатый). Технические условия |
[3] | ТУ 301-07-27-91 | Водород газообразный высокой чистоты. Технические условия |
[4] | ТУ 6-09-922-76 | Пентан для хроматографии. Технические условия |
[5] | ТУ 6-09-4521-77 | Гексан для хроматографии. Технические условия |
[6] | ТУ 2631-003-05807999-98 | н-Гексан. Технические условия |
[7] | ТУ 6-09-4520-77 | Гептан для хроматографии. Технические условия |
[8] | ТУ 6-09-661-76 | Октан для хроматографии. Технические условия |
[9] | ТУ-6-09-660-76 | Нонан для хроматографии. Технические условия |
[10] | ТУ 6-09-659-77 | Декан для хроматографии. Технические условия |
[11] | ТУ-6-09-4518-77 | Додекан для хроматографии. Технические условия |
[12] | ТУ 6-09-3705-74 | Тетрадекан. Технические условия |
[13] | ТУ 6-09-3689-74 | Пентадекан. Технические условия |
[14] | ТУ-6-09-3659-74 | Гексадекан. Технические условия |
[15] | ТУ 6-09-3660-74 | Гептадекан. Технические условия |
[16] | ASTM D 2887-15 | Стандартный метод определения распределения фракций нефти по температурам кипения методом газовой хроматографии |
ASTM D 2887-15 | (Standard test method for boiling range distribution of petroleum fractions by gas chromatography) | |
[17] | ИСО 3924:2010 | Нефтепродукты. Определение распределения компонентов по температурам кипения. Метод газовой хроматографии |
ISO 3924:2010 | (Petroleum products - Determination of boiling range distribution - Gas chromatography method) | |
[18] | IP 344:1988 | Определение легких углеводородов в стабилизированных сырых нефтях. Метод газовой хроматографии |
IP 344:1988 | (Determination of light hydrocarbons in stabilized crude oils - Gas chromatography method) | |
[19] | Рекомендации по межгосударственной стандартизации РМГ 76-2004 | Государственная система обеспечения единства измерений. Внутренний контроль качества результатов количественного химического анализа |
УДК 661.715:543.544.3:006.354 |
| ОКС 75.160.30 |
Ключевые слова: нефтепродукты, стабильный газовый конденсат, определение фракционного состава, метод газовой хроматографии | ||
Электронный текст документа
и сверен по:
, 2019

{kind=link}