ГОСТ 7047-55
Группа Р19
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
ВИТАМИНЫ А, С, Д, В, В
и Р
Отбор проб, методы определения витаминов и испытания качества витаминных препаратов*
_______________
* В части витаминных препаратов отменен.
В части разд.III для консервированных плодоовощных консервов действует ГОСТ 24556-81 с 01.01.1982 г.
В части разд.II, в части определения витамина А в печени рыб, морских млекопитающих и морских беспозвоночных действует ГОСТ 7636-85.
Дата введения 1956-02-01
УТВЕРЖДЕН Комитетом стандартов, мер и измерительных приборов при Совете Министров Союза ССР 28 ноября 1955 г. Срок введения установлен 01.02.56
Проверен в 1991 г. Постановлением Госстандарта СССР от 29.12.91 N 2330 снято ограничение срока действия
ВЗАМЕН ГОСТ 7047-54
ПЕРЕИЗДАНИЕ. Август 1994 г.
I. ОТБОР ПРОБ
1. Витаминное драже, таблетки и порошки. При расфасовке в пробирки, банки и флаконы отбирают 10% всех тарных мест, но не менее трех мест, и из каждого отобранного тарного места берут такое количество пробирок, банок, флаконов и коробок, чтобы общая масса пробы составляла не менее 600 г.
Отобранные единицы расфасовки с порошком, таблетками и драже вскрывают после оценки внешнего вида и проверки укупорки и выделяют среднюю пробу следующим образом: содержимое коробок, банок, флаконов и пробирок высыпают в отдельную чистую, сухую банку, тщательно перемешивают и выделяют среднюю пробу в количестве 400 г.
При расфасовке в барабаны, бочонки, картонную или бумажную литую тару отбирают 10% всех упаковочных мест, но не менее трех мест.
Затем из разных мест барабана, бочонка, картонной и бумажной литой тары отбирают по 100-200 г продукта в отдельную чистую, сухую банку и после тщательного перемешивания выделяют среднюю пробу в количестве 400 г.
Примечания:
1. При наличии в одной партии пробирок, банок, барабанов и т.д. выделенные для каждого вида упаковки пробы соединяют и затем после тщательного перемешивания выделяют среднюю пробу в количестве 400 г.
2. Пробу поливитаминного драже отбирают в количестве 600 г.
2. Кристаллические витаминные препараты
а) При расфасовке в пробирки, банки, коробки и флаконы отбирают 10% всех тарных мест, но не менее трех, и из каждого отобранного тарного места в зависимости от величины партии берут от одной до трех пробирок или по одному флакону, банке и коробке. Отобранные пробирки, флаконы, коробки или банки вскрывают, содержимое их помещают в чистую, сухую банку, тщательно перемешивают и выделяют среднюю пробу в количестве от 20 до 100 г.
б) При расфасовке в барабаны или коробки из разных (но не менее трех) мест барабана или коробки отбирают 200 г продукта в отдельную чистую, сухую банку и после тщательного перемешивания выделяют среднюю пробу в количестве от 20 до 100 г.
3. Жидкие витаминные препараты
а) При расфасовке в флаконы вместимостью до 50 см включительно отбирают 4% всех тарных мест, но не менее трех.
б) При расфасовке в флаконы и бутылки вместимостью свыше 50 см отбирают 5% всех тарных мест, но не менее трех.
Из отобранных тарных мест выделяют пробу:
при расфасовке в флаконы вместимостью до 50 см включительно не менее 300 см
- от всех отобранных тарных мест;
при расфасовке в флаконы, банки, коробки и бутылки вместимостью свыше 50 см - по одному флакону, банке, коробке или по одной бутылке от каждого отобранного тарного места.
в) При расфасовке в бочки, бидоны, цилиндры (барабаны) и стеклянные банки (вместимостью свыше 3 дм) отбирают 10% всех тарных мест, но не менее трех.
Среднюю пробу выделяют следующим образом: содержимое отдельных флаконов выливают в отдельную чистую, сухую банку и затем после тщательного перемешивания отбирают среднюю пробу в плотно закрывающуюся склянку в количестве 200 см.
Из отобранных флаконов, бутылок и банок вместимостью свыше 50 см после тщательного перемешивания их содержимого отливают по 50 см
в отдельную чистую, сухую банку, из которой после тщательного перемешивания отбирают среднюю пробу в плотно закрывающуюся склянку, в количестве 200 см
.
Примечание. При наличии в одной партии флаконов, банок и бутылок, выделенные для каждого вида упаковки пробы соединяют и затем после тщательного перемешивания выделяют среднюю пробу в количестве 200 см.
г) Из бочек, бидонов, цилиндров (барабанов) и стеклянных баллонов пробы отбирают при помощи стеклянной трубки или сифона; после тщательного перемешивания жидкости выливают в отдельную чистую, сухую склянку в количестве от 50 до 100 см.
Содержимое склянки тщательно перемешивают и из нее отбирают среднюю пробу в количестве 200 см в хорошо закрывающуюся склянку.
4. Жидкие витаминные препараты в ампулах. Отбирают 10% всех тарных мест, но не менее трех. Из каждого отобранного места отбирают такое количество ампул, чтобы общий объем их содержимого составлял не менее 200 см.
5. Отобранную согласно пп.1, 2 и 3 среднюю пробу делят пополам; одна часть предназначается для отправки на анализ, другая часть хранится в лаборатории завода в течение времени, предусмотренного гарантийным сроком хранения данного препарата, но не более 6 мес.
Обе пробы помещают в чистые, сухие склянки, герметически укупоренные, опечатывают смолкой или сургучом с оттиском печати завода и снабжают этикеткой с указанием наименования продукта, даты выпуска и отбора пробы.
Примечание. Отбор проб на продукты, не предусмотренные настоящим стандартом, производится в соответствии с действующими стандартами на данные продукты.
6. Подготовка средней пробы к анализу. Подготовка проб к анализу производится непосредственно перед анализом.
а) Витаминное драже и таблетки. Из средней пробы драже и таблеток отбирают 30-50 шт., взвешивают их и определяют вес одной штуки, затем тщательно растирают и перемешивают в фарфоровой ступке.
б) Порошки и кристаллические витаминные препараты. Среднюю пробу порошкообразных веществ (например, сухое молоко, порошок шиповника и т.д.) отбирают в количестве не менее 50 г. Перед взятием пробы порошок тщательно перемешивают. Пробу кристаллических витаминных препаратов отбирают в количестве 5-50 г и тщательно перемешивают.
в) Жидкие витаминные препараты. Жидкие витаминные препараты в количестве 50-100 см тщательно перемешивают путем многократного взбалтывания или переворачивания склянки, содержащей пробу.
Примечание. При анализе на содержание витамина С перемешивание производят осторожно, избегая аэрации.
г) Растворы витаминных препаратов в ампулах. Все отобранные ампулы вскрывают и содержимое их выливают в чистую, сухую склянку с притертой пробкой и тщательно перемешивают.
д) Витаминизированные кондитерские изделия (конфеты, пряники и печенье). Пробу штучных изделий берут в количестве не менее 200 г (в зависимости от содержания витаминов), взвешивают, определяют вес единицы изделия, а затем подвергают размельчению и растиранию в ступке.
е) Твердые жиры и масла. Среднюю пробу твердых жиров и масел в количестве не менее 200 г расплавляют в склянке на водяной бане при 45-50 °С с перемешиванием во избежание расслоения, затем охлаждают и отбирают навеску для анализа.
ж) Жидкие жиры и масла. Отбор средней пробы см. п.3.
з) Молоко. Молоко в количестве не менее 200 см тщательно перемешивают.
и) Твердые ткани и органы животных (анализы на витамин А и Д). Твердые ткани и органы животных в количестве не менее 200 г перед взятием навески измельчают ножницами или в мясорубке и перемешивают в ступке.
к) Яйца. От десяти яиц отделяют желтки, взвешивают для установления средней массы желтка, затем хорошо смешивают их, избегая взбалтывания, и отбирают пробу для анализа.
л) Сухие плоды, овощи и травы. Сухие плоды, овощи и травы в количестве не менее 50 г измельчают на лабораторной мельнице, мясорубке или ножницами и полученную пробу тщательно перемешивают. Измельченный материал по мере раздробления ссыпают в банку с притертой пробкой.
м) Свежие травы. Свежие травы берут в количестве не менее 100 г, измельчают ножницами, перемешивают в ступке и тотчас же используют для анализа.
и) Свежие плоды, овощи и ягоды. Пробы овощей и плодов вырезают ножом из нержавеющей стали в форме сегментов и немедленно используют для анализа. Ягоды и мелкие сочные плоды предварительной обработке не подвергаются. Общая масса пробы, поступающей на анализ, должна быть не менее 200 г.
о) Консервы. Консервы после вскрытия банок немедленно переносят в ступку и тщательно и быстро измельчают и размешивают или измельчают на мясорубке.
п) Обеды (при анализе на содержание витамина С). Пробы первых блюд (супы) взвешивают, затем пробы помещают на мелкое волосяное сито или марлю и отделяют жидкую часть от твердой, далее определяют массу жидкой части путем взвешивания и массу оставшейся твердой части - по разности масс. Твердую часть быстро растирают в ступке до однородной консистенции. Определение витамина С в обеих частях производится в отдельности.
Суп-пюре анализируют без разделения на жидкую и твердую часть.
Пробы вторых блюд анализируют так же, как плотную часть первых блюд.
Третьи блюда анализируются соответственно своему характеру, как первые или вторые блюда.
Общие замечания
1) Из подготовленных для анализа проб, как указано в п.6, берут навески согласно прописи методов анализа.
2) В жидких пробах (сиропы, концентраты) после размешивания определяют по общим правилам плотность для перечисления содержания витамина на единицу объема.
3) Из измельченной и перемешанной пробы берут 2-3 навески. Навески жидких проб (соки, настои, растворы и т.п.) отбирают пипеткой, навески проб густой консистенции (густые сиропы, концентраты и пр.), плохо стекающие из пипетки, берут весовым путем подобно твердым продуктам.
II. МЕТОДЫ ОПРЕДЕЛЕНИЯ ВИТАМИНА А
7. Применяемые реактивы
Кали едкое х.ч.
Натр едкий х.ч.
Натрий сернокислый (кристаллический) или безводный х.ч.
Кислота серная ч.
Ангидрид уксусный ч.д.а.
Кобальт азотнокислый ч.д.а.
Медь сернокислая ч.д.а.
Калий марганцевокислый х.ч.
Спирт этиловый ректификат.
Бензидин-основание ч.
Углекислый газ (в баллоне или получаемый в аппарате Киппа) или азот.
Хлороформ для наркоза или ч.д.а.*
_______________
* Текст соответствует оригиналу. - .
Сурьма треххлористая.
Эфир этиловый (серный).
Калий йодистый х.ч.
Все применяемые реактивы должны соответствовать требованиям действующих стандартов или технических условий.
8. Приготовление и очистка некоторых реактивов. Реактивы, применяемые при определении витамина А, подлежат очистке и предварительной обработке.
Натрий сернокислый кристаллический прокаливают для обезвоживания, а безводный просушивают при 105-110 °С.
Углекислый газ или азот для очистки от примесей пропускают через поглотители с водой, щелочным раствором марганцевокислого калия и серной кислотой.
Хлороформ промывают 5-6 раз дистиллированной водой (в отношении 2:1), высушивают безводным сернокислым натрием, перегоняют в склянку темного стекла и хранят в прохладном месте. Хлороформ необходимо проверять на отсутствие фосгена по качественной реакции с бензидином.
Сурьма треххлористая. Продажную треххлористую сурьму промывают хлороформом, пока не будет стекать прозрачный и бесцветный раствор, и высушивают в эксикаторе над серной кислотой в течение 1-2 суток. Насыщенный раствор треххлористой сурьмы в очищенном хлороформе приготовляют при 20 °С.
Эфир этиловый (серный). В склянку с корковой пробкой помещают 500 см эфира, 50 см
4%-ного раствора KMnO
, 5 см
40%-ного раствора NaOH или KOH, взбалтывают и оставляют на сутки в темноте. Смесь переносят в делительную воронку, нижний слой сливают, а эфир промывают 5-6 раз дистиллированной водой (в отношении 2:1), высушивают в течение суток сернокислым натрием, перегоняют и сохраняют в темноте. Эфир необходимо проверять на отсутствие перекисей по качественной реакции с йодистым калием.
9. Подготовка к колориметрированию
а) Жиры рыб и морских млекопитающих (натуральные и витаминизированные)
1) Навеску жира в количестве от 0,5 до 1 г омыляют 0,5 н. спиртовым раствором едкого кали (10 см) в течение 30 мин - 1 ч на водяной бане при 85-90 °С с обратным холодильником.
2) Омыленный раствор разбавляют 20 см воды. Неомыляемую фракцию трижды экстрагируют в делительной воронке серным эфиром, свободным от перекиси: 1-й раз - 50 см
, 2-й и 3-й - по 25 см
эфира. Во избежание образования стойкой эмульсии, экстракцию эфиром производят лишь в хорошо охлажденном растворе и путем осторожного перемешивания содержимого воронки.
3) Соединенные эфирные вытяжки промывают 3-4 раза водой (по 20 см) до нейтральной реакции промывных вод на лакмус.
4) В промытую эфирную вытяжку добавляют 6-8 г сернокислого натрия и высушивают в течение 30 мин, периодически взбалтывая, затем фильтруют и отгоняют эфир в токе инертного газа.
5) Неомыляемый остаток растворяют в хлороформе, количество которого берут в зависимости от предполагаемого содержания витамина А (10-25 см), переносят в мерную колбу и раствор доводят до объема хлороформом. Дальнейший ход анализа см. п.10 А и Б.
б) Концентраты витамина А (полуфабрикаты). Навеску концентрата в количестве от 0,2 до 0,5 г омыляют 0,5 и спиртовым раствором KОН (15 см) в течение 1 ч на водяной бане. Дальнейший ход анализа - см. п.9а, подпункты 2-5.
в) Концентраты витамина А (очищенные). Навеску концентрата в количестве около 0,2 г растворяют в хлороформе в мерной колбе емкостью 25 см.
Из полученного раствора отбирают от 1 до 10 см (в зависимости от активности концентрата) и производят второе разбавление хлороформом в мерной колбе вместимостью 25 см
. Дальнейший ход анализа см. п.10 А и Б.
г) Сливочное и топленое масло и маргарин (натуральные и витаминизированные) и растительные витаминизированные масла. Навеску масла или маргарина в количестве от 10 до 20 г омыляют 20-40 см (в зависимости от взятой навески) 20%-ного спиртового раствора KОН в течение 1-2 ч на водяной бане при 85-90 °С. К охлажденному раствору прибавляют двукратный объем воды и проводят экстракцию неомыляемой фракции эфиром, используя на первую экстракцию не менее 75 см
эфира, на вторую и третью - по 25 см
. Далее поступают, как указано в п.9а, подпункты 3 и 4. Неомыляемый остаток растворяют в 5-10 см
хлороформа. Дальнейший ход анализа см. п.10 А и Б.
д) Молоко. От 100 до 200 см молока смешивают с
объема водного 60%-ного раствора KОН и 20-40 см
этилового спирта и ставят на 48 ч в темное место при температуре 20-25 °С, периодически взбалтывая содержимое колбы. Экстракцию неомыляемого остатка проводят в делительной воронке эфиром отдельными порциями 4 раза: 1-й раз - 75 см
, последующие - по 30-40 см
. Далее поступают, как указано в п.9а, подпункты 3 и 4. Неомыляемый остаток растворяют в 2-10 см
хлороформа. Дальнейший ход анализа см. п.10 А и Б.
е) Печень. К навеске печени в количестве 3 г добавляют 1 см 60%-ного водного раствора KОН и 10-20 см
этилового спирта и нагревают на водяной бане при 85-90 °С с обратным холодильником не менее 2 ч до полного растворения ткани. Если ткань полностью не растворяется, добавляют еще 10-20 см
спирта и продолжают омыление до полного растворения ткани. После омыления к раствору добавляют 10-20 см
спирта и двойное (по отношению к общему объему спирта) количество воды; неомыляемую фракцию трижды экстрагируют эфиром: 1-й раз - 50 см
, 2-й и 3-й раз - по 25 см
. Далее поступают, как указано в п.9а, подпункты 3 и 4. Неомыляемый остаток растворяют в 10 см
хлороформа. Дальнейший ход анализа см. п.10 А и Б.
ж) Яйца. К навеске яичного желтка в количестве около 20 г добавляют 6 см 60%-ного водного раствора KОН и 10-20 см
этилового спирта и нагревают на водяной бане при 85-90 °С с обратным холодильником не менее 2 ч. По окончании омыления к раствору добавляют 10-20 см
спирта и двойное (по отношению к общему объему спирта) количество воды; неомыляемую фракцию трижды экстрагируют эфиром: 1-й раз - 50 см
, 2-й и 3-й раз - по 25 см
. Далее поступают, как указано в п.9а, подпункты 3 и 4. Не-омыляемый остаток растворяют в 10 см
хлороформа. Дальнейший ход анализа см. п.10 А и Б.
з) Драже и таблетки с витамином А. Навеску драже или таблеток в количестве от 1 до 2 г омыляют 10 см 0,5 н спиртового раствора KOH на водяной бане при температуре 85-90 °С в течение 30 мин с обратным холодильником. Далее поступают, как указано в п.9а, подпункты 2-5.
и) Витаминизированные кондитерские изделия (конфеты, пряники и пр.). Навеску витаминизированных кондитерских изделий в количестве 20-50 г экстрагируют эфиром холодным способом, затрачивая на каждую экстракцию по 20-50 см эфира. Эфирные вытяжки фильтруют, соединяют и отгоняют эфир в токе инертного газа. Остаток после отгонки эфира омыляют 0,5 н спиртовым раствором едкого кали (10 см
в течение 30 мин - 1 ч на водяной бане при 85-90 °С с обратным холодильником). В дальнейшем поступают, как указано в п.9а, подпункты 2-5.
к) Определение витамина А в продуктах, содержащих витамин А и каротин. В продуктах, содержащих, кроме витамина А, каротин (сливочное и топленое масло, яйца, молоко, печень и др.), ход анализа изменяется следующим образом. Высушенный эфирный экстракт неомыляемых веществ делят на две части: в одной части определяют после отгонки эфира и растворения остатка в хлороформе витамин А, в другой после отгонки эфира и растворения остатка в петролейном эфире определяют каротин.
10. Колориметрирование. Для колориметрирования применяют упрощенный прибор или электрофотоколориметр.
А. КОЛОРИМЕТРИРОВАНИЕ В УПРОЩЕННОМ ПРИБОРЕ
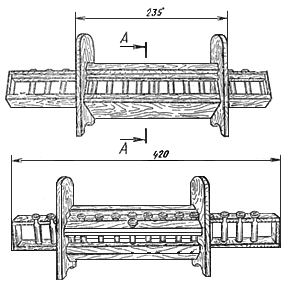
а) Описание прибора
Упрощенный прибор (см. чертеж) представляет собой штатив с передвижной шкалой, в которую вставлено 15 пробирок-эталонов из бесцветного стекла. В неподвижной части штатива имеется отверстие для пробирки с испытуемым раствором и окошко, через которое сравнивают окраску последнего с растворами в пробирках-эталонах.
А-А
В пробирках-эталонах, имеющих диаметр 1 см и высоту 6 см, содержатся типовые растворы, представляющие собой смесь водных растворов сернокислой меди и азотнокислого кобальта.
Пробирки-эталоны должны быть закрыты пробками с прокладками из пергаментной бумаги и залиты менделеевской замазкой.
Размеры прибора:
длина подвижной шкалы с эталонами | 420 мм |
ширина " " " " | 25 " |
высота " " " " | 55 " |
Размеры окошка - 40x35 мм.
б) Приготовление типовых растворов
Типовые растворы приготовляют следующим образом: в 500 мл воды растворяют 75 г сернокислой меди и 3,5 г азотнокислого кобальта; из основного раствора, содержащего витамин А, путем разбавления водой приготовляют эталоны согласно приведенной ниже табл.1.
Типовые растворы для пробирок-эталонов
Таблица 1
Номер пробирок | Количество, см | Число синих единиц | |
основного раствора | воды | ||
1 | 20 | 0,4 | 9 |
2 | 20 | 1,6 | 8,5 |
3 | 20 | 3 | 8 |
4 | 20 | 4,5 | 7,5 |
5 | 20 | 6,3 | 7 |
6 | 20 | 8,7 | 6,5 |
7 | 20 | 11,6 | 6 |
8 | 20 | 14,6 | 5,5 |
9 | 20 | 19 | 5 |
10 | 20 | 24,4 | 4,5 |
11 | 20 | 31,6 | 4 |
12 | 20 | 41,2 | 3,5 |
13 | 20 | 55,4 | 3 |
14 | 20 | 74,2 | 2,5 |
15 | 20 | 104,2 | 2,0 |
16 | 20 | 131 | 1,75 |
17 | 20 | 175 | 1,5 |
18 | 20 | 190 | 1,25 |
19 | 20 | 240 | 1 |
20 | 20 | 330 | 0 ,75 |
21 | Дистиллированная вода | ||
Для объектов, содержащих мало витамина А, например, молоко, рекомендуется пользоваться пробирками с N 7 до N 21.
в) Проведение определений
Из приготовленного для колориметрирования хлороформенного раствора витамина А в пробирку, предназначенную для испытуемого раствора, отбирают 0,2 см испытуемого раствора, добавляют туда же 1-3 капли уксусного ангидрида и 2 см
хлороформенного раствора треххлористой сурьмы и колориметрируют не позднее чем через 5-10 с.
При определении сравнивают окраску пробирки с испытуемым раствором с окраской пробирок-эталонов через окошечко в передней части прибора, помещая его на белую бумагу.
Пробирку с испытуемым раствором устанавливают в неподвижной части прибора и подводят последовательно к ней пробирки-эталоны путем передвижения шкалы.
Определение считают законченным, когда окраска испытуемого раствора совпадет с окраской раствора в одной из пробирок-эталонов.
Содержание витамина А в испытуемом растворе будет соответствовать числу синих единиц, обозначенному на совпавшей по окраске пробирке-эталону.
Если окраска испытуемого раствора лежит между окрасками двух смежных пробирок-эталонов, берут среднее из значений синих единиц, обозначенных на этих пробирках.
Наиболее точные результаты получают при колориметрировании раствора в пределах от 4 до 6 синих единиц. Концентрированные растворы разбавляют соответствующим образом хлороформом и проводят определение повторно.
Примечание. При необходимости получения большего объема колориметрируемого раствора можно увеличить его вдвое, втрое и больше, соответственно сохраняя указанное соотношение количества испытуемого раствора и треххлористой сурьмы.
г) Вычисление содержания витамина А
Количество витамина A () в граммах на 1 г продукта вычисляют по формуле
![]() ,
,
где - число синих единиц, установленное при колориметрировании;
- объем раствора хлороформа, см
;
- навеска, г;
![]() - коэффициенты пересчета в граммы витамина А;
- коэффициенты пересчета в граммы витамина А;
- коэффициент пересчета в миллиграммы.
Количество витамина А () в ин. ед. (интернациональных единицах) на 1 г продукта вычисляют по формуле
![]() ,
,
где - коэффициент перевода числа грамм витамина А в ин. ед.
Остальные обозначения те же, что и в предыдущей формуле.
Б. КОЛОРИМЕТРИРОВАНИЕ В ЭЛЕКТРОФОТОКОЛОРИМЕТРЕ
а) Требования, предъявляемые к электрофотоколориметру. Электрофотоколориметры, применяемые для определения витамина А, должны соответствовать следующим требованиям:
обеспечить проведение измерения (отсчета) в течение 5-10 с;
обеспечить хорошую воспроизводимость результатов и пропорциональность отсчетов концентрации витамина А;
иметь достаточно монохроматический светофильтр с максимумом пропускания 620 м;
иметь хорошо отрегулированный гальванометр с коротким периодом отклонений;
иметь набор одинаковых пробирок (кювет) из бесцветного стекла с внутренним диаметром 1 см.
Соответствующими вышеуказанным требованиям являются приборы конструкции Шипалова и фотоэлектрические колориметры ВНИВИ КФЭ-1.
Допускается пользование электрофотоколориметром ФЭК-М с компенсационной схемой.
б) Калибрование электрофотоколориметра. Точную навеску стандартизованного препарата витамина А растворяют в хлороформе. Указанный раствор, являющийся исходным, должен содержать 100 ин. ед. витамина А в 1 см. Из него готовят затем 5-6 последовательных разведений с таким расчетом, чтобы во взятых для цветной реакции пробах отсчеты по гальванометру укладывались в пределах 40-65 делений шкалы гальванометра (
0,22-0,45).
Электрофотоколориметр устанавливают в рабочее положение подведением стрелки гальванометра к нулевой экстинкции по раствору треххлористой сурьмы с попеременным использованием двух пробирок, обладающих одинаковым пропусканием.
Примечание. Если электрофотоколориметр построен по компенсационной схеме, то раствор треххлористой сурьмы вносят в обе пробирки.
В одну из пробирок вносят точно отмеренное количество хлороформенного стандартного раствора, пробирку вставляют в гнездо прибора и быстро добавляют раствор треххлористой сурьмы. Показание гальванометра отмечают не позднее чем через 5-10 с после приливания раствора треххлористой сурьмы. Каждое определение проводят два раза с сохранением соотношения исследуемого раствора и треххлористой сурьмы, как 1:9. Суммарный объем жидкости устанавливают в соответствии с объемом кюветы и высотой световой щели.
в) Составление калибровочной кривой. Для составления калибровочной кривой откладывают по оси ординат найденные значения экстинкции (), по оси абсцисс - соответствующие им количества витамина А в интернациональных единицах. Калибровочная кривая должна практически представить собой прямую линию.
Примечание. Если калибровочная кривая имеет изгиб к оси абсцисс, то нельзя пользоваться тем отрезком кривой, в котором она сильно отклоняется от прямой линии, идущей от начала координат.
г) Определение витамина А в исследуемом растворе. Определение производят, как описано в п.10 Б, подпункт б "Калибрование электрофотоколориметра"; концентрацию раствора, взятого на определение, подбирают в зависимости от концентрации витамина А в анализируемом материале с таким расчетом, чтобы показание гальванометра (Д) укладывалось в пределы 40-65 делений шкалы.
Расчет содержания витамина А производят по калибровочной кривой. По показаниям гальванометра (Д) находят соответствующее значение экстинкции () на таблице, прилагаемой в инструкциях к пользованию электрофотоколориметрами. С помощью калибровочной кривой непосредственно находят соответствующее найденной экстинкции содержание витамина А в интернациональных единицах. Полученное значение пересчитывают на разведение и на взятую навеску.
III. МЕТОДЫ ОПРЕДЕЛЕНИЯ ВИТАМИНА С
Настоящий раздел стандарта распространяется на методы определения витамина С в растительных объектах, витаминных препаратах (кроме синтетической аскорбиновой кислоты по нормативному документу по стандартизации), пищевых продуктах и готовой пище.
11. Навески
Драже, таблетки, порошок и концентраты шиповника, концентраты из хвои | 1-2 г |
Таблетки витамина С с глюкозой | 2-3 г |
Сироп из плодов шиповника | 5 г |
Чай витаминизированный плиточный | 2-5 г |
Витаминизированные конфеты и чай (кирпичный) | 10-50 г |
Настои (хвои, шиповника) | 10-20 см |
Соки и экстракты | 1-50 см |
Плоды шиповника очищенные, пюре шиповника с сахаром, хвоя | 5 г |
Плоды шиповника целые, картофель сушеный, картофель сушеный сульфитированный и другие сушеные сульфитированные продукты | 10 г |
Консервы | 5-50 г |
Свежие растительные продукты (фрукты, плоды, ягоды, овощи и зелень) | 10-50 г |
Блюда готовые: | |
твердая часть первого блюда и второе блюдо | 20-50 г |
жидкая часть первого блюда | 20-50 см |
Молоко | 5 см |
Примечания:
1. Для арбитражного анализа навески очищенных плодов шиповника берут из хорошо измельченной средней пробы (100 г) в количестве 10 г, в целых плодах - 20 г.
2. Те или иные величины навесок различных продуктов берутся в зависимости от большего или меньшего содержания в них витамина С.
3. Взвешивание навесок производят с точностью до 0,01 г, кроме навесок готовых блюд, которые взвешивают с точностью до 1 г.
12. Применяемые реактивы, приготовление их и установка титров
Кислота уксусная; 80%-ный раствор; 5%-й раствор (58 см 80%-ной уксусной кислоты доводят дистиллированной водой до 1 дм
).
Свинец уксуснокислый; насыщенный раствор; 5%-ный раствор на 5%-ной уксусной кислоте (недостаточно чистая соль свинца должна быть трижды перекристаллизована).
Кислота серная ч.; раствор 1:2 (по объему); раствор 1:4 (по объему); 0,02 н раствор (0,56 см серной кислоты х.ч. плотностью 1,84 доводят дистиллированной водой до 1 дм
).
Кислота соляная ч.; раствор 1:1 (по объему); 2%-ный раствор (45,1 см соляной кислоты плотностью 1,19 доводят дистиллированной водой до 1 дм
).
Кислота соляная техническая; раствор 1:1 (по объему).
Кальций углекислый.
Железо сернистое ч.д.а.
Аммоний щавелевокислый х.ч.; насыщенный раствор; 0,01 н раствор (0,062 г щавелевокислого аммония растворяют в воде и объем доводят до 100 см дважды перегнанной водой).
Натрий щавелевокислый х.ч.; насыщенный раствор; 0,01 н раствор (0,067 г щавелевокислого натрия растворяют в воде и объем доводят до 100 см дважды перегнанной водой).
Натрий серноватистокислый (гипосульфит); 0,1 н раствор; 0,01 н раствор. Для приготовления 0,1 н раствора навеску 24,82 г растворяют в свежепрокипяченной дистиллированной воде и доводят объем до 1 дм. Титр раствора серноватистокислого натрия устанавливают по 0,1 н раствору марганцевокислого калия, который проверяют по точной навеске щавелевокислого аммония или щавелевокислого натрия по общим правилам объемного анализа.
Калий марганцовокислый; 0,1 н раствор; 0,01 и раствор (для приготовления 0,1 н раствора навеску 3,158 г растворяют в дистиллированной воде и объем доводят до 1 дм).
Калий йодистый; 1%-ный раствор.
Соль натриевая 2,6-дихлорфенолиндофенола; 0,001 н раствор (в тексте настоящего стандарта обозначается далее как 2,6-дихлорфенолиндофенол).
Двойная сернокислая соль закиси железа и аммония FeSO (NH
)
SO
·6H
O (соль Мора); 0,01 н раствор. Для получения 0,01 н раствора соли Мора навеску 3,92 г соли растворяют в 1 дм
0,02 н раствора серной кислоты. Раствор соли Мора должен храниться в склянке из темного стекла, и титр его должен проверяться каждые 3-4 недели. Установку титра соли Мора ведут по титрованному 0,01 н раствору марганцевокислого калия по общим правилам объемного анализа.
Кали едкое х.ч.; 10%-ный раствор.
Натр едкий х.ч.; 10%-ный раствор.
Калий иодноватокислый; 0,001 н раствор.
Крахмал, 1%-ный и 0,5%-ный растворы.
Мрамор в кусках или углекислый газ в баллоне.
Буферная смесь (однозамещенный фосфат калия и двузамещенный фосфат натрия с рН около 7).
Стеклянный порошок, приготовленный из чистого, не зеленого, лабораторного стекла.
Все применяемые реактивы должны соответствовать требованиям действующих стандартов или технических условий.
Для приготовления растворов, а также для проведения анализов применяют дистиллированную воду, перегнанную в аппаратуре из стекла. Для приготовления точных растворов щавелевокислого аммония или щавелевокислого натрия применяют дважды перегнанную воду; при ее получении в перегонную колбу вносят 0,1 г марганцевокислого калия и несколько капель серной кислоты х.ч. плотностью 1,84 на 1 дм воды.
Приготовление 0,001 н раствора 2,6-дихлорфенолиндофенола: 0,2 г 2,6-дихлорфенолиндофенола растворяют в 600 см воды при энергичном взбалтывании (допускается применение буферной смеси). Раствор фильтруют и доводят водой до 1 дм
. Срок годности раствора не более 7 дней при условии хранения в темном месте. Титр раствора проверяют ежедневно.
Установка титра раствора 2,6-дихлорфенолиндофенола производится по одному из следующих методов:
а) По соли Мора. В коническую колбу наливают 10 см раствора 2,6-дихлорфенолиндофенола, а в микробюретку 0,01 н раствора соли Мора; к раствору 2,6-дихлорфенолиндофенола прибавляют 5 см
насыщенного раствора щавелевокислого аммония или щавелевокислого натрия и титруют, пока синяя окраска раствора не сменится соломенно-желтой (нерезкая перемена окраски указывает на порчу реактива).
Титр раствора соли Мора устанавливают, как указано выше.
Поправку () на титр раствора 2,6-дихлорфенолиндофенола вычисляют по формуле
![]() ,
,
где - количество соли Мора, используемое на титрование 10 см
данного раствора 2,6-дихлорфенолиндофенола, в см
;
- количество раствора марганцовокислого калия, используемое на титрование 10 см
соли Мора, см
;
- количество раствора марганцовокислого калия, используемое на титрование 10 см
точно 0,01 н раствора щавелевокислого аммония или щавелевокислого натрия, см
.
1 см точно 0,001 н раствора 2,6-дихлорфенолиндофенола соответствует 0,088 мг аскорбиновой кислоты,
б) По иодату. Несколько кристаллов аскорбиновой кислоты растворяют в 50 см 2%-ной х.ч. серной кислоты.
Из полученного раствора отбирают одной и той же пипеткой 5 см для титрования рабочим (приблизительно 0,001 н) раствором 2,6-дихлорфенолиндофенола и 5 см
для титрования точно 0,001 н раствором KJO
.
Титрование раствором KJO ведется в присутствии нескольких кристаллов (1-2 мг) KJ и двух-трех капель 1%-ного раствора крахмала до появления голубого окрашивания; титрование раствором 2,6-дихлорфенолиндофенола производят до появления розового окрашивания, не исчезающего в течение 3 мин.
Примечание. Титрование удобно производить в фарфоровой чашке.
Расчет титра раствора 2,6-дихлорфенолиндофенола () по аскорбиновой кислоте производят по следующей формуле, исходя из того, что 1 см
0,001 н иода, а, следовательно, и иодата, эквивалентен 0,088 мг аскорбиновой кислоты
![]() ,
,
где - количество 0,001 н раствора KJO
, используемое на титрование, см
;
- количество раствора 2,6-дихлорфенолиндофенола, используемое на титрование, см
.
Таким образом узнают, какому количеству аскорбиновой кислоты в миллиграммах соответствует 1 см раствора 2,6-дихлорфенолиндофенола.
Приготовление 0,1 н раствора иодноватокислого калия. Растворяют точно отвешенные 3,567 г х.ч. иодноватокислого калия в воде, доводят объем до 1 дм и получают 0,1 н раствор, из которого по мере надобности путем разведения в 100 раз получают 0,001 н раствор. При отсутствии х.ч. иодноватокислого калия применяют перекристаллизованный и высушенный при 105 °С до постоянного веса реактив.
Приготовление 0,1 н раствора иода. 12,692 г свежевозогнанного иода растворяют в 20 см водного раствора иодистого калия, содержащего 20 г KJ в указанном объеме. После полного растворения иода раствор переводят в мерную колбу вместимостью 1 дм
и содержимое колбы доводят водой до метки. Из 0,1 н раствора по мере надобности путем разведения в 10 раз получают 0,01 н раствор, титр которого устанавливают по общим правилам объемного анализа.
Приготовление раствора крахмала. 0,5 г растворимого крахмала, взвешенного с точностью до 0,01 г, растирают в ступке с 5 см воды до получения однородной кашицы; смесь медленно вливают при постоянном размешивании в 100 см
кипящей воды и кипятят 2-3 мин до получения прозрачной или слабо опалесцирующей жидкости. Раствор должен храниться на холоде не более 2-3 дней.
Приготовление буферной смеси. 11,876 г NaHPO
·2H
O растворяют в литре дистиллированной воды, получают раствор I; 9,078 г KH
РО
растворяют также в 1 дм
дистиллированной воды - раствор II. Смешивают 7 частей раствора I и 3 части раствора II; рН раствора соответствует 7.
Получение сероводорода. Вместо аппарата Киппа для получения сероводорода возможно применять следующую упрощенную установку. Берут небольшую банку вместимостью до 250 см с широким горлом, в которое вставляют пробку. На дно банки кладут сернистое железо слоем в 2-3 см. В пробке делают два отверстия: одно для небольшой делительной воронки, куда наливают техническую соляную кислоту (крепкая кислота разводится 1:1), другое - для отводной стеклянной трубки, согнутой под углом. Эта трубка соединяется с маленькой колбой или предохранительной склянкой (играющими роль клапана) с водой, которые, в свою очередь, соединены с сосудом, содержащим анализируемый экстракт. Выделяющийся сероводород отводят в склянку (коническую колбу) с 5-10% едкой щелочью, где он и поглощается. Чтобы избежать лишней траты соляной кислоты, в промежутке между анализами соляную кислоту сливают с сернистого железа в специальную склянку с притертой пробкой и сернистое железо заливают водой.
Очистка кварцевого песка. Песок просеивают через сито с диаметром отверстий 4-5 мм и отмучивают водопроводной водой. Затем приливают к песку соляную кислоту (крепкая кислота разводится вдвое), перемешивают и оставляют стоять на ночь. Далее песок тщательно промывают водопроводной и дистиллированной водой до исчезновения реакции на хлор (проба с раствором азотнокислого серебра) и высушивают. Песок вновь просеивают через сито с диаметром отверстий 1-1,5 мм и прокаливают для удаления органических веществ. Очищенный песок должен быть проверен на отсутствие следов железа (проба с 5%-ной соляной кислотой и роданидом с учетом результатов параллельной контрольной пробы на содержание железа в соляной кислоте).
Очищенный песок хранят в чистой и плотно закрытой стеклянной банке.
13. Условия применения методов определения
а) Методы определения витамина С подразделяются на арбитражные и контрольные.
Арбитражные методы применяют в тех случаях, когда требуется наибольшая точность определения (в том числе и в спорных случаях).
Контрольные методы применяют на предприятиях и в лабораториях, когда требуется быстрота определений и допускается точность анализа в пределах ±10%.
При массовых анализах готовых блюд и консервов применяют контрольный (упрощенный) метод, точность которого для данных продуктов лежит в пределах ±20%;
б) в зависимости от характера проб и назначения анализа применяют методы определения витамина С, указанные в табл.2.
Таблица 2
Наименование продукта | Арбитражный метод с H | Модификация арбитражного метода без H | Контрольные методы | ||
упрощенный | иодатный | ||||
1 | Драже и таблетки с витамином С | - | Х | Х | Х |
2 | Драже и таблетки с витамином СB | - | Х | Х | Х |
3 | Драже поливитаминное | - | X | X | X |
4 | Таблетки - витамин С с глюкозой | - | X | X | X |
5 | Сироп из плодов шиповника, витаминизированный | - | Х | Х | - |
6 | Порошок и таблетки из плодов шиповника | - | X | X | X |
7 | Концентрат витамина С из плодов шиповника в порошке | - | X | Х | X |
8 | Жидкие концентраты из плодов шиповника | - | X | X | - |
9 | Пюре из плодов шиповника с сахаром | - | X | X | - |
10 | Конфеты витаминизированные | - | X | X | - |
11 | Плоды шиповника | - | X | X | X |
12 | Настой шиповника | - | X | X | X |
13 | Хвоя и свежеприготовленный настой из хвои | X | - | X | X |
14 | Настой из хвои, подвергшийся хранению | X | - | - | - |
15 | Концентрат из хвои | - | X | X | - |
16 | Соки консервированные | - | X | X | - |
17 | Соки консервированные, интенсивно окрашенные, и соки свежеотжатые | X | - | - | - |
18 | Консервы (растительные) | X | - | X | - |
19 | Свежие плоды и овощи, и другие свежие растительные продукты | X | - | X | - |
20 | Сушеные продукты | X | - | - | - |
21 | Сушеный сульфитированный картофель | X | - | - | - |
22 | Чай витаминизированный | X | - | - | - |
23 | Блюда готовые | X | - | X | - |
24 | Молоко | - | - | X | - |
Примечания:
1. Препараты: 40%-ный раствор глюкозы с 5%-ной аскорбиновой кислотой в ампулах и 40%-ный раствор глюкозы с 0,2%-ной аскорбиновой кислотой в ампулах - анализируются с применением раствора (в соответствии с техническими условиями).
2. Не допускается анализировать свежие растительные продукты упрощенным методом, если продукты содержат большое количество дубильных веществ.
3. Для свежеотжатых томатного, мандаринового и апельсинового соков допускается применение упрощенного метода.
4. Предварительную подготовку и анализ не вошедших в таблицу продуктов производят по навеске и методу, принятому для соответствующего по характеру объекта, включенного в табл.2.
При анализе всякого нового продукта обязательно применение арбитражного метода (с применением сероводорода) параллельно с другими методами; в зависимости от результатов в дальнейшем анализ проводят по одному из соответствующих методов, указанных в табл.2.
В том случае, когда в табл.2 для данного продукта указан только арбитражный метод, этот метод следует применять и как контрольный.
в) Сероводород применяют:
когда имеется основание предполагать наличие в исследуемом материале обратимо окисленной формы витамина С - дегидроаскорбиновой кислоты (в свежих растительных продуктах, а также в продуктах, подвергшихся термической обработке и хранению);
для освобождения экстракта от пигментов, не устраняемых действием уксуснокислого свинца;
для устранения мутности в экстрактах (при анализе сульфитированного картофеля, некоторых крахмалистых изделий и т.п.), если определение без применения сероводорода невозможно.
г) Упрощенный и иодатный методы применяют для всех продуктов, за исключением сушеных (кроме шиповника), сульфитированных, интенсивно окрашенных или содержащих дегидроаскорбиновую кислоту в количестве 10% и более при условии соблюдения п.13а настоящего стандарта; кроме того, иодатный метод не применяется при анализе готовых блюд и молока.
Наличие дегидроаскорбиновой кислоты определяют сравнением результатов, полученных при параллельном анализе данного продукта арбитражным методом (с HS) и его модификацией (без H
S).
д) При каждом анализе проводят не менее двух параллельных определений (с двумя навесками), причем повторные титрационные числа в каждом определении не должны разниться между собой более чем на 0,03 см; из них берут среднее арифметическое. Титрование должно продолжаться не более 2 мин, и количество соответствующего раствора 2,6-дихлорфенолиндофенола или иодноватокислого калия, пошедшее на одно титрование, должно быть в пределах 1-2 см
.
е) Результаты параллельных определений (из двух навесок) не должны отличаться друг от друга более чем на ±5%.
14. Описание определения
Арбитражный метод (с применением сероводорода)
а) Навеску жидких продуктов и продуктов густой консистенции разводят 5%-ным раствором уксусной кислоты до определенного объема (в мерной колбе вместимостью 50-100 см); получают так называемый первоначальный раствор.
Первоначальные растворы для настоев хвои, жидкой части первых блюд, соков и прочих продуктов с невысоким (не выше 20 мг %) содержанием витамина С получают путем разведения без применения мерных колб. В этом случае в зависимости от содержания витамина С уксусную кислоту берут к массе навески в отношении 1:1 или больше; 10 см первоначального раствора вносят пипеткой в коническую колбу или в центрифужный стакан вместимостью 60-80 см
и прибавляют 0,4 г х.ч. углекислого кальция, слегка встряхивая. Затем приливают пипеткой 5 см
5%-ного раствора уксуснокислого свинца, взбалтывают и тотчас центрифугируют в течение 1-2 мин или фильтруют через складчатый фильтр.
Примечание. В случае анализа слабых витаминоносителей употребляют большее количество анализируемой жидкости и берут 20 см первоначального раствора (вместо 10 см
), пропорционально увеличивая количество углекислого кальция и раствора уксуснокислого свинца.
б) Навеску твердых продуктов тщательно растирают, применяя, в случае надобности, 5-10 г стеклянного порошка, и приливают постепенно 5%-ный раствор уксусной кислоты в кратном к навеске отношении (не менее 3 см на 1 г навески). При использовании в ходе анализа мерной посуды стеклянный порошок не употребляют.
После растирания смесь оставляют в ступке настаиваться в течение 10 мин.
После настаивания таких продуктов, как драже, таблетки или порошок шиповника содержимое ступки количественно переносят в мерную колбу вместимостью 50-100 см и отмеренную для настаивания кислоту расходуют не полностью - часть кислоты оставляют для промывки ступки и пестика и доведения жидкости в колбе до метки. Допускается непосредственный перенос навесок хорошо растворяющихся драже и таблеток в мерную колбу для растворения в ней и настаивания.
Перед переносом в центрифужные стаканы содержимое колбы перемешивают путем переворачивания.
При экстракции прочих твердых продуктов 5%-ный раствор уксусной кислоты наливают в ступку строго по расчету выбранной кратности к навеске; по окончании настаивания жидкость из ступки вместе с нерастворившийся частью разливают по центрифужным стаканам.
Содержимое центрифужных стаканов центрифугируют до просветления.
При отсутствии центрифуги центрифугирование заменяют фильтрованием через гигроскопическую вату или сложенную в несколько слоев марлю.
Примечание. При применении контрольных методов стеклянный порошок, в случае его отсутствия, может быть заменен кварцевым песком.
в) После центрифугирования жидкость из центрифужных стаканов сливают в один сосуд, перемешивают и получают так называемый первоначальный экстракт.
При применении фильтрования содержимое ступки после настаивания тщательно перемешивают и отфильтровывают не всю жидкость, а только некоторую часть ее, в таком количестве, чтобы в случае надобности возможно было многократное повторение анализа первоначального экстракта.
Из первоначального экстракта берут 10 см и обрабатывают углекислым кальцием и раствором уксуснокислого свинца согласно п.14а настоящего стандарта для растворов, получающихся разведением жидких продуктов.
г) После того как первоначальный раствор или экстракт обработан углекислым кальцием и раствором уксуснокислого свинца, а затем отцентрифугирован или отфильтрован от образовавшегося осадка, через раствор или экстракт пропускают в течение 5 мин быстрый ток сероводорода.
Для более быстрого образования осадка сернистого свинца раствор или экстракт тотчас же после начала пропускания сероводорода энергично взбалтывают. Выпавший осадок отфильтровывают и через фильтрат пропускают для удаления сероводорода ток углекислоты, предварительно пропущенный через дистиллированную воду, налитую в склянку Тищенко.
Полноту удаления сероводорода из раствора или экстракта контролируют фильтровальной бумагой, смоченной насыщенным раствором уксуснокислого свинца, путем сравнения с фильтровальной бумагой, смоченной простой водой.
После удаления сероводорода производят титрование жидкости.
Примечание. Работу с сероводородом необходимо производить в вытяжном шкафу через небольшие форточки, сделанные в дверцах шкафа. Для поглощения отработанного сероводорода употребляют 10%-ный раствор щелочи.
д) 1-10 см жидкости (в зависимости от содержания витамина С) вносят пипеткой в коническую колбу, куда заранее прилит 1 см
2%-ного раствора соляной кислоты, добавляют туда же воду до объема 15 см
и титруют из микробюретки раствором 2,6-дихлорфенолиндофенола, слегка взбалтывая, до появления розового окрашивания, удерживающегося 0,5-1 мин. В случае титрования слабого витаминоносителя раствор 2,6-дихлорфенолиндофенола приливают из микробюретки по каплям; при титровании сильного витаминоносителя вначале приливают по нескольку капель раствора 2,6-дихлорфенолиндофенола сразу. По окончании титрования после записи отсчета по бюретке дополнительно приливают две контрольные капли раствора 2,6-дихлорфенолиндофенола, и если они дадут интенсивное розовое окрашивание, то это указывает, что конец титрования определен правильно.
Примечание. При отсутствии микробюретки допускается применение при титровании градуированной через 0,01 см пипетки.
е) Если при первом титровании пойдет раствора 2,6-дихлорфенолиндофенола больше, чем указано в п.13д настоящего стандарта, надо раствор, из которого отбирают 10 см для обработки углекислым кальцием и уксуснокислым свинцом, развести раствором 5%-ной уксусной кислоты вдвое или более, сколько требуется по первому титрованию. Это вторичное разведение принимают во внимание при конечном подсчете результатов путем введения в числитель формулы дополнительного множителя 2 или более, в зависимости от фактического разведения.
Примечание. Вместо разведения допускается брать для титрования меньшие количества жидкости.
ж) Для каждого анализа делают поправку на реактивы (контрольный опыт). В коническую колбу наливают 1 см 2%-ной соляной кислоты и такое количество воды, чтобы получился объем, который был при титровании исследуемого объекта; затем прибавляют из микробюретки по каплям раствор 2,6-дихлорфенолиндофенола до первого появления розовой окраски. Количество израсходованного раствора 2,6-дихлорфенолиндофенола в миллилитрах является поправкой на реактивы. Поправка обычно составляет для титруемого объема (15 см
) 0,04-0,06 см
и вычитается из объема раствора 2,6-дихлорфенолиндофенола, используемого на титрование экстракта. При увеличении титруемого объема до 20-30 см
производят соответствующее определение поправки.
з) При анализе готовых блюд применяют следующие приемы. Уксуснокислые экстракты картофельных изделий, дающих при анализе мельчайшие взвеси, и экстракты супов-пюре подвергают для получения первоначальных экстрактов центрифугированию в течение не менее 15 мин. Если при обработке сероводородом готовых блюд осадок сернистого свинца проходит через фильтр, допускается разводить первоначальный экстракт вдвое, втрое и более 5%-ной уксусной кислотой и брать для анализа соответственно большее количество разбавленного экстракта (вместо 10 мл), а также пропорционально большее количество углекислого кальция и уксуснокислого свинца с последующим применением для титрования больших количеств жидкости.
и) При анализе сушеных сульфитированных продуктов (картофеля и пр.) навеску переносят в ступку, куда помещают стеклянный порошок в количестве не свыше 10 г. После тщательного растирания в ступке с раствором уксусной кислоты (п.14б настоящего стандарта), взятой не менее чем в 10-кратном, количестве к навеске, и настаивания экстракт центрифугируют (10 или более минут) или фильтруют. Из центрифугатов или фильтратов, обычно очень мутных, берут по 20 см, прибавляют последовательно 1,6 г х.ч. углекислого кальция и 20 см
5%-ного раствора уксуснокислого свинца. После центрифугирования в течение 1-2 мин или фильтрования центрифугат или фильтрат обрабатывают сероводородом, как указано в п.14г настоящего стандарта. Далее поступают, как указано в п.14д настоящего стандарта.
к) Содержание аскорбиновой кислоты (витамина С) (), мг %, вычисляют по формуле
![]() ,
,
где - количество рабочего раствора 2,6-дихлорфенолиндофенола, используемое на титрование, за вычетом поправки на реактивы, см
;
- поправка на титр раствора 2,6-дихлорфенолиндофенола для перевода на точно 0,001 н раствор;
- объем, до которого доведена навеска при прибавлении к ней экстрагирующей жидкости, см
;
- объем анализируемой жидкости, взятой для титрования, см
;
- навеска, г или объем см
;
- объем первоначального раствора или первоначального экстракта (10 см
), взятый для анализа после прибавления к нему 5%-ного раствора уксуснокислого свинца;
- объем (10 см
) первоначального раствора или первоначального экстракта, взятый для анализа, перед его обработкой 5%-ным раствором уксуснокислого свинца;
- количество аскорбиновой кислоты, соответствующее 1 см
точно 0,001 н раствора 2,6-дихлорфенолиндофенола, мг.
Примечание. При применении в ходе анализа вторичного разведения первоначального раствора или первоначального экстракта в числителе проставляют множитель в соответствии с п.14е настоящего стандарта.
Модификация арбитражного метода (без применения сероводорода)
л) Начальную стадию анализа исследуемого объекта производят так, как это изложено в п.14а, б и в настоящего стандарта.
После того как первоначальный раствор или экстракт обработан углекислым кальцием и раствором уксуснокислого свинца и затем отцентрифугирован или отфильтрован от образовавшегося осадка, следует проводить титрование жидкости по способу, изложенному в п.14д настоящего стандарта.
м) В случае анализа окрашенных объектов, когда в результате обработки 5%-ным раствором уксуснокислого свинца согласно п.14а, б и в настоящего стандарта не получаются достаточно обесцвеченные центрифугаты или фильтраты, применяют последовательно один из следующих приемов:
увеличивают объем титруемой жидкости до 30 см вместо 15 см
путем добавления воды;
увеличивают количество уксуснокислого свинца вдвое, втрое или даже вчетверо (10; 15; 20 см) и соответственно увеличивают количество углекислого кальция, т.е. берут 0,8; 1,2; 1,6 г с последующим пропусканием через центрифугат или фильтрат сероводорода (п.14г настоящего стандарта), если обесцвечивание при данных условиях не было достигнуто.
и) Вычисление результатов анализа производят по формуле, приведенной в п.14к настоящего стандарта.
Упрощенный метод
о) Первоначальные растворы и первоначальные экстракты готовят согласно п.14 а, б и в настоящего стандарта с заменой уксусной кислоты раствором 2%-ной соляной кислоты или в случае анализа плодов шиповника и молока - дистиллированной водой. Из таких растворов или экстрактов отбирают пипеткой 1-10 см (в зависимости от содержания витамина С), вносят в коническую колбу вместимостью 50-100 см
, куда заранее налито столько воды, чтобы общий объем жидкости равнялся 15 см
и титруют 0,001 н раствором 2,6-дихлорфенолиндофенола, согласно п.14д настоящего стандарта, без предварительной обработки углекислым кальцием и уксуснокислым свинцом.
Примечание. При анализе нерезко окрашенных продуктов титрование проводят в объеме не 15, а 30 см, изменяя количество добавляемой воды. В этом случае титруют, сравнивая окраску титруемой жидкости с экстрактом, приготовленным для титрования.
п) При анализе сухих плодов шиповника навеску измельченных плодов количественно переносят в ступку с заранее внесенным туда стеклянным порошком (около 5 г), осторожно, тщательно и возможно мельче растирают, подливая небольшими порциями 300 см дистиллированной воды комнатной температуры, и настаивают 10 мин. Из полученной взвеси после тщательного размешивания центрифугируют или фильтруют некоторую часть через слой ваты, вложенный в воронку.
В зависимости от содержания витамина С, руководствуясь первым титрованием, отбирают пипеткой oт 1 до 5 см центрифугата или фильтрата и переносят в колбу вместимостью 50-100 см
, куда заранее прилит 1 см
раствора 2%-ной соляной кислоты, затем прибавляют столько воды, чтобы общий объем жидкости в колбе был 15 см
, и титруют раствором 2,6-дихлорфенолиндофенола согласно п.14д настоящего стандарта.
В случае окрашенности центрифугата или фильтрата, мешающей уловить появление розового окрашивания, а также при высоком содержании витамина С центрифугаты или фильтраты разводят перед титрованием водой вдвое или более.
Примечания:
1. В случае анализа незрелых плодов шиповника вместо воды для экстракции применяют 2%-ную соляную кислоту.
2. При анализе плодов шиповника контрольными методами употребляют не 300 см воды, а вдвое меньше.
р) При анализе молока 5 см молока разводят в 3 раза дистиллированной водой (1:2). При разведении молоко отмеривают пипеткой, а дистиллированную воду наливают из бюретки вместимостью 25-50 см
. 5 см
полученного раствора вносят пипеткой в коническую колбу вместимостью 25-50 см
, куда заранее наливают 1 см
2%-ной соляной кислоты и дистиллированной воды до общего объема в 15 см
; затем, осторожно взбалтывая, содержимое колбы титруют из микробюретки 0,001 и раствором 2,6-дихлорфенолиндофенола, приливая последний каплями до появления слаборозового окрашивания, удерживающегося 0,5-1 мин. Для повторного титрования отбирают пробу из той же порции разведенного молока.
Примечания:
1. Молозиво разводят в 3-6 раз.
2. В случае необходимости хранения молоко должно быть помещено в склянку темного стекла, закрытую пробкой.
с) Содержание аскорбиновой кислоты (витамина С) (), мг %, вычисляют по формуле
![]() ,
,
где обозначения те же, что и в п.14к, настоящего стандарта.
Иодатный метод
т) Экстракцию витамина С из навески производят по методу, указанному в п.14а, б и в настоящего стандарта, с заменой уксусной кислоты раствором 2%-ной соляной кислоты.
В случаях анализа продуктов твердой или густой консистенции из центрифугата или фильтрата солянокислых экстрактов отбирают 1-5 см, в зависимости от содержания витамина С, определяемого первым титрованием, вносят в коническую колбу емкостью 50-100 см
, куда заранее налито 0,5 см
1%-ного раствора иодистого калия, 2 см
0,5%-ного раствора крахмала и столько воды, чтобы общий объем жидкости равнялся 10 см
; затем титруют из микробюретки 0,001 н раствором иодноватокислого калия до появления стойкого слабосинего окрашивания.
Жидкие пробы разводят непосредственно перед титрованием соляной кислотой или водой или титруют без разведения; в последних случаях титрование производят в присутствии 1 см 2%-ной соляной кислоты.
Поправка на реактивы (контрольный опыт) производится следующим образом: в коническую колбу наливают 0,5 см 1%-ного раствора иодистого калия, 2 см
0,5%-ного раствора крахмала, 1 см
2%-ной HCl и столько воды, чтобы общий объем равнялся 10 см
и титруют 0,001 н раствором KJO
до появления слабосинего окрашивания.
Допускается также применение при титровании 0,01 н раствора иодноватокислого калия.
у) Содержание аскорбиновой кислоты (витамина С) (), мг %, вычисляют по формуле
![]() ,
,
где - количество 0,001 н раствора иодноватокислого калия, используемое на титрование, за вычетом поправки на реактивы, см
;
- объем, до которого доведена навеска при прибавлении к ней экстрагирующей жидкости, см
;
- количество экстракта, взятое для титрования, см
;
- поправка на титр иодноватокислого калия для перевода его на 0,001 н раствор, равная 1 при употреблении точно 0,001 н раствора;
- навеска, г или объем, см
;
- количество аскорбиновой кислоты, соответствующее 1 см
точно 0,001 н раствора иодноватокислого калия, мг.
IV. МЕТОДЫ ОПРЕДЕЛЕНИЯ ВИТАМИНА Д
(Биологический метод)
15. Предварительная обработка исследуемого объекта
а) Жидкие продукты (обогащенный витамином рыбий жир, препараты витамина
в масле и в спирте). Среднюю пробу жидких продуктов разводят в растительном масле, не содержащем витамин
(в подсолнечном масле). Для первого разведения берут пипеткой не менее 0,5 см
. Из первого разведения приготовляют путем дальнейшего разведения раствора, предназначенные для непосредственного скармливания крысам.
б) Твердые штучные изделия (драже с витамином Д, поливитаминное драже и т.д.).
Из средней пробы берут для анализа навеску на технохимических весах в количестве 20 г для поливитаминного драже, 15 г для граммового драже и 5 г мелкого драже.
Из навески извлекают витамин Д с помощью серного эфира, используя для этого 1-й раз - 100 см, 2-й, 3-й и 4-й раз - по 50 см
эфира, после чего эфир отгоняют в токе углекислоты. Сухой остаток растворяют в подсолнечном масле.
Из полученного Д-витаминного раствора приготовляют соответствующие растворы, согласно выбранным рабочим дозам.
Разводки хранят в леднике в склянках, закрытых пробками. Каждую склянку снабжают этикеткой с указанием скармливаемой дозы исследуемого препарата.
16. Подготовка крыс
а) Опыты проводятся на белых крысах массой 40-50 г; крысы каждой семьи по возможности равномерно распределяются по группам в количестве не менее 10 в каждой группе.
б) Каждую крысу взвешивают при поступлении на опыт и на 16-й день опыта; при анализе результатов взвешиваний исключаются животные, потерявшие в течение опыта в массе.
в) Каждую крысу метят путем окрашивания различных участков тела.
г) В течение опыта все крысы получают один из следующих рационов:
Рацион N 1 | в процентах |
Пшеничная мука 30 или 72% | 90 |
Сухие пивные или пекарские дрожжи | 5 |
Очищенный мел | 3 |
Пищевая соль | 2 |
Рацион N 2 | в процентах |
Пшеничная мука 30 или 72% | 50 |
Овсяная мука | 45 |
Очищенный мел | 4 |
Пищевая соль | 1 |
Составные части рациона тщательно смешивают с водой, из смеси выпекают хлеб; каждая крыса получает примерно по 20 г хлеба в день.
д) Стандартным раствором витамина Д в опытах служит раствор кальциферола в подсолнечном масле, проверенный по международному эталону.
е) Испытуемые и эталонные растворы приготовляются в четырех разведениях из расчета 3-6-9-12 ин. ед. в 0,05 см (или в 0,1 см
) и скармливаются в этих дозах крысам на 1-й и 8-й день опыта.
ж) Для скармливания животным испытуемого и эталонного растворов применяют пипетку длиной 8-10 см, в которую насасывают масло и выпускают посредством находящегося на одном конце небольшого резинового баллона. Пипетку в зависимости от скармливаемой суточной дозы калибруют на 0,05 или 0,1 см весовым путем.
17. Методы определения степени рахита
а) Рентгенографический метод. Производят рентгенографическую съемку задних конечностей живых или убитых крыс; конечности при этом необходимо несколько вытягивать. Степень рахита определяют по ширине хрящевой зоны метафизов большеберцовых костей.
б) Проба на черту. При отсутствии рентгеновской установки у убитых крыс вырезают большеберцовые кости, которые после очистки от мышц помещают в 4%-ный раствор формалина на 18-24 ч. Промыв кости в течение 5-10 мин водой, их рассекают продольно по медиальной линии при помощи острого лезвия и погружают в 0,5%-ный раствор азотнокислого серебра. После этого кости выставляют на свет (солнечный или яркой электролампы) на 10-20 мин и, как только появится на поверхности разреза серая окраска средней интенсивности, быстро промывают водой и переносят в 5%-ный раствор гипосульфита на 10-15 мин; вновь быстро промывают водой. На поверхности разреза в области зоны эпифизарного окостенения на темном фоне костной ткани резко выделяется светлая полоса хряща. Степень рахита определяют по ширине этой хрящевой зоны при помощи бинокулярной лупы с рисовальным аппаратом Аббе. С каждой кости на отдельную карточку зарисовывают карандашом контуры хрящевой зоны верхнего эпифиза и производят оценку степени рахита.
18. Определение минимальной профилактической дозы. Под минимальной профилактической дозой как исследуемой пробы, так и эталона витамина Д понимается минимальное количество препарата, предохраняющее от рахита не менее 80% крыс данной группы.
Определив, какая доза исследуемой пробы эталона витамина Д соответствует минимальной профилактической, вычисляют содержание витамина Д в ин. ед. () в 1 см
(или 1 г) исследуемой пробы по следующей формуле
![]() ,
,
где - минимальная профилактическая доза исследуемой пробы, см
или г;
- минимальная профилактическая доза эталона витамина Д в ин. ед.
V. МЕТОД ОПРЕДЕЛЕНИЯ ВИТАМИНА Д
(Химический метод)
19. Применяемые реактивы
а) Хлороформ химически чистый, не содержащий примесей. Хлороформ промывают три-четыре раза дистиллированной водой, отделяют от воды, высушивают свежепрокаленным сернокислым натром или хлористым кальцием в течение суток и отгоняют на водяной бане при температуре 61 °С. Хранить в склянках темного стекла с притертой пробкой не более 1 мес.
Хлороформ подвергают испытанию на его пригодность для анализа при помощи следующих реакций:
1) Реакция на наличие свободного хлора в хлороформе: 10 см хлороформа взбалтывают в 40 см
дистиллированной воды. Водный слой отделяют. 10 см
водного слоя не должны окрашиваться в синий цвет при добавлении 0,5 см
бесцветного 10%-ного раствора иодата калия и 0,5 см
раствора крахмала.
2) Реакция на содержание спирта: хлороформ, не содержащий спирта, не восстанавливает 0,1%-ный раствор перманганата калия (при проведении реакции дают постоять 15 мин).
3) Реакция на содержание воды: при охлаждении до 3-4 °С 10 см хлороформа не должны давать мути.
4) Реакция на содержание органических примесей: 15 см хлороформа и 10 см
концентрированной серной кислоты помещают в склянку с притертой пробкой (склянка предварительно ополаскивается 2 раза хлороформом и 2 раза серной кислотой); после стояния в течение одного часа при 15 °С в слое серной кислоты не должно появляться окрашивания.
б) Раствор треххлористой сурьмы. Продажную треххлористую сурьму промывают очищенным хлороформом, пока не будет стекать прозрачный и бесцветный раствор и высушивают в эксикаторе над серной кислотой в течение 1-2 сут в темноте. Насыщенный раствор треххлористой сурьмы в очищенном хлороформе приготовляют при 20 °С.
в) Серный эфир. Очистка эфира от перекисей производится следующим образом: в склянку помещают 500 см эфира, 50 см
4%-ного раствора KMnO
, 5 см
40%-ного раствора NaOH или KOH, взбалтывают и оставляют на сутки в темноте. Смесь переносят в делительную воронку, нижний слой сливают, эфир промывают 5-6 раз дистиллированной водой (в отношении 2:1), высушивают в течение суток сернокислым натрием, перегоняют и сохраняют в темноте. Эфир необходимо проверять на отсутствие перекисей по качественной реакции с раствором йодистого калия.
г) Ацетилхлорид. Температура кипения 51 °С. Сохранять в склянке или капельнице из темного стекла с притертой пробкой.
д) Спирт этиловый. Для освобождения спирта от альдегидов настаивают в течение 10-12 ч над твердым NaOH (5-10 г на 1 дм спирта) и перегоняют.
е) Натрий сернокислый кристаллический, безводный х.ч. или свежепрокаленный.
ж) Кали едкое, 10%-ный спиртовой раствор.
з) Фенолфталеин, 1%-ный спиртовой раствор.
и) Углекислый газ, полученный в аппарате Киппа или из баллона, или азот.
к) Иодистый калий, 10%-ный раствор.
Все применяемые реактивы должны соответствовать требованиям действующих стандартов или технических условий.
20. Необходимая аппаратура
Электроколориметр или штуфенфотометр.
21. Составление калибровочной кривой
Для построения калибровочной кривой применяют чистый, кристаллический кальциферол, растворенный в хлороформе, содержащий 10000 ин. ед. витамина Д в 10 см (1 ин. ед. витамина Д соответствует 0,025
г чистого кальциферола). Хлороформенный раствор витамина Д хранится не более одного дня.
Измерение интенсивности окраски хлороформенного раствора кристаллического кальциферола с треххлористой сурьмой проводят в электрофотоколориметре со светофильтром 480-530 м или штуфенфотометре.
Для приведения электрофотоколориметра к нулю пользуются хлороформенным раствором треххлористой сурьмы. Для получения точек калибровочной кривой берут 1,0; 0,75; 0,5 и 0,25 см стандартного раствора витамина Д, доводят хлороформом до объема 1 см
, добавляют по 3 капли ацетилхлорида и 6 см
раствора треххлористой сурьмы. Отношение испытуемого раствора к раствору треххлористой сурьмы составляет 1:6.
Общий объем реакционной смеси должен составлять 7 см.
Точно через 4 мин после добавления последнего реактива измеряют получаемые окраски и величины экстинкции откладывают на графике по оси ординат, а соответствующие концентрации витамина Д - по оси абсцисс.
В пределах содержания витамина Д от 200 до 1000 ин. ед. в 1 см на графике получается прямая линия.
22. Определение витамина Д в масляных концентратах с омылением
Навеску концентрата в количестве 0,5-2 г, в зависимости от содержания витамина Д, взятую на аналитических весах, помещают в колбу, снабженную обратным холодильником, и подвергают омылению на водяной бане при помощи 10 см 10%-ного спиртового раствора KOH в течение 1 ч при температуре кипения спирта.
По охлаждении смеси к ней добавляют двойной объем дистиллированной воды, переносят в делительную воронку, смывают из колбы небольшими порциями воды и неомыляемую фракцию извлекают трижды серным эфиром, свободным от перекиси, используя первый раз 50 см, второй и третий раз по 25 см
эфира.
Объединенные эфирные вытяжки тщательно промывают водой до полного удаления щелочи (проба на лакмусовую бумагу или фенолфталеин), отделяют водный слой, к эфирному экстракту добавляют 5-7 г свежепрокаленного сернокислого натрия и взбалтыванием колбы с содержимым высушивают вытяжку до полной прозрачности (в течение 15-20 мин).
Эфирную вытяжку фильтруют через бумажный фильтр; колбу и фильтр ополаскивают небольшим количеством эфира, сливая последний через тот же фильтр, и отгоняют эфир на водяной бане в токе углекислого газа или азота до получения сухого остатка. Остаток растворяют в чистом сухом хлороформе в мерной колбе подходящего объема с таким расчетом, чтобы в 1 см раствора содержалось не менее 200 и не более 1000 ин. ед. витамина Д.
Из полученного раствора отбирают точно 1 см в кювету электрофотоколориметра или штуфенфотометра, туда же добавляют 3 капли ацетилхлорида и 6 см
хлороформенного раствора треххлористой сурьмы (отношение 1:6) и точно через 4 мин после добавления последнего реактива измеряют интенсивность образовавшейся окраски с указанным выше светофильтром.
В качестве контроля для установки прибора на нуль используют раствор, состоящий из 1 см хлороформа, 6 см
раствора треххлористой сурьмы и 3 капель ацетилхлорида.
Расчет содержания витамина Д в 1 см концентрата (
) в ин. ед. производят по формуле
![]() ,
,
где - найденное по калибровочной кривой количество витамина Д в 1 см
раствора, ин. ед.;
- разведение см
;
- навеска концентрата, г;
- плотность концентрата.
При определении витамина Д в масляных растворах с содержанием до 10000 ин. ед. в 1 см надо вносить поправку на вещества, содержащиеся в неомыляемой фракции рафинированного подсолнечного масла, дающие такую же, как витамин Д, окраску с треххлористой сурьмой. При вычислении содержания витамина Д в вышеуказанных масляных растворах уменьшают полученные данные на 25%.
23. Определение витамина Д в спиртовых концентратах
Высокоактивные спиртовые концентраты предварительно разбавляют спиртом с таким расчетом, чтобы в 1 см содержалось не более 10000 ин. ед. витамина Д. Из разбавленного спиртового раствора отбирают пипеткой 1 см
, помещают в небольшую колбу Вюрца и отгоняют растворитель на водяной бане досуха в токе углекислого газа или азота.
Полученный остаток растворяют в хлороформе и переносят в мерную колбу такого объема, чтобы в 1 см раствора содержалось от 200 до 1000 ин. ед. витамина Д.
Из полученного раствора отбирают 1 см, помещают в кювету электрофотоколориметра или штуфенфотометра, туда же добавляют 3 капли ацетилхлорида и 6 см
хлороформенного раствора треххлористой сурьмы. Точно через 4 мин после добавления последнего реактива измеряют интенсивность образовавшейся окраски с указанным выше светофильтром.
В качестве контроля для установки прибора на нуль используют раствор, состоящий из 1 см хлороформа, 6 см
раствора треххлористой сурьмы и 3 капель ацетилхлорида.
Расчет содержания витамина Д в 1 см концентрата (
) в ин. ед. производят по формуле
![]() ,
,
где - найденное по калибровочной кривой количество витамина Д в 1 см
раствора, ин. ед.;
- разведение, см
;
- количество взятого на анализ концентрата, см
.
VI. МЕТОДЫ ОПРЕДЕЛЕНИЯ ВИТАМИНА В
24. Применяемые реактивы
а) Тиаминхлорид-гидрохлорид 100%-ной чистоты с температурой плавления 246-250 °С. Если нужно, его перекристаллизовывают 2 раза из 80%-ного этилового спирта, высушивают в вакуум-эксикаторе над серной кислотой и хранят в эксикаторе, в темноте.
б) Красная кровяная соль. После перекристаллизации из дистиллированной воды из нее перед анализом приготовляют 1%-ный водный раствор.
в) Натр едкий х.ч. 30%-ный водный раствор; 15%-ный водный раствор.
г) Окислительная смесь: перед анализом смешивают 4 см 1%-ного раствора красной кровяной соли с 96 см
15%-ного раствора едкого натра.
д) Изобутиловый спирт с температурой кипения 107-108 °С, изоамиловый спирт с температурой кипения 130 °С или бутиловый спирт с температурой кипения 117 °С (употребляется любой из указанных спиртов).
е) Этиловый спирт, ректификованный 96%-ный.
Примечание. В случае наличия флуоресценции спирты перед употреблением подвергают очистке: к 1 дм спирта прибавляют 15-20 г активированного угля, встряхивают 15 мин, оставляют на сутки, повторяя несколько раз встряхивание. Декантируют, фильтруют, высушивают над хлористым кальцием и перегоняют при соответствующей температуре (перегонку изобутилового, изоамилового и бутилового спирта ведут на глицериновой или песчаной бане, а этилового - на водяной бане).
ж) Натрий сернокислый безводный, прокаленный.
з) Калий марганцовокислый, х.ч.
и) Серебро азотнокислое, 10%-ный раствор.
Все применяемые реактивы должны соответствовать требованиям действующих стандартов или технических условий.
25. Необходимая аппаратура
Флуороскоп: ртутно-кварцевая лампа ПРК-2 или ПРК-4, помещенная в кожух с черным никелевым светофильтром с набором пробирок одинакового диаметра из однородного нефлуоресцирующего стекла.
26. Описание определения
Флуороскопический визуальный метод
Приготовление шкалы стандартных растворов
100 мг чистого кристаллического тиаминхлорида-гидрохлорида растворяют в дистиллированной воде в мерной колбе, вместимостью 1 дм. Раствор хранят в склянке темного стекла с притертой пробкой на холоде не более 1 мес. Из этого раствора приготовляют путем разведения стандартные рабочие растворы в мерных колбах вместимостью 100 см
по табл.3.
Таблица 3
Номер стандартного раствора | Количество основного раствора, см | Количество добавленной воды, см | Содержание витамина |
1 | 0,1 | до 100 | 0,1 |
2 | 0,2 | " 100 | 0,2 |
3 | 0,3 | " 100 | 0,3 |
4 | 0,4 | " 100 | 0,4 |
5 | 0,5 | " 100 | 0,5 |
6 | 0,6 | " 100 | 0,6 |
7 | 0,7 | " 100 | 0,7 |
8 | 0,8 | " 100 | 0,8 |
9 | 0,9 | " 100 | 0,9 |
10 | 1,0 | " 100 | 1,0 |
11 | 1,2 | " 100 | 1,2 |
12 | 1,4 | " 100 | 1,4 |
13 | 1,6 | " 100 | 1,6 |
14 | 1,8 | " 100 | 1,8 |
15 | 2,0 | " 100 | 2,0 |
Из каждого рабочего стандартного раствора отбирают по 1 см в капельные или делительные воронки вместимостью 50 см
или в цилиндры с притертой пробкой и добавляют при помощи градуированных пипеток по 4 см
дистиллированной воды, по 3 см
окислительной смеси, сильно встряхивают, прибавляют 12 см
изобутилового или другого рекомендованного спирта из бюретки и встряхивают в течение 2 мин. После разделения водной и спиртовой фаз, водную нижнюю фазу удаляют, а в спиртовую фазу добавляют около 0,5 г сернокислого натрия или 2 см
этилового спирта. После перемешивания и отстаивания отбирают 10 см
этого раствора и переносят в пробирку для последующего определения флуоресценции. Пробирку закрывают корковой пробкой и сохраняют в темноте.
Примечание. Стандартные растворы тиохрома могут храниться в темноте на холоде в течение двух дней.
Определение в кристаллическом препарате, в драже и таблетках
При анализе витаминных препаратов (кристаллический витамин , драже и таблетки) вначале необходимо выяснить природу галоида, входящего в состав молекулы витамина; для этого проводят качественную реакцию на ионы брома и хлора с раствором азотнокислого серебра; при наличии хлорида выпадает белый, при наличии бромида - желтоватый осадок.
Примечание. В случае анализа витаминных препаратов, содержащих аскорбиновую кислоту, реакцию проводят после предварительного озоления препарата в присутствии поташа или концентрированной серной кислоты.
Навеску кристаллического препарата, предварительно высушенного в вакуум-эксикаторе над концентрированной серной кислотой в течение не менее 12 ч, берут на аналитических весах в количестве 100 мг и растворяют в 1 дм дистиллированной воды.
При анализе драже или таблеток взвешивают 30-50 шт. и определяют массу одной штуки как среднее арифметическое. Отобранную пробу тщательно растирают в ступке и из растертой массы берут на аналитических весах две параллельные навески, около 1 г каждая. Навески количественно переносят в мерные колбы вместимостью на 100 см, растворяют в воде и доводят раствор до метки; при наличии мути раствор фильтруют.
Из полученных основных растворов (кристаллического препарата, драже или таблеток) приготовляют путем дополнительного разведения рабочие растворы с таким расчетом, чтобы в растворе, поступающем на окисление, содержалось от 1 до 2 витамина
в 1 см
. Из полученных растворов отбирают три пробы по 1 см
в делительные или капельные воронки или в цилиндры с притертыми пробками, туда же добавляют по 4 см
дистиллированной воды и перемешивают. К двум пробам добавляют по 3 см
окислительной смеси, к третьей (контрольной) - 3 см
15%-ного раствора щелочи.
Пробы встряхивают, добавляют в каждую по 12 см изобутилового, изоамилового или бутилового спирта и встряхивают 2 мин, после чего спиртовому и водному слою дают отстояться. Водную нижнюю фазу удаляют, а в спиртовую добавляют около 0,5 г сернокислого натрия или 2 см
этилового спирта и после перемешивания отбирают 10 см
в пробирки для определения флуоресценции. Окисленные растворы необходимо предохранять от яркого света и тотчас проводить измерение интенсивности флуоресценции.
Пробирки с испытуемыми растворами помещают перед светофильтром ртутно-кварцевой лампы и сравнивают интенсивность флуоресценции со шкалой стандартов: для этого одну из пробирок с испытуемым раствором помещают между двумя соседними пробирками со стандартными растворами, останавливаясь на той, которая по интенсивности флуоресценции совпадает с испытуемой. Если интенсивность флуоресценции окажется между интенсивностью флуоресценции соседних стандартных пробирок, то берется средняя величина.
Аналогично поступают с растворами контрольного опыта (пробирки с раствором без окислителя).
Содержание (чистота) витамина в кристаллических препаратах (
) в процентах вычисляют по формуле
![]() ,
,
где - количество витамина
в 1 см
стандартного раствора в
, флуоресценция которого совпадает с флуоресценцией испытуемого раствора;
- количество витамина
в 1 см
стандартного раствора в
, флуоресценция которого совпадает с флуоресценцией контрольного опыта;
- навеска, г.;
- объем, в котором растворена навеска, см
;
- объем, взятый для приготовления рабочего раствора, см
;
- конечный объем рабочего раствора, см
;
- объем рабочего раствора, взятый на окисление, см
;
- пересчет на проценты.
Содержание витамина на одну штуку драже или таблеток (
), мг, вычисляют по формуле
![]() ,
,
где - средняя масса одной штуки драже или таблеток, г;
- коэффициент пересчета, мг.
(Остальные обозначения см. выше).
Точность метода ±5%.
Примечание. В случае анализа препаратов с тиаминбромидом-гидробромидом необходимо ввести коэффициент 1,29, умножая на него полученный результат.
VII. МЕТОДЫ ОПРЕДЕЛЕНИЯ ВИТАМИНА В
27. Применяемые реактивы
а) Рибофлавин кристаллический.
б) Стандартный раствор рибофлавина (А): 40 мг кристаллического рибофлавина растворяют в воде в мерной колбе вместимостью 1 дм при нагревании (или 10 мг на 250 см
), по охлаждении раствор доводят до метки. 1 см
раствора содержит 40
рибофлавина. Раствор хранят в темноте на холоде не более 1 мес.
Перед анализом приготовляют рабочий раствор (Б), для чего 20 см раствора А помещают в мерную колбу вместимостью на 100 см
и доводят водой до метки.
В 1 см раствора Б содержится 8
рибофлавина.
28. Необходимая аппаратура:
Колориметр типа Дюбоска или электрофотоколориметр.
29. Описание определения
Колориметрический метод определения в кристаллических препаратах, драже и таблетках
При анализе кристаллических препаратов берут навеску препарата в количестве 40 мг на 1 дм (или 10 мг на 250 см
) дистиллированной воды и растворяют так, как указано при приготовлении стандартных растворов.
При анализе драже или таблеток взвешивают 30-50 шт. и определяют массу одной штуки, как среднее арифметическое. Отобранную пробу тщательно растирают в ступке и из растертой массы берут на аналитических весах 2 параллельные навески, около 1 г каждая. Навески растворяют в дистиллированной воде в мерных колбах вместимостью 100 или 250 см (в зависимости от предполагаемого содержания витамина
) с таким расчетом, чтобы в 1 см
раствора содержалось около 8
витамина
. Растворы доводят до метки водой, фильтруют и интенсивность окраски измеряют в колориметре типа Дюбоска, сравнивая со стандартным раствором витамина
, или в электрофотоколориметре.
При использовании электрофотоколориметра применяют светофильтр 470 м и составляют калибровочную кривую по стандартному раствору, разведенному дистиллированной водой до различных концентраций по принципу, изложенному в разделе II (1
, 2
, 3
, 4
, 5
, 6
и 8
в 1 см
).
Содержание (чистота) витамина в кристаллических препаратах (
) в процентах вычисляют по формуле:
При использовании колориметра типа Дюбоска
![]() ,
,
где - количество витамина
в 1 см
стандартного раствора,
;
- высота столба стандартного раствора, мм;
- высота столба испытуемого раствора, мм;
- навеска, г:
- объем раствора, в котором растворена навеска, см
;
- пересчет на проценты.
При использовании электрофотоколориметра
![]() ,
,
где - содержание рибофлавина,
, найденное по калибровочной кривой для испытуемого раствора. (Остальные обозначения см. выше).
Содержание витамина на 1 шт. драже или таблеток (
), мг, вычисляют по формуле
При использовании колориметра типа Дюбоска
![]() ,
,
где - средняя масса драже или таблеток, г;
- пересчет в миллиграммы.
(Остальные обозначения см. выше).
При использовании электрофотоколориметра
![]() .
.
(Обозначения см. выше).
Точность метода ±5%.
VIII. МЕТОД ОПРЕДЕЛЕНИЯ ВИТАМИНА РР
30. Применяемые реактивы
а) Стандартный раствор витамина РР: 20 мг чистой никотиновой кислоты растворяют в 100 см дистиллированной воды. 1 см
раствора содержит 0,2 мг никотиновой кислоты. Дальнейшую обработку раствора ведут параллельно с испытуемым раствором, как описано ниже.
б) Стандартный раствор никотинамида, содержащий 0,2 мг в 1 см. Приготовляется так же, как предыдущий раствор.
в) Натр едкий х.ч., 0,5 н раствор и 0,1 н раствор, приготовленные по общим правилам объемного анализа.
г) Фенолфталеин, 1%-ный спиртовый раствор.
д) Бромтимоловый синий, 1%-ный раствор.
е) Медь сернокислая CuSO·5H
O, перекристаллизованная, 10%-ный раствор.
ж) Кислота соляная ч., уд. в. 1,19, разведенная водой в отношении 1:1.
з) Калий иодистый х.ч.
и) Гипосульфит натрия, 0,1 н раствор, приготовленный по общим правилам объемного анализа.
к) Крахмал, 0,5%-ный раствор.
л) Кислота серная ч., 0,1 н раствор, приготовленный по общим правилам объемного анализа.
м) Натр едкий х.ч., 40%-ный раствор.
н) Метиловый оранжевый, 0,5%-ный раствор.
о) Роданбромидный раствор: 4 см брома вносят в 100 см
дистиллированной воды, энергично встряхивают и отстоявшийся раствор декантируют. К охлажденной на льду бромной воде (взятой в количестве, нужном для анализа) прибавляют по каплям 10%-ный раствор роданистого аммония или роданистого калия до светло-желтого окрашивания, затем 1%-ный раствор до полного обесцвечивания. К обесцвеченному раствору добавляют небольшими порциями 20-50 мг углекислого кальция до прекращения выделения СО
и образования мути. Раствор фильтруют в склянку темного стекла с притертой пробкой и сохраняют на холоде. Годен в день приготовления.
п) Калий роданистый или аммоний роданистый, 10%-ный раствор.
р) Кальций углекислый х.ч.
с) Бром х.ч., плотность 3,2 (хранить с особой предосторожностью).
т) Анилин, спиртовой раствор. Один объем свежеперегнанного над цинковой пылью анилина растворяют в шести объемах 96%-ного этилового спирта. Раствор должен быть бесцветным (хранят в склянке темного стекла с притертой пробкой в темноте на холоде).
у) Спирт этиловый 96%-ный ректификованный.
Все применяемые реактивы должны соответствовать требованиям действующих стандартов или технических условий.
31. Необходимая аппаратура
Колориметр типа Дюбоска или электрофотоколориметр.
32. Описание определения
Перед анализом необходимо определить, содержит ли испытуемый препарат никотиновую кислоту или никотинамид. Для этого небольшое количество испытуемого кристаллического препарата или мелко измельченного в ступке драже помещают в пробирку, добавляют 1-2 см воды,
объема 40%-ного раствора едкого натра и содержимое пробирки нагревают на кипящей водяной бане около 2 мин. В присутствии никотинамида появляется запах аммиака. При наличии никотиновой кислоты определение проводят по прописи, указанной в подпункте а, при наличии никотинамида определение проводят согласно подпункту б.
а) Объемный метод определения никотиновой кислоты в кристаллическом препарате, в драже и в таблетках
При анализе драже и таблеток взвешивают 30-50 шт. и определяют вес 1 шт. как среднее арифметическое. Отобранную пробу тщательно растирают в ступке.
Точную навеску около 0,5 г кристаллической никотиновой кислоты или соответствующую навеску растертого драже или таблеток, содержащую около 0,5 г витамина РР, растворяют в 25 см свежепрокипяченной горячей дистиллированной воды, по охлаждении нейтрализуют 0,5 н и под конец 0,1 н раствором едкого натра, переводят в мерную колбу вместимостью 100 см
и прибавляют 10 см
10%-ного раствора сернокислой меди, доводят водой до метки, перемешивают и через 10-15 мин фильтруют.
Нейтрализацию раствора никотиновой кислоты и драже с витамином РР проводят по бромтимоловому синему до первого изменения цвета.
В коническую колбу с притертой пробкой отбирают 50 см фильтрата, прибавляют 10 см
разведенной соляной кислоты (1:2), 2 г иодистого калия, колбу закрывают пробкой и оставляют в темноте на 10 мин.
Выделившийся иод титруют 0,1 н раствором гипосульфита натрия (индикатор - крахмал). В другую коническую колбу с притертой пробкой отбирают 5 см 10%-ного раствора CuSO
прибавляют 10 см
разведенной НСl (1:2), 2 г KJ и через 10 мин выделившийся иод титруют 0,1 н раствором гипосульфита (индикатор - крахмал).
Содержание (чистота) никотиновой кислоты в кристаллическом препарате () в процентах вычисляют по следующей формуле
![]() ,
,
где - объем точно 0,1 н раствора гипосульфита, используемый на титрование контрольной пробы, см
;
- объем точно 0,1 н раствора гипосульфита, используемый на титрование испытуемой пробы, см
;
![]() - количество никотиновой кислоты, соответствующее 1 см
- количество никотиновой кислоты, соответствующее 1 см 0,1 н раствора гипосульфита, г;
- навеска, г;
- объем, до которого доведена навеска, см
;
- количество раствора, взятое для титрования, в см
;
- пересчет на проценты.
Содержание никотиновой кислоты на 1 шт. драже или таблеток (), мг, вычисляют по следующей формуле
![]() ,
,
где - средняя масса 1 шт. драже или таблеток, г;
- объем, до которого доведена навеска, см
;
- количество раствора, взятого на титрование, в см
;
![]() - количество никотиновой кислоты, соответствующее 1 см
- количество никотиновой кислоты, соответствующее 1 см 0,1 н раствора гипосульфита, мг.
(Остальные обозначения см. выше).
Точность метода ±1%.
б) Определение никотинамида в кристаллическом препарате, в драже и таблетках
Навеску кристаллического препарата около 0,2 г или соответствующую навеску мелко измельченного драже, содержащую около 0,2 г никотинамида, взятую на аналитических весах, переносят в круглодонную колбу, вместимостью 100-120 см, прибавляют 75 см
воды, колбу закрывают резиновой пробкой с двумя отверстиями, в которые вставлены каплеуловитель и капельная воронка. Каплеуловитель соединяют с холодильником Либиха, на форштосс которого надета трубка, погруженная в точно отмеренное в приемной колбе количество (25 см
) 0,1 н раствора серной кислоты с 1-2 каплями метилового оранжевого.
Через капельную воронку в колбу для отгона наливают 25 см 40%-ного раствора едкого натра, быстро закрывают кран воронки, и содержимое колбы нагревают до кипения. Отгонку жидкости в колбе ведут до
первоначального объема.
Избыток внесенной в приемник серной кислоты оттитровывают 0,1 н раствором едкого натра до появления соломенно-желтого окрашивания.
Содержание (чистота) никотинамида в исследуемом препарате () в процентах вычисляют по следующей формуле:
![]() ,
,
где - объем точно 0,1 н раствора серной кислоты, взятый в приемник, см
;
- объем точно 0,1 н раствора едкого натра, используемый на титрование, см
;
![]() - количество никотинамида, соответствующее 1 см
- количество никотинамида, соответствующее 1 см 0,1 н раствора едкого натра, г;
- навеска, г;
- пересчет на проценты.
Содержание никотинамида на 1 шт. драже или таблеток (), мг, вычисляют по следующей формуле
![]() ,
,
где - количество никотинамида, соответствующее 1 см
0,1 н раствора едкого натра, мг;
- средняя масса драже, г.
(Остальные обозначения см. выше).
Точность метода ±1%.
в) Определение никотиновой кислоты или никотинамида в поливитаминном драже
Взвешивают на технохимических весах 30-50 шт. драже для установления средней массы 1 шт. и растирают в ступке. На аналитических весах берут навеску измельченной массы в количестве около 2 г, помещают ее в стаканчик и растворяют при нагревании в небольшом количестве воды. По охлаждении раствор переносят в мерную колбу вместимостью 50 см, доводят до метки водой, перемешивают и фильтруют.
В фильтрате определяют содержание рибофлавина (см. метод определения витамина ).
Установленную концентрацию рибофлавина создают в стандартном растворе никотиновой кислоты, который будет употреблен при анализе, внося в него соответствующее количество рибофлавина.
После этого приступают к определению никотиновой кислоты. 5 см стандартного раствора с прибавленным рибофлавином и 5 см
фильтрата помещают в конические колбы с притертыми пробками или в пробирки с притертыми пробками, вместимостью 20-50 см
, подогревают на водяной бане до 60-70 °С и прибавляют в каждую из них по 2 см
роданбромидного раствора, продолжая подогрев в течение 5-10 мин, после охлаждения в каждую пробирку прибавляют по 7 см
анилина, хорошо перемешивают и оставляют стоять в темноте в течение 30 мин. По истечении указанного времени содержимое колб или пробирок профильтровывают или осторожно сливают с осадка и образовавшуюся желтую окраску колориметрируют в колориметре типа Дюбоска или в электрофотоколориметре.
Содержание никотиновой кислоты на 1 шт. драже (), мг, вычисляют по следующей формуле:
При пользовании колориметром типа Дюбоска
![]() ,
,
где - количество витамина РР в 1 см
стандартного раствора, мг;
- высота столба стандартного раствора, мм;
- высота столба испытуемого раствора, мм;
- навеска, г;
- объем раствора, в котором растворена навеска, см
;
- средняя масса 1 шт. драже;
- пересчет в миллиграммы.
При пользовании электрофотоколориметром
![]() ,
,
где - содержание никотиновой кислоты или никотинамида, мг, найденное по калибровочной кривой для испытуемого раствора.
(Остальные обозначения см. выше).
Точность метода ±5%.
Примечание. Роданбромидный метод может быть распространен на кристаллические препараты никотиновой кислоты и никотинамида.
При анализе ампулированных препаратов, содержащих витамин и никотиновую кислоту или никотинамид, применяют тот же метод.
IX. МЕТОДЫ ИСПЫТАНИЯ КАЧЕСТВА ВИТАМИННЫХ ПРЕПАРАТОВ
33. Настоящий раздел стандарта распространяется на методы испытания органолептических и физико-химических показателей качества таблеток и драже, содержащих витамины.
К ним относятся сахарные таблетки с витаминами синтетическими и из естественного сырья, глюкозные таблетки с синтетическими витаминами и драже с синтетическими витаминами из естественного сырья.
34. Определение цвета, запаха и вкуса (органолептически).
Определение цвета производят при дневном освещении на белой поверхности.
Для оценки запаха чистую склянку с притертой пробкой заполняют на растертым продуктом, закрывают пробкой и через 30 мин, открыв склянку, определяют запах на уровне края горла склянки.
Вкус определяют в нерастертой пробе продукта.
35. Определение влажности
а) В глюкозных таблетках
2-3 г измельченной средней пробы таблеток отвешивают на аналитических весах в чистую, предварительно высушенную и взвешенную бюксу и помещают в сушильный шкаф. Высушивание навески производят в течение 2 ч при температуре 55 °С и в течение 1 ч при температуре 60-70 °С, после чего бюксу с содержимым охлаждают в эксикаторе и взвешивают.
Повторное высушивание производят в течение 1 ч при 60-70 °С и вновь взвешивают. Все последующие взвешивания производят через каждые 30 мин высушивания. Постоянная масса считается достигнутой, если разница между двумя взвешиваниями не будет превышать 0,002 г.
Содержание влаги () в процентах вычисляют по формуле
![]() ,
,
где - масса бюксы с навеской до высушивания, г;
- масса бюксы с навеской после высушивания, г;
- навеска, г.
б) В сахарных драже и таблетках
В чистую, сухую бюксу помещают 7-10 г очищенного прокаленного кварцевого песка или стеклянного порошка и стеклянную палочку и высушивают в сушильном шкафу в течение 1 ч при температуре 103-105 °С. Высушенную бюксу охлаждают в эксикаторе и взвешивают с точностью до 0,001 г. Затем в бюксу помещают около 5 г измельченной средней пробы, закрывают крышкой и вновь взвешивают с той же точностью.
После взвешивания открывают крышку бюксы и навеску тщательно смешивают с песком при помощи палочки.
Открытую бюксу с содержимым помещают в сушильный шкаф и высушивают в течение 3-4 ч при температуре 95-100 °С.
После высушивания бюксу вынимают из сушильного шкафа, предварительно закрыв крышкой, охлаждают в эксикаторе и взвешивают. Последующие взвешивания производят через каждые 1-2 ч высушивания, пока разница между двумя взвешиваниями не будет менее 0,005 г.
Содержание влаги () в процентах вычисляют по формуле
![]() ,
,
где - масса бюксы с песком, палочкой и навеской до высушивания, г;
- масса бюксы с песком, палочкой и навеской после высушивания, г;
- масса бюкса с песком и палочкой, г.
36. Определение содержания золы, нерастворимой в 10%-ной соляной кислоте
Отвешивают на аналитических весах пробу в количестве 10-15 г и помещают ее в предварительно прокаленный и взвешенный фарфоровый тигель. Навеску в тигле осторожно обугливают и озоляют (для ускорения сжигания добавляют несколько капель 30%-ной перекиси водорода).
Полученную золу растворяют в том же тигле в 20 см 10%-ной соляной кислоты.
Раствор выпаривают на кипящей водяной бане досуха, остаток высушивают в течение одного часа в сушильном шкафу или на песчаной бане при температуре 105-130 °С. К высушенному в тигле остатку вновь добавляют 20 см 10%-ной соляной кислоты и нагревают в течение 10 мин на водяной бане, растирая стеклянной палочкой. Раствор фильтруют через беззольный фильтр, промывают сначала три раза горячей разбавленной соляной кислотой (одна часть кислоты плотностью 1,19 на 6 частей воды) и затем горячей дистиллированной водой до исчезновения реакции на хлор с азотнокислым серебром.
Фильтрат и промывные воды сливают вместе и сохраняют для определения солей тяжелых металлов. Фильтр же с остатком сжигают в том же тигле и затем прокаливают до постоянного веса в тигельной или муфельной печи при красном калении, охлаждают в эксикаторе и взвешивают.
Количество золы, нерастворимой в 10%-ной соляной кислоте, () в процентах вычисляют по формуле
![]() ,
,
где - масса тигля с остатком золы, нерастворимой в 10%-ной соляной кислоте, г;
- масса пустого прокаленного тигля, г;
- масса тигля с навеской до сжигания, г.
37. Определение наличия солей тяжелых металлов (меди, олова, свинца)
Солянокислую вытяжку и промывные воды, собранные при определении содержания золы, помещают в фарфоровую чашку и выпаривают на водяной бане досуха. Сухой остаток растворяют в 10 см 1,5%-ного водного раствора соляной кислоты, продолжая нагревание на водяной бане 10 мин. Горячий раствор переносят в пробирку, добавляют 2-3 см
свежеприготовленной сероводородной воды, затем пробирку закрывают пробкой, встряхивают и оставляют на 10 мин.
По истечении указанного срока в случае отсутствия тяжелых металлов в пробирке не должно появляться окрашивания жидкости или образование осадка сульфидов.
Электронный текст документа
и сверен по:
М.: Издательство стандартов, 1994

{kind=link}