ГОСТ Р 50002-92
(ИСО 894-77)
Группа Л29
ГОСУДАРСТВЕННЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ВЕЩЕСТВА ПОВЕРХНОСТНО-АКТИВНЫЕ
Алкилсульфаты натрия первичные технические. Методы анализа
Surface active agents. Primary technical sodium alkvlsulphates. Methods of analysis
ОКСТУ 2409
Дата введения 1993-07-01
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. ПОДГОТОВЛЕН И ВНЕСЕН ТК 193 "Кислоты жирные синтетические, высшие жирные спирты, поверхностно-активные вещества"
2. УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Госстандарта России от 15.07.92 N 692
Настоящий стандарт подготовлен методом прямого применения международного стандарта ИСО 894-77* "Вещества поверхностно-активные. Алкилсульфаты натрия первичные технические. Методы анализа" и полностью ему соответствует
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
3. Срок первой проверки - 1997 г.
Периодичность проверки - 5 лет
4. ВВЕДЕН ВПЕРВЫЕ
5. ССЫЛОЧНЫЕ НОРМАТИВНО-ТЕХНИЧЕСКИЕ ДОКУМЕНТЫ
Обозначение НТД, на который дана ссылка | Номер пункта, подпункта |
ГОСТ 4204-77 | 6.8.4 |
ГОСТ 6732.2-89 | 3; 4 |
ГОСТ 14870-77 | 3; 6.2.1; 6.2.2 |
ГОСТ 20292-74 | 6.8.3.5 |
ГОСТ 22386-77 | 3; 6 3; 6.4 |
ГОСТ 22567.5-77 | 3; 6.3; 6.4 |
ГОСТ 25336-82 | 6.5.4.3; 6.5.4.4; 6.5.4.5; 6.6.3 |
ГОСТ 28478-90 | 6.7 |
0. ВВЕДЕНИЕ
Частица "prim" (первичные), которая предшествует общему названию веществ, служит для отделения их от веществ, которые могут быть названы, согласно принятым в настоящее время машинным способам обработки, как вторичные технические алкилсульфаты натрия; первичные, как это показывает общая формула, которая приводится ниже, являются производными от первичных спиртов, тогда как вторичные можно рассматривать как производные от вторичных спиртов.
Таким образом, именно первичные и являются объектом данного стандарта. Обычно они называются технические первичные сульфаты спиртов жирного ряда.
Для того, чтобы упростить текст стандарта и избежать излишних повторений, слово "первичный" в тексте опущено, но следует понимать, что здесь рассматривают только "первичные алкилсульфаты натрия".
Общая формула продуктов, которые являются предметом настоящего стандарта, такова:
R-CH-О-SО
-Na,
где R - алифатический радикал.
1. НАЗНАЧЕНИЕ
Настоящий стандарт определяет метод анализа технических алкилсульфатов натрия. Он включает в себя следующие анализы:
измерение рН;
определение воды;
определение несвязанной щелочности, в случае необходимости несвязанной кислотности;
определение общей щелочности;
определение веществ, которые могут быть экстрагированы петролейным эфиром;
определение веществ, которые могут быть экстрагированы диэтиловым эфиром после кислотного гидролиза (комбинированные технические спирты жирного ряда);
определение сульфата натрия;
определение хлорида натрия.
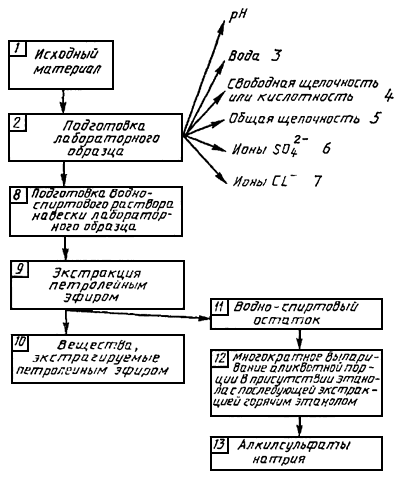
Кроме того, стандарт устанавливает общую схему анализа (приложение).
2. ОБЛАСТЬ ПРИМЕНЕНИЯ
Настоящий стандарт применяется только для технических алкилсульфатов натрия в виде порошков, паст или жидкостей (водных растворов), которые не содержат каких-либо посторонних веществ, не связанных с их получением.
3. ССЫЛКИ
ГОСТ 6732.2 | "Красители органические, продукты промежуточные для красителей, вещества текстильно-вспомогательные. Методы отбора проб". |
ГОСТ 4204 | "Кислота серная. Технические условия". |
ГОСТ 22386 | "Кислоты жирные синтетические. Метод определения кислотного числа". |
ГОСТ 22567.5 | "Средства моющие синтетические порошкообразные. Метод определения концентрации водородных ионов". |
ГОСТ 14870 | "Реактивы. Методы определения содержания воды". |
4. ОТБОР ПРОБ
Лабораторный образец весом около 300 г готовят и хранят в соответствии с указаниями ГОСТ 6732.2.
5. ОБЩИЙ ПРИНЦИП*
_______________
* См.общую схему анализа (приложение).
Получение водно-спиртового раствора отобранной пробы, из которого удаляются вещества, которые могут быть экстрагированы петролейным эфиром.
Испарение досуха оставшейся водно-спиртовой жидкости в присутствии этанола. Кислотный гидролиз сухого остатка при нагревании и выделение из реакционноспособной жидкости с кислотными свойствами всех веществ, которые могут быть экстрагированы с помощью диэтилового эфира (комбинированных технических спиртов жирного ряда).
На выделенных пробах для испытаний проводят:
измерение рН;
определение воды;
определение несвязанной щелочности; иногда несвязанной кислотности;
определение общей щелочности;
определение сульфата натрия;
определение хлорида натрия.
6. МЕТОД АНАЛИЗА
6.1. Измерение рН
Согласно методу, описанному в ГОСТ 22567.5, провести измерение рН раствора лабораторного образца для испытаний с массовой долей 10%.
Примечание. Если рН меньше 7,0, то образец и партия, из которой он взят, являются нестабильными. Как следствие, результаты большей части опытов будут изменяться со временем. В этих условиях партию обычно отклоняют без проведения других анализов.
6.2. Определение воды
В зависимости от содержания воды в веществе ее определяют по одному из двух методов:
а) методу Карла Фишера, применим к веществам, имеющим менее 10% (мольное отношение) воды;
б) методу азеотропного разделения, применим только к веществам, имеющим более 5% (мольное отношение) воды.
6.2.1. Метод Карла Фишера
Согласно методу, описанному в ГОСТ 14870, определяют содержание воды.
6.2.2. Метод азеотропного разделения
Согласно методу, описанному в ГОСТ 14870, определяют содержание воды.
6.3. Определение несвязанной щелочи, при необходимости несвязанной кислотности
Согласно методу, описанному в ГОСТ 22386, определяют несвязанную щелочность или несвязанную кислотность.
6.4. Определение общей щелочности
Этот случай может иметь место, когда при измерении рН согласно п.6.1 получают значение, существенно превышающее 7, или когда в результате определения щелочности согласно п.6.3 получают индекс щелочности, существенно превышающий 0,3. В таких случаях следует провести определение общей щелочности по методу, описанному в ГОСТ 22386.
6.5. Определение веществ, которые могут быть экстрагированы петролейным эфиром
6.5.1. Введение
К веществам, которые могут быть экстрагированы петролейным эфиром, относятся вещества, которые не содержат серу, а также, в некоторых случаях и вещества, содержащие серу, но не ионизирующиеся в водном растворе.
6.5.2. Принцип
Экстракция веществ, указанных в п.6.5.1, с помощью петролейного эфира из водно-спиртового раствора отобранной пробы, учитывая при этом летучесть рассматриваемых веществ.
6.5.3. Реактивы
В процессе проведения анализа следует использовать только реактивы известного аналитического качества и дистиллированную воду или воду эквивалентной чистоты.
6.5.3.1. Сульфат натрия безводный. .
6.5.3.2. Этанол, 96%-ный раствор (по объему).
6.5.3.3. Этанол, 50%-ный раствор (по объему).
6.5.3.4. Петролейный эфир, дистиллят температурного диапазона 40-60°С.
Остаток после испарения не должен превышать 0,002% (мольное отношение).
6.5.3.5. Гидроксид натрия, раствор молярной концентрации приблизительно (NaOH)=0,1 моль/дм
.
6.5.3.6. Фенолфталеин, раствор в этаноле 1 г/дм.
6.5.4. Оборудование
Обычное лабораторное оборудование и
6.5.4.1. Колба круглодонная вместимостью 250 см с притертым горлом.
6.5.4.2. Разделительная колонка длиной 20 см на стержнях (нижний диаметр около 10 мм), снабженная на нижнем конце соединением на шлифе, на которое может быть насажено притертое горло колбы (п.6.5.4.1).
6.5.4.3. Прямоточный холодильник по ГОСТ 25336 номинальной длиной 160 мм.
6.5.4.4. Три колбы для декантации по ГОСТ 25336 вместимостью 500 см со стеклянной притертой пробкой.
6.5.4.5. Коническая колба по ГОСТ 25336 вместимостью 250 см.
6.5.5. Методика проведения операций
6.5.5.1. Отбор пробы
В стакане вместимостью 100 см взвесить с точностью до 0,01 г такое количество лабораторного образца для испытаний, которое содержало бы около 4 г алкилеульфата натрия.
6.5.5.2. Определение
Отобранную пробу растворить или разбавить в зависимости от необходимости в 50 см горячей воды (около 70°С), перемешивая при этом стеклянной палочкой, затем добавить 50 см
раствора этанола (п.6.5.3.2), продолжая перемешивание. Поместить полученный раствор в одну из колб для декантации (п.6.5.4.4) (А). Поочередно промыть стакан небольшими равными порциями горячей воды и раствором этанола (п.6.5.3.2) до тех пор, пока окончательный объем станет равным приблизительно 300 см
.
Проверить с помощью раствора фенолфталеина (п.6.5.3.6), является ли раствор слабо щелочным, и при необходимости увеличить щелочность с помощью раствора гидроксида натрия (п.6.5.3.5) до изменения окраски индикатора до бледно-розовой.
Взболтать, чтобы растворы хорошо перемешался. Дать охладиться.
Добавить 50 см петролейного эфира (п.6.5.3.4).
Хорошо взболтать в течение около 30 с и дать декантироваться. Добавить минимальное количество раствора этанола (п.6.5.3.2), необходимого для устранения образующейся в некоторых случаях эмульсии.
Перелить нижний слой во вторую колбу для декантации (п.6.5.4.4) (В). Вновь провести экстракцию 50 см петролейного эфира. Собрать нижний слой в третью колбу для декантации (п.6.5.4.4) (С), а верхний слой перелить в первую колбу для декантации.
Провести экстракцию водно-спиртовой фазы еще три раза, используя всякий раз 50 см петролейного эфира.
Собрать углеводородные фазы в колбу для декантации (А) и перелить водно-спиртовую фазу после последней экстракции в стакан вместимостью 400 см. Три раза промыть колбы для декантации (В) и (С), используя всякий раз 10 см
раствора этанола (п.6.5.3.3).
Добавить промывную жидкость к водно-спиртовой фазе, находящейся в стакане.
Промыть углеводородный экстракт порциями раствора этанола (п.6.5.3.3) до исчезновения щелочной реакции и добавить эту промывную жидкость к водно-спиртовой фазе.
Этот раствор служит для определения содержания комбинированных технических спиртов жирного ряда.
Полностью перелить содержащий углеводороды слой в коническую колбу (п.6.5.4.5), содержащую около 10 г сульфата натрия (п.6.5.3.1). Взболтать, дать отстояться в течение 30 мин, затем профильтровать через бумажный фильтр в колбу (п.6.5.4.1), в которой находятся предварительно взвешенные шарики. Промыть коническую колбу, сульфат натрия и фильтр пять раз, используя каждый раз 10 см петролейного эфира. Особое внимание следует обратить на края фильтровальной бумаги, на которых не должно быть жирных пятен.
Подготовить разделительную колонку (п.6.5.4.2) и холодильник (п.6.5.4.3), поместить колбу на нагревательную плитку или в баню с кипящей водой и провести дистилляцию почти до полного улетучивания растворителя, Убрать разделительную колонку, дать охладиться приблизительно до 30 °С и удалить последние следы растворителя путем продувки сухого и холодного воздуха. Для этого надо продувать воздух вдали от нагревательной плитки или бани с кипящей водой и вручную вращать колбу в наклонном положении вокруг собственной оси. В таком случае жидкость в колбе распределяется по стенкам в виде тонкой пленки, что облегчает удаление последних следов растворителя.
Низшие спирты, такие как лауриновый спирт, являются достаточно летучими, особенно в присутствии следов влаги, что может привести в некоторых случаях к потерям, которыми нельзя пренебречь. Таким образом, необходимо соблюдать особую предосторожность при испарении растворителя, особенно в процессе продувки воздухом. Для этого необходимо провести первое взвешивание охлажденной до комнатной температуры и тщательно протертой колбы тогда, когда еще ощущается запах растворителя. Отметить массу, затем вновь нагреть приблизительно до 30 °С с тем, чтобы осуществить сжижение содержимого и осуществить вновь продувку в течение 1 мин. После охлаждения и протирки колбы взвесить ее снова и отметить массу.
Повторяя эти операции и нанося результаты последовательных взвешиваний на график на миллиметровке, можно будет увидеть, что после быстрого спада кривая достигает нижней практически горизонтальной площадки. Второе взвешивание, результат которого попадает на площадку, считается окончательным, а отмеченная масса рассматривается как конечный сухой остаток. Разность результатов двух последних взвешиваний должна проявляться только в третьей значащей цифре.
6.5.6. Обработка результатов
6.5.6.1. Метод расчета
Массовую долю веществ, которые могут быть экстрагированы петролейным эфиром, в процентах рассчитывают по формуле:
![]() ,
,
где - масса полученного остатка, г;
- масса отобранной пробы (п.6.5.5.1), г.
6.5.6.2. Воспроизводимость
Разница между результатами, полученными в двух различных лабораториях для одного и того же образца, не должна превышать 1%.
6.6. Определение веществ, которые могут быть экстрагированы диэтиловым эфиром после кислотного гидролиза (технические комбинированные спирты жирного ряда)
6.6.1. Принцип
Испарение водно-спиртовой жидкости , полученной в п.6.5.5.2, до достижения пятой части от своего объема, добавление этанола, испарение досуха.
Растворение сухого остатка в воде, затем после добавления соляной кислоты кислотный гидролиз при кипячении. После завершения гидролиза - экстракция на холоде с помощью диэтилового эфира всех образовавшихся несвязанных жирных веществ.
Совокупность этих жирных веществ состоит почти исключительно из комбинированных спиртов жирного ряда, но тем не менее может содержать в некоторых случаях обычно в малых количествах сульфоновые производные (действительно сульфоповые) и жирные кислоты.
Примечание. Очень важно, чтобы в процессе этого определения раствор оставался щелочным.
6.6.2. Реактивы
В процессе проведения анализа следует использовать только реактивы известного аналитического качества и дистиллированную воду или воду эквивалентной чистоты.
6.6.2.1. Сульфат натрия безводный.
6.6.2.2. Этанол, 96%-ный раствор (по объему), проявляющий слабо щелочную реакцию после добавления раствора гидроксида натрия молярной концентрации (NaOH)=0,1 моль/дм
в присутствии фенолфталеина в качестве индикатора.
6.6.2.3. Диэтиловый эфир.
6.6.2.4. Соляная кислота, раствор, не содержащий сульфат-ионов, =1,19 г/см
.
6.6.2.5. Соляная кислота, раствор приблизительно молярной концентрации (HCl)=1 моль/дм
.
6.6.3. Оборудование
Обычное лабораторное оборудование и
6.6.3.1. Колба для омыления вместимостью 250 см.
6.6.3.2. Три колбы для декантации по ГОСТ 25336 вместимостью 500 см.
6.6.3.3. Колба с круглым дном вместимостью 250 см с взаимозаменяемым шлифом.
6.6.3.4. Разделительная колонка длиной 20 см на стержнях (нижний диаметр около 10 мм), имеющая на своей нижней части соединение на шлифе, на которое может насаживаться шлиф колбы (п.6.6.3.3).
6.6.3.5. Прямоточный холодильник по ГОСТ 25336 номинальной длиной 160 мм.
6.6.4. Методика проведения анализа
6.6.4.1. Отбор проб
Использовать водно-спиртовый раствор , полученный при определении веществ, которые могут быть экстрагированы петролейным эфиром (см. п.6.5).
6.6.4.2. Определение
Довести отобранную пробу (п.6.6.4.1) примерно до одной пятой части от ее объема путем испарения на бане с кипящей водой в токе воздуха. Добавить 20 см раствора этанола (п.6.6.2.2) и испарить досуха.
Растворить в случае необходимости при нагревании в 50 см воды и полностью перелить в колбу омыления (п.6.6.3.1) с помощью воды и довести до конечного объема приблизительно 100 см
. Добавить два стальных шарика, затем осторожно 35 см
раствора соляной кислоты (п.6.6.2.4). Следует иметь в виду, что раствор может перелиться через край из-за пены, которая возникает при разложении карбонатов, образующихся в некоторых случаях в процессе испарения досуха.
Присоединить холодильник (п.6.6.3.5) и кипятить с образованием флегмы в течение по крайней мере 4 ч. Охладить и полностью перенести в одну из колб для декантации (п.6.6.3.2) (А). Тщательно полностью промыть холодильник и колбу диэтиловым эфиром (п.6.6.2.3) и водой и добавить промывную жидкость к содержимому колбы для декантации.
Взболтать для улучшения перемешивания. Добавить 30 см диэтилового эфира. Как следует взболтать и оставить декантироваться.
Перелить нижний слой во вторую колбу для декантации (п.6.6.3.2) (В). Вновь осуществить экстракцию 30 см диэтилового эфира. Собрать нижний слой в третью колбу для декантации (п.6.6.3.2) (С) и перелить верхний слой в мерную колбу для декантации (А).
Провести экстракцию водной фазы еще три раза, используя всякий раз 30 см диэтилового эфира.
Собрать эфирные фазы в колбу для декантации (А) и трижды промыть эфирный экстракт, используя всякий раз 15 см раствора соляной кислоты (п.6.6.2.5).
Полностью перелить эфирный экстракт в коническую колбу вместимостью 250 см, содержащую около 10 г сульфата натрия (п.6.6.2.1). Взболтать, дать отстояться в течение 30 мин, затем профильтровать через бумажный фильтр в колбу (п.6.6.3.3), содержащую несколько предварительно взвешенных стеклянных шариков. Промыть коническую колбу, сульфат натрия и фильтр пять раз, используя всякий раз 10 см
диэтилового эфира. Особое внимание обратить на края бумажного фильтра, на котором больше не должно быть жирных пятен.
Присоединить разделительную колонку (п.6.6.3.4) и холодильник (п.6.6.3.5), поместить колбу на баню с кипящей водой и провести дистилляцию почти до полного удаления растворителя. Убрать разделительную колонку и удалить последние следы растворителя путем короткой продувки сухим и холодным воздухом. Для этого продолжить продувку воздуха и вдали от бани с кипящей водой вращать колбу в наклонном положении вокруг своей оси. При этом жидкость в колбе будет распределяться по стенкам колбы в виде тонкой пленки, что облегчает удаление последних следов растворителя.
Необходимо помнить, что низшие спирты, такие как лауриновый, являются довольно летучими, особенно в присутствии следов влаги, что может в некоторых случаях служить причиной потерь, которыми нельзя пренебречь. Таким образом, необходимо проявлять особую осторожность при испарении растворителя, особенно при продувке воздухом. Для этого необходимо осуществлять первое взвешивание охлажденной до комнатной температуры и тщательно протертой колбы тогда, когда ощущаешь запах растворителя. Отметить массу, затем вновь нагреть колбу приблизительно до 30 °С с тем, чтобы произошло сжижение содержимого, и вновь провести продувку в течение 1 мин. После охлаждения и протирки колбы ее нужно взвесить вновь и отметить массу.
Повторяя эти операции и нанося результаты последовательных взвешиваний на график на миллиметровке, можно увидеть, что после быстрого спада кривая достигает нижней практически горизонтальной площадки. Второе взвешивание, результаты которого попадают на площадку, считается окончательным, а отмеченная масса рассматривается как конечный сухой остаток. Разность результатов двух последних взвешиваний должна проявляться только в третьей значащей цифре.
6.6.5. Обработка результатов
6.6.5.1. Метод расчета
Массовая доля веществ, которые могут быть экстрагированы диэтиловым эфиром после кислотного гидролиза, в процентах рассчитывают по формуле
![]() ,
,
где - масса полученного остатка, г;
- масса отобранной пробы (п.6.5.5.1), г.
6.6.5.2. Воспроизводимость
Разница между двумя результатами, полученными на одном и том же образце в двух разных лабораториях, не должна превышать 1%.
Примечания:
1. Для технических комбинированных спиртов жирного ряда следует определять показатели щелочности и кислотности по ГОСТ 22386.
2. Показатель кислотности должен быть не больше 2.
6.7. Определение сульфата натрия
Провести определение сульфата натрия согласно методу, описанному ГОСТ 28478.
6.8. Определение хлорида натрия
6.8.1. Принцип
Потеициометрическое титрование хлорид-ионов с помощью титрованного раствора нитрата серебра в азотнокислой среде, контролируемое с помощью электродов: серебряного электрода (измерительный электрод) и электродом ртуть-сульфат ртути (1) (электрод сравнения).
6.8.2. Реактивы
В процессе проведения анализа использовать только реактивы известного аналитического качества и дистиллированную воду или воду эквивалентной чистоты.
6.8.2.1. Азотная кислота, раствор =1,33 г/см
.
6.8.2.2. Азотная кислота, раствор приблизительной молярной концентрации (НNO
)=6 моль/дм
.
6.8.2.3. Нитрат серебра, титрованный раствор, приблизительной молярной концентрации 0,1 моль/дм, готовят следующим образом: в мерной колбе вместимостью 500 см
растворить 8,5 г нитрата серебра, довести объем раствора водой до метки и перемешать.
Раствор хранить в бутыли из стекла.
6.8.2.4. Хлорид калия, калибровочный раствор сравнения молярной концентрации (KСl)=0,1 моль/дм
, готовят следующим образом: 3,728 г хлорида калия, предварительно просушенного при 105°С в течение 2 ч и охлажденного в эксикаторе, взвесить с точностью 0,001 г. Растворить в небольшом количестве воды и полностью перелить раствор в мерную колбу вместимостью 500 см
. Довести объем колбы водой до метки и перемешать.
6.8.2.5. Метилоранж, раствор массовой концентрации 1 г/дм.
6.8.3. Оборудование
Обычное лабораторное оборудование и
6.8.3.1. Потенциометр чувствительностью 2 мВ (потенциал от -500 до +500 мВ).
6.8.3.2. Серебряный электрод.
6.8.3.3. Ртуть (1), ртуть сульфатный электрод (с раствором сульфата калия в качестве соединительной жидкости).
Примечание. Если такого электрода сравнения нет, вместо него можно использовать электрод каломель-насыщенный раствор KCl, но он должен соединяться мостиком из нитрата калия - агара со стаканом, в котором проводится титрование и куда опускается измерительный электрод (Ag).
Такой мостик очень легко может быть изготовлен следующим образом: насытить 100 см воды приблизительно 32 г нитрата калия, затем добавить 4 г агара; нагреть при 70°С до полного растворения; заполнить капиллярную U-образную трубку (с внутренним диаметром от 2 до 3 мм, длиной колен от 8 до 10 см и расстоянием между коленами около 6 см), сохраняя температуру около 70°С, последним раствором, затем дать охладиться. Приг охлаждении свободные концы колен должны быть погружены к раствор.
6.8.3.4. Электромеханическая мешалка.
6.8.3.5. Бюретка емкостью 50 см, соответствующая ГОСТ 20292.
6.8.4. Методика проведения операций
С целью уменьшения эффектов термического и электрического гистерезиса сделать так, чтобы температуры электродов, используемых для промывания воды, калибровочных растворов и испытываемого раствора были бы как можно более близкими одни по отношению к другим. Температуры калибровочных растворов и испытываемого раствора не должны отличаться более чем на 1°С. Рекомендуемая температура измерения должна, на сколько это возможно, быть равной 20°С.
Примечание. Метод определения содержания соответствует методу, который описан ГОСТ 4204, а также общему методу определения содержания хлоридов путем потенциометрии, разработанному техническим комитетом ИСО 1047.
6.8.4.1. Отбор проб
В стакане вместимостью 250 см взвесить около 2 г лабораторного образца с точностью 0,001 г.
6.8.4.2. Калибровка раствора нитрата серебра
6.8.4.2.1. Подготовка оборудования
Сделать соответствующие подключения и подать напряжение на аппарат. Дать ему возможность работать в течение времени, которое требуется для хорошей электрической стабилизации сети перед началом проведения измерений согласно инструкциям изготовителя. Заметить температуру калибровочного раствора сравнения, осуществить соответствующие регулировки в цепи корректировки температуры и проверить ноль аппарата. Эти регулировки в процессе последующих операций не должны изменяться.
6.8.4.2.2. Титрование
Отобрать соответственно 5,00 см и 10,00 см
калибровочного раствора сравнения хлорида калия (п.6.8.2.4) и поместить их в два чистых и сухих стакана соответствующей вместимости (например 150 см
). Провести для каждого раствора следующее титрование.
После подкисления раствора азотной кислотой (п.6.8.2.2) добавить объем воды, достаточный для получения объема около 100 см.
Обеспечить перемешивание полученного раствора и погрузить в него электрод из серебра (п.6.8.3.2), а также свободный конец мостика; подключить электроды к потенциометру (п.6.8.3.1) и отметить исходное значение потенциала после того, как проверен ноль аппарата.
С помощью бюретки (п.6.8.3.5) добавить частями по 1 см за один раз раствор нитрата серебра (п.6.8.2.3) и после каждого добавления выжидать стабилизацию потенциала.
Отметить в двух первых колонках таблицы величины постепенно добавляемых объемов, а также соответствующие величины потенциалов. При приближении к точке равновесия продолжать добавление раствора нитрата серебра частями по 0,05 см.
В третьей колонке отметить последовательные приращения потенциала (
). В четвертой колонке отметить положительные или отрицательные разности
между приращениями потенциала
.
Окончание титрования соответствует добавлению 0,05 см раствора нитрата серебра, которое дает максимальную величину
.
Для расчета точного объема () раствора нитрата серебра, соответствующего окончанию реакции, использовать формулу
![]() ,
,
где - объем раствора нитрата серебра (п.6.8.2.3), самый близкий снизу к объему, который дает максимальное приращение
, см
;
- объем раствора нитрата серебра (п.6.8.2.3), соответствующий последней добавленной части (0,05 см
), см
;
- последнее положительное значение
;
- сумма абсолютных значений последнего положительного значения
и первого отрицательного значения
.
6.8.4.2.3. Расчет титра раствора
Титр () раствора нитрата серебра рассчитывают по формуле
![]() ,*
,*
где - титр калибровочного раствора сравнения хлорида калия (п.6.8.2.4);
- значение
, соответствующее титрованию 10 см
калибровочного раствора сравнения хлорида калия (п.6.8.2.4), см
;
* - значение
, соответствующее титрованию 5 см
калибровочного раствора сравнения хлорида калия (п.6.8.2.4), см
;
5 - разница между двумя отобранными калибровочными растворами сравнения хлорида калия (п.6.8.2.4), см.
______________
* Формула и экспликация к ней соответствуют оригиналу. - .
6.8.4.3. Определение
Растворить или разбавить, смотря по необходимости, отобранную пробу (п.6.8.4.1) в 100 см воды. Добавить 10 см
раствора азотной кислоты (п.6.8.2.1), продолжить определение согласно пп.6.8.4.2.1 и 6.8.4.2.2.
6.8.5. Представление результатов
6.8.5.1. Способ расчета
Массовую долю хлорид-ионов в процентах по хлориду натрия рассчитывают по формуле
![]() ,
,
где - титр раствора нитрата серебра, рассчитанный согласно п.6.8.4.2.8;
- значение
, соответствующее определению содержания (п.6.8.4.3), см
;
- масса отобранной пробы (п.6.8.4.1), г.
6.8.5.2. Воспроизводимость
Разница между результатами, полученными для одного и того же образца в двух различных лабораториях, не должна превышать 0,2%
Примечание: Если массовая доля хлорида натрия меньше 0,1% (мольное отношение), указать "следы".
7. ПРОТОКОЛ ИСПЫТАНИЙ
В протоколе испытаний должны содержаться следующие сведения:
а) все сведения, необходимые для полной идентификации образца;
б) ссылку на использованный метод;
в) результаты, а также форму, по которой они представлены;
г) условия проведения испытаний;
д) все детали, не предусмотренные в настоящем стандарте или дополнительные, а также все возможные происшествия, способные повлиять на результаты.
Электронный текст документа
и сверен по:
М.: Издательство стандартов, 1992

{kind=link}