УДК 668.5 : 543.544 : 006.354 Группа Н69
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
МАСЛА ЭФИРНЫЕ, ВЕЩЕСТВА ДУШИСТЫЕ И ПОЛУПРОДУКТЫ ИХ СИНТЕЗА
Газохроматографический метод анализа
Essential oils, aromatics and their intermediates. Method of analysis by gas chromatography
ОКСТУ 9151, 9152, 9154
ГОСТ
14618.5—78
Срок действия с 01.01.80
до 04.01.95
Настоящий стандарт распространяется на эфирные масла, душистые вещества и полупродукты их синтеза и устанавливает газохроматографический метод анализа этих веществ.
1. АППАРАТУРА, МАТЕРИАЛЫ И РЕАКТИВЫ
Хроматограф газовый с детектором по теплопроводности или ионизации в пламени, обеспечивающий работу в режиме 50—300°С.
Колонка газохроматографнческая U-образная или спиральная из нержавеющей стали или стекла длиной 100—300 см, внутренним диаметром 0,3—0,4 см.
Микрошприц типа МШ-10, вместимостью ЫО-2 см3 (10 мкл) с ценой деления 0,2-10~3 см3 (0,2 мкл).
Микрошприц типа МШ-1 или Газохром-101, вместимостью 1 -10—3 см3 (1 мкл) с ценой деления 0,02-10_3 см3 (0,02 мкл).
Линейка логарифмическая.
Лупа измерительная по ГОСТ 25706—83.
Интегратор цифровой автоматический для обработки хроматограмм типа И-02.
Газ-носитель— азот, сжатый в баллоне по ГОСТ 9293—74 (для детектора ионизации в пламени), или гелий газообразный высшей очистки 99—99,5% (для детектора по теплопроводности).
Водород технический марки А по ГОСТ 3022—80 или электролитический, получаемый от генератора водорода типа СГС-2.
Воздух технический по ГОСТ 17433—80.
Носитель твердый — хромосорб W-AW или W-AW-ДМСЗ, частицами размером 0,20—0,25 мм (60—80 меш) или 0,16—0,20 мм (80—100 меш).
Издание официальное
Перепечатка воспрещена
Хроматон N-AW или N-AW-ДМСЭ, частицами размером 0,16—0,20 или 0,20—0,25 мм.
В особых случаях допускается применение других носителей или готовых насадок.
Фаза неподвижная — апиезон Л, полидиэтиленгликольадипинат, связанный с пентаэритритом (LAC-2R—446), полиэтиленгликоль с молекулярной массой 20000 (ПЭГ 20М, карбовакс 20М), силикон SE-30, силикон ХЕ-60, полипропиленгликольадипинат (реоплекс-400). Для полиэфирных фаз (LAC-2R-446, реоплекс-400) допустимое кислотное число — не более 3.
В особых случаях допускается применение других неподвижных фаз или готовых насадок.
Растворители для неподвижных фаз — петролейный эфир (для апиезона Л), хлороформ по ГОСТ 20015—88 (для SE-30 и ПЭГ 20М), ацетон по ГОСТ 2603—79 (для реоплекса-400, ХЕ-60, LAC-2R-446).
Спирт этиловый ректификованный по ГОСТ 5962—67 или по ГОСТ 18300—87.
Толуол по ГОСТ 5789—78.
Эфир этиловый медицинский.
Колба КП-1—500—29/32 ТС по ГОСТ 25336—82.
Линалилацетат чистотой не менее 94,0%.
(Измененная редакция, Изм. № 1, 2).
2. ПОДГОТОВКА К АНАЛИЗУ
2.1. Приготовление насадки
Для газохроматографической колонки насадку приготовляют следующим образом: в колбу КП-1—500 засыпают твердый носитель, затем приливают раствор неподвижной фазы в соответствующем растворителе. Растворитель берут в таком количестве, чтобы он полностью покрыл носитель. Колбу закрывают ватным тампоном и оставляют на 2 ч при периодическом встряхивании. Далее под вакуумом (15—20 мм рт. ст.) и при нагревании на водяной бане отгоняю г растворитель. Готовая насадка должна быть сыпучей и не иметь комков. Указанный процент неподвижной фазы эквивалентен числу граммов неподвижной фазы, отнесенной к 100 г носителя. Результат взвешивания в граммах записывают до первого десятичного знака.
Количество неподвижной фазы от массы твердого носителя составляет от 2 до 20%.
(Измененная редакция, Изм. № 1, 2).
2.2. Приготовление колонки
Колонку перед заполнением промывают последовательно толуолом, ацетоном, водой, этиловым спиртом и этиловым эфиром.
После окончания промывки колонку высушивают под вакуумом (15—20 мм рт. ст.) и при нагревании при 60—80°С. Для заполнения U-образной колонки приготовленную насадку засыпают через воронку небольшими порциями попеременно в оба конца колонки при периодическом постукивании. При заполнении спиральной колонки один ее конец закрывают тампоном из стеклянной ваты и присоединяют его к вакуум-насосу (15—20 мм рт. ст.). В другой конец колонки через воронку небольшими порциями присыпают насадку, уплотняя ее постукиванием по колонке резиновым шлангом. После заполнения концы колонки закрывают тампонами из стеклянной ваты.
Количество насадки, израсходованное на заполнение колонки длиной 100 см и внутренним диаметром 0,3 см, составляет (2,1±0,2) г при использовании хромосорба W-AW и W-AW-ДМСЗ и (2,6±0,2) г при использовании хроматона N—AW и
N-AW-ДМСБ.
Монтаж, наладку и вывод хроматографа на рабочий режим проводят в соответствии с инструкцией, прилагаемой к прибору.
(Измененная редакция, Изм. № 1).
2.3. Определение показателей работы колонки
2.3.1. Определение степени разделения
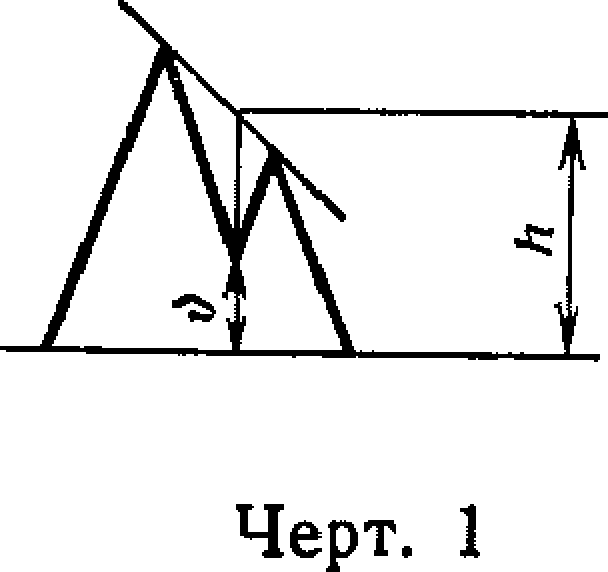
Для определения степени разделения двух пиков (Р) проводят линию, соединяющую максимумы пиков. Из минимума между двумя пиками (черт. 1) проводят перпендикуляр к нулевой линии и продолжают его до пересечения с линией, соединяющей вершины пиков. Величину (Р) в процентах вычисляют по формуле
р 100-(h—v)
Г~ h
где v — высота в точке минимума;
h — высота в точке минимума, продолженная до пересечения с прямой, соединяющей максимумы пиков.
Степень разделения Р определяют в случае неполного разделения пиков, соответствующих основным веществам, для новой колонки. Далее значение Р проверяют периодически перед каждой новой серией анализов. Значение Р должно быть не менее 85%.
(Измененная редакция, Изм. № 1).
2.3.2. О п р е д е л е н и е эффективности колонки Эффективность колонки (п) характеризуют числом теоретических тарелок (т. т.) и вычисляют по формуле
где dx—расстояние от максимума пика гексана до максимума пика вещества, мм;
Ь\ — ширина пика вещества на половине высоты, мм.
В расчет принимают ширину линий, очерчивающей пик (черт. 2).
Значение величины п определяют по основному анализируемому веществу в изотермическом режиме. Значение п на 100 см длины колонки должно быть в пределах 500—1000 теоретических тарелок (т.т.).
(Измененная редакция, Изм. № 1).
2.3.3. Проверка колонки на инертность
Для анализа лабильных соединений (например, некоторых соединений терпенового ряда) используют стеклянные колонки, предварительно проверенные на инертность. Для этого при 130°С хроматографируют линалилацетат. На хроматограмме должны отсутствовать дополнительные пики, свидетельствующие о его разложении.
(Введен дополнительно, Изм. № 1).
2.4. Приготовление пробы
Пробы жидких продуктов вводят в хроматограф; твердые продукты предварительно растворяют в соответствующем растворителе.
(Измененная редакция, Изм. № 1).
2.4.1. При определении относительного калибровочного коэффициента (Ki) пробу готовят следующим образом: взвешивают 0,5—1 г анализируемого вещества и добавляют к нему примерно равное количество эталонного вещества. В качестве последнего используют вещество, близкое по времени удерживания к времени удерживания анализируемого вещества. Результат взвешивания в граммах записывают до четвертого десятичного знака.
2.4.2. При использовании метода «внутреннего эталона» пробу
п=5,54
2

Черт. 2
готовят аналогичным образом (п. 2.4.1), добавляя эталон к анализируемой смеси в количестве примерно равном определяемому компоненту.
2.4.1, 2.4.2. (Введены дополнительно, Изм. № 1).
2.5. Ввод пробы в хроматограф
Пробу анализируемого продукта вводят в хроматографическую колонку микрошприцем через испаритель, прокалывая резиновую мембрану. Иглу шприца вводят на полную длину и быстро впрыскивают такое количество, чтобы пики основных продуктов; занимали не менее 2/з ширины бумаги.
(Измененная редакция, Изм. № 1).
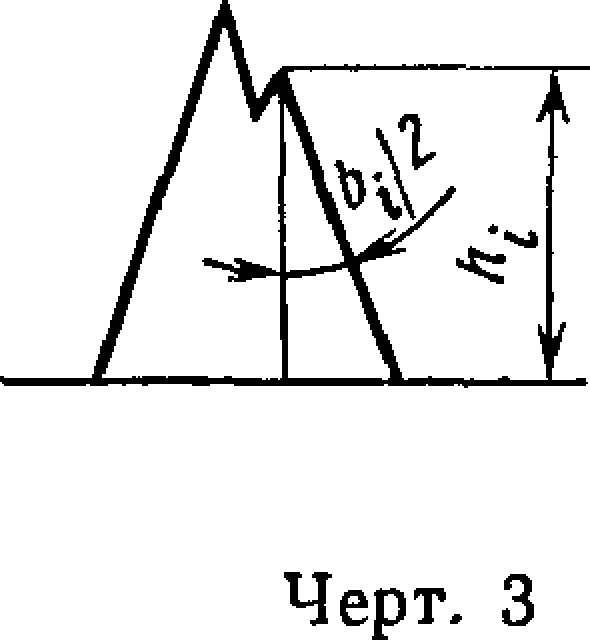
2.6. Площадь пика (St) на хроматограмме измеряют интегратором или вычисляют по формуле
Si=fti* Ьи
где Ы—высота пика, измеренная логарифмической линейкой, мм;
Ь\ — ширина пика на половине высоты, измеренная логарифмической линейкой (при ширине 5 мм и более) или измерительной лупой (при ширине менее 5 мм), мм.
При неполном разделении пиков (черт. 3) за ширину пика при-
ьг
нимают удвоенную полуширину пика -g- , которая измеряется на половине высоты.
В расчет принимают ширину линии, очерчивающей пик (см. черт. 2).
2.7. Относительный калибровочный коэффициент (/G) вычисляют по формуле
mi-S эт
/77а т *-5i
9
где т\— масса искомого компонента, г;
/Пэт— масса «внутреннего эталона», г;
Sst — площадь пика «внутреннего эталона», мм2; Si — площадь пика искомого компонента, мм2.
Относительный калибровочный коэффициент «внутреннего эталона» принимают равным единице.
Относительный калибровочный коэффициент определяют анализом искусственной смеси. Пробу готовят по п. 2.4.1, Относительный калибровочный коэффициент является средней арифметической величиной, полученной для двух искусственных смесей, анализируемых 10 раз. Определяют среднее квадратичное отклонение (Sn ) и погрешность (ек) при jV=20 и доверительной вероятности Р=0,95 по формулам
V N— 1
где Xi — измеряемое значение;
X — среднее значение;
N— число измерений.
e"=±HL-
тIN
Значение ек для относительного калибровочного коэффициента (К) не должно превышать ±0,03 при К от 0,80 до 1,20.
Суммарную погрешность калибровки (бк) вычисляют по формуле
где бас — относительная погрешность приготовления калибровочной смеси, основными составляющими которой являются погрешность от содержания примесей в исходных веществах и погрешность от операции взвешивания (суммируются составляющие в соответствии с требованиями ГОСТ 8.207—76).
(Измененная редакция, Изм, № 1, 2).
2.8. Массовую долю каждого компонента анализируемого продукта (С) в процентах методом «внутренней нормализации» вычисляют по формуле
^ Si-iCi-lOO
2 SK '
где Si — площадь пика определяемого компонента, мм2;
Я. — относительный калибровочный коэффициент определяемого компонента;
— сумма произведений площадей пиков на соответствующие относительные калибровочные коэффициенты всех компонентов анализируемого продукта, мм2.
2.9. Массовую долю каждого компонента анализируемого продукта (С) в процентах методом «внутреннего эталонам вычисляют по формуле
р тэт* 100
mrS*r
где тэт — масса «внутреннего эталона», г;
St — площадь пика искомого компонента, мм2;
Ki — относительный калибровочный коэффициент; тi — масса навески вещества, г;
S9t — площадь пика «внутреннего эталона», мм2.
(Измененная редакция, Изм. № 1).
2.10. Для метода «внутренней нормализации» (п. 2.8) и метода «внутреннего эталона» (п. 2.9) определяют среднее квадратическое отклонение Sn (при ЛГ=10) для полученной величины С. Погрешность анализа (е) для двух параллельных измерений рассчитывают при доверительной вероятности Р—0,95 по формуле
_2,26-Sn
За результат анализа принимают среднее арифметическое двух параллельных измерений, допускаемое расхождение между которыми не должно превышать 4 Sы.
Суммарную погрешность анализа вычисляют по ГОСТ 8.207—76 с учетом систематической составляющей, равной погрешности калибровки (при условии полного разделения хроматографических пиков).
(Измененная редакция, Изм. № 1, 2).
Разд. 3. (Исключен, Изм. № 1).
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. РАЗРАБОТАН И ВНЕСЕН Министерством пищевой промышленности СССР
РАЗРАБОТЧИКИ
С. А. Войткевич, канд. хим. наук; Т. А. Рудольфи, канд. хим. наук; М. М. Щедрина, канд. хим. наук; М. А. Дуброва; Е. В. Свиридова, Г. Д. Каюкова
2. УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Государственного комитета СССР по стандартизации от 29.11.78 NS 3169
3. В стандарт введен международный стандарт ИСО 7359—85
4. ВЗАМЕН ГОСТ 14618.5—69
5. ССЫЛОЧНЫЕ НОРМАТИВНО-ТЕХНИЧЕСКИЕ ДОКУМЕНТЫ
Обозначение НТД, на который дана ссылка | Помер пункта |
ГОСТ 8.207—76 | 2,7; 2.10 |
ГОСТ 2603—79 | 1 |
ГОСТ 3022—80 | 1 |
ГОСТ 5789—78 | 1 |
ГОСТ 5962—67 | 1 |
ГОСТ 9293—74 | 1 |
ГОСТ 17433—80 | 1 |
ГОСТ 18300—87 | 1 |
ГОСТ 20015—88 | 1 |
ГОСТ 25336—82 | 1 |
ГОСТ 25706—83 | 1 |
6. СРОК ДЕЙСТВИЯ ПРОДЛЕН до 01.01.95 г. Постановлением Госстандарта СССР от 16.06.89 № 1680
7. ПЕРЕИЗДАНИЕ (декабрь 1990 г.) с Изменениями № 1, 2, утвержденными в октябре 1984 г., июне 1989 г. (ИУС 2—85, 9—89)

{kind=link}