ГОСТ Р 55357-2012
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ФОРМАТЫ ОПИСАНИЯ И НОРМИРОВАНИЯ ТРЕБОВАНИЙ
Система информации о показателях и требованиях к медицинской технике
Descriptive and normative requirement formats. Information system on indicators and requirements for medical technics
ОКС 25.040.40
Дата введения 2014-01-01
Предисловие
1 РАЗРАБОТАН Автономной некоммерческой организацией (АНО "Международная академия менеджмента и качества бизнеса")
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 100 "Стратегический и инновационный менеджмент"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 29 ноября 2012 года N 1726-ст
4 ВВЕДЕН ВПЕРВЫЕ
5 ПЕРЕИЗДАНИЕ. Август 2020 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
По данным Всемирной торговой организации (ВТО), современная экономика несет значительные потери из-за большого количества технических барьеров в торговле, которые возникают вследствие несоответствия систем технического регулирования различных стран мира.
Несмотря на требования Соглашения по ТБТ ВТО о максимальном устранении технических барьеров в торговле, в силу сложившейся практики и учета национальных интересов стран, условия доступа продукции на их внутренние рынки остаются во многом различными, что создает серьезные препятствия для экспорта/импорта продукции. Кроме того, нужную информацию о требованиях, содержащихся в целом ряде документов (обязательные технические регламенты, добровольные стандарты и др.), как правило, достаточно трудно идентифицировать и получить.
Это приводит к серьезным затруднениям в работе различных участников национальных или региональных систем технического регулирования, которым по роду своих обязанностей необходимо обеспечить требуемое качество и эффективность технических регламентов, стандартов и процедур оценки соответствия. Далее приведены основные задачи, которые необходимо решать участникам систем технического регулирования.
При написании технических регламентов или стандартов регулирующим органам необходимо:
- оценить уровень снижения риска за счет мер, устанавливаемых в технических регламентах;
- определить, являются ли зависимыми или корректируемыми требования, включаемые в технические регламенты или стандарты;
- определить перечень стандартов, подтверждающих соответствие требованиям технических регламентов;
- определить наилучшие форматы требований;
- определить эквивалентные форматы требований для оценки соответствия и обеспечения взаимного признания продукции.
Чтобы обеспечить безопасность продукции, производители должны:
- оценивать уровень снижения рисков, используя схемы распространения опасностей;
- повышать уровень безопасности продукции за счет использования дополнительных добровольных мер безопасности;
- доказывать соответствие своей продукции требованиям технических регламентов;
- использовать принципы эквивалентности для оценки своих экспортных возможностей;
- разрабатывать инструкции для пользователей и обеспечивать меры защиты.
Пользователи при применении продукции имеют право:
- принимать дополнительные меры по повышению безопасности продукции;
- получать общие сведения о возможной опасности продукции;
- доказывать в компетентных инстанциях наличие опасных свойств продукции;
- выбирать наиболее безопасные виды продукции из имеющейся на рынке, используя информацию об этой продукции, связанную с аспектами безопасности и качества;
- рекомендовать производителям, каким образом можно повысить безопасность и качество их продукции.
Органы по оценке (подтверждению) соответствия при проведении оценки соответствия продукции установленным требованиям должны:
- выбирать эквивалентные форматы оценки безопасности;
- выбирать эквивалентные методы оценки соответствия;
- выбирать наилучшие возможности применения стандартов с целью оценки соответствия;
- помогать производителям оценивать эквивалентность требований в отношении экспорта продукции;
- оценивать снижение уровня риска, если это предписано техническими регламентами или применяемыми стандартами.
Органам контроля и надзора и регистрирующим органам при осуществлении своей непосредственной деятельности необходимо:
- оценивать уровни риска от использования продукции для планирования проверок;
- соотносить случаи причинения вреда с нарушениями требований технических регламентов;
- оценивать правильность предоставления информации о продукции на этикетках и в инструкциях в отношении требований технических регламентов;
- разрабатывать арбитражные методы оценки соответствия требованиям продукции;
- инициировать применение технических регламентов для снижения уровня риска.
На современном этапе развития мировой торговли создание системы, позволяющей сравнивать требования и получать информацию, является чрезвычайно актуальной задачей, которая вызывает большой интерес у производителей и потребителей продукции во всем мире.
Наличие комплекса стандартов, призванных создать систему форматов описания и нормирования требований (ФОНТ), особенно важно для региональных систем технического регулирования, нацеленных на упрощение процедур обращения продукции за счет максимального сокращения технических барьеров в торговле, без существенного снижения уровня безопасности выпускаемой продукции.
Для обеспечения всех заинтересованных лиц и организаций соответствующей информацией в области технического регулирования необходимо, чтобы в каждой стране или организации, заинтересованной в обмене такой информацией на основе настоящего стандарта (далее - участники), существовали информационные источники, содержащие:
- требования к конкретной продукции;
- условия доступа продукции на рынки участников;
- сведения об отличиях требований и условий доступа на рынки участников.
Для этого участники должны иметь унифицированные механизмы сбора и хранения данных и обмена информацией с целью сравнения требований.
Для того чтобы заинтересованные организации своевременно получали такую информацию, необходимо:
- поддерживать информационные ресурсы в данной области в своей стране;
- наладить прямую связь с другими аналогичными участниками;
- осуществлять взаимный обмен информацией;
- осуществлять перевод этой информации на национальный язык;
- обеспечивать доступ к этой информации всех заинтересованных пользователей, как внутренних, так и внешних;
- использовать единую или совместимую программную основу для комплекса стандартов ФОНТ.
Это позволит приблизиться к реализации принципа эквивалентности, при котором участники торговых отношений признают, что соблюдение требований к продукции, установленных в различных документах, обеспечивает одинаковый результат, а именно - необходимый уровень безопасности.
Для проведения сравнения следует пользоваться шаблоном и структурировать информацию таким образом, чтобы можно было сопоставлять наличие или отсутствие конкретных требований и устанавливать их идентичность или эквивалентность.
В качестве такого шаблона может использоваться модель обеспечения безопасности для конкретного объекта регулирования (продукции или технологии).
Полученная на основе такой модели и доступная всем заинтересованным пользователям информация может применяться для оценки:
- снижения степени риска благодаря применению положений технических регламентов;
- возможности признания эквивалентности требований технических регламентов и стандартов на основе оценки уровня снижения рисков;
- эффективности применения процедур оценки соответствия;
- эффективности и планирования государственного контроля и надзора.
1 Область применения
Настоящий стандарт является одним из комплекса стандартов на форматы описания и нормирования требований (ФОНТ).
Комплекс стандартов ФОНТ создается для нормативно-методического обеспечения разработки технических регламентов и стандартов в различных областях промышленности и экономики, а также для информационного обеспечения и более широкого применения менеджмента знаний и проектного менеджмента, включая процедуры надлежащей (добросовестной) практики, в том числе в государственном секторе.
Основные цели комплекса стандартов ФОНТ заключаются в том, чтобы:
- разработать методологию и создать условия для обмена знаниями и информацией в соответствующих предметных областях;
- предложить методологию для создания информационного фонда с целью накопления знаний и технических решений в конкретных областях деятельности;
- обеспечить возможность сравнения производственных показателей при проведении сравнительных оценок (бенчмаркинга) с другими предприятиями;
- определить требуемую для этого терминологию;
- установить шаблоны для кодификации знаний и информации о требованиях в данных предметных областях;
- создать условия для признания эквивалентности требований различных технических регламентов и результатов оценки соответствия;
- содействовать проведению оценки результирующего воздействия технических регламентов и стандартов в данных предметных областях;
- обеспечить обмен данными о технических регламентах и стандартах или других документах, используемых для регулирования конкретных областей и требований.
Применение комплекса национальных стандартов ФОНТ позволит:
- определить форматы описания требований, в первую очередь касающихся аспектов безопасности и качества продукции и услуг для жизни, здоровья, имущества и окружающей среды, что расширит применение стандартов в сфере технического регулирования, обеспечивающих соблюдение положений соответствующих технических регламентов, техническую и информационную совместимость, взаимозаменяемость продукции и процедур оценки соответствия;
- увязать требования и положения комплексов технических регламентов и стандартов;
- оценить гармонизацию или эквивалентность требований национальных технических регламентов и стандартов с международными и региональными, а также национальными техническими регламентами и стандартами промышленно развитых стран.
Комплекс национальных стандартов ФОНТ создаст основу для повышения уровня консолидации и применения знаний в различных сферах экономической деятельности, в первую очередь в сфере технического регулирования, а также для расширения информационного обеспечения с целью устранения технических барьеров в торговле и содействия экспортным возможностям отечественной продукции.
Данный комплекс стандартов может быть использован специалистами как для разработки технических регламентов и стандартов на конкретные объекты технического регулирования, так и при принятии решения об идентичности или эквивалентности требований, в том числе:
- экспертами - для проведения экспертизы технических регламентов и стандартов;
- экспертами в области оценки соответствия - при проведении процедур оценки соответствия или принятии решения о возможности взаимного признания результатов оценки соответствия;
- государственными органами - при осуществлении надзора за рынками;
- производителями - для повышения качества и безопасности продукции, особенно при экспорте ее в другие страны;
- производителями или специалистами компетентных органов - при закупках продукции или услуг и проведении соответствующих тендеров.
В отношении требований к медицинской технике настоящий стандарт описывает наиболее общие обязательные для применения и исполнения требования к медицинской технике или связанным с требованиями к медицинской технике процессам производства, использования, хранения, перевозки (транспортирования), реализации и утилизации, а также правила и формы оценки соответствия, правила идентификации и классификации, требования к терминологии, упаковке, маркировке или этикеткам и правилам их нанесения.
В данном стандарте не делается различия в отношении применения терминов "медицинская техника" и "медицинское изделие", несмотря на то, что понятие "медицинская техника" рассматривается на практике как менее общее, чем "медицинское изделие", и распространяется на технические медицинские изделия, включая медицинские приборы и устройства.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ Р ИСО 13485-2011* Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования
ГОСТ Р ИСО 14971-2011** Изделия медицинские. Применение менеджмента риска к медицинским изделиям.
________________
* Вероятно, ошибка оригинала. Следует читать: ГОСТ ISO 13485-2011, здесь и далее по тексту;
** Вероятно, ошибка оригинала. Следует читать: ГОСТ ISO 14971-2011. - Примечания изготовителя базы данных.
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя "Национальные стандарты" за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями.
3.1 безопасность медицинской техники (изделия): Состояние, при котором отсутствует недопустимый риск причинения вреда жизни или здоровью граждан, имуществу физических или юридических лиц, государственному или муниципальному имуществу, окружающей среде, жизни или здоровью животных, а потенциальный риск применения медицинского изделия ниже предельно допустимого риска причинения вреда при использовании в условиях, предусмотренных изготовителем.
Примечание - Безопасность медицинского изделия обеспечивается сохранением его эксплуатационных свойств.
3.2 ввод в эксплуатацию: Этап жизненного цикла, на котором изделие готово к первому применению на территории Российской Федерации в соответствии с предусмотренным назначением.
3.3 выпуск в обращение: Первая платная или бесплатная передача изделия, предназначенного не для клинических исследований, с целью распространения и/или применения на территории Российской Федерации, независимо от того, новое это изделие или модифицированное.
3.4 изготовитель: Физическое или юридическое лицо, ответственное за разработку, изготовление, упаковку и/или маркирование этого изделия, сборку системы или модификацию изделия перед выпуском на рынок под его наименованием независимо от того, выполняются ли указанные действия самим упомянутым лицом или от его имени третьей стороной.
Примечание - Изготовителями не являются лица, которые осуществляют сборку или модификацию изделий для конкретного пациента, при условии, что такие изделия уже введены в обращение.
3.5 изделие для клинических исследований/испытаний: Изделие, предназначенное изготовителем для проведения клинических испытаний в медицинских организациях либо частнопрактикующим врачом соответствующей квалификации для проведения в надлежащей медицинской обстановке надлежащих исследований.
3.6 изделие индивидуального назначения: Изделие, изготовленное в соответствии с техническим заданием, в котором должным образом квалифицированный врач либо другое лицо с соответствующей квалификацией и полномочиями в письменном виде, под свою ответственность предъявляет специальные требования для проектирования или изготовления.
Примечание - Такое изделие должно быть предназначено исключительно для конкретного пациента.
3.7 клинические исследования/испытания: Любое разработанное и запланированное систематическое исследование с участием человека в качестве субъекта клинических испытаний, предпринятое для оценки безопасности и/или эксплуатационных свойств конкретного медицинского изделия.
3.8 медицинская техника: Совокупность технических средств, используемых в медицине в целях профилактики, диагностики, лечения, реабилитации, проведения санитарно-гигиенических, противоэпидемических и других медицинских мероприятий, а также работ по приготовлению лекарств.
3.9 медицинские изделия: Любые инструменты, аппараты, приборы, устройства, программное обеспечение, материалы или иные изделия, используемые по отдельности или в сочетании между собой, включая программное обеспечение, необходимое для их применения по назначению, и предназначенные изготовителем для человека посредством реализации их функций.
Примечание - Медицинские изделия используются с целью:
- профилактики, диагностики, наблюдения, лечения или облегчения заболеваний;
- диагностики, наблюдения, лечения, облегчения или компенсации нарушенных или утраченных физиологических функций;
- исследования, замещения или изменения анатомического строения или физиологических процессов;
- предотвращения или прерывания беременности;
- получения информации в диагностических или медицинских целях путем исследования образцов биологического материала человека.
Необходимым условием является то, что принципиальное воздействие медицинских изделий не основывается на фармакологическом, иммунологическом или метаболическом эффектах применения, но они могут способствовать введению в организм или доставке к поверхности тела человека средств, вызывающих вышеуказанные эффекты.
3.10 пользователь: Лицо, осуществляющее применение медицинского изделия в целях, установленных в пункте 1 статьи 3 настоящего стандарта.
3.11 потенциальный риск применения: Минимально возможный риск причинения вреда при применении по назначению сохранившего эксплуатационные свойства медицинского изделия, который нельзя изменить мерами по ограничению рисков.
3.12 предусмотренное назначение: Применение медицинского изделия в соответствии с информацией изготовителя, указанной на этикетке, в инструкции по применению или руководстве по эксплуатации.
3.13 принадлежности: Предметы, которые при отдельном применении не являются медицинскими изделиями, но специально предназначены изготовителем для использования совместно с ними.
3.14 серийно выпускаемое изделие: Изделие, изготовленное или подвергающееся модификации в соответствии со специфическими требованиями квалифицированного врача либо другого лица с соответствующей квалификацией и полномочиями и не являющееся изделием индивидуального назначения.
3.15 эксплуатационные свойства, характеристики: Сочетание клинической результативности изделия и его технических характеристик. Эксплуатационные свойства тесно связаны с безопасностью.
4 Общие положения
Требования, предъявляемые к продукции в различных технических регламентах и стандартах, можно структурировать в соответствии со следующими используемыми в международной практике уровнями, представленными в таблице 1. Эта структура отражает иерархию установления требований в различных странах, а разные уровни можно интерпретировать в качестве показателей структуры технического регулирования в стране. В таблице 1 также содержатся и некоторые значения показателей, действующих в области медицинской техники.
Таблица 1 - Показатели структуры системы технического регулирования в России в области медицинской техники
Показатели | Системы и документы |
Общая терминология и соответствующие терминам определения | Стандарты ИСО, МЭК, ГОСТ и ГОСТ Р |
Использование международных, региональных или национальных классификаций | Международный классификатор стандартов (МКС), ОКП и ТНВЭД ТС |
Объектная область распространения требований, ограничения области требований и исключения из данной области требований | Определена в соответствующих технических регламентах, действующих на территории России, и стандартах ГОСТ и ГОСТ Р |
Применимость международных протоколов, договоров и соглашений в сфере технического регулирования | - |
Применимость региональных соглашений в сфере технического регулирования | Соглашения Таможенного союза и стран СНГ |
Наличие региональных систем технического регулирования и их применимость для данного вида продукции | Система регулирования Таможенного союза |
Применимость двусторонних или многосторонних соглашений о взаимном признании | В рамках Соглашений МГС СНГ и Таможенного союза |
Национальное членство в международных и региональных организациях по стандартизации | ИСО, МЭК, МГС |
Наличие основополагающих международных и региональных стандартов | ГОСТ ИСО 13485 |
Наличие и применимость для данного вида продукции международных или региональных систем оценки соответствия | Система МЭК для взаимного признания протокола испытаний и сертификатов - схема СВ (частично) |
Устройство и структура применяемой в стране системы технического регулирования (механизм технического регулирования в стране) | Федеральный закон "О техническом регулировании" |
Требования к продукции и связанным с ней процессам | Технические регламенты Таможенного союза в области электромагнитной совместимости и низковольтного оборудования, национальные стандарты ГОСТ и ГОСТ Р |
Используемые формы и схемы (процедуры) оценки соответствия | Технические регламенты Таможенного союза в области электромагнитной совместимости и низковольтного оборудования, национальные стандарты ГОСТ и ГОСТ Р |
Обобщенная структура описания систем технического регулирования приведена в ГОСТ Р "Форматы описания и нормирования требований. Руководство по разработке и применению" на рисунке 2.
В настоящем стандарте на рисунке 1 представлена общая связь между элементами, характеризующими объекты регулирования, которые используются в системе описания (характеризации) требований, предъявляемых к объектам регулирования. Показатели - это элементы и параметры объектов, с помощью которых описывают их характеристики или свойства, которые могут быть измерены или оценены. Значения показателей - это количественная оценка показателей или их измеренные значения. Диапазоны показателей - это диапазоны значений, которые соответствуют разрешенным значениям или значениям, которые соответствуют установленным требованиям.
|
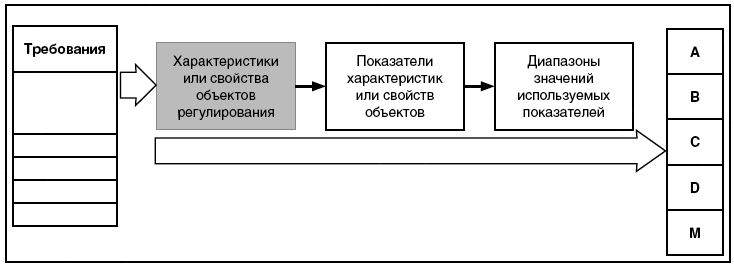
Рисунок 1 - Связь между элементами, характеризующими объекты регулирования
Данное разделение требований на классы полезно в том случае, если система регулирования имеет много разноуровневых документов, устанавливающих требования к продукции, и в документах разных уровней эти требования устанавливаются не только в отношении самой продукции, но и в отношении результатов и аспектов данного воздействия. В этом случае возможны различные дальнейшие детализации требований и особенности в отношении оценки соответствия этим требованиям.
5 Требования и показатели
Используемое в настоящем стандарте разделение основных видов существующих требований представлено в таблице 2.
Таблица 2 - Общее представление требований для снижения рисков проявления опасностей и опасных событий
Требование | Классификация требований | |||
Требования к продукции или связанным процессам | Класс требований А: общие требования к классу (виду) продукции или связанным процессам | Класс требований В: частные требования к отдельным видам (группам) продукции или связанным процессам | Класс требований С: виды опасных воздействий или критические события | Класс требований D: виды подверженности человека или окружающей среды (влияние дозы и воздействия) |
Требования к мерам безопасности | Класс требований М: меры, применяемые для повышения безопасности при эксплуатации или использовании продукции | |||
Далее в качестве примера приведено возможное отнесение различных требований к категориям, представленным в таблице 2.
К классу требований А, как правило, относят требования:
- к показателям качества и безопасности медицинской техники (назначение, применение и др.);
- зависимости применения от эксплуатационных характеристик;
- срокам хранения и применения;
- потребительским (эксплуатационным) характеристикам;
- различным физическим, химическим и другим опасным свойствам продукции;
- опасностям, связанным с устройством данной конкретной группы продукции и влияющим на потребительские (эксплуатационные) характеристики;
- опасностям, непосредственно связанным с устройством продукции;
- неотъемлемым (не потребительским) характеристикам продукции;
- всему вышеназванному, устанавливаемому в рамках проектирования;
- системам управления и контроля;
- системам измерений и калибровки;
- системам интерпретации результатов;
- мобильности и портативности продукции;
- применению в сочетании с другими устройствами;
- в отношении причин, приводящих к разрушению частей;
- в отношении разрушения частей.
К классу требований В, как правило, относят требования:
- к материалам, из которых изготовлена данная продукция;
- конструкционным характеристикам данной продукции;
- программному обеспечению для конкретной продукции.
К классу требований С, как правило, относят требования:
- к видам контакта человека с продукцией;
- воздействиям на окружающую среду (но не к результирующему ущербу);
- обработке продукции (стерилизация, очищение);
- влиянию расходных материалов;
- интерфейсам между продукцией и человеком (в плане влияния на проявление возможных негативных событий).
К классу требований D, как правило, относят требования:
- к видам физического, химического или биологического действия на человека;
- передаче энергии пользователю или пациенту;
- вводу (выводу) вещества в организм (из организма) пользователя или пациента;
- другим видам вреда, причиняемого человеку или животным;
- вреду, наносимому окружающей среде.
К классу требований М, как правило, относятся требования:
- к производственным процессам;
- компетенции или обучению персонала;
- влиянию управления продукцией;
- защитным ограждениям;
- маркировке и аспектам информирования;
- демонтажу и утилизации;
- пользовательскому интерфейсу (в плане инициирования действий пользователя в результате регистрации неисправностей или угроз, а также правильного применения);
- отвлекающим факторам, которые могут привести к ошибке применения;
- самому процессу и качеству проектирования;
- в отношении мер, препятствующих разлету разрушившихся частей.
Для выбора наиболее общих требований и показателей, на основе которых формируются структуры (шаблоны) требований, прежде всего необходимо определить:
- какие отношения должны существовать между требованиями, относящимися к различным классам требований;
- каким образом и какие виды требований следует выбирать для создания структур формализованных требований (шаблонов).
Ниже приведено несколько основных правил для осуществления вышеуказанного выбора.
1. Совокупность требований должна быть максимально независимой. Фактически это означает, что суммарный риск должен быть равен сумме рисков по каждой группе требований или отдельным требованиям.
2. Суммарный риск должен оцениваться как совокупность рисков, проявляющихся в виде различных воздействий на человека и окружающую среду.
3. При наличии различных требований, которые регулируют риск (безопасность) в цепи последовательных событий (схемах или сценариях), приводящих к негативному воздействию или негативным последствиям, необходимо для оценивания выбирать такие схемы или сценарии и такие требования, риск от которых (или от невыполнения которых) оценивается как максимальный.
Требования к продукции могут устанавливаться не только в отношении безопасности, но и в отношении качества продукции, которое непосредственно влияет на снижение уровня безопасности.
Показатели, которые рассматривают как определяющие в различной степени качество и безопасность продукции, представлены в таблице 3.
Таблица 3 - Показатели качества и безопасности медицинской техники
Показатель | Пример |
Назначение | Рабочее давление, потребляемая мощность, напряжение питания, время выхода на режим рабочей температуры, диапазон рабочих температур |
Безопасность | Герметичность, стерильность, температура наружной поверхности изделия, класс защиты от поражения электрическим током |
Интероперабельность (функциональная совместимость) | Показатель электромагнитной совместимости |
Совместимость | Совместное применение с веществами, материалами и газами при взаимодействии |
Надежность | Установленная безотказная наработка, вероятность безотказной работы |
Унификация | Коэффициент повторяемости, коэффициент применяемости |
Экологичность | Доля отходов при производстве, возможность утилизации отходов производства, энергопотребление, утилизация испорченной |
Материалоемкость | Коэффициент использования рациональных материалов, удельная материалоемкость |
Энергоемкость | Расход энергии при заданном режиме работы |
Прослеживаемость качества и безопасности системы на протяжении ее жизненного цикла | Система отвечает установленным требованиям на этапах жизненного цикла |
Транспортабельность | Габаритные размеры, устойчивость к климатическим воздействиям при транспортировании и хранении, устойчивость к механическим воздействиям при транспортировании |
Эргономические показатели | Показатель соответствия изделия функциональным возможностям человека, соответствие изделия форме тела человека |
Сопротивляемость внешним воздействиям | Устойчивость к климатическим воздействиям при транспортировании и хранении, устойчивость к механическим воздействиям при транспортировании |
Автоматизируемость | Коэффициент сборности, удельная трудоемкость изготовления |
Эксплуатационная пригодность | Удобная регулировка, дистанционное управление, гибкость при эксплуатации; простота в обслуживании и эксплуатации |
Эстетические показатели | Показатель рациональности формы, показатель совершенства производственного исполнения, показатель информационной выразительности |
Технологичность | Коэффициент сборности, коэффициент эффективности взаимозаменяемости, удельная трудоемкость изготовления |
Компетентность персонала | Образование, стаж работы, уровень квалификации, класс мастерства |
Менеджмент качества | Наличие системы менеджмента качества или интегрированной системы менеджмента |
Однородность (погрешность) | Нижняя и верхняя границы погрешности измерений |
6 Достижение регулирующего сотрудничества
Индустрия производства медицинской техники (изделий, приборов), включающей как имплантируемые кардиостимуляторы, так и шприцы, иглы, имплантаты для замены сустава, кресла-каталки, бурно развивается и является строго регулируемой. Стандарты играют важную роль в этом процессе и в то же время удовлетворяют требованиям общественности и регулирующих органов к безопасности и соответствию назначению.
Ключевыми характеристиками этой деятельности являются:
- принятие стандартов как основного элемента в регулирующей инфраструктуре, обеспечивающей производство медицинской техники: стандарты не являются обязательными, но обеспечивают соответствие определенным регулирующим требованиям;
- участие регулирующих органов совместно с производителями медицинской техники и потребителями в процессе разработки международных стандартов;
- тесное сотрудничество между ИСО, МЭК и другими организациями в разработке стандартов в области медицинской техники.
6.1 Участники
К основным участникам относится целевая группа по глобальной гармонизации.
Целевая группа по глобальной гармонизации (Global Harmonization Task Force, GHTF; ЦГГГ) не является регулирующим органом. Она представляет собой форум национальных регулирующих органов и представителей промышленности, содействующий международной согласованности регулирующих требований и практической деятельности. В частности, целью ЦГГГ является содействие обеспечению безопасности, эффективности, результативности и качества медицинских приборов в интересах технологического инновационного процесса и укрепления международной торговли. Она также выполняет функции форума по обмену информацией, посредством которого страны с развивающимися регулирующими системами по производству медицинских приборов могут знакомиться с опытом стран, уже имеющих функционирующие системы. Такая деятельность осуществляется через разработку руководящих документов и рекомендованных процедур, целью которых является конвергенция регулирующих систем ее членов по производству медицинских приборов в рамках их законодательных и организационных ограничений.
Участие в ЦГГГ включает работу регулирующих органов, ответственных за производство медицинских приборов, и представителей регулирующей отрасли промышленности из стран и регионов, которые имеют опыт в разработке регламентов на медицинские приборы. В состав членов ЦГГГ входят представители из Европы, США, Канады, Японии и Австралии.
6.2 ИСО и МЭК
Многие технические комитеты ИСО и МЭК активно занимаются разработкой стандартов, имеющих большое значение для регулирования медицинских приборов:
ИСО/ТК 121 "Анестезирующее и дыхательное оборудование";
ИСО/ТК 150 "Имплантаты в хирургии";
ИСО/ТК 194 "Биологическая оценка медицинских изделий";
ИСО/ТК 198 "Стерилизация продукции для здравоохранения";
ИСО/ТК 210 "Менеджмент качества и соответствующие общие аспекты медицинских изделий";
МЭК/ТК 62 "Электрооборудование в медицинской практике".
6.3 Пути достижения регулирующего сотрудничества в этой области
ИСО сотрудничает с ЦГГГ как организация, обеспечивающая общую координацию деятельности, и участвует в открытых сессиях исполнительного комитета ЦГГГ.
Кроме того, меморандумы о взаимопонимании заключены между отдельными исследовательскими группами ЦГГГ (ЦГГГ/ИГ) и техническими комитетами ИСО (ИСО/ТК).
ИСО/ТК 194 и ЦГГГ подготовили и согласовали меморандум о взаимопонимании, определяющий обязанности каждой организации в процессе сотрудничества в области стандартизации и гармонизации в секторе медицинских приборов, особенно в отношении работы ЦГГГ/ИГ 5, которая содействует сближению регулирующих требований, обеспечивающих подтверждение клинической безопасности и результативности медицинских приборов.
ИСО/ТК 210 успешно и активно сотрудничает с ЦГГГ/ИГ 3 в области систем менеджмента качества. ИСО 13485:2003* (межгосударственный стандарт ГОСТ ISO 13485-2011) был официально признан ЦГГГ как приемлемая модель для системы менеджмента качества, используемая для регулирующих целей в отношении медицинских приборов. Кроме того, эксперты - члены ИСО/ТК 210 и МЭК/ТК 62/ПК 62А СРГ 1 "Применение менеджмента рисков к медицинским приборам" - работали как технические эксперты в ЦГГГ/ИГ 3 над подготовкой руководящего документа ЦГГГ "Внедрение принципов менеджмента рисков и работы в рамках системы менеджмента качества". Помимо работы ИСО/ТК 210 над общими аспектами, относящимися к медицинским приборам, другие технические комитеты, включая комитеты, указанные выше, разработали самые различные стандарты на продукцию и процессы, которые получили широкое признание и пользуются хорошей репутацией у регулирующих органов, ответственных за безопасность и результативность медицинских приборов.
_______________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Эти усилия подтвердили свою успешность и эффективность в результате объединения опыта участников процесса и предупреждения любого дублирования работ.
6.4 Сравнение требований
На рисунке 2 приведена детализированная структура требований к безопасности медицинской техники. Следует отметить, что деление не всегда является точным и некоторые требования могут быть отнесены сразу к нескольким позициям. Далее приведен шаблон требований для медицинской техники, соответствующий представленной на рисунке 2 структуре шаблона.
Требования могут быть структурированы по следующим областям:
- экологические аспекты;
- исходные данные;
- выпуск в обращение;
- использование и эксплуатация;
- управление и функционирование.
|
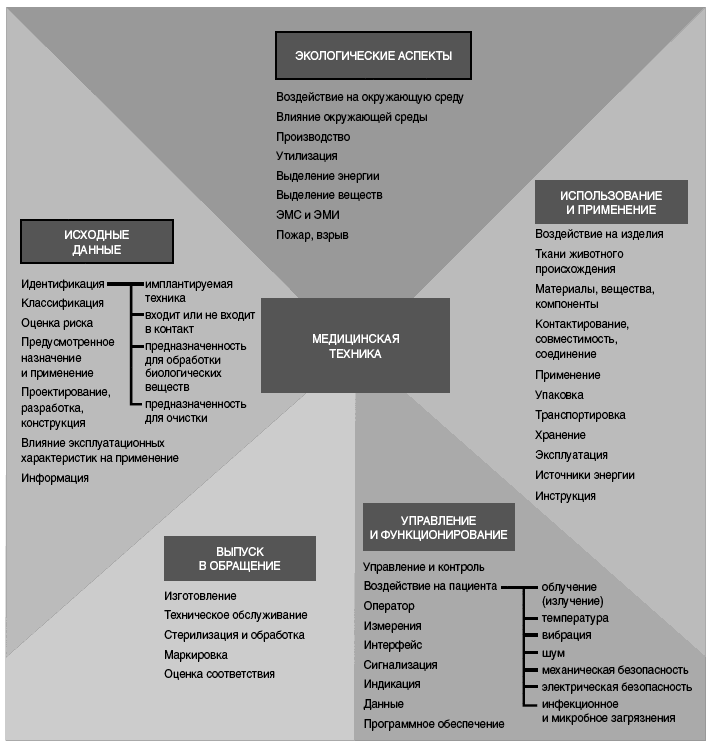
Рисунок 2 - Детализированная структура шаблона для требований к медицинской технике
7 Шаблон основных требований
В таблице 4 приведены основные разделы (виды) требований, предъявляемых к пищевой продукции, и их идентификаторы (условные обозначения), которые предложено использовать для кодификации информации о требованиях в информационных базах данных, разрабатываемых на основе системы стандартов ФОНТ. При большом уровне детализации возможно использование комбинаций этих или других индикаторов (через знак "/" или запятую).
Таблица 4 - Основные виды требований к пищевой продукции и их идентификаторы
Объекты и аспекты, в отношении которых устанавливаются требования | Идентификаторы требований |
Экологические аспекты | ЕА |
Воздействие на окружающую среду | АЕ |
Влияние окружающей среды | EI |
Производство | MF |
Утилизация | UT |
Выделение энергии | ER |
Выделение веществ | SR |
ЭМС, ЭМИ | ЕС, ЕЕ |
Пожар, взрыв | FR, EX |
Исходные данные | OD |
Идентификация: | II |
- имплантируемая техника | IT |
- входит или не входит в контакт | IC, OC |
- предназначенность для обработки биологических веществ | ВТ |
- предназначенность для очистки | СТ |
Классификация | CF |
Оценка риска | RA |
Предусмотренное назначение и применение | FS, FA |
Проектирование | DS |
Влияние эксплуатационных характеристик на применение | IA |
Информация | IN |
Выпуск в обращение | PB |
Изготовление | PD |
Техническое обслуживание | MN |
Стерилизация и обработка | ST, TR |
Маркировка | MR |
Оценка соответствия | CA |
Использование и эксплуатация | UT, EX |
Воздействие на технику | ТА |
Ткани животного происхождения | AT |
Материалы, вещества, компоненты | MT, SB, CN |
Контактирование, совместимость, соединения | CT, CM |
Применение | АР |
Эксплуатация | ЕХ |
Упаковка | РК |
Транспортировка | TN |
Хранение | SO |
Энергетические источники | ES |
Инструкции | IN |
Управление и функционирование | MG, FN |
Управление и контроль | MG, CT |
Воздействие на пациента: | PI |
- облучение | IR |
- температура | TM |
- вибрация | VB |
- шум | NS |
- механическая безопасность | MS |
- электрическая безопасность | ES |
- инфекционное и микробное загрязнение | IС, MC |
- ионизирующее излучение | IE |
Оператор | OP |
Измерения, интерпретация данных | MS, DI |
Интерфейс | IN |
Сигнализация | SG |
Индикация | IС |
Данные | DT |
Программные средства | SW |
8 Примеры отнесения требований
Ниже приведены примеры отнесения требований к различным позициям шаблона для структуры требований, представленной на рисунке 2. Для каждой позиции шаблона приведены примеры требований (если таковые существуют), заложенных в Директиве Совета 93/42/ЕЭС по медицинским приборам (Пример А), а также в ГОСТ Р ИСО 14971-2009 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" (Пример Б), что позволяет обеспечить соотнесение требований, установленных в этих документах. Примеры Б заимствованы из Приложения С ГОСТ Р ИСО 14971-2009, содержащего вопросы для определения характеристик медицинского изделия, влияющих на безопасность применения. На данные вопросы необходимо ответить для соблюдения требований.
Данные примеры представлены исключительно с целью демонстрации отнесения требований к различным позициям (разделам) шаблона, и поэтому на них не рекомендуется ссылаться как на конкретные требования к медицинской технике.
8.1 Воздействие на окружающую среду
В данном разделе описываются требования в отношении воздействия медицинской техники на окружающую среду.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
9. Качество конструкции и окружение
9.2 Приборы должны разрабатываться и изготавливаться таким образом, чтобы устранить или минимизировать по мере возможности:
- риски, связанные с предполагаемыми условиями окружающей среды, такими как магнитные поля, внешние электрические воздействия, электростатические разряды, давление или колебания давления и ускорение".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.10 Влияет ли медицинское изделие на среду, окружающую пациента?
Рассматриваемые факторы:
- температура;
- влажность;
- состав атмосферных газов;
- давление;
- освещенность".
8.2 Влияние окружающей среды
В данном разделе описываются требования в отношении влияния окружающей среды на безопасность медицинской техники.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.15 Чувствительно ли медицинское изделие к воздействию окружающей среды?
Рассматриваемые факторы: производственная среда, транспортные средства и условия хранения. К ним относят: освещенность, температуру, вибрации, утечки, чувствительность к изменениям в энергоснабжении и охлаждении, электромагнитные помехи".
8.3 Производство
В данном разделе описываются требования в отношении производства медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"I. Общие требования
3. Приборы должны обладать функциональными свойствами, предусмотренными изготовителем, и разрабатываться, производиться и упаковываться таким образом, чтобы соответствовать одной или нескольким функциям, указанным в статье 1 (2) (а), как это определено изготовителем".
8.4 Утилизация
В данном разделе описываются требования в отношении утилизации медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.25 Необходимы ли безопасный демонтаж или утилизация медицинского изделия?
Рассматриваемые факторы: отходы, возникающие при утилизации медицинского изделия. Например, следует ответить на вопрос, содержит ли данное изделие токсичные или опасные вещества или вещества, пригодные для переработки".
8.5 Выделение энергии
В данном разделе описываются требования к выделению энергии медицинскими изделиями.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля".
8.6 ЭМС и ЭМИ
В данном разделе описываются требования в отношении электромагнитных излучений и совместимости медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12. Требования к медицинским приборам, подключаемым к источнику энергии или оборудованным источником энергии
12.5 Приборы должны разрабатываться и изготавливаться таким образом, чтобы минимизировать образование электромагнитных полей, которые могут нарушить работу другого прибора или оборудования в обычных условиях".
8.7 Пожар, взрыв
В данном разделе описываются требования в отношении пожарной и взрывобезопасности медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I. Основные требования:
"II. Требования к разработке и конструкции
9. Качество конструкции и окружения
9.3 Приборы должны разрабатываться и изготавливаться таким образом, чтобы минимизировать риск пожара или взрыва при обычном применении и в условиях единичного отказа. Особое внимание должно уделяться приборам, использование которых по назначению предполагает воздействие воспламеняющихся веществ или веществ, способных вызывать возгорание".
8.8 Идентификация
В данном разделе описываются требования в отношении идентификации медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, ст.1:
"1. Настоящая Директива должна применяться к медицинским приборам и принадлежностям к ним. В настоящей Директиве принадлежности трактуются как равноправные медицинские приборы. В дальнейшем как медицинские приборы, так и принадлежности будут называться приборами.
2. В данной Директиве используются следующие определения:
а) "медицинский прибор" означает любой инструмент, аппарат, устройство, материал или другой предмет, используемый самостоятельно или в сочетании, включая компьютерные программы, необходимые для надлежащего его применения, и предназначенный изготовителем для применения на людях в целях:
- диагностики, профилактики, наблюдения, лечения или облегчения заболевания;
- диагностики, наблюдения, лечения, облегчения или компенсации травмы или увечья;
- исследования, реплантации или модификации анатомии или физиологических процессов;
- контроля зачатия, а также не достигающее своей основной предполагаемой цели в организме человека или на нем с помощью фармакологических, иммунологических или метаболических средств, но функции которого данные средства могут содействовать".
8.8.1 Имплантируемая техника
В данном разделе описываются требования к идентификации имплантируемых медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.2 Предусмотрена ли имплантация медицинского изделия?
Рассматриваемые факторы: местонахождение имплантата; характеристики предполагаемых пользователей: возраст, вес, физическая активность; влияние старения на рабочие характеристики имплантата; предполагаемый срок действия имплантата; возможность извлечения имплантата".
8.8.2 Входит или не входит в контакт
В данном разделе описываются требования по идентификации медицинских изделий по их контактности или отсутствию контактов.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.3 Предусмотрен ли контакт медицинского изделия с пациентом или другими лицами?
Рассматриваемые факторы: характер предполагаемого контакта (поверхностный контакт, инвазивный контакт или имплантация) и в каждом случае - длительность и частота контакта".
8.8.3 Предназначенность для обработки биологических веществ
В данном разделе описываются требования к идентификации медицинских изделий по их предназначению для обработки биологических веществ.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.7 Проводят ли в медицинском изделии обработку биологических веществ для их последующего использования, трансфузии или трансплантации?
Рассматриваемые факторы: вид обработки и обрабатываемое(ые) вещество(а) (например, аутотрансфузия, диализ, обработка компонентов крови или клеточная терапия)".
8.8.4 Предназначенность для очистки
В данном разделе описываются требования к идентификации медицинских изделий по их предназначению для очистки.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.9 Предназначено ли медицинское изделие для рутинной очистки и дезинфекции, выполняемых пользователем?
Рассматриваемые факторы: виды применяемых чистящих и дезинфицирующих средств, а также любые ограничения числа циклов очистки. Конструкция медицинского изделия может влиять на результативность рутинной очистки и дезинфекции. Кроме того, следует учитывать влияние чистящих и дезинфицирующих средств на безопасность или характеристики медицинского изделия".
8.9 Классификация
В данном разделе описываются требования в отношении классификации медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС:
"1. Оборудование должно подразделяться на классы I, IIa, lIb и III. Классификация осуществляется в соответствии с Приложением IX.
2. В случае возникновения спора между изготовителем и соответствующим нотифицированным органом по поводу применения правил классификации вопрос должен передаваться для решения в компетентный орган, субъектом которого является этот нотифицированный орган.
3. Правила классификации, установленные в Приложении IX, могут быть изменены в соответствии с процедурой, определенной в статье 7 (2) в свете технического прогресса и другой информации, которая доступна в информационной системе, предусмотренной в статье 10.
Приложение IX. Классификационные критерии
II. Правила реализации
2. Применяемые правила
2.1 Применение правил классификации должно определяться предназначенным использованием приборов.
2.2 Если прибор предназначается для использования с другим прибором, правила классификации должны применяться к каждому прибору отдельно. Принадлежности классифицируются самостоятельно, отдельно от прибора, с которым они совместно используются.
2.3 Программное обеспечение, с помощью которого осуществляется управление прибором и которое влияет на применение прибора, автоматически относится к тому же классу.
2.4 Если прибор не предназначен для применения исключительно или преимущественно в определенной части организма, он должен рассматриваться и классифицироваться в соответствии с наиболее значимым вариантом установленного применения.
2.5 Если несколько правил применимо к одному и тому же прибору, то на основании рабочих характеристик, указанных изготовителем, должны применяться наиболее жесткие правила, предусматривающие самую высокую классификацию".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.23 Является ли медицинское изделие мобильным или портативным?
Рассматриваемые факторы: необходимые зажимы, рукоятки, колеса, тормоза, механическая устойчивость и прочность медицинского изделия".
8.10 Оценка риска
В данном разделе описываются требования в отношении оценки риска для медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"I. Общие требования
1. Приборы должны разрабатываться и изготавливаться таким образом, чтобы при использовании при установленных условиях и назначении они не подвергали риску клиническое состояние или безопасность пациентов, или безопасность и здоровье пользователей, или, возможно, других лиц при условии, что любые риски, которые могут быть связаны с их применением, являются допустимыми рисками, соизмеримыми с положительным результатом для пациента, и были совместимы с высоким уровнем защиты здоровья и безопасности.
2. Решения, принимаемые изготовителем при разработке и конструировании приборов, должны соответствовать принципам безопасности с учетом современного технического уровня.
3. При выборе наиболее приемлемых вариантов решения изготовитель должен применять следующие принципы в следующем порядке:
- по возможности устранять или снижать риски (обеспечивать безопасность уже на этапе разработки и конструкции);
- по возможности принимать адекватные меры защиты, включающие при необходимости аварийную сигнализацию в случае рисков, которые невозможно исключить;
- информировать пользователей об остаточных рисках при недостаточности принятых мер защиты".
8.11 Предусмотренное назначение и применение
В данном разделе описываются требования в отношении предусмотренного назначения и применения медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
13. Информация, предоставляемая изготовителем
13.4 Если предназначенное применение прибора не является очевидным для пользователя, изготовитель обязан четко указывать его на этикетке и в инструкции по применению".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.1 Каково предусмотренное применение и как следует применять медицинское изделие?
Рассматриваемые факторы:
- роль медицинского изделия:
- в диагностике, профилактике, мониторинге, лечении или облегчении заболевания;
- компенсации травм или физических недостатков;
- замещении или модификации частей тела или контроле зачатия;
- показания к применению (например, предполагаемые группы пользователей);
- предназначено ли медицинское изделие для сохранения или поддержания жизни;
- необходимость вмешательства специалистов при отказе медицинского изделия.
С.2.24 Предназначено ли медицинское изделие для однократного применения?
Рассматриваемые факторы: возможность самоликвидации медицинского изделия после применения; наличие указаний на то, что данное изделие уже было применено.
С.2.29.2 Используют ли медицинское изделие в среде, в которой отвлекающие факторы могут привести к ошибкам применения?
Рассматриваемые факторы:
- последствия ошибок применения;
- сведения о том, являются ли отвлекающие факторы привычными для пользователя;
- вероятность появления редко случающихся отвлекающих факторов.
С.2.29.7 Будет ли медицинское изделие применяться лицами с особыми потребностями?
Рассматриваемые факторы: предполагаемый пользователь, его умственные способности и физические возможности, навыки и подготовка, эргономические аспекты, среда применения, требования к установке/монтажу, способность пациента управлять применением медицинского изделия или воздействовать на данное применение. Особое внимание следует уделять пользователям со специфическими потребностями, таким как лица с ограниченными возможностями, пожилые люди и дети. К специфическим потребностям можно также отнести потребность в дополнительной помощи другого лица в процессе использования медицинского изделия. Следует рассмотреть возможность использования медицинского изделия лицами с разным уровнем подготовки и культурными особенностями.
С.2.31 Каковы способы неправильного применения медицинского изделия?
Рассматриваемые факторы: неправильное применение переключателей; выход из строя системы сигнализации или элементов, обеспечивающих безопасность применения; пренебрежение рекомендациями изготовителя в отношении технического обслуживания".
8.12 Проектирование
В данном разделе описываются требования в отношении проектирования медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы гарантировать характеристики и функциональные свойства, указанные в разделе I "Общие требования". Особое внимание необходимо уделять:
- выбору используемых материалов, особенно в части токсичности и, в соответствующих случаях, воспламеняемости;
- совместимости между используемым материалом и биологическими тканями, клетками и жидкостями организма с учетом предназначенного применения прибора.
7.2 Приборы должны разрабатываться, изготавливаться и упаковываться таким образом, чтобы минимизировать риск, который несут с собой загрязняющие вещества и остатки для лиц, связанных с транспортировкой, хранением и применением приборов, и для пациентов с учетом предназначенного применения изделия. Особое внимание должно уделяться незащищенным тканям, а также продолжительности и периодичности воздействия.
7.3 Приборы должны разрабатываться и изготавливаться таким образом, чтобы они могли безопасно использоваться вместе с материалами, веществами и газами, с которыми они вступают в контакт при нормальном применении или во время обычных процедур; если приборы предназначены для ввода лекарственных препаратов, они должны разрабатываться и изготавливаться таким образом, чтобы быть совместимыми с соответствующими лекарственными препаратами согласно положениям и ограничениям, регулирующим эти препараты, и чтобы их функциональные свойства поддерживались в соответствии с предназначенным применением.
7.4 Если прибор включает в качестве неотъемлемой части вещество, которое при его самостоятельном применении может рассматриваться как медицинский препарат, определенный в статье 1 Директивы Совета 65/65/ЕЭС, и которое дополняет действие прибора, то безопасность, качество и пригодность этого вещества с учетом назначения прибора должны быть проверены по аналогии соответствующими методами, определенными в Директиве Совета 75/318/ЕЭС.
7.5 Приборы должны разрабатываться и изготавливаться таким образом, чтобы минимизировать риск, который создают вещества при их утечке из прибора.
7.6 Приборы должны разрабатываться и изготавливаться таким образом, чтобы в максимальной степени минимизировать риск, вызываемый случайным попаданием в прибор веществ с учетом самого прибора и характера среды, в которой его предполагается применять".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.28 Будут ли необходимы разработка и внедрение новых производственных процессов?
Рассматриваемые факторы: новые технологии или новые масштабы производства".
8.13 Влияние эксплуатационных характеристик на применение
В данном разделе описываются требования в отношении влияния эксплуатационных характеристик на применение медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы гарантировать характеристики и функциональные свойства, указанные в разделе I "Общие требования"".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.34 Зависит ли применение медицинского изделия от его важнейших эксплуатационных характеристик?
Рассматриваемые факторы: выходные характеристики изделий, обеспечивающих жизнеобеспечение организма; срабатывание системы сигнализации.
В разделе рассмотрены важнейшие эксплуатационные характеристики медицинских электрических изделий и медицинских электрических систем".
8.14 Информация
В данном разделе описываются требования в отношении информации о медицинских изделиях.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
13. Информация, предоставляемая изготовителем
13.1 Каждый прибор должен сопровождаться информацией, необходимой для его безопасного применения и для идентификации изготовителя с учетом уровня подготовки и знаний потенциальных пользователей.
Эта информация содержит подробные данные, приводимые на этикетке и в инструкциях пользователя.
Информация, необходимая для безопасного применения прибора, должна, насколько это возможно и уместно, размещаться на самом приборе и/или на упаковке каждого прибора, или в соответствующих случаях на торговой упаковке. Если индивидуальная упаковка каждой единицы прибора неосуществима, то информация должна размещаться на листке-вкладыше, поставляемом с одним или несколькими приборами.
Инструкции по применению должны помещаться в упаковку каждого прибора. В порядке исключения такие инструкции по применению не требуются для приборов классов I или IIа, если они могут безопасно использоваться без подобных инструкций.
13.2 По возможности данную информацию следует приводить в виде символов. Любой используемый символ или идентификационный цвет должен соответствовать гармонизированным стандартам. В тех областях, где не имеется стандартов, описание символов и цветов должно содержаться в документации, поставляемой с прибором.
13.3 Этикетка должна содержать следующие данные:
- наименование или фирменное название изготовителя. Для приборов, импортируемых в Сообщество с целью распространения в Сообществе, этикетка или их внешняя упаковка, или инструкции по применению должны дополнительно содержать наименование и адрес либо ответственного лица, указанного в статье 14 (2), либо уполномоченного представителя изготовителя в Сообществе, либо импортера, признанного в Сообществе, в зависимости от конкретной ситуации;
- данные, абсолютно необходимые пользователю для идентификации прибора и содержания упаковки;
- где это уместно, слово "СТЕРИЛЬНО";
- где это уместно, код партии с размещаемым перед ним словом "ПАРТИЯ" или порядковым номером;
- где это уместно, указание даты, до которой допускается безопасное применение прибора, выраженное в годах и месяцах;
- где это уместно, указание того, что данный прибор предназначен для одноразового использования;
- если прибор индивидуального исполнения, на нем следует указывать "прибор индивидуального исполнения";
- если прибор предназначен для клинических исследований, на нем следует указывать "исключительно для клинических исследований";
- любые специальные условия хранения и/или обращения;
- любые специальные рабочие инструкции;
- любые предупреждения и/или меры предосторожности, которые необходимо соблюдать;
- год изготовления для активных приборов, отличных от тех, которые указаны в пункте (е). Это указание может включаться в номер партии или порядковый номер;
- где это уместно, способ стерилизации.
13.4 Если предназначенное применение прибора не является очевидным для пользователя, изготовитель обязан четко указывать его на этикетке и в инструкции по применению".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.27 Какая информация по безопасному применению будет предоставлена пользователю?
Рассматриваемые факторы:
- сведения о том, будет ли информация предоставлена конечному пользователю непосредственно изготовителем или будут задействованы другие лица, например специалисты по монтажу/установке, поставщики медицинских услуг, медицинские работники или фармацевты, и как это повлияет на обучение;
- информация о пусконаладочных работах (вводе в эксплуатацию) и передаче изделия конечному пользователю, а также о целесообразности/возможности монтажа лицами, не имеющими специальных навыков;
- сведения о необходимости переобучения или переаттестации операторов или обслуживающего персонала, исходя из предполагаемого срока службы изделия".
8.15 Изготовление
В данном разделе описываются требования в отношении изготовления медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы гарантировать характеристики и функциональные свойства, указанные в разделе I "Общие требования". Особое внимание необходимо уделять:
- выбору используемых материалов, особенно в части токсичности и, в соответствующих случаях, воспламеняемости;
- совместимости между используемым материалом и биологическими тканями, клетками и жидкостями организма с учетом предназначенного применения прибора.
7.2 Приборы должны разрабатываться, изготавливаться и упаковываться таким образом, чтобы минимизировать риск, который несут с собой загрязняющие вещества и остатки для лиц, связанных с транспортировкой, хранением и применением приборов, и для пациентов с учетом предназначенного применения изделия. Особое внимание должно уделяться незащищенным тканям, а также продолжительности и периодичности воздействия.
7.3 Приборы должны разрабатываться и изготавливаться таким образом, чтобы они могли безопасно использоваться вместе с материалами, веществами и газами, с которыми они вступают в контакт при нормальном применении или во время обычных процедур; если приборы предназначены для ввода лекарственных препаратов, они должны разрабатываться и изготавливаться таким образом, чтобы быть совместимыми с соответствующими лекарственными препаратами согласно положениям и ограничениям, регулирующим эти препараты, и чтобы их функциональные свойства поддерживались в соответствии с предназначенным применением.
7.4 Если прибор включает в качестве неотъемлемой части вещество, которое при его самостоятельном применении может рассматриваться как медицинский препарат, определенный в статье 1 Директивы Совета 65/65/ЕЭС, и которое дополняет действие прибора, то безопасность, качество и пригодность этого вещества с учетом назначения прибора должны быть проверены по аналогии соответствующими методами, определенными в Директиве Совета 75/318/ЕЭС.
7.5 Приборы должны разрабатываться и изготавливаться таким образом, чтобы минимизировать риск, который создают вещества при их утечке из прибора.
7.6 Приборы должны разрабатываться и изготавливаться таким образом, чтобы в максимальной степени минимизировать риск, вызываемый случайным попаданием в прибор веществ, с учетом самого прибора и характера среды, в которой его предполагается применять.
8. Инфекционное и микробное загрязнения
8.5 Приборы, предназначенные быть стерильными, должны изготавливаться под соответствующим контролем (например, в контролируемых условиях среды)".
8.16 Техническое обслуживание
В данном разделе описываются требования именно в отношении самого технического обслуживания медицинских изделий, а не влияние правильности его проведения на здоровье пациентов.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.18 Необходимы ли техническое обслуживание или калибровка медицинского изделия?
Рассматриваемые факторы включают сведения о том:
- должны ли техническое обслуживание и/или калибровка выполняться оператором, пользователем или специалистом;
- необходимы ли специальные вещества и/или оборудование для надлежащего технического обслуживания или калибровки".
8.17 Стерилизация и обработка
В данном разделе описываются требования в отношении стерилизации и обработки медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
8. Инфекционное и микробное загрязнения
8.3 Приборы, поставляемые в стерильном состоянии, должны изготавливаться и стерилизоваться соответствующими подтвержденными методами".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.8 Стерильно ли поставляемое медицинское изделие, или оно предназначено для стерилизации пользователем, или необходимы другие виды микробиологической обработки?
Рассматриваемые факторы:
- предназначено ли медицинское изделие для однократного или многократного применения;
- данные об условиях хранения;
- ограничение числа повторных применений;
- способы стерилизации;
- воздействие способов стерилизации, не предусмотренных изготовителем".
8.18 Маркировка
В данном разделе описываются требования в отношении маркировки медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
8. Инфекционное и микробное загрязнения
8.7 Упаковка и/или маркировка прибора должны обеспечивать различие между идентичными или схожими изделиями, продаваемыми как в стерильном, так и в нестерильном состоянии".
8.19 Оценка соответствия
В данном разделе описываются требования в отношении оценки соответствия медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
9. Качество конструкции и окружение
9.1 Если приборы предназначены для применения вместе с другими приборами или оборудованием, то весь набор, включая систему соединения, должен быть безопасным и не снижать установленных функциональных качеств прибора. Любые ограничения по применению должны быть указаны на этикетке или в инструкции пользователя".
8.20 Воздействие на технику
В данном разделе описываются требования в отношении воздействия на медицинские изделия.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.22 Какие механические силы могут воздействовать на медицинское изделие?
Рассматриваемые факторы: сведения о том, управляет ли механическими силами, оказывающими воздействие на медицинское изделие, только пользователь или пользователь совместно с другими лицами".
8.21 Ткани животного происхождения
В данном разделе описываются требования в отношении тканей животного происхождения, применяемых в области медицины.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
8. Инфекционное и микробное загрязнения
8.2 Ткани животного происхождения должны быть получены от животных, прошедших ветеринарный контроль и надзор с учетом предполагаемого использования тканей.
Нотифицированные органы должны хранить у себя информацию о географическом происхождении животных.
Переработка, хранение, испытание и обработка тканей, клеток и веществ животного происхождения должны производиться таким образом, чтобы обеспечить максимальную безопасность. В частности, необходимо предпринимать меры безопасности в отношении вирусов и других переносимых возбудителей заразных заболеваний путем реализации утвержденных методов уничтожения или вирусной инактивации в ходе процесса изготовления".
8.22 Материалы, вещества, компоненты
В данном разделе описываются требования в отношении материалов, веществ и компонентов, используемых в области медицины.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы гарантировать характеристики и функциональные свойства, указанные в разделе I "Общие требования". Особое внимание необходимо уделять:
- выбору используемых материалов, особенно в части токсичности и, в соответствующих случаях, воспламеняемости.
7.2 Приборы должны разрабатываться, изготавливаться и упаковываться таким образом, чтобы минимизировать риск, который несут с собой загрязняющие вещества и остатки для лиц, связанных с транспортировкой, хранением и применением приборов, и для пациентов с учетом предназначенного применения изделия. Особое внимание должно уделяться незащищенным тканям, а также продолжительности и периодичности воздействия.
7.3 Приборы должны разрабатываться и изготавливаться таким образом, чтобы они могли безопасно использоваться вместе с материалами, веществами и газами, с которым они вступают в контакт при нормальном применении или во время обычных процедур; если приборы предназначены для ввода лекарственных препаратов, они должны разрабатываться и изготавливаться таким образом, чтобы быть совместимыми с соответствующими лекарственными препаратами согласно положениям и ограничениям, регулирующим эти препараты и чтобы их функциональные свойства поддерживались в соответствии с предназначенным применением.
7.4 Если прибор включает в качестве неотъемлемой части вещество, которое при его самостоятельном применении может рассматриваться как медицинский препарат, определенный в статье 1 Директивы Совета 65/65/ЕЭС, и которое дополняет действие прибора, то безопасность, качество и пригодность этого вещества с учетом назначения прибора должны быть проверены по аналогии соответствующими методами, определенными в Директиве Совета 75/318/ЕЭС.
7.5 Приборы должны разрабатываться и изготавливаться таким образом, чтобы минимизировать риск, который создают вещества при их утечке из прибора.
7.6 Приборы должны разрабатываться и изготавливаться таким образом, чтобы в максимальной степени минимизировать риск, вызываемый случайным попаданием в прибор веществ, с учетом самого прибора и характера среды, в которой его предполагается применять".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.4 Какие материалы или компоненты входят в состав медицинского изделия, используются совместно либо контактируют с ним?
Рассматриваемые факторы:
- совместимость с рассматриваемыми веществами;
- совместимость с тканями или биологическими жидкостями;
- характеристики, относящиеся к безопасности;
- наличие в составе медицинского изделия материалов животного происхождения.
С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в том случае, если они остаются на изделии.
Другие рассматриваемые факторы, связанные с веществами: выведение химических веществ, продуктов жизнедеятельности и биологических жидкостей.
С.2.17 Имеются ли расходные материалы или принадлежности, связанные с медицинским изделием?
Рассматриваемые факторы: технические требования к расходным материалам или принадлежностям, связанным с медицинским изделием, и любые ограничения для пользователей в выборе данных материалов или принадлежностей".
8.23 Контактирование, совместимость, соединения
В данном разделе описываются требования в отношении влияния контактирования, совместимости и соединений медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы гарантировать характеристики и функциональные свойства, указанные в разделе I "Общие требования". Особое внимание необходимо уделять:
- совместимости между используемым материалом и биологическими тканями, клетками и жидкостями организма с учетом предназначенного применения прибора".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.29.3 Имеет ли медицинское изделие соединительные части или принадлежности?
Рассматриваемые факторы: возможность неправильных соединений, сходство с соединениями в других изделиях, прочность соединения, обратная связь при повреждении соединения, а также слишком прочное или недостаточно прочное соединение".
8.24 Применение
В данном разделе описываются требования, связанные с правильностью и последствиями применения медицинской техники.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.21 Существуют ли отсроченные или длительные последствия применения медицинского изделия?
Рассматриваемые факторы: эргономические и кумулятивные эффекты. К ним относят: воздействие коррозии на насосы для физиологического раствора, механическую усталость, ослабление узлов или креплений, воздействие вибрации, истирание или отклеивание маркировок, ухудшение свойств материалов при долгосрочном применении медицинского изделия.
Чем определяется срок службы медицинского изделия?
Рассматриваемые факторы: старение изделия и истощение элементов питания".
8.25 Эксплуатация
В данном разделе описываются требования в отношении эксплуатации медицинской техники. Этот раздел отличается от предыдущего тем, что в нем описываются требования, связанные с правильной эксплуатацией, в отношении безопасности пациентов и сотрудников.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"I. Общие требования
1. Приборы должны разрабатываться и изготавливаться таким образом, чтобы при использовании при установленных условиях и назначении они не подвергали риску клиническое состояние или безопасность пациентов, или безопасность и здоровье пользователей, или, возможно, других лиц при условии, что любые риски, которые могут быть связаны с их применением, являются допустимыми рисками, соизмеримыми с положительным результатом для пациента, и совместимы с высоким уровнем защиты здоровья и безопасности".
8.26 Упаковка
В данном разделе описываются требования в отношении упаковки для медицинской техники.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
8. Инфекционное и микробное загрязнения
8.3 Приборы, поставляемые в стерильном состоянии, должны разрабатываться, изготавливаться и упаковываться в одноразовую упаковку и/или по соответствующим процедурам должна обеспечиваться их стерильность при поставке на рынок и сохраняться при установленных условиях хранения и транспортировки, пока не будет нарушена или вскрыта защитная упаковка.
8.6 Системы упаковки для нестерильного оборудования должны обеспечивать сохранность продукции на установленном уровне чистоты и, если оборудование должно стерилизоваться перед применением, - сводить к минимуму риск микробного загрязнения; система упаковки должна быть приемлемой с учетом метода стерилизации, указанного изготовителем.
8.7 Упаковка и/или этикетирование оборудования должны обеспечивать различение идентичных или схожих видов продукции, реализуемых как в стерильных, так и в нестерильных условиях".
8.27 Транспортировка
В данном разделе описываются требования в отношении транспортировки медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
7. Химические, физические и биологические свойства
7.2 Приборы должны разрабатываться, изготавливаться и упаковываться таким образом, чтобы минимизировать риск, который несут с собой загрязняющие вещества и остатки для лиц, связанных с транспортировкой, хранением и применением приборов, и для пациентов с учетом предназначенного применения изделия. Особое внимание должно уделяться незащищенным тканям, а также продолжительности и периодичности воздействия.
8. Инфекционное и микробное загрязнения
8.3 Приборы, поставляемые в стерильном состоянии, должны разрабатываться, изготавливаться и упаковываться в одноразовую упаковку и/или по соответствующим процедурам должна обеспечиваться их стерильность при поставке на рынок и сохраняться при установленных условиях хранения и транспортировки, пока не будет нарушена или вскрыта защитная упаковка".
8.28 Хранение
В данном разделе описываются требования в отношении хранения медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.20 Имеет ли медицинское изделие ограниченный срок хранения?
Рассматриваемые факторы: соответствующая маркировка или индикаторы, а также сведения об утилизации медицинского изделия по истечении срока годности".
8.29 Энергетические источники
В данном разделе описываются требования в отношении энергетических источников для медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12. Требования к медицинским приборам, подключаемым к источнику энергии или оборудованным источником энергии
12.2 Приборы, при использовании которых безопасность пациентов зависит от внутреннего источника энергии, должны быть оборудованы средствами определения состояния источника питания.
12.3 Приборы, при использовании которых безопасность пациентов зависит от внешнего источника энергии, должны иметь систему сигнализации для предупреждения о любом отказе энергоснабжения.
12.7 Защита от механических и термических рисков
12.7.4 Клеммы и соединительные звенья в случае с электроэнергией, средства подачи газа, гидравлические или пневматические источники энергии, с которыми пользователь должен соприкасаться, следует разрабатывать и конструировать таким образом, чтобы минимизировать все возможные риски.
12.7.5 Доступные части приборов (за исключением частей или поверхности, предназначенной для подачи тепла или достижения установленной температуры) и прилегающие к ним участки не должны иметь потенциально опасную температуру при обычном применении.
12.8 Защита от рисков для пациента от источников энергии или веществ
12.8.2 Приборы должны оснащаться средствами для предотвращения и/или индикации любых несоответствий в интенсивности подачи, которые могут представлять опасность. Приборы должны включать в себя средства, пригодные для предотвращения случайного выделения опасного количества энергии из источника энергии и/или вещества".
8.30 Инструкции
В данном разделе описываются требования в отношении инструкций по применению или эксплуатации медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
11.4 Инструкции
11.4.1 Инструкции по эксплуатации прибора, испускающего излучение, должны содержать подробную информацию о характере излучения, средствах защиты пациента или пользователя и методах недопущения неправильного применения и исключения риска, связанного с установкой".
8.31 Управление и контроль
В данном разделе описываются требования в отношении управления и контроля медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.29.6 Имеет ли медицинское изделие меню управления?
Рассматриваемые факторы: сложность и число уровней меню, осведомленность пользователя о состоянии меню, расположение настроек, способ перемещения между объектами, число шагов в одном действии, проблемы четкой последовательности действий и запоминания, соотношение важности и доступности функции управления, а также влияние, оказываемое отклонениями от установленных операционных процедур".
8.32 Воздействие на пациента
В данном разделе описываются требования в отношении воздействия медицинских изделий на пациента.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
9. Качество конструкции и окружение
9.2 Приборы должны разрабатываться и изготавливаться таким образом, чтобы устранить или минимизировать по мере возможности:
- риск травмы, связанной с их физическими свойствами, включая соотношение объема и давления, габаритные и, в соответствующих случаях, эргономические свойства.
12.7 Защита от механических и термических рисков
12.7.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы защитить пациента и пользователя от механических повреждений, связанных, например, с прочностью, устойчивостью и наличием движущихся деталей".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.5 Может ли энергия быть передана пациенту или выработана пациентом?
Рассматриваемые факторы:
- вид передаваемой энергии;
- управление энергией, качество, количество, интенсивность и длительность воздействия;
- являются ли уровни энергии выше, чем в уже применяемых подобных изделиях.
С.2.6 Вводят ли пациенту или выводят из него какие-либо вещества?
Рассматриваемые факторы:
- сведения о введении или выведении веществ;
- сведения о том, одно это вещество или группа веществ;
- данные о максимальной и минимальной скорости введения/выведения вещества и управлении этим процессом".
8.32.1 Облучение
В данном разделе описываются требования безопасности в отношении облучения при применении медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
11. Защита от излучения (облучения)
11.1 Общие положения
11.1.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы воздействие излучения на пациентов, пользователей и других лиц было по мере возможности уменьшено до уровня, совместимого с предназначенным применением, но при этом не ограничивая применение соответствующим образом определенных уровней для терапевтических и диагностических целей.
11.2 Назначенное облучение
11.2.1 Если приборы предназначаются для излучения опасных уровней радиации, необходимых для конкретной медицинской цели, положительный эффект которых превышает риски, связанные с облучением, необходимо, чтобы пользователь имел возможность контролировать такое излучение. Подобные приборы должны разрабатываться и изготавливаться таким образом, чтобы обеспечивать воспроизводимость и толерантность соответствующих изменяемых параметров.
11.2.2 Если приборы предназначены для испускания опасного видимого и/или невидимого излучения, оно должно быть по мере возможности отображено визуально и/или снабжено звуковыми предупреждениями об излучении.
11.3 Непредусмотренное облучение
11.3.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы воздействие облучения на пациентов, пользователей и других лиц непредусмотренного, блуждающего или рассеянного излучения было уменьшено по мере возможности".
8.32.2 Температура
В данном разделе описываются требования в отношении температуры медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12.7 Защита от механических и термических рисков
12.7.5 Легкодоступные детали оборудования (за исключением деталей или участков, предназначенных для подачи тепла или достижения установленной температуры) и окружающее их пространство не должны развивать потенциально опасную температуру при обычных условиях эксплуатации.
12.8 Защита пациента от рисков при поступлении энергии или веществ
12.8.1 Приборы, применяемые для подачи энергии и веществ пациенту, должны разрабатываться и конструироваться таким образом, чтобы интенсивность подачи могла устанавливаться и поддерживаться с достаточной точностью, гарантирующей безопасность пациента и пользователя".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в случае, если они остаются на изделии".
8.32.3 Вибрация
В данном разделе описываются требования в отношении вибрации и шума медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12.7 Защита от механических и термических рисков
12.7.2 Приборы должны разрабатываться и изготавливаться таким образом, чтобы снизить до самого низкого возможного уровня риск, возникающий при вибрации, создаваемой прибором, с учетом технического прогресса и средств, с помощью которых можно ограничить вибрацию, особенно около ее источника, если вибрация не является частью установленных функциональных качеств".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в случае, если они остаются на изделии".
8.32.4 Шум
В данном разделе описываются требования в отношении шума при использовании медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС:
"12.7 Защита от механических и термических рисков
12.7.3 Приборы должны разрабатываться и изготавливаться таким образом, чтобы снизить до самого низкого возможного уровня риск, возникающий генерируемым шумом, с учетом технического прогресса и средств, с помощью которых можно ограничить шум, особенно около его источника, если шум не является частью установленных функциональных качеств".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в случае, если они остаются на изделии".
8.32.5 Электрическая безопасность
В данном разделе описываются требования в отношении электрической безопасности медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12.6 Защита от поражения электрическим током
12.6.1 Приборы должны разрабатываться и изготавливаться таким образом, чтобы по возможности избежать риска случайного поражения электрическим током при обычном использовании и при единичном отказе при условии, что прибор установлен правильно".
8.32.6 Инфекционное и микробное загрязнения
В данном разделе описываются требования к механической безопасности медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
8. Инфекционное и микробное загрязнения
8.1 Приборы и процессы изготовления должны разрабатываться таким образом, чтобы устранять или минимизировать риск инфицирования пациента, пользователя и третьих лиц. При разработке необходимо предусматривать простоту в обращении и минимизировать возможность загрязнения прибора пациентом и наоборот во время его применения".
8.32.7 Ионизирующее излучение
В данном разделе описываются требования безопасности в отношении ионизирующих излучений.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
11.5 Ионизирующее излучение
11.5.1 Приборы, генерирующие ионизирующее излучение, должны разрабатываться и изготавливаться таким образом, чтобы обеспечить, где это осуществимо, возможность изменения и контроля количества, геометрии и особенностей излучения с учетом предназначенного применения прибора.
11.5.2 Приборы, генерирующие ионизирующее излучение и предназначенные для диагностической радиологии, должны разрабатываться и изготавливаться таким образом, чтобы получить соответствующее изображение и/или качество результатов для намеченной медицинской цели при минимальном воздействии излучения на пациента и пользователя.
11.5.3 Приборы, генерирующие ионизирующее излучение и предназначенные для терапевтической радиологии, должны разрабатываться и изготавливаться таким образом, чтобы обеспечивать возможность надежного контроля и регулирования получаемой дозы облучения, типа луча и энергии и при возможности качества излучения".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.14 Происходит ли нежелательное выделение энергии или веществ?
Рассматриваемые факторы, связанные с энергией: шум и вибрация, тепло, излучение (в том числе ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное), температура на контактных поверхностях, токи утечки, электрические или магнитные поля.
Рассматриваемые факторы, связанные с веществами: вещества, используемые при изготовлении, очистке и испытаниях и оказывающие нежелательное физиологическое воздействие в том случае, если они остаются на изделии".
8.33 Оператор
В данном разделе описываются требования к безопасности оператора при работе с медицинскими изделиями.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.26 Необходимы ли специальное обучение или специальные навыки для монтажа или применения медицинского изделия?
Рассматриваемые факторы: новизна медицинского изделия; навыки и знания, необходимые для монтажа данного изделия".
8.34 Измерения, интерпретация данных
В данном разделе описываются требования в отношении измерений и интерпретации данных в области медицины.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
10. Приборы с функциями измерения
10.1 Приборы с функциями измерения должны разрабатываться и изготавливаться таким образом, чтобы обеспечивать достаточную точность и устойчивость в соответствующих пределах точности и с учетом предназначенного применения прибора. Пределы точности должны быть указаны изготовителем.
10.2 Шкалы для измерения, контроля и индикации должны разрабатываться в соответствии с принципами эргономики с учетом предназначенного применения прибора.
10.3 Измерения, выполняемые прибором с функциями измерения, должны выражаться в стандартных единицах измерения, соответствующих положениям Директивы Совета 80/181/ЕЭС".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.11 Предназначено ли медицинское изделие для проведения измерений?
Рассматриваемые факторы: измеряемые переменные, правильность и точность результатов измерений.
С.2.12 Является ли медицинское изделие интерпретирующим?
Рассматриваемые факторы: способность медицинского изделия давать заключение на основе входных или полученных в процессе применения данных, использованных алгоритмов и доверительных интервалов. Особое внимание следует уделить непредусмотренному применению данных или алгоритмов".
8.35 Интерфейс
В данном разделе описываются требования к интерфейсу медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.29 Зависит ли успешное применение медицинского изделия от человеческого фактора, такого как пользовательский интерфейс?
С.2.29.1 Могут ли особенности конструкции пользовательского интерфейса привести к ошибкам применения?
Рассматриваемые факторы: особенности конструкции пользовательского интерфейса, которые могут привести к ошибкам применения. К ним относят: элементы управления или индикаторы, используемые символы, эргономические свойства, дизайн изделия и схему расположения элементов управления, иерархию действий, меню для изделий с программным управлением, наглядность предупреждений, хорошую слышимость сигналов тревоги, стандартные цветовые коды.
С.2.29.4 Имеет ли медицинское изделие управляющий интерфейс?
Рассматриваемые факторы: пространственное распределение элементов управления, их кодирование, группировка, схема пространственного распределения, режимы обратной связи, грубые ошибки, незначительные промахи, дифференциация управления, качество изображений, направление активирования или изменения, дискретность или непрерывность функционирования элементов управления, обратимость настроек или действий.
С.2.29.8 Можно ли использовать пользовательский интерфейс для инициирования действий пользователя?
Рассматриваемые факторы: возможность инициирования намеренных действий пользователя для входа в управляемый рабочий режим (что увеличивает риски для пациента); осведомленность пользователя в данных обстоятельствах".
8.36 Сигнализация
В данном разделе описываются требования в отношении сигнализации для медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12. Требования к медицинским приборам, подключаемым к источнику энергии или оборудованным источником энергии
12.4 Приборы, предназначенные для контроля одного или нескольких клинических параметров пациента, должны быть оборудованы соответствующими системами сигнализации для предупреждения пользователя о ситуациях, которые могут привести к гибели пациента или серьезному ухудшению состояния его здоровья.
12.8 Защита от рисков для пациента от источников энергии или веществ
12.8.2 Приборы должны оснащаться средствами для предотвращения и/или индикации любых несоответствий в интенсивности подачи, которые могут представлять опасность.
12.9 На приборе должны быть четко указаны функции управления и индикаторы. Если на прибор нанесены инструкции, необходимые для его эксплуатации, или указываются рабочие или регулировочные параметры с помощью визуальных систем, такая информация должна быть понятной пациенту и в соответствующих случаях пользователю".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.30 Используют ли в медицинском изделии систему сигнализации?
Рассматриваемые факторы: риск подачи ложных сигналов тревоги, несрабатывание системы сигнализации, отсоединение системы сигнализации, ненадежность удаленных систем сигнализации, а также возможность понимания медицинским персоналом принципов работы системы сигнализации".
8.37 Индикация
В данном разделе описываются требования в отношении индикации для медицинских изделий.
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.29.5 Имеет ли медицинское изделие функцию отображения информации?
Рассматриваемые факторы: качество изображения в разных условиях; ориентация, зрительные способности пользователя; степень заполнения экрана монитора и перспективное изображение; четкость представленной информации; единицы отображения информации; цветовые коды; доступ к наиболее важной информации".
8.38 Данные
В данном разделе описываются требования в отношении данных для медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
14. В случае если соответствие основным требованиям должно быть основано на клинических данных, как указано в разделе I (6), подобные данные должны быть определены в соответствии с Приложением X".
8.39 Программные средства
В данном разделе описываются требования в отношении программных средств для медицинских изделий.
Пример А - Директива Совета 93/42/ЕЭС, Приложение I:
"II. Требования к разработке и конструкции
12. Требования к медицинским приборам, подключаемым к источнику энергии или оборудованным источником энергии
12.1 Приборы, включающие электронные программируемые системы, должны разрабатываться так, чтобы обеспечить повторяемость, надежность и эффективность этих систем в соответствии с предназначенным применением. В случае единичного отказа (в системе) следует принимать соответствующие меры для исключения или снижения возможных последующих рисков".
Пример Б - ГОСТ Р ИСО 14971-2009:
"С.2.19 Входит ли в состав медицинского изделия программное обеспечение?
Рассматриваемые факторы: сведения о том, должно ли программное обеспечение быть установлено, верифицировано, модифицировано или заменено оператором, пользователем или специалистом".
УДК 006.354:615.47:006.354 | ОКС 25.040.40 | |
Ключевые слова: медицинская техника, изделие, оборудование, приборы, пациент, информация, данные, производство. | ||
Электронный текст документа
и сверен по:
, 2020

{kind=link}