ГОСТ Р ИСО 14708-7-2016
Группа Р29
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИМПЛАНТАТЫ ДЛЯ ХИРУРГИИ
Активные имплантируемые медицинские изделия
Часть 7
Частные требования к системам кохлеарной имплантации
Implants for surgery. Active implantable medical devices. Part 7. Particular requirements for cochlear implant systems
ОКС 11.040.40
ОКП 944480
Дата введения 2017-10-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "Научно-технический центр "МЕДИТЭКС" (ООО "НТЦ "МЕДИТЭКС") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 011 "Медицинские приборы, аппараты и оборудование"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 21 октября 2016 г. N 1489-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14708-7:2013* "Имплантаты для хирургии. Активные имплантируемые медицинские изделия. Часть 7. Частные требования к системам кохлеарной имплантации" (ISO 14708-7:2013 "Implants for surgery - Active implantable medical devices - Part 7: Particular requirements for cochlear implant systems", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации и действующие в этом качестве межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего документа соответствующее уведомление будет опубликовано в ближайшем выпуске информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт определяет частные требования к АКТИВНЫМ ИМПЛАНТИРУЕМЫМ МЕДИЦИНСКИМ ИЗДЕЛИЯМ, предназначенным для лечения ухудшения слуха посредством электростимуляции (например, системы кохлеарной или слуховой стволомозговой имплантации) в целях обеспечения безопасности использования как для пациентов, так и для специалистов.
СИСТЕМЫ КОХЛЕАРНОЙ ИМПЛАНТАЦИИ и СИСТЕМЫ СЛУХОВОЙ СТВОЛОМОЗГОВОЙ ИМПЛАНТАЦИИ являются активными имплантируемыми МЕДИЦИНСКИМИ ИЗДЕЛИЯМИ и состоят из имплантируемых и НЕИМПЛАНТИРУЕМЫХ ЧАСТЕЙ (внешние части). Источник питания изделия может быть как внешним, так и внутренним (внутренняя батарея). Имплантируемая система предназначена для восстановления слуха посредством электростимуляции слуховых путей. Внешне или внутренне обработанная акустическая информация преобразовывается в сигналы электростимуляции, передающиеся через один или несколько электродов. Рабочие параметры изделия могут быть скорректированы через неимплантируемые принадлежности.
Действие настоящего стандарта распространяется на все части ИМПЛАНТИРУЕМЫХ СИСТЕМ, включая принадлежности.
Требования стандарта дополняют или изменяют требования ИСО 14708-1 "Имплантаты хирургические. Активные имплантируемые медицинские изделия. Часть 1. Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем".
Дополнительные рисунки и таблицы к ИСО 14708-1 пронумерованы начиная со 101; дополнительные приложения обозначены буквами АА, ВВ и т.д.
Термины, определенные в разделе 3 настоящего стандарта, набраны прописными буквами. Если термин используется в ином значении, он набран строчными буквами.
1 Область применения
Настоящий стандарт распространяется на АКТИВНЫЕ ИМПЛАНТИРУЕМЫЕ МЕДИЦИНСКИЕ ИЗДЕЛИЯ, предназначенные для лечения нарушений слуха посредством электростимуляции слуховых путей. Настоящий стандарт не распространяется на изделия, используемые для лечения нарушений слуха не посредством электростимуляции.
Методы испытаний, приведенные в настоящем стандарте, являются типовыми для оценки соответствия образцов изделий.
Настоящий стандарт также распространяется на НЕИМПЛАНТИРУЕМЫЕ ЧАСТИ и принадлежности систем кохлеарной имплантации (см. примечание).
Электрические характеристики ИМПЛАНТИРУЕМОЙ ЧАСТИ определяются либо методом, регламентированным настоящим стандартом, либо другим, не менее точным методом. В случае спорной ситуации применяется метод настоящего стандарта.
Примечание - Активное имплантируемое медицинское изделие может быть как отдельным устройством, так и набором устройств с одной или более принадлежностями. Не все из этих компонентов должны быть полностью или частично имплантированы, но при этом необходимо определить требования к неимплантируемым частям и принадлежностям, если они могут повлиять на безопасность или функциональные характеристики имплантируемой части.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на стандарты и другие нормативные документы*, которые необходимо учитывать при его использовании. Для датированных ссылок применяют только указанные издания. Для недатированных ссылок применяют самые последние издания (включая любые изменения и поправки).
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ISO 10993-1, Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process (Биологическая оценка медицинских устройств - Часть 1. Оценка и проверка в процессе управления рисками)
ISO 11607-1, Packaging for terminally sterilized medical devices - Part 1: Requirements for materials, sterile barrier systems and packaging systems (Упаковка медицинских изделий, подлежащих финишной стерилизации. Часть 1. Требования к материалам, барьерным системам для стерилизации и упаковочным системам)
ISO 14155, Clinical investigation of medical devices for human subjects - Good clinical practice (Клинические исследования медицинских изделий для людей. Надлежащая клиническая практика)
ISO 14971, Medical devices - Application of risk management to medical devices (Изделия медицинские. Применение менеджмента риска к медицинским изделиям)
IEC 60068-2-27, Environmental testing - Part 2-27: Tests - Test Ea and guidance: Shock (Испытания на воздействие внешних факторов. Часть 2-27. Испытания. Тестирование Еa и руководство: Удар)
IEC 60068-2-31, Environmental testing - Part 2-31: Tests - Test Еc: Rough handling shocks, primarily for equipment-type specimens (Испытания на воздействие внешних факторов. Часть 2-31. Испытания. Испытание Еc: Воздействия при грубом обращении, в основном, с образцами аппаратуры)
IEC 60068-2-47, Environmental testing - Part 2-47: Test - Mounting of specimens for vibration, impact and similar dynamic tests (Испытания на воздействие внешних факторов. Часть 2-47. Монтаж элементов, аппаратуры и других изделий для испытаний на вибрацию, удар и подобных динамических испытаний)
IEC 60068-2-64, Environmental testing - Part 2-64: Tests - Test Fh: Vibration, broadband random and guidance (Испытания на воздействие внешних факторов. Часть 2-64. Испытание Fh. Широкополосная случайная вибрация (цифровое управление) и руководство)
IEC 60068-2-75, Environmental testing - Part 2-75: Tests - Test Eh: Hammer tests (Испытания на воздействие внешних факторов. Часть 2-75. Испытание Eh: Ударные испытания)
IEC 60118-6, Hearing aids - Part 6: Characteristics of electrical input circuits for hearing aids (Аппараты слуховые. Часть 6. Характеристики входных электрических цепей слуховых аппаратов)
IEC 60601-1:2006, Medical electrical equipment - Part 1: General requirements for basic safety and essential performance (Изделия медицинские электрические. Часть 1. Общие требования безопасности с учетом основных функциональных характеристик)
IEC 60601-1-2, Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral standard: Electromagnetic compatibility - Requirements and tests (Изделия медицинские электрические. Часть 1-2. Общие требования безопасности с учетом основных функциональных характеристик. Параллельный стандарт. Электромагнитная совместимость. Требования и испытания)
IEC 61000-4-2, Electromagnetic compatibility (EMC) - Part 4-2: Testing and measurement techniques - Electrostatic discharge immunity test (Электромагнитная совместимость. Часть 4-2. Методики испытаний и измерений. Испытание на невосприимчивость к электростатическому разряду)
IEC 62304, Medical device software - Software life cycle processes (Изделия медицинские. Программное обеспечение. Процессы жизненного цикла)
EN 1593, Non-destructive testing - Leak testing - Bubble emission techniques (Испытание на прочность - Испытание на герметичность - Пузырьковые методы эмиссии)
EN 13185, Non-destructive testing - Leak testing - Tracer gas method (Испытание на прочность - Испытание на герметичность - Проверка герметичности с использованием пробного газа)
3 Термины и определения
В настоящем стандарте применены термины по ИСО 14708-1, а также следующие термины с соответствующими определениями:
3.3.1 система кохлеарной имплантации (cochlear implant system; CIS): Активное имплантируемое медицинское изделие, включающее в себя имплантируемые и НЕИМПЛАНТИРУЕМЫЕ ЧАСТИ, позволяющее компенсировать потерю слуха посредством электрической стимуляции улитки внутреннего уха.
3.3.2 система слуховой стволомозговой имплантации (auditory brainstem implant system; BIS): АКТИВНОЕ ИМПЛАНТИРУЕМОЕ МЕДИЦИНСКОЕ ИЗДЕЛИЕ, включающее в себя имплантируемые и НЕИМПЛАНТИРУЕМЫЕ ЧАСТИ, позволяющее компенсировать потерю слуха путем электрической стимуляции слуховых центров ствола головного мозга.
3.3.3 имплантируемая система (implant system): СИСТЕМА КОХЛЕАРНОЙ ИМПЛАНТАЦИИ или СИСТЕМА СЛУХОВОЙ СТВОЛОМОЗГОВОЙ ИМПЛАНТАЦИИ.
3.3.4 неимплантируемая часть (non-implantable part): Внешняя часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ.
Примечание - Например, речевой процессор, микрофон, катушка передатчика или источник питания.
3.3.5 стимулятор (stimulator): Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, содержащая электронную схему для электростимуляции.
3.3.6 переносная часть (body-worn): НЕИМПЛАНТИРУЕМАЯ ЧАСТЬ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, переносимая на теле пациента (например, на поясе или на уровне уха).
3.5.1 электродный контакт (electrode contact): Часть изделия, электрический проводник, разработанный для формирования интерфейса с внутренними тканями тела пациента или жидкостью в организме.
3.5.2 электродный массив (electrode array): ДИСТАЛЬНАЯ часть проводника, содержащего более одного электродного контакта.
3.5.3 электрод сравнения (reference electrode): Электрически проводящая часть, разработанная для прохождения обратного электрического тока стимуляции.
3.5.4 дистальный (distal): Расположенный дальше от места расположения СТИМУЛЯТОРА.
3.5.5 проксимальный (proximal): Расположенный ближе к месту расположения СТИМУЛЯТОРА.
3.9.1 обозначение модели (model designation): Наименование и/или комбинация букв и чисел, используемых изготовителем для различения изделий по функциям или типам.
3.9.2 серийный номер (serial number): Уникальная комбинация букв и/или цифр, выбранных изготовителем для различения изделий с одинаковым ОБОЗНАЧЕНИЕМ МОДЕЛИ друг от друга.
3.20.1 выходной сигнал (output signal): Аналоговый или импульсный электрический выходной сигнал ИМПЛАНТИРУЕМОЙ СИСТЕМЫ для стимуляции слуховых путей.
3.20.2 импульс (pulse): Определенный электрический ВЫХОДНОЙ СИГНАЛ (напряжения или тока) с заданными амплитудой и длительностью.
3.20.3 бифазный импульс (biphasic pulse): ИМПУЛЬС, имеющий и отрицательные, и положительные фазы.
3.22.1 срок годности (use-before-date): Дата, после которой изготовитель не рекомендует ИМПЛАНТИРОВАТЬ СИСТЕМУ.
3.22.2 магнит (magnet): Компонент, который создает внешний магнитный поток.
4 Обозначения и сокращения
В настоящем стандарте не определены специальные требования к обозначениям и сокращениям. Это не ограничивает использование обозначений, определенных в других стандартах или в сопроводительной документации.
5 Общие требования к неимплантируемым частям
5.1 Применяют соответствующий раздел стандарта ИСО 14708-1.
5.2 Замена:
Программное обеспечение АКТИВНОГО ИМПЛАНТИРУЕМОГО МЕДИЦИНСКОГО ИЗДЕЛИЯ или программное обеспечение, попадающее под это определение, должно быть разработано и утверждено в соответствии с жизненным циклом изделия согласно МЭК 62304.
6 Осмотр и измерение
Если в настоящем стандарте упоминается проверка документации по анализу конструкции, предоставленной изготовителем, необходимо также провести проверку файла менеджмента риска согласно ИСО 14971.
6.1 Измерение характеристик выходного сигнала
Измерение требуется проводить с имплантируемой частью ИМПЛАНТИРУЕМОЙ СИСТЕМЫ при температуре (37±2)°С. ИМПЛАНТИРУЕМАЯ СИСТЕМА должна быть настроена на использование максимального числа выходов, и каждый выход следует запрограммировать на максимальное значение (амплитуды и длительности импульса). Входной сигнал, эквивалентный уровню звукового давления 70 дБ, должен быть подан на вход микрофона. Если это применимо, то связь через кожу должна осуществляться на расстоянии (5±1) мм. Если ИМПЛАНТИРУЕМАЯ СИСТЕМА обеспечивает различные выходные сигналы, то каждый необходимо измерить и описать отдельно. Для упрощения соединения испытательный образец может быть собран частично. Принимая во внимание все ошибки, погрешность измерений не должна превышать 5%.
6.2 Измерение амплитуды выходного сигнала и длительности импульса
Каждый выход представленного образца ИМПЛАНТИРУЕМОЙ СИСТЕМЫ необходимо соединить с нагрузочным сопротивлением, равным 1 кОм (±1%) (см. рисунок 101), а систему настроить согласно 6.1. Осциллограф необходимо настроить таким образом, чтобы отобразить весь выходной сигнал с максимальным разрешением. Измерение проводят на пиках ВЫХОДНЫХ СИГНАЛОВ. Каждый выход поочередно подключают к осциллографу для измерения амплитуды и длительности импульсов. Фиксируют среднее значение амплитуды и длительности импульса, а также их диапазон, результат выражают в мкА и мкс.
6.3 Точность измерения полного сопротивления
Если ИМПЛАНТИРУЕМАЯ СИСТЕМА позволяет измерить полное сопротивление (с помощью дистанционных или прямых измерений), изготовитель должен определить точность измерения полного сопротивления нагрузки 10 кОм. В местах, где имплантируемая система позволяет измерить полное сопротивление (с помощью дистанционных или прямых измерений), изготовитель должен установить точность его измерения для нагрузочного сопротивления, равного 10 кОм. Условия измерений при этом аналогичны обычным условиям клинической практики. Измерить следует каждый выходной сигнал (см. рисунок 101). Точность измерения полного сопротивления выражают в процентах.
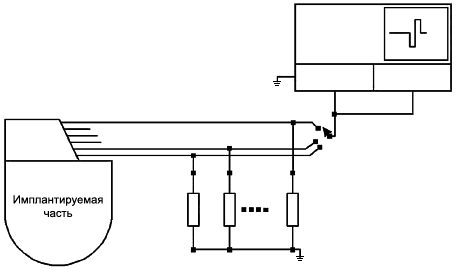
Земля соединена с внешним электродом сравнения, если он используется.
Рисунок 101 - Измерение амплитуды выходного сигнала и полного сопротивления нагрузки
7 Общие требования к упаковке
7.1 По ИСО 14708-1.
7.2 По ИСО 14708-1.
8 Общие требования к маркировке активных имплантируемых медицинских изделий
8.1 По ИСО 14708-1.
8.2 По ИСО 14708-1.
9 Маркировка на товарной упаковке
9.1 По ИСО 14708-1.
9.2 По ИСО 14708-1, за исключением следующего.
Замена:
На ТОВАРНОЙ УПАКОВКЕ должны быть указаны наименование и адрес изготовителя, как минимум город и страна. На ТОВАРНОЙ УПАКОВКЕ должны быть указаны наименование и адрес официального представителя, если у изготовителя нет зарегистрированного места осуществления хозяйственной деятельности на территории Российской Федерации.
Соответствие проверяют в ходе осмотра.
9.3 Замена:
Если ИМПЛАНТИРУЕМАЯ СИСТЕМА поставляется в виде упакованных сборочных узлов, на каждой индивидуальной ТОВАРНОЙ УПАКОВКЕ нужно указать содержимое, номер модели или детали, а также номер партии или серии, если он есть.
Соответствие проверяют в ходе осмотра.
9.4 По ИСО 14708-1.
9.5 По ИСО 14708-1.
9.6 По ИСО 14708-1.
9.7 Замена:
На ТОВАРНОЙ УПАКОВКЕ имплантируемых частей АКТИВНОГО ИМПЛАНТИРУЕМОГО МЕДИЦИНСКОГО ИЗДЕЛИЯ также необходимо указать СРОК ГОДНОСТИ согласно 9.6.
Соответствие проверяют в ходе осмотра.
9.8 По ИСО 14708-1.
9.9 По ИСО 14708-1.
9.10 По ИСО 14708-1.
9.11 По ИСО 14708-1.
9.12 Дополнительный подпункт: Если имплантируемая система поставляется в виде упакованных сборочных узлов, на каждой индивидуальной товарной упаковке необходимо указать содержимое, номер модели или детали, а также номер партии или серии, если он есть.
Соответствие проверяют в ходе осмотра.
10 Конструкция товарной упаковки
10.1 По ИСО 14708-1.
10.2 По ИСО 14708-1.
10.3 По ИСО 14708-1.
Примечание - Сменные этикетки, содержащие дополнительные данные помимо информации, определенной в разделе 9, не могут тестироваться методом, описанным в 10.3.
11 Маркировка на стерильном пакете
11.1 По ИСО 14708-1.
11.2 По ИСО 14708-1.
11.3 По ИСО 14708-1.
11.4 По ИСО 14708-1.
11.5 По ИСО 14708-1.
11.6 По ИСО 14708-1.
11.7 По ИСО 14708-1.
11.8 По ИСО 14708-1.
11.9 По ИСО 14708-1.
11.10 По ИСО 14708-1.
Примечание - Требования настоящего подпункта можно выполнить с использованием общепринятого обозначения.
12 Конструкция одноразовой упаковки
12.1 По ИСО 14708-1, за исключением следующего.
Замена:
ОДНОРАЗОВАЯ УПАКОВКА должна соответствовать ИСО 11607-1
Соответствие проверяют в ходе осмотра изделия и анализа документации, предоставленной изготовителем.
12.2 По ИСО 14708-1.
12.3 По ИСО 14708-1.
13 Маркировка активных имплантируемых медицинских изделий
13.1 По ИСО 14708-1.
13.2 По ИСО 14708-1.
13.3 Замена:
В случае необходимости имплантируемые части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должны быть однозначно идентифицируемы (особенно обозначение модели) без хирургического вмешательства.
Соответствие должно быть подтверждено в ходе осмотра в порядке, установленном изготовителем в инструкции по применению (см. 28.6).
13.4 По ИСО 14708-1.
14 Защита от непреднамеренных биологических воздействий, вызванных активным имплантируемым медицинским изделием
14.1 По ИСО 14708-1.
14.2 Замена:
Любая имплантируемая часть АКТИВНОГО ИМПЛАНТИРУЕМОГО МЕДИЦИНСКОГО ИЗДЕЛИЯ, которая при реальном использовании контактирует с жидкими средами организма человека, должна быть проверена на выделение твердых частиц в приемлемом количестве.
Методика испытаний. Имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ асептически извлекают из ОДНОРАЗОВОЙ УПАКОВКИ. Имплантируемую часть погружают в нейтральном стеклянном контейнере в инъекционный раствор соли 9 г/л. Объем солевого раствора должен быть примерно в (5±0,5) раз больше численного значения площади поверхности имплантируемой части, выраженной в квадратных сантиметрах. Контейнер накрывают стеклянной пластиной и оставляют при температуре примерно (37±2)°С на 8-18 ч, периодически встряхивая. Затем готовят контрольный раствор аналогичного объема с солью из той же партии, поддерживают те же условия, что и в контейнере с образцом. Образцы жидкости из обычного и контрольного контейнеров сравнивают, применяя аппаратуру для измерения размеров частиц, работающую по принципу оптического затенения (метод V.5.7.1 Европейской Фармакопеи) или по принципу электрического зондирования (см. принцип Культера, приложение XIII из Британской Фармакопеи).
Среднее число частиц, выделяющихся из образца, по отношению к контрольному образцу не должно превышать 100 частиц/мл для частиц размерами более 5,0 мкм и 5 частиц/мл для частиц размерами более 25 мкм.
14.3 Замена:
По ИСО 14708-1 и ИСО 10993-1.
14.4 По ИСО 14708-1.
15 Защита пациента или пользователя от вреда, вызванного внешними физическими особенностями активного имплантируемого медицинского изделия
15.1 По ИСО 14708-1.
15.2 Замена:
Не допускается наличие в имплантируемых частях ИМПЛАНТИРУЕМОЙ СИСТЕМЫ конструктивных особенностей, таких как острые углы или края, которые могут вызвать чрезмерную реакцию или воспаление, помимо вызванных непосредственно процедурой имплантации, также недопустимо наличие грубых поверхностей, если это не обусловлено необходимостью обеспечения правильного функционирования изделия.
Соответствие должно быть подтверждено информацией, представленной изготовителем, в которой указано, что безопасность физических характеристик проверена соответствующими методами.
16 Защита пациента от вреда, вызванного электрическим током
16.1 Замена:
Электрические звуковые входные сигналы в НЕИМПЛАНТИРУЕМЫХ ЧАСТЯХ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должны соответствовать требованиям электробезопасности слухового аппарата согласно МЭК 60118-6. Другие электрические входы или выходы НЕИМПЛАНТИРУЕМЫХ ЧАСТЕЙ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, связывающие их с питающей электрической сетью или изделием, работающим от сети, не удовлетворяющие требованиям изоляции МЭК 60601-1, должны содержать разделительное устройство, которое соответствует применимым пунктам в части изоляции МЭК 60601-1 (разделительное устройство согласно определению МЭК 60601-1:2006, подраздел 16.5).
Примечание - Разделительное устройство не требуется, если одиночное изделие работает от батареи.
Соответствие проверяется по МЭК 60601-1 (если применимо) путем анализа документации, предоставленной изготовителем.
16.2 Замена:
Имплантируемые части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ в контакте с телом пациента должны быть электрически нейтральными, за исключением использования по целевому назначению. Во время использования изделия необходимо исключить в любом из проводящих путей утечку постоянного тока, превышающую 0,1 мкА.
Соответствие проверяют в ходе проверки испытательных процедур и на основе результатов, предоставленных изготовителем.
16.3 По ИСО 14708-1.
17 Защита пациента от вреда, вызванного тепловыделениями
17.1 По ИСО 14708-1.
17.2 Настоящий подпункт будет проработан в следующих изданиях.
18 Защита от ионизирующего излучения, выделяемого или испускаемого активным имплантируемым медицинским изделием
18.1 По ИСО 14708-1.
18.2 По ИСО 14708-1.
18.3 По ИСО 14708-1.
19 Защита от непреднамеренных воздействий, вызванных изделием
Примечание - См. также 28.20.
19.1 По ИСО 14708-1.
19.2 Замена:
Если имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ включает в себя батарею, изготовитель должен предусмотреть индикатор, который заблаговременно предупреждает врача и пользователя о низком заряде изделия.
Соответствие подтверждается контролем проведенного изготовителем анализа конструкции с необходимыми расчетами и данными испытаний.
19.3 По ИСО 14708-1.
19.4 По ИСО 14708-1, за исключением следующего.
Замена оценки:
Побочные эффекты и преимущества изделия определяются на основе современной медицинской практики с применением аналогичных изделий или в ходе клинических исследований в соответствии с ИСО 14155.
Дополнительные подпункты:
19.5 Физическая, биологическая и геометрическая целостность имплантируемых частей ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должна нарушаться при замене на изделие того же изготовителя или удалении. Это необходимо учитывать в конструкции изделия.
Соответствие подтверждают анализом представленного изготовителем проекта, по возможности подтвержденного соответствующими испытаниями и клиническими данными, в том числе постпродажного контроля в части замены изделий.
19.6 Корпус имплантируемого СТИМУЛЯТОРА ИМПЛАНТИРУЕМОЙ СИСТЕМЫ при реальном использовании находится в контакте с внутренней средой организма пациента, поэтому должен быть герметичным, чтобы жидкость не могла проникнуть в корпус СТИМУЛЯТОРА.
Метод испытания. Испытания на малые и большие течи проводят на герметичном корпусе СТИМУЛЯТОРА ИМПЛАНТИРУЕМОЙ СИСТЕМЫ в соответствии с ЕН 13185 и ЕН 1593. Если используются методы из группы А стандарта ЕН 13185, то испытание на большие течи не требуется; если используются методы группы В, то сначала необходимо провести испытания на малые течи, затем - на большие.
Примечание - Изготовитель должен провести необходимые испытания для проверки герметичности на этапе производства.
Соответствие подтверждается контролем процедур проведения испытаний и предоставленных изготовителем результатов. Интенсивность протечки изделия не должна превышать 5·10 Па м
/с для испытаний на малые течи и без потока пузырьков или двух и более пузырьков в том же месте корпуса СТИМУЛЯТОРА при испытании на большие течи.
20 Защита изделия от повреждений, вызванных применением наружных дефибрилляторов
Примечание - См. также 28.12.
20.1 Пункт ИСО 14708-1 не применяется для настоящего стандарта.
20.2 По ИСО 14708-1.
21 Защита изделия от изменений, вызванных электрическими полями высокой энергии, воздействующими непосредственно на пациента
Примечание - См. также 28.12 и 28.13.
21.1 Замена:
Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна быть сконструирована так, чтобы исключить повреждение изделия длительным воздействием высокочастотного тока от электрохирургического оборудования (электрохирургия), проходящего через пациента. Это условие обязательно для соблюдения, если ИМПЛАНТИРУЕМАЯ СИСТЕМА не размещена непосредственно между электродом для рассечения и возвратным электродом (земля) (см. также требование для предупреждения 28.13).
Методика испытания. Необходимо использовать сигнальный генератор с полным выходным сопротивлением 50 Ом (R1). Частота испытательного сигнала синусоиды составляет 500 кГц, размах напряжения тестового сигнала разомкнутого контура равен 20 В.
ИМПЛАНТИРУЕМАЯ СИСТЕМА должна быть выключена. Каждый выход имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ необходимо соединить через резистор (R) 4,7 кОм с общей точкой, связанной с выходом сигнального генератора (см. рисунок 102). ЭЛЕКТРОД СРАВНЕНИЯ имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ нужно соединить через резистор 100 Ом (R3) с землей сигнального генератора.
Необходимо использовать 10 тестовых импульсов длительностью 1 с с перерывом 5 с.
Соответствие подтверждается, если после выполнения тестовых процедур и перезапуска характеристики ИМПЛАНТИРУЕМОЙ СИСТЕМЫ будут соответствовать заявленным изготовителем спецификациям.
21.2 Настоящий пункт будет проработан в следующих изданиях.
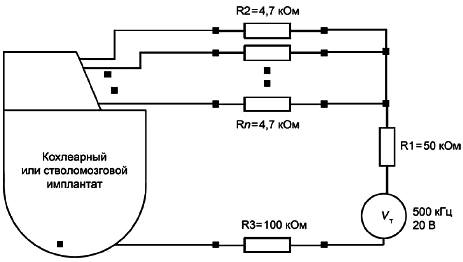
Рисунок 102 - Испытательный стенд для проверки
22 Защита активного имплантируемого медицинского изделия от изменений, вызванных применением других видов лечения
Примечание - См. также 28.12, 28.14 и 28.15.
22.2 По ИСО 14708-1, раздел 22.
Дополнительные подпункты:
22.2* Изготовитель должен идентифицировать безопасность имплантируемых частей ИМПЛАНТИРУЕМОЙ СИСТЕМЫ при проведении магнитно-резонансной томографии (MPT) (см. 28.8), указать (см. 28.12) условия (в том числе конкретные напряженности поля), в которых была проверена безопасность использования MPT, перечислить риски размагничивания и искажения изображения, а также привести инструкции для безопасного проведения MPT исследования, если оно допустимо.
__________________
* Нумерация соответствует оригиналу. - .
Риски для пациента с ИМПЛАНТИРУЕМОЙ СИСТЕМОЙ во время МРТ-исследования можно сгруппировать по следующим категориям: усилие, создаваемое в магнитном поле, тепловыделение, случайный выходной сигнал изделия и повреждения имплантата. Каждый из этих факторов должен быть проверен следующим образом:
a) Сила
Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должна наносить пациенту вред путем воздействия механических сил, которые могут возникнуть во время МРТ-сканирования.
Методика испытания. Силу рассчитывают по напряженности магнитного поля установки МРТ, магнитным свойствам ферромагнитных или парамагнитных материалов, входящих в состав имплантируемой части, силе любого внутреннего магнита и геометрии имплантируемой части, содержащей магнит. Также силу можно измерить.
Соответствие подтверждается, если максимальная сила в случае наихудшей ориентации будет ниже 10 Н или не будет выявлено смещение имплантата или магнита.
b) Тепловыделение
Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должна выделять излишнее количество тепла во время процедуры МРТ.
Методика испытания. Необходимо выбрать два одинаковых закрытых пластиковых контейнера объемом, достаточным для погружения всей имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ в раствор. Объем раствора NaCI равен (3±0,3) объемам имплантируемой части. Общий объем имплантата и раствора соли в одном контейнере должен быть идентичен объему раствора в другом контейнере. Имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ необходимо поместить в контейнер на 24 ч при температуре, аналогичной месту проведения процедуры МРТ. Оба контейнера заполняют 9 г/л раствора NaCI, находившимся в течение предыдущих 24 ч в том же помещении. Температура раствора NaCI каждого контейнера регистрируется с помощью цифрового термометра с точностью 0,1°С. Необходимо также замерить температуру в помещении. Оба контейнера помещают в аппарат для проведения МРТ в месте, предполагающем максимальную мощность радиочастотного излучения. Проводят испытательную процедуру МРТ-сканирования, представляющую наихудший случай клинического исследования (наиболее высокая степень поглощения), длительностью не менее 15 мин. Немедленно после завершения сканирования контейнеры убирают из камеры МРТ и снова регистрируют температуры каждого контейнера. Также можно использовать стандарт ASTM F2182 (American Society for Testing and Materials - американское общество испытаний и материалов) для испытания повышения температуры имплантата и проводника.
Соответствие подтверждается, если разница температур контейнеров или повышение температуры на имплантате или наконечнике электрода составляет менее 2°С.
c) Случайный выходной сигнал
Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должна генерировать вредный для пациента выходной сигнал во время процедуры МРТ.
Методика испытания. Имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ помещают в аппарат МРТ. Необходимо проверить две схемы включения ИМПЛАНТИРУЕМОЙ СИСТЕМЫ: первая - ИМПЛАНТИРУЕМАЯ СИСТЕМА имеет дополнительный измерительный резистор R1, подключенный последовательно с ЭЛЕКТРОДОМ СРАВНЕНИЯ, с доступом к обоим концам R1, вторая - имплантируемая система с доступом к напряжению питания имплантата. Приемопередающая волоконно-оптическая схема и осциллограф подключаются к резистору R1, как показано на рисунке 103. Для этого испытания необходимо использовать экранированный кабель типа витой пары и пассивный фильтр нижних частот. Рекомендованные резисторы: R1 =10 кОм, R2 = R3 =22 кОм. Эти три резистора должны быть смонтированы в область менее 1 см. Фильтр нижних частот формируется резисторами R2-R5 и конденсатором С1. При определении значения R4 и R5 необходимо принять во внимание входное полное сопротивление волоконно-оптического элемента. Частота среза составляет около 10 кГц. Все компоненты должны быть сконструированы с использованием технологии поверхностного монтажа и сделаны из немагнитных материалов. Необходимо использовать измерительное оборудование, которое не влияет на МРТ. В противном случае осциллограф следует размещать за пределами помещения для проведения МРТ. Имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, включая ЭЛЕКТРОДНЫЙ МАССИВ и ЭЛЕКТРОД СРАВНЕНИЯ, помещают в контейнер, заполненный солевым раствором 9 г/л или гелеобразным фантомным материалом с подобной проводимостью, в положение, характерное для имплантируемого изделия. В ходе испытания моделируется самое сложное сканирование на МРТ. Выходной заряд определяется по напряжению на измерительном резисторе.
Соответствие подтверждается, если заряд за фазу не превысит 10 нКл.
d) Повреждение имплантата
Во время сканирования МРТ не допускается повреждение имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ.
Методика испытания. Данное испытание применяют для каждого значения напряженности магнитного поля, определенного изготовителем имплантата как безопасное при МРТ-сканировании. Испытательный образец имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ полностью погружают в неметаллический контейнер, заполненный 9 г/л раствора NaCI. Для проведения самого сложного сканирования [согласно описанию в перечислении b)] контейнер размещают в центре аппарата МРТ.
Соответствие подтверждается, если после сканирования изделие будет соответствовать спецификациям изготовителя. Уменьшение силы внутреннего магнита является приемлемым, если изготовитель обеспечивает альтернативный метод фиксации и указывает соответствующую информацию в маркировке изделия (см. 28.12).
22.3 Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна выдержать уровни терапевтического ионизирующего излучения, определенные изготовителем имплантата.
Методика испытания. Три образца имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ облучают с помощью фотонного излучения дозой 5 Гр до максимальной накопленной дозы, определенной изготовителем. Облучение необходимо проводить с интервалом 24 ч, по крайней мере четыре раза в неделю. После каждой процедуры изделие должно включаться в реальных клинических условиях. Перед каждым облучением амплитуду ВЫХОДНОГО СИГНАЛА следует проверять согласно 6.1 и 6.2. Пока амплитуда выходного сигнала каждого образца остается в пределах 10% своего исходного значения перед первым облучением, применяют следующую дозу облучения. Изготовитель должен указать среднюю дозу для трех образцов, при которой ВЫХОДНОЙ СИГНАЛ на последнем испытании отвечал критерию, упомянутому выше. В маркировке (см. 28.12) необходимо указать дозу облучения с запасом 20% от данной.
Соответствие проверяется анализом предоставленной изготовителем документации и результатов испытаний.
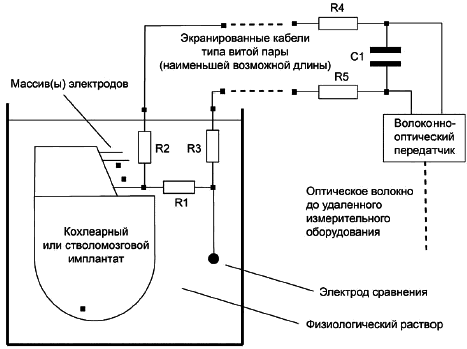
Рисунок 103 - Испытательная установка для проверки защиты от вредного выходного сигнала во время процедуры МРТ
23 Защита активного имплантируемого медицинского изделия от воздействия механических сил
23.1 Замена:
Конструкция НЕИМПЛАНТИРУЕМЫХ ЧАСТЕЙ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, которые при обычном использовании представляют собой ручное портативное или НОСИМОЕ НА ТЕЛЕ оборудование массой не более 10 кг, должна иметь повышенную ударопрочность.
Методика испытания. Указанное выше оборудование должно выдерживать свободное падение в соответствии с МЭК 60068-2-31 при следующих условиях:
a) испытательная поверхность: твердое дерево плотностью не менее 630 кг/м и толщиной от 50 до 55 мм;
b) высота падения:
- 1 м - для ручного оборудования;
- 50 мм - для портативного оборудования;
- 1,5 м или высота реального использования, с которой падение наиболее вероятно, - для носимого на теле оборудования;
c) положение, при котором падает образец, соответствует положению при использовании в реальных условиях.
Соответствие подтверждается, если после проведения испытания, описанного выше, испытавшая падение часть изделия функционирует согласно спецификации изготовителя.
23.2 Замена:
Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна быть сконструирована таким образом, чтобы выдерживать воздействие механических сил, которые могут возникать в реальных условиях использования, включая период до имплантации.
Методика испытания. Имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ устанавливают в соответствии с рекомендациями МЭК 60068-2-47 на испытательном оборудовании для оценки вибрационных воздействий в соответствии с МЭК 60068-2-64, тест Fh, при следующих условиях:
a) частотный диапазон: от 5 до 550 Гц;
b) спектральная плотность мощности: 0,7 (м/с)
/Гц;
c) форма спектральной плотности мощности: плоская горизонтальная от 5 до 500 Гц;
d) продолжительность тестирования: 30 мин по каждой из трех взаимно перпендикулярных осей.
Соответствие подтверждается, если после проведения испытания значения характеристик имплантируемой системы соответствуют спецификации изготовителя.
23.3 Замена:
Имплантируемые ПРОВОДНИКИ, которые находятся за пределами СТИМУЛЯТОРА, должны выдерживать силы натяжения, возникающие во время или после имплантации, без разрыва проводника и ухудшения функциональной электрической изоляции.
Для проведения испытаний необходимы два образца:
- имплантируемая часть в том же состоянии, в котором она доставляется заказчику (образец А). Если необходимо, проводники перед испытанием можно соединить в соответствии с инструкцией изготовителя;
- имплантируемый проводник без СТИМУЛЯТОРА (образец В).
Методика испытания. Необходимо использовать контейнер с раствором NaCI 9 г/л при температуре (37±5)°С, контрольный прибор для измерения натяжения и вольтметр или осциллограф. Продолжительность нахождения образцов в контейнере - минимум 10 дней. Непосредственно перед испытанием проводник необходимо промыть в дистиллированной или деионизированой воде и затем вытереть насухо.
Изготовитель должен определить тот участок ПРОВОДНИКА, который при имплантации может подвергнуться растяжению, и предусмотреть соответствующий метод фиксации ПРОВОДНИКА, включая растянутую часть.
a) Методика испытаний для образца А:
Образец A подключают одним зажимом к стимулятору или к блоку соединения, если он используется. Другой зажим необходимо присоединить к ДИСТАЛЬНОЙ части ПРОВОДНИКА, который подвергается растяжению. Затем замеряют расстояние между точками крепления.
ПРОВОДНИК подвергают растяжению минимум на 15 мм или силе растяжения минимум 1 Н, в зависимости от того, что наступит раньше. Растяжение производится в течение как минимум одной минуты до момента снятия нагрузки. Так необходимо проверить каждый ПРОВОДНИК. Затем образец как минимум на 1 ч погружается в ванну перед повторным испытанием.
Необходимо проверить отсутствие разрыва электрической цепи каждого проводника (испытание на обрыв цепи) и изоляции (испытание на короткое замыкание) между каждой парой ПРОВОДНИКОВ (если это возможно).
Соответствие подтверждается, если испытание на образце А не выявило постоянное функциональное повреждение (например, отсутствие обрыва цепи или короткого замыкания).
b) Методика испытания изоляции образца В:
Образец В следует так же испытать на растяжение, как и образец А. Отличие в том, что оба конца проводника необходимо зафиксировать. После испытания на растяжение изоляция должна быть подвергнута испытательному напряжению. Испытательный сигнал представляет собой меандр частотой 1 кГц с размахом напряжения в два раза больше, чем размах выходного напряжения ИМПЛАНТИРУЕМОЙ СИСТЕМЫ. Испытательный сигнал следует подавать минимум в течение 15 с между каждой комбинацией электропроводящих пар в проводнике. Измеряют полное сопротивление между каждой парой.
Соответствие подтверждается, если не будет выявлено повреждений проводника в результате испытания на растяжение и сопротивление между каждой парой проводников превысит 100 кОм.
23.4 По ИСО 14708-1.
23.5 Замена:
ПРОВОДНИКИ электродов должны выдержать без излома любого проводника испытание на изгиб, который может возникнуть во время и после имплантации. Необходимо проверить три образца методиками испытаний 1 и 2.
Методика испытаний 1: испытательные образцы должны быть предоставлены на испытание в том виде, в котором они поставляются заказчику. Испытания выполняются в сухих условиях и при комнатной температуре.
Каждый образец ПРОВОДНИКА необходимо закрепить подходящим мягким зажимным механизмом (таким образом, чтобы ПРОВОДНИК был надежно зафиксирован во время испытания) на расстоянии (10±2) мм от ПРОКСИМАЛЬНОГО ЭЛЕКТРОДНОГО КОНТАКТА (см. рисунок 104). СТИМУЛЯТОР должен находиться на одной высоте с зажимом, как можно ближе к нему. Стимулятор отпускают пять раз.
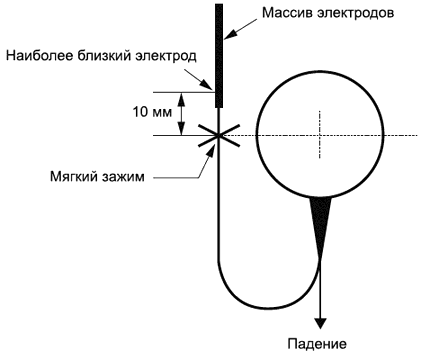
Рисунок 104 - Испытание стимулятора на падение
Соответствие должно быть подтверждено, если измеренное сопротивление каждого проводящего пути каждого образца соответствует спецификации изготовителя и каждый проводник функционально исправен согласно этим спецификациям.
Методика испытаний 2: испытание проводят в той части ПРОВОДНИКА, где после имплантации может произойти сгибание из-за микродвижений. Испытательные образцы должны находиться в том же состоянии, что и полностью собранное и готовое к отправке изделие. Испытание выполняют в сухих условиях и при комнатной температуре.
Необходимо использовать зажимное приспособление, изготовленное из твердого материала, чтобы (см. рисунок 105) зафиксировать СТИМУЛЯТОР.
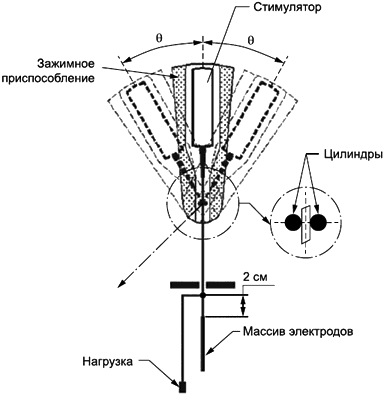
Рисунок 105 - Испытательный стенд сгибания
Крепежное приспособление устанавливают в вибрационную машину, которая может согнуть ПРОВОДНИК в любую сторону от прямого направления. Крепеж позволяет натянуть ПРОВОДНИК в направлении, противоположном креплению к СТИМУЛЯТОРУ. ПРОВОДНИК необходимо пропустить между двумя цилиндрами так, чтобы оба цилиндра касались его. Точка излома должна быть расположена посередине отрезка между центрами цилиндров. Диаметр цилиндров должен быть равным двум диаметрам ПРОВОДНИКА. Если в СТИМУЛЯТОР входит больше одного ПРОВОДНИКА, то каждый следует проверить отдельно.
Нагрузку прочно присоединяют к ПРОВОДНИКУ на расстоянии (2±0,2) см от ПРОКСИМАЛЬНОГО ЭЛЕКТРОДА. Ожидаемый уровень суммарной нагрузки - (0,03±0,01) Н.
Крепежное приспособление должно отклоняться на угол 15° (или на больший, определенный изготовителем) в каждую сторону с частотой примерно 2 Гц в течение не менее 100000 (ста тысяч) циклов.
Также можно провести аналогичное испытание, когда СТИМУЛЯТОР остается неподвижным, а ПРОВОДНИК колеблется. Важно, чтобы были соблюдены все другие условия испытания.
Соответствие подтверждается, если после испытания измеренное сопротивление каждого проводящего пути отвечает спецификации изготовителя и каждый проводник функционально исправен согласно этой спецификации.
23.6 Замена:
Имплантируемые соединители, предназначенные для соединения имплантируемых частей врачами, необходимо пометить соответствующей маркировкой (см. 8.2 и 9.9). Изготовитель также должен установить (см. 28.4) предполагаемую производительность после имплантации. Недопустимо ухудшение качества соединения по мере использования. При повторном соединении также исключается ухудшение производительности изделия.
Соответствие должно быть подтверждено предоставленным изготовителем анализом конструкции и данными испытаний.
Дополнительные подпункты:
23.7 Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна быть сконструирована таким образом, чтобы вызванные процедурой имплантации легкие толчки не повреждали изделие.
Методика испытания. Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна выдержать механическое испытание на ударопрочность в соответствии с МЭК 60068-2-27, испытание Еа при следующих условиях:
a) форма импульса толчка: полусинусоида или гаверсинус;
b) интенсивность: максимальное ускорение: 5000 м/с (500 г);
c) продолжительность толчков: 1 мс;
d) направление и число толчков: один толчок в каждом направлении вдоль трех взаимно перпендикулярных осей (общее число - шесть толчков).
Соответствие подтверждается, если после проведения испытаний характеристики имплантируемой системы (см. раздел 6) соответствуют спецификации изготовителя.
23.8 Имплантируемая часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна быть сконструирована таким образом, чтобы воздействия во время реальной эксплуатации не повреждали изделие.
Методика испытания. Если СТИМУЛЯТОР ИМПЛАНТИРУЕМОЙ СИСТЕМЫ может быть подвергнут механическому воздействию во время имплантации из-за своего расположения, необходимо зафиксировать его в испытательной установке в соответствии с МЭК 60068-2-75, испытание Eha или Ehc, при следующих условиях:
a) энергия удара в Дж (±5%): 1,5 Дж после публикации настоящего стандарта и 2,5 Дж через три года от даты публикации настоящего стандарта;
b) число воздействий: одно за испытание (защитный материал и имплантат);
c) тип испытательного аппарата: маятниковый молот (МЭК 60068-2-75, испытание Eha) или вертикальный молот (МЭК 60068-2-75, испытание Ehc). Ударный элемент: 5 Дж в соответствии с МЭК 60068-2-75 (таблица 1);
d) монтаж образца, проходящего тест: образец присоединяют к твердой и плоской опорной поверхности так, чтобы сторона, обращенная к черепной кости во время реальной эксплуатации (in situ), равномерно прилегала к опорной поверхности. Во время воздействия следует равномерно распределить кусок силикона (толщина: 3 мм, размеры: 1010 см
, твердость по Шору: от 40° до 60°) по имплантату между ним и измерительной точкой (защитный материал). Силиконовую прокладку необходимо заменять перед каждым испытанием.
Примечание 1 - "Наконечник" ударного элемента определен в МЭК 60068-2-75:1997, пункт 4.1.1;
е) предварительная обработка: нет;
f) первоначальные измерения: функции образца согласно его спецификации должны быть проверены и подтверждены;
g) позиция и зоны воздействия: имплантат необходимо присоединить так, чтобы поверхность, которая во время реальной эксплуатации (in situ) обращена к коже, сформировала область воздействия, на которой защитный материал прекращает движение. Направление движения ударного элемента задается перпендикулярно к поверхности имплантата. Ударный элемент должен воздействовать на защитный материал в центре поверхности, которая во время реальной эксплуатации (in situ) обращена к коже. Во втором испытании с новым СТИМУЛЯТОРОМ и новым защитным материалом ударный элемент должен воздействовать на корпус имплантата не в центре, а там, где предположительно находится самое слабое место СТИМУЛЯТОРА.
Примечание 2 - Выходящие из стимулятора проводники не считаются частью корпуса имплантата;
h) закрепление опорных пластин, облицовки и подобных частей: особых требований не предъявляется. При выполнении удара необходимо избегать повторного воздействия (к примеру, отскока);
i) режим работы и контроль функций: мониторинг функций имплантата во время испытания на удар не осуществляется, имплантат должен быть выключен;
j) критерии оценки: требования были удовлетворены, если после испытания на удар оба образца продолжают соответствовать спецификациям 28.8.1, перечисление b), и выполняют требования герметичности в соответствии с 19.6 (испытание на большие течи). После испытания на удар допускается прекращение функционирования имплантируемого микрофона или другого датчика. Это приемлемо, если отказ микрофона или датчика не требует замены имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ;
k) последующая обработка: нет;
I) заключительные измерения: измерения, необходимые для анализа спецификаций ИМПЛАНТИРУЕМОЙ СИСТЕМЫ и герметичности, проверяют в соответствии с 19.6 для испытаний на большие течи;
m) испытательный протокол должен содержать: наименование стандарта и спецификации, дату и время испытания, точное описание образца, процедуру испытания на воздействие (маятниковый или вертикальный молот), конкретную позицию точки воздействия (например, изобразить на чертеже), тип используемого силикона (например, наименование продукта, источник продукта, механические свойства), точное описание испытаний на соответствие спецификации до и после воздействия, результаты испытания на соответствие спецификации, результаты испытания на герметичность, результаты всего испытания.
Соответствие должно быть подтверждено предоставленными изготовителем результатами испытаний согласно перечислению j).
24 Защита активного имплантируемого медицинского изделия от повреждений, вызванных электростатическим разрядом
24.1 Замена:
Имплантируемая и НЕИМПЛАНТИРУЕМАЯ ЧАСТИ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должны быть сконструированы таким образом, чтобы электростатический разряд, который может произойти в реальных условиях, не вызывал необратимых изменений.
Методика испытания. Имплантируемую часть при комнатной температуре полностью погружают в неметаллический контейнер, заполненный 9 г/л раствора NaCI. НЕИМПЛАНТИРУЕМУЮ ЧАСТЬ подключают на расстоянии (5±1) мм от имплантируемой части. Настройки работы ИМПЛАНТИРУЕМОЙ СИСТЕМЫ необходимо установить в соответствии с инструкциями изготовителя. Имплантируемая и НЕИМПЛАНТИРУЕМАЯ ЧАСТИ ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должны выдержать испытание электростатическим разрядом, приложенным к внешним компонентам согласно МЭК 61000-4-2 (с климатическими условиями, установленными в 8.1.1). Испытательное напряжение при этом составляет 2 кВ в случае воздействия разряда на проводящие поверхности и 8 кВ в случае воздействия воздушного разряда на изолирующие поверхности. Необходимо применить как минимум 10 разрядов с испытательным напряжением 2 кВ и пять разрядов с испытательным напряжением 8 кВ.
Соответствие подтверждается, если ИМПЛАНТИРУЕМАЯ СИСТЕМА работает в безопасном режиме и обеспечивает выполнение всех заявленных в спецификации изготовителя функций после испытания.
Примечание - При необходимости настройки могут быть сброшены до заводских путем выключения и включения имплантируемой системы.
24.2 Настоящий подпункт зарезервирован для использования в следующих изданиях.
25 Защита активного имплантируемого медицинского изделия от повреждений, вызванных изменениями атмосферного давления
25.1 По ИСО 14708-1.
25.2 Имплантируемые части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должны быть сконструированы таким образом, чтобы выдерживать предсказуемое увеличение давления, которое может возникнуть во время реальной эксплуатации.
Методика испытания. Изделие необходимо поместить в подходящую водяную компрессионную камеру и 20 раз подвергнуть воздействию давления: от нормального давления окружающей среды до максимального, в 1,5 раза превышающего давление, указанное в документации изготовителя (см. 28.21). Скорость изменения давления составляет не менее 100 кПа в минуту. Максимальное давление должно поддерживаться в течение не менее 1 мин.
Соответствие должно быть подтверждено в ходе проверки процедуры испытания и предоставленных изготовителем результатов.
26 Защита активного имплантируемого медицинского изделия от повреждений, вызванных изменениями температуры
26.1 По ИСО 14708-1.
26.2 По ИСО 14708-1.
27 Защита активного имплантируемого медицинского изделия от электромагнитного неионизирующего излучения
27.1 Замена:
При любых условиях, которые могут возникнуть в общественных местах, имплантируемые части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должны наносить ВРЕДА через сбои в работе, повреждения, нагрев изделия или локальное увеличение плотности индуцированного электрического тока в теле пациента, причиной которых является восприимчивость к электрическим воздействиям внешних электромагнитных полей.
Все требования касательно защиты 27.3 и 27.4 распространяются на все настройки ИМПЛАНТИРУЕМОЙ СИСТЕМЫ. Это не означает, что все комбинации настроек должны быть рассмотрены. Должен быть рассмотрен вариант "наихудшего" случая: ИМПЛАНТИРУЕМАЯ СИСТЕМА должна быть настроена для непрерывного выходного сигнала максимального значения, определенного в 6.2, по крайней мере на двух выходных электродах. Если это возможно, то чувствительность микрофона следует настроить на нормальные клинические условия.
Соответствие подтверждается, если после воздействия наибольшей интенсивности, приведенной в 27.3 и 27.4, ИМПЛАНТИРУЕМОЙ СИСТЕМЕ не нанесен необратимый ущерб и если во время воздействия поля выходные токи в теле пациента не превышали максимального значения выходного сигнала, определенного в 6.2. Соответствие должно быть подтверждено результатами испытаний или проверкой теоретического моделирования, проведенного изготовителем, сопровождаемого вычислением и данными результатов испытаний. В случае если выходной ток невозможно прямо или косвенно измерить и при этом присутствует сигнал помехи, необходим дополнительный анализ устройства электронной схемы, чтобы продемонстрировать, что ИМПЛАНТИРУЕМАЯ СИСТЕМА не может обеспечить более высокие выходные сигналы, чем определено в 6.2.
27.2 Замена:
Функции ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не должны зависеть от внешних электромагнитных полей, которые обычно присутствуют в повседневной жизни. Отсутствие зависимости означает, что не должно быть длительного дискомфорта, при этом допускается некоторое ухудшение сигнала во время воздействия электромагнитного поля.
Все требования незначительного воздействия на функционирование согласно 27.3 и 27.4 обязательны для всех настроек ИМПЛАНТИРУЕМОЙ СИСТЕМЫ. Это не означает, что все комбинации настроек должны быть проверены, но как минимум необходимо рассмотреть "наихудший" случай: изделие должно быть настроено для получения непрерывного выходного сигнала между 25% ("пороговое значение") и 50% ("уровень комфорта") от максимального значения выходного сигнала, определенного в 6.2, по крайней мере на двух выходных электродах. Если это применимо, чувствительность микрофона должна быть настроена на нормальные клинические условия. Можно акустически блокировать порты микрофона и выключить индукционную катушку, если это возможно. Для испытания требуется настроить изделие на диапазон входных частот, доступный пользователю.
Соответствие подтверждается, если во время воздействия любой выходной сигнал остается ниже "уровня комфорта" - на более низком уровне, указанном в 27.3 и 27.4. Во время испытания ИМПЛАНТИРУЕМАЯ СИСТЕМА может иногда пропускать сигналы стимуляции. В случае если изделие полностью останавливает стимуляцию до достижения более низких уровней 27.3 и 27.4, изготовитель должен указать тот уровень, на котором происходит остановка (см. 28.22.1). Соответствие должно быть подтверждено результатами испытаний или проверкой проведенного изготовителем теоретического моделирования, сопровождаемого вычислениями и данными результатов испытаний.
Дополнительные подпункты:
27.3 Сигнал помехи для частот 16,6 Гц f <10 МГц
Характер сигнала помехи определен в 27.5. Время в нерабочем состоянии =10 мс и время импульса
дано в таблицах 101 и 102.
Таблица 101 - Пиковая напряженность магнитного поля
Частота | Пиковая напряженность магнитного поля | |||
Нижний уровень | Время импульса | Верхний уровень | Время импульса | |
16,6 Гц | 340 А/м | Непрерывная волна | 480 А/м | Непрерывная волна |
50 Гц | 110 А/м | Непрерывная волна | 1200 А/м | Непрерывная волна |
1,66 кГц | 7,0 А/м | 10 мс | 150 А/м | 10 мс |
5 кГц | 7,0 А/м | 10 мс | 150 А/м | 10 мс |
16,6 кГц | 7,0 А/м | 10 мс | 150 А/м | 10 мс |
50 кГц | 7,0 А/м | 10 мс | 150 А/м | 10 мс |
166 кГц | 7,0 А/м | 10 мс | 110 А/м | 10 мс |
500 кГц | 4,0 А/м | 3 мс | 26 А/м | 1,5 мс |
1,66 МГц | 2,0 А/м | 1 мс | 5,5 А/м | 200 мкс |
5 МГц | 0,15 А/м | 500 мкс | 2,9 А/м | 50 мкс |
Примечание - Поля могут не быть однородными. | ||||
27.4 Сигнал помехи для частот 10 МГц f <3000 МГц
Сигнал помехи определен в 27.5.
Таблица 102 - Пиковая напряженность электрического поля
Частота | Пиковая напряженность электрического поля | |||
Нижний уровень | Время импульса | Верхний уровень | Время импульса | |
10 МГц | 40 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
33 МГц | 40 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
100 МГц | 40 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
450 МГц | 40 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
900 МГц | 58 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
1800 МГц | 82 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
2450 МГц | 86 В/м | 10 мс или непрерывная волна | 200 В/м | 400 мкс |
Примечание - Поля могут не быть однородными. | ||||
27.5 Описание сигнала помехи
На частотах 16 и 50 Гц сигнал помехи является синусоидальным (непрерывная волна).
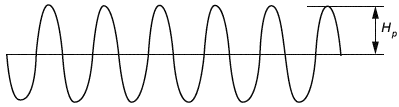
Рисунок 106 - Сигнал помехи на 16 и 50 Гц
На всех других частотах сигнал помехи без несущей частоты.
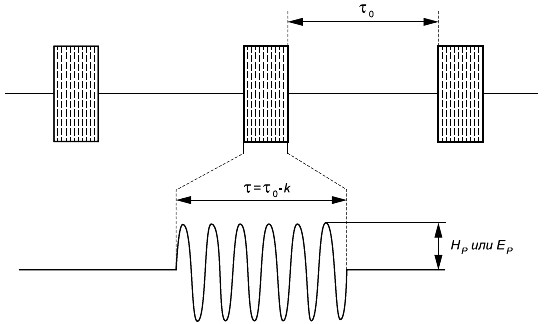
Рисунок 107 - Сигнал помехи на частотах выше 1 кГц
28 Сопроводительная документация
В соответствии с ИСО 14708-1, за исключением следующего.
28.1 Замена:
Сопроводительная документация должна включать наименование и адрес (почтовый адрес и телефонный номер) изготовителя или наименование и адрес официального представителя, если у изготовителя нет зарегистрированного места осуществления хозяйственной деятельности на территории Российской Федерации.
Соответствие проверяют в ходе осмотра.
28.2 По ИСО 14708-1.
28.3 По ИСО 14708-1.
28.4 Замена:
Если упаковка содержит имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, предназначенную для подключения к другому имплантируемому изделию или принадлежности, то сопроводительная документация должна содержать информацию о технических характеристиках разъемов, инструкцию по сборке и работе блока соединения согласно 23.6.
Соответствие проверяют в ходе осмотра.
28.5 По ИСО 14708-1.
28.6 По ИСО 14708-1.
28.7 По ИСО 14708-1.
28.8 Дополнительные подпункты:
28.8.1 Сопроводительная документация должна включать следующую соответствующую информацию для имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ:
a) Описание изделия:
- общее описание, краткое объяснение функционального назначения, доступных способов стимуляции;
- перечисление и краткое описание других функций (измерение полного сопротивления и т.д.);
- масса (в граммах);
- основные размеры (в миллиметрах);
- объем без ПРОВОДНИКОВ (в кубических сантиметрах);
- перечисление материалов, которые вступают в контакт с тканями человека.
b) Технические характеристики:
- амплитуда и длительность импульса ВЫХОДНОГО СИГНАЛА на сопротивлении 1 кОм (согласно 6.2);
- точность измерения полного сопротивления (согласно 6.3);
- уровень безопасности МРТ (согласно 22.2);
- заводские настройки ИМПЛАНТИРУЕМОЙ СИСТЕМЫ по умолчанию, если имеются;
- рекомендуемые методы для определения нормального функционирования имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ (например, измерение полного сопротивления).
c) Спецификация и характеристики для каждого ПРОВОДНИКА и ЭЛЕКТРОДНОГО МАССИВА:
- электрические конфигурации (униполярный, число электрически независимых электродных контактов и т.д.);
- форма и другие характеристики (перимодиолярные, с введением лекарственного средства и т.д.);
- перечисление материалов, используемых для проводников, ЭЛЕКТРОДНЫХ КОНТАКТОВ и изоляции ПРОВОДНИКОВ;
- декларация изготовителя о наличии в составе ПРОВОДНИКА ЛЕКАРСТВЕННОГО ВЕЩЕСТВА как неотъемлемого компонента с указанием наименования данного ЛЕКАРСТВЕННОГО ВЕЩЕСТВА;
- физические размеры, включая (номинальное значение):
- длину ПРОВОДНИКА (в миллиметрах);
- поперечные размеры ЭЛЕКТРОДНОГО МАССИВА на ПРОКСИМАЛЬНОМ и ДИСТАЛЬНОМ концах (в миллиметрах);
- геометрическую площадь поверхности наименьших и наибольших стимулирующих КОНТАКТОВ ЭЛЕКТРОДА (в квадратных миллиметрах);
- расстояние(я) между ЭЛЕКТРОДНЫМИ КОНТАКТАМИ и расстояние между ПРОКСИМАЛЬНЫМ и ДИСТАЛЬНЫМ стимулирующими КОНТАКТАМИ ЭЛЕКТРОДА (в миллиметрах);
- размеры и форму соединителя (длина и диаметры в миллиметрах), если применимо, или ссылку на изданные стандарты на соединители, включая любые обозначения и маркировки.
Соответствие проверяют в ходе осмотра.
28.9 По ИСО 14708-1.
28.10 По ИСО 14708-1.
28.11 По ИСО 14708-1.
28.12 Замена:
Сопроводительная документация также должна содержать информацию о предполагаемом применении и нормальном функционировании, а также предупреждение о возможном риске помех от или для имплантируемого изделия во время других клинических процедур или оказания медицинской помощи. Примеры таких процедур указаны, помимо этого списка, в 20.2, примечании к 21.1, 22.2, 22.3 и разделе 27. Если требуются ограничения во время проведения лечения (например, близость или мощность энергетических полей), изготовитель также должен указать на маркировке и/или в инструкциях те условия, за пределами которых может присутствовать риск для пациента.
Соответствие проверяют в ходе осмотра.
28.13 По ИСО 14708-1.
28.14 По ИСО 14708-1.
28.15 По ИСО 14708-1. Также см. 28.12.
28.16 По ИСО 14708-1.
28.17 По ИСО 14708-1.
28.18 По ИСО 14708-1.
28.19 Замена:
Если ИМПЛАНТИРУЕМАЯ СИСТЕМА содержит встроенный источник энергии, то сопроводительная документация должна включать информацию о его сроке годности для двух случаев: когда ИМПЛАНТИРУЕМАЯ СИСТЕМА будет использоваться в соответствии с базовыми клиническими настройками, определенными изготовителем, и при настройках для наихудших условий эксплуатации.
Соответствие проверяют в ходе осмотра.
28.20 По ИСО 14708-1.
28.21 По ИСО 14708-1.
28.22 По ИСО 14708-1.
Дополнительный подпункт:
28.22.1 Информация, касающаяся характеристик электромагнитных помех изделия в соответствии с 27.2, предоставляется по запросу.
28.23 По ИСО 14708-1.
Приложение АА
(справочное)
Соответствие между фундаментальными принципами ИСО/ТО 14283 [8] и разделами настоящего стандарта
Таблица АА.1
Фундаментальные принципы | Разделы ИСО 14708-1 | Разделы настоящего стандарта и охватываемые аспекты |
3 Общие принципы | ||
3.1 Имплантаты должны быть разработаны и сконструированы таким образом, чтобы при использовании в условиях и с целью, для которых они созданы, не возникало угрозы безопасности пациентов или безопасности и жизни пользователей, а также других лиц, гарантируя тем самым, что любой риск, который может быть связан с их использованием, является приемлемым наряду с пользой для пациента или сопоставим с высоким уровнем защиты его здоровья и безопасности | 8.1 Требуется, чтобы предупреждения были заметными | Действует |
3.2 Решения, принятые изготовителем для разработки и конструирования имплантатов, должны соответствовать принципам безопасности с учетом общего уровня знаний и технического развития. При выборе наиболее подходящего решения изготовитель должен руководствоваться перечисленными ниже принципами в следующем порядке: | (Настоящий принцип является фундаментальным по отношению ко всем активным имплантируемым медицинским изделиям, для которых предназначен ИСО 14708. Данный подход в основном относится к содержанию требований разделов 14, 19 и 21) | Действует |
3.3 Имплантаты должны достигать рабочих характеристик, предусмотренных изготовителем, и должны быть разработаны, изготовлены и упакованы таким образом, чтобы они были пригодны для выполнения одной или более функций, приведенных в 2.1 (ИСО/ТО 14283), как указано изготовителем | 10.4 Требуется, чтобы сопровождающая информация была нанесена на изделие | Действует |
3.4 На характеристики и изделия, приведенные в 3.1, 3.2 и 3.3, не должно быть негативного влияния в такой степени, чтобы нарушались клинические условия и безопасность пациентов и, где применимо, других лиц, | 19.2 Требуется наличие индикатора истощения источника энергии | Заменен |
в течение срока службы имплантата, указанного изготовителем, при воздействии на имплантат нагрузок, возникающих при нормальном использовании имплантата | 19.3 Требуется определить метод подтверждения того, что условия единичного отказа не опасны | Действует |
23.1 Требуется провести испытание неимплантируемых частей на падение | Заменен | |
23.2 Требуется провести испытания на вибрационную прочность частей, носимых пациентами | Заменен | |
23.3 Требуется провести испытание на определение разрыва (проводники и т.д.) | Заменен | |
23.4 Требуется защита от неблагоприятного внешнего нагружения (проводники и т.д.) | Действует | |
23.5 Требуется защита от усталостного разрушения (проводники и т.д.) | Заменен | |
23.6 Устанавливает требования к надежности соединений | Заменен | |
26.1 Требуется защита от нагрева при использовании неимплантируемых частей, работающих от электропривода | Действует | |
28.4 Требуется наличие информации о максимальной доказанной прочности соединителей (коннекторов) | Заменен | |
28.23 Требуются нанесение предупреждающих надписей о запрете входа пациентов в окружающую среду с опасными условиями | Действует | |
3.5 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы на их характеристиках и работе в течение периода предполагаемого использования отрицательно не сказывались транспортирование и хранение, принимая | 7.2 Требуется, чтобы стерильная упаковка была защищена товарной упаковкой | Действует |
во внимание инструкции и информацию, указанную изготовителем | 10.1 Требуется, чтобы упаковка была долговечной | Действует |
10.2 Требуется, чтобы упаковка была защищена от последствий влияния влажности | Действует | |
10.3 Требуется, чтобы маркировки на товарной упаковке были нестираемыми | Действует | |
12.3 Требуется, чтобы маркировка на стерильной упаковке была нестираемой | Действует | |
26.2 Требуется, чтобы изделие было защищено от изменения температурных воздействий | Действует | |
3.6 В отношении любого нежелательного побочного эффекта необходимо оценить приемлемость риска относительно предполагаемых рабочих характеристик | 19.3 Требуется определить методику подтверждения того, что условия единичного отказа не вызывают причинения вреда | Действует |
19.4 Требуется проведение исследования о непредусмотренных эффектах, связанных с изделием | Действует 19.5, 19.6 дополнительные требования | |
4 Специальные принципы, касающиеся дизайна конструкции | ||
4.1 Химические, физические и биологические свойства | ||
4.1.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы гарантировать характеристики и производительность, требуемые разделом 3 "Основные принципы". Особое внимание необходимо уделить: | ||
а) выбору используемых материалов, в частности оценке токсичности и, где применимо, свойствах возгораемости; | 14.3 Требуется провести оценку биосовместимости. | Действует |
b) совместимости используемых материалов и биологических тканей, клеток и тканевых жидкостей, принимая во внимание предполагаемое назначение имплантата | 14.3 Требуется провести оценку биосовместимости | Действует |
4.1.2 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы минимизировать риск, вызванный загрязнением и наличием остаточных веществ, для лиц, занятых | 14.2 Требуется провести оценку на загрязнение частицами | Заменен |
транспортированием, хранением и использованием имплантатов, и пациентов, принимая во внимание предполагаемое назначение изделия. Особое внимание необходимо уделить незащищенным тканям и продолжительности и частоте воздействия | 14.3 Требуется провести оценку биосовместимости | Действует |
4.1.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы они могли быть использованы безопасно с материалами, веществами и газами, с которыми они контактируют при нормальном использовании или при рутинных процедурах; если предполагается использование имплантатов для доставки лекарственных веществ, то они должны быть разработаны и изготовлены таким образом, чтобы быть совместимыми с лекарственным веществом в соответствии с положениями и ограничениями для этих веществ и чтобы их рабочие характеристики поддерживались в соответствии с предполагаемым использованием | 19.5 Требуется доказать совместимость с лекарственными веществами | Заменен |
4.1.4 В случае если в имплантаты включено как неотъемлемая часть вещество, которое, если применяется отдельно, может быть рассмотрено как лекарственное вещество согласно определению 2.4 (ИСО/ТО 14283) и которое может воздействовать на организм дополнительно к влиянию, оказываемому изделием, его безопасность должна быть подтверждена, принимая во внимание предполагаемое использование имплантированного изделия | 14.4 Требуется оценить качество и безопасность содержащихся в имплантате лекарственных веществ | Действует |
4.1.5 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы свести к минимуму риск, вызванный утечкой вещества из имплантата | 25 Требуется, чтобы имплантируемые части выдерживали изменения давления | Действует 25.2 Дополнительные требования |
4.1.6 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы уменьшить, насколько возможно, риски, вызванные случайным проникновением вещества в имплантат, принимая во внимание имплантат и характер окружающей среды, в которой предполагается его использовать | 25 Требуется, чтобы имплантируемые части выдерживали изменения давления | Действует 25.2 Дополнительные требования |
4.1.7 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски пациента или пользователя системы, включая программное обеспечение | 19.3 Требуется провести анализ дизайна разработки и выбрать методику анализа | Действует |
4.2 Инфекция и микробное заражение | ||
4.2.1 Имплантаты и процесс их изготовления должны быть разработаны таким образом, чтобы уменьшить или устранить, насколько возможно, риск инфекции для пациента, пользователя или третьего лица. Конструкция должна позволять легкость манипулирования и, если необходимо, минимизировать инфицирование имплантата пациентом или, наоборот, во время использования | 14.1 Требуется, чтобы изделие поставлялось стерильным | Действует |
4.2.2 Ткани животного происхождения должны быть поставлены от животных, подвергнутых ветеринарным контролю и надзору, адаптированным к предполагаемому применению ткани. | (Не применяется к активным имплантируемым медицинским изделиям) | |
4.2.3 Имплантаты, поставляемые стерильными, должны быть разработаны, изготовлены и упакованы в защитную упаковку, которая создает антимикробный барьер, для обеспечения их стерильности при | 7.1 Требуется поставка изделия в одноразовой упаковке | Действует |
поступлении на рынок и при условиях хранения и транспортирования, оговоренных изготовителем, до тех пор, пока упаковка не будет вскрыта или повреждена | 7.2 Требуется защита стерильной упаковки с помощью товарной упаковки | Действует |
10.1 Требуется, чтобы упаковка была долговечной | Действует | |
10.2 Требуется защита упаковки от воздействия влаги | Действует | |
11.7 Требуется, чтобы содержимое стерильной упаковки имело наименование или было легко различимо через нее | Действует | |
11.9 Требуется, чтобы на стерильную упаковку была нанесена инструкция по ее вскрытию | Действует | |
12.1 Требуется применение требований ИСО 11607 к одноразовой упаковке | Действует | |
12.2 Требуется, чтобы было очевидно, что упаковка вскрывалась | Действует | |
14.1 Требуется, чтобы изделие поставлялось стерильным | Действует | |
4.2.4 Имплантаты, поставляемые стерильными, должны быть изготовлены и стерилизованы, если применимо, валидированным методом | 14.1 Требуется, чтобы изделие поставлялось стерильным | Действует |
4.2.5 Имплантаты, для которых предполагается стерилизация, должны быть изготовлены в соответствующих контролируемых условиях (например, окружающей среды) | 14.1 Требуется, чтобы изделие поставлялось стерильным | Действует |
14.2 Требуется провести испытание на наличие загрязняющих частиц | Пересмотрен | |
4.2.6 Система упаковки нестерильных имплантатов должна поддерживать на оговоренном уровне чистоты изделие без ухудшения и, если имплантаты стерилизуют перед использованием, минимизировать риск микробного заражения; система упаковки должна соответствовать методу стерилизации, указанному изготовителем | (Не применимо, поскольку в подпункте требуется, чтобы имплантируемые части активного имплантируемого медицинского изделия поставлялись стерильными) | Те же |
4.2.7 Упаковка и/или этикетка имплантата должны устанавливать различие между идентичными или похожими изделиями в стерильном и нестерильном состояниях | (Не применимо, поскольку в подпункте требуется, чтобы имплантируемые части активного имплантируемого медицинского изделия поставлялись стерильными) | Те же |
4.3 Свойства конструкции и окружающей среды | ||
4.3.1 Если имплантат предназначен для использования совместно с другим изделием или оборудованием, вся комбинация, включая систему соединений, должна быть безопасной и не должна ухудшать специфических свойств изделия. Любые | 9.9 Требуется, чтобы соединения имплантируемого изделия были идентифицированы | Действует |
ограничения по использованию должны быть указаны на этикетке или в инструкции по применению | 11.8 Требуется, чтобы имплантируемые соединения были идентифицированы на стерильной упаковке | Действует |
23.6 Требуется определить усилие фиксации соединения | Заменен | |
28.4 Требуется привести подробные данные: раскрытие информации о максимальной достоверной величине прочности фиксации соединения | Заменен | |
28.5 Требуется обеспечить сведения по принадлежностям, которые могут потребоваться для облегчения предполагаемого использования изделия | Действует | |
4.3.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы устранить или минимизировать, насколько это возможно: | ||
а) риск травмы с учетом их физических особенностей, таких как соотношение объем/давление, размерами и, если применимо, эргономическими особенностями; | 15.1 Требуется установить требования к качеству поверхностей неимплантируемых частей | Действует |
15.2 Требуется, чтобы имплантируемые части имели соответствующую конструктивную форму | Заменен | |
b) риски, связанные с разумными, предсказуемыми условиями окружающей среды, такими как магнитные поля, воздействиями внешнего электричества, электростатического разряда, давления, температуры или изменений давления и ускорения; | 23.1 Требуется провести испытание неимплантируемых частей на падение | Заменен |
23.2 Требуется провести вибрационные испытания частей, носимых пациентами | Заменен | |
24 Требуется провести испытание на действие электростатического разряда неимплантируемых частей | Заменен | |
25 Требуется установить, выдерживают ли имплантируемые части изменения давления | Действует 25.2 дополнительные требования | |
26.2 Требуется убедиться в том, что при экстремальных температурах транспортирование не приводит к повреждениям имплантируемых изделий | Действует | |
27 Требуется установить требования по электромагнитной совместимости | Заменен 27.2, 27.3, 27.4, 27.5 дополнительные требования | |
с) риски обоюдных помех при использовании с другими изделиями (такими, как дефибрилляторы или высокочастотные хирургические инструменты), обычно используемыми для исследований или при данном виде лечения; | 20.1 Требуется защита внешних электродов ЭКГ от дефибрилляции | Не применимо к кохлеарным имплантатам |
20.2 Требуется провести испытание, подтверждающее защищенность имплантируемого изделия от дефибрилляции | Действует | |
21 Требуется защита от диатермии и пр. | Заменен | |
22 Требуется защита от воздействий ультразвука в диагностическом диапазоне | Действует 22.2, 22.3 дополнительные требования | |
28.12 Требует наличия предупреждающих надписей | Заменен | |
28.13 Требуется указать на необходимость мониторинга изделия при диатермии и пр. | Действует | |
28.14 Требуется предупреждать о том, что изделие нельзя подвергать воздействию терапевтического уровня ультразвука | Действует | |
28.15 Требуется предупреждать о влиянии терапевтической дозы радиации на имплантируемое изделие | Действует | |
d) риски, которые могут возникнуть при условии, что обслуживание и калибровка невозможны, включая (если применимо): чрезмерное увеличение тока утечки, старение материалов, усиление нагрева, генерируемого имплантатом, снижение точности | 17 Требуется оценить воздействие локального нагрева, вызванного отказом имплантируемого изделия | Действует |
любого измерительного или контрольного механизма | 19.1 Требуется анализ дизайна конструкции имплантата | Действует |
19.2 Требуется наличие индикатора истощения источника энергии | Заменен | |
4.3.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски возгорания или взрыва при нормальных условиях эксплуатации или в случае единичного отказа. Риски "при нормальных условиях или в случае единичного отказа" означают риски, которые можно оценить путем анализа рисков. Особое внимание следует обратить на имплантаты, предполагаемое использование которых включает воздействие огнеопасных веществ или веществ, которые могут вызвать возгорание | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Действует |
4.4 Имплантаты с функцией измерения | ||
4.4.1 Имплантаты с функцией измерения должны быть разработаны и изготовлены таким образом, чтобы обеспечить установленную точность и стабильность в заданных пределах точности с учетом предполагаемого использования имплантата. Пределы точности должны быть установлены изготовителем | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.4.1.1 Методы выполнения измерений с помощью шкалы на дисплее или в режиме наблюдения должны быть разработаны в соответствии с принципами эргономики, с учетом предполагаемого использования имплантата | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.4.1.2 Если на имплантате или его принадлежностях приведены инструкции по работе с имплантатом или работе индикации, или параметрам настройки с помощью визуальной системы, эта информация | 13.4 Требования к индикаторам визуального типа | Действует |
должна быть понятна пользователю и, если необходимо, пациенту | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.4.2 Измерения, осуществляемые с помощью имплантатов, снабженных функцией измерения, должны быть выражены в единицах, соответствующих положениям серии ИСО 31 | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.5 Защита от излучения | ||
4.5.1 Общие положения | (См. более подробную информацию ниже) | |
4.5.2 Предусмотренное излучение | (В настоящее время не применимо к активным имплантируемым медицинским изделиям) | Те же |
4.5.2.1 В случае если имплантаты предназначены для испускания опасного уровня излучения, необходимого для специфических медицинских целей, когда польза предполагается превышающей риски от излучения, имплантаты должны быть спроектированы и изготовлены так, чтобы обеспечить воспроизводимость и надлежащий уровень точности соответствующих переменных параметров | - | - |
4.5.2.2 В случае если излучение имплантатов потенциально опасное, видимое или невидимое, имплантаты должны быть снабжены, где возможно, визуальными дисплеями и/или звуковыми предупреждающими сигналами осуществления такого излучения | - | - |
4.5.3 Непредусмотренное излучение | 9.1 Требуется использовать маркировку с предупреждениями о любых радиоактивных веществах | Действует |
непредусмотренного, рассеянного или побочного излучений было как можно более снижено | 18.1 Требуется установить требования к закрытым источникам | Действует |
18.2 Требуется обосновать значение дозы облучения для пациента | Действует | |
18.3 Требуется уменьшить дозу облучения, насколько возможно | Действует | |
28.2 Требуется предоставить информацию о радиоактивных веществах | Действует | |
4.5.4 Инструкции | (В настоящее время не применимо к активным имплантируемым медицинским изделиям) | Те же |
4.6 Ионизирующее излучение | (В настоящее время не применимо к активным имплантируемым медицинским изделиям) | Те же |
4.6.1 Имплантаты, предназначенные для испускания ионизирующего излучения, должны быть спроектированы и изготовлены так, чтобы гарантировать, где это возможно, что структуру и качество излучения можно измерять и контролировать с учетом предполагаемого назначения | - | - |
4.6.2 Имплантаты ионизирующего излучения, предназначенные для диагностической радиологии, должны быть спроектированы и изготовлены таким образом, чтобы достичь удовлетворительного качества изображения и/или выходных данных для предполагаемого медицинского назначения при минимальном радиационном облучении пациента и пользователя | - | - |
4.6.3 Имплантаты ионизирующего излучения, предназначенные для диагностической радиологии, должны быть спроектированы и изготовлены таким образом, чтобы обеспечить надежный мониторинг и контроль доставляемой дозы | - | - |
4.7 Принципы, касающиеся имплантатов, подсоединенных или оборудованных источником энергии | ||
4.7.1 Имплантаты со встроенными электронными программируемыми системами должны быть разработаны таким образом, чтобы обеспечивать воспроизводимость, надежность и эффективность этих систем в соответствии с предполагаемым использованием. При возникновении рисков (рисков системы), установленных при анализе рисков для данного изделия/системы, должны быть приняты соответствующие меры по уменьшению или устранению риска, насколько возможно | 19.3 Требуется анализ дизайна конструкции и выбор метода анализа | Действует |
4.7.2 Имплантаты, в которых безопасность пациента зависит от внутреннего источника энергии, должны быть оснащены средствами индикации состояния источника питания | 19.2 Требуется наличие индикатора истощения источника питания | Заменен |
4.7.3 На имплантатах, насколько возможно, должен быть нанесен код, с помощью которого тип имплантата или изготовители могут быть идентифицированы (в | 13.3 Требование установлено и расширено | Заменен |
частности, в соответствии с типом имплантата); при необходимости код должен быть читаем без хирургического вмешательства | 28.6 Требуется, чтобы вместе с изделием были представлены пояснения к кодам | Действует |
4.7.4 Для имплантатов, в которых безопасность пациента зависит от внешнего источника энергии, внешний источник энергии должен включать в себя систему сигнализации для подачи сигнала в случае отказа питания | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.5 Внешние изделия, предназначенные для мониторинга одного или более клинических параметров имплантата, должны быть оснащены системой сигнализации для подачи сигнала пользователю в ситуации, которая может привести к смерти или серьезному ухудшению состояния пациента | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.6 Защита от электрических рисков | ||
4.7.6.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы избежать, насколько возможно, риска случайного поражения электрическим током при нормальных условиях и в случаях отказа, которые могут возникнуть, несмотря на правильную установку имплантата. В понятие "риски | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
при нормальных условиях и в случаях отказа" входят риски, которые определяются анализом рисков для данного вида изделия | 16.1 Требуется установить безопасные пределы утечки тока от неимплантируемых частей | Заменен |
4.7.6.2 Активные имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, связанные с использованием источников энергии, в первую очередь при использовании электроэнергии, уделяя особое | 16.2 Требуется установить безопасные пределы утечки тока от имплантируемых частей | Заменен |
внимание изоляции, токам утечки и перегреву изделия | 16.3 Требуется проведение испытания электрической изоляции (электродов и пр.) | Не применяется к кохлеарным имплантатам |
17 Требуется оценить воздействие локального нагрева, вызванного отказом имплантируемого изделия | Действует | |
26.1 Требуется защита от нагрева от неимплантируемых частей, снабженных источником питания | Действует | |
4.7.7 Защита от механических рисков | ||
4.7.7.1 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы защитить пользователя и пациента от механических рисков, связанных, например, с сопротивлением, устойчивостью, движущимися частями изделий | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.7.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие от вибрации, генерируемой изделием, принимая во внимание технический прогресс и средства, доступные для снижения вибрации, особенно источника энергии, если только вибрация не является частью специальных рабочих характеристик изделий | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.7.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие из-за шумов, принимая во внимание технический прогресс и средства, доступные для снижения шумов, особенно от источника энергии, если только шумы не являются частью специальных рабочих характеристик изделий | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.7.4 Терминалы и коннекторы для источников электрической, газовой или гидравлической и пневматической энергии, с которыми приходится работать пользователю, должны быть разработаны и сконструированы таким образом, чтобы минимизировать все возможные риски | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.8 Защита пациента от рисков, вызванных источником энергии или веществами | ||
4.7.8.1 Имплантаты должны быть разработаны и сконструированы таким образом, чтобы собственно функционирование программируемых и контрольных систем, включая программное обеспечение, не ухудшало безопасность пациента и пользователя, принимая во внимание предусмотренные назначением изделия условия применения | 19.3 Требуется анализ дизайна конструкции и выбор методики этого анализа | Действует |
4.7.8.2 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы таким образом, чтобы параметры потока могли устанавливаться и поддерживаться достаточно точно для минимизации риска для пациента | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Действует |
4.7.8.3 Имплантаты, разработанные для введения лекарственных веществ, должны включать в себя соответствующие средства предотвращения и/или индикации любой некорректной скорости потока, которая может привести к опасности | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.7.8.4 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы со встроенными средствами минимизации рисков случайного превышения опасного уровня энергии или лекарственного вещества | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия | Заменен |
4.8 Информация, предоставляемая изготовителем | ||
4.8.1 Каждый имплантат должен поставляться с сопровождающей информацией, необходимой для его безопасного использования и идентификации изготовителя, с учетом уровня подготовки и знаний потенциального пользователя. | 10.4 Требуется, чтобы сопровождающая документация была нанесена на изделие | Действует |
| 12.3 Требуется, чтобы любая маркировка была нестираемой | Действует |
4.8.2 Если применимо, информацию следует представлять в виде символов. Любой символ или цветовая индикация должны соответствовать международным стандартам. Если стандарта не существует, символ должен быть описан в документации, поставляемой с имплантатом | 4 Разрешается использование символов, аббревиатур и цветовой индикации | Действует |
4.8.3 Этикетка должна содержать следующие сведения: | ||
а) наименование или фирменный знак и адрес изготовителя; | 5 Требуется соблюдение требований к этикеткам МЭК 60601-1 для неимплантируемых частей | Заменен |
9.2 Требуется указать наименование и адрес изготовителя на товарной упаковке | Действует | |
11.1 Требуется указать наименование и адрес изготовителя на стерильной упаковке | Действует | |
b) подробные сведения, необходимые пользователю для идентификации имплантата и содержимого упаковки; | 9.3 Требуется поместить описание изделия и обозначение модели на товарной упаковке | Заменен |
9.4 Требуется при маркировке указать характеристики, достаточные для идентификации изделия | Действует | |
9.8 Требуется поместить на товарной упаковке информацию о поставляемых принадлежностях | Действует | |
9.10 Требуется дополнительное описание, если требования 9.3 и 9.4 не соблюдены в полной мере | Действует | |
11.6 Требуется поместить на стерильной упаковке описания изделия и обозначения модели | Действует | |
11.7 Требуется идентифицировать содержимое стерильной упаковки | Действует | |
с) если применимо, индикацию, что содержимое упаковки стерильно (например, "СТЕРИЛЬНО"); | 9.5 Требуется нанесение утверждения, что упаковка была простерилизована | Действует |
11.2 Требуется заявление, что упаковка и ее содержимое были стерилизованы | Действует | |
11.3 Требуется разместить символ "СТЕРИЛЬНО" | Действует | |
d) если применимо, номер партии или серийный номер, которому предшествует соответствующая идентификация (например, "LOT" или "SN"); | 9.3 Требуется поместить описание изделия и обозначение модели на товарной упаковке | Заменен |
11.6 Требуется поместить на стерильной упаковке описания изделия и обозначения модели | Действует | |
е) если применимо, дата, до которой имплантат может быть использован; | 9.7 Требуется нанесение маркировки "использовать до" | Заменен |
11.5 Требуется нанесение маркировки "использовать до" | Действует | |
f) индикацию, что имплантаты для одноразового использования; | 28.18 Требуется выбрать вид предупреждающей надписи о повторном использовании изделия | Действует |
g) если применимо, указание на специальное назначение (например, "изделие, сделанное на заказ" или "только для клинических исследований"); | 9.12 Требуется нанесение маркировки специального назначения | Действует |
11.10 Требуется нанесение маркировки специального назначения | Действует | |
h) любые специальные условия хранения и/или транспортирования; | 9.11 Требуется нанесение маркировки с информацией о любых специальных условиях окружающей среды или ограничениях по транспортированию | Действует |
i) любые специальные рабочие инструкции; | (Для имплантируемых частей активного имплантируемого медицинского изделия все рабочие инструкции приводятся в сопровождающей документации) | |
j) любые предупреждения или меры предосторожности, которые следует предпринять; | (В общем случае предупреждения и меры предосторожности, за исключением тех, что связаны со специальными условиями транспортирования [см. 4.8.3, перечисление h)], должны быть описаны в сопровождающей документации, а не на этикетке) | |
k) для активных имплантатов - месяц и год производства; | 9.6 Требуется нанесение маркировки с выбором формата | Действует |
11.4 Требуется нанесение маркировки с выбором формата | Действует | |
l) если применимо, метод стерилизации | 11.2 Требуется, чтобы на маркировке был указан метод стерилизации | Действует |
4.8.4 Если предполагаемое использование имплантата не очевидно для пользователя, изготовитель должен четко указать назначение изделия на этикетке и в инструкции по применению | 9.10 Требуется дополнительное описание, если требования 9.3 и 9.4 не соблюдены в полной мере | Действует |
4.8.5 Если это разумно и применимо, имплантаты и съемные компоненты должны быть идентифицированы по возможности в терминах | 8.2 Требуется отслеживать имплантируемые части | Действует |
серийного номера или номера партии для обеспечения возможности проведения всех надлежащих действий после установления любых потенциальных рисков, обусловленных применением как имплантата, так и съемных компонентов | 13.1 Требуется идентифицировать изготовителя, модель и т.д. непосредственно на изделии | Действует |
13.2 При возможности использования различных источников энергии следует идентифицировать используемый источник | Заменен | |
4.8.6 Если применимо, инструкция по применению должна содержать следующие сведения: | ||
а) детали, упомянутые в 4.8.3, за исключением перечислений d), е) и k); | 28.1 Требуется указать наименование и адрес изготовителя | Заменен |
28.3 Требует описания изделия | Действует | |
28.16 Требуется утверждение о том, что имплантируемые части были стерилизованы | Действует | |
28.18 Требуется определить форму предупреждения о повторном использовании изделия | Действует | |
28.21 Требуется нанесение маркировки с информацией о любых особых условиях транспортирования | Действует | |
b) рабочие характеристики, приведенные в ИСО/ТО 14283, подраздел 3.3, и любые нежелательные побочные эффекты; | 28.8 Требуется разместить информацию о предполагаемом использовании и характеристиках, а также о возможных побочных эффектах | Действует 28.8.1 дополнительный подпункт |
с) если имплантат используется в сочетании с другими медицинскими изделиями или оборудованием, как требует его предполагаемое использование, необходимые подробности о его характеристиках для определения корректных используемых имплантатов или оборудования для получения безопасной | 28.4 Требуется указать сведения о максимальной достоверной величине усилия фиксации соединения | Заменен |
комбинации; | 28.5 Требуется указать сведения о принадлежностях, которые могут потребоваться для облегчения предполагаемого использования изделия | Действует |
28.9 Требуется указать сведения о возможности выбора изделия, принадлежностей и изделий | Действует | |
d) всю информацию, необходимую для подтверждения того, что имплантат используется надлежащим образом и может работать правильно и безопасно, а также, где применимо, сведения, позволяющие оценить срок службы источника энергии; | 28.10 Требуется предоставить полную инструкцию по применению | Действует |
е) если применимо, информацию, необходимую для предотвращения специальных рисков, связанных с имплантацией имплантата; | 28.11 Требуется предоставить сведения по предотвращению опасностей во время имплантации | Действует |
f) информацию, относящуюся к рискам перекрестного влияния, вызванного присутствием имплантата во время проведения специфического лечения или обследований; | 28.12 Требуется нанести предупреждающие надписи об опасностях, возникающих при взаимодействии | Заменен |
g) необходимые инструкции на случай повреждения стерильной упаковки и, если применимо, подробные сведения о допустимом методе повторной стерилизации; | 28.17 Требуется указать меры предосторожности при работе со вскрытой или поврежденной стерильной упаковкой | Действует |
h) если имплантаты поставляются с условием, что они будут стерилизованы перед использованием, инструкция по очистке и стерилизации должна быть такой, что при ее соблюдении имплантат будет соответствовать требованиям ИСО/ТО 14283, раздел 3 | 28.17 Требуется указать в инструкции особенности стерилизации принадлежностей, поставляемых нестерильными | Действует |
i) детали последующей обработки или обслуживания, требуемые до использования имплантата (например, стерилизация, окончательная сборка и т.д.); | (Неприменимо, т.к. в 14.1 требуется, чтобы активные имплантируемые медицинские изделия поставлялись стерильными) | |
j) в случае если имплантат испускает радиоактивное излучение с лечебными целями, сведения о характере, типе, интенсивности и распространении этого излучения | (Неприменимо к активным имплантируемым медицинским изделиям) | |
В инструкцию по применению также должны быть включены подробные сведения, позволяющие медицинскому персоналу оповестить пациента о любых противопоказаниях и любых необходимых мерах предосторожности. Эти сведения должны, в частности, охватывать: | ||
k) меры предосторожности, соблюдаемые в случае изменения рабочих характеристик имплантата; | 28.19 Требуется указать сведения, позволяющие оценить срок службы источника энергии | Заменен |
28.20 Требуется указать сведения по мерам предосторожности, которые необходимо предпринять, чтобы предотвратить негативное влияние изменений рабочих характеристик изделия | Действует | |
l) меры предосторожности в ответ на воздействие в разумно предсказуемых условиях окружающей среды, например, магнитного поля, внешних источников электроэнергии, электростатических разрядов, давления или перепада давлений, ускорения, источников тепла и т.д.; | 28.22 Требуется указать сведения по мерам предосторожности, которые необходимо предпринять, чтобы защититься от негативного влияния окружающей среды | Действует 28.22.1 дополнительный подпункт |
m) достаточный объем информации, касающийся лекарственных веществ или веществ, которые должны доставляться с помощью данного имплантата, включая любые ограничения при выборе веществ, которые необходимо доставлять; | 28.7 Требуется информация о лекарственных веществах, для введения которых разработано изделие | Действует |
n) меры предосторожности, предпринимаемые против особых, необычных рисков, связанных с удалением имплантата; | 28.24 Требуется информация о правильной утилизации устройства | Действует |
о) лекарственные вещества, включенные в имплантат как неотъемлемая часть в соответствии с ИСО/ТО 14283:2004, подраздел 4.1.4; | 28.8 Требуется предоставить сведения о предполагаемом использовании изделия, характеристиках, а также о возможных побочных эффектах | Действует 28.8.1 дополнительный подпункт |
р) уровень точности измерений, заявленный для имплантатов, снабженных функцией измерения | Не применяется | |
4.9 Клиническая оценка | ||
a) сбора данных из соответствующей научной литературы, доступной в настоящее время, по назначению изделия, предусмотренного изготовителем, либо | 19.4 Требуется оценить нежелательные воздействия, вызванные изделием | Действует |
b) результатов всех клинических исследований, проводимых таким образом, чтобы защищать пациентов и обеспечивать при этом научный характер исследования | 19.4 Требуется оценить нежелательные воздействия, вызванные изделием | Действует |
Приложение ВВ
(справочное)
Связь между разделами настоящего стандарта и основными принципами, перечисленными в приложении АА
Таблица ВВ.1
Подраздел | Соответствующий основной принцип | Подраздел | Соответствующий основной принцип |
4 | 4.8.2 | 16.2 | 4.7.6.2 |
5 | 4.4.1, 4.4.1.1, 4.4.1.2, 4.4.2, 4.7.4, | 16.3 | 4.7.6.2 |
4.7.5, 4.7.7.1, 4.7.7.2, 4.7.7.3, 4.7.7.4, | 17 | 4.7.6.2 и 4.3.2 (d) | |
4.7.8.2, 4.7.8.3, 4.7.8.4, 4.8.3 и | 18.1 | 4.5.3 | |
4.8.6. (р) | 18.2 | 4.5.3 | |
7.1 | 4.2.3 | 18.3 | 4.5.3 |
7.2 | 3.5 и 4.2.3 | 19.1 | 4.3.2 (d) |
8.1 | 3.1 | 19.2 | 3.4, 4.3.2 (d) и 4.7.2 |
8.2 | 4.8.5 | 19.3 | 3.4, 3.6, 4.1.7, 4.7.1 и 4.7.8.1 |
9.1 | 4.5.3 | 19.4 | 3.6, 4.9 (а) и 4.9 (b) |
9.2 | 4.8.3 (a) | 19.5 | 4.1.3 |
9.3 | 4.8.3 (b) и 4.8.3 (d) | 20.1 | 4.3.2 (c) |
9.4 | 4.8.3 (b) | 20.2 | 4.3.2 (c) |
9.5 | 4.8.3 (c) | 21 | 4.3.2 (c) |
9.6 | 4.8.3 (k) | 23.1 | 3.4 и 4.3.2 (b) |
9.7 | 4.8.3 (e) | 23.2 | 3.4 и 4.3.2 (b) |
9.8 | 4.8.3 (b) | 23.3 | 3.4 |
9.9 | 4.3.1 | 23.4 | 3.4 |
9.10 | 4.8.3 (b) и 4.8.4 | 23.5 | 3.4 |
9.11 | 4.8.3 (h) | 23.6 | 3.4 и 4.3.1 |
9.12 | 4.8.3 (g) | 24 | 4.3.2 (b) |
10.1 | 3.5 и 4.2.3 | 25 | 4.3.2 (b) |
10.2 | 3.5 и 4.2.3 | 26.1 | 3.4 и 4.7.6.2 |
10.3 | 3.5 | 26.2 | 3.5 и 4.3.2 (b) |
10.4 | 3.3 и 4.8.1 | 27 | 4.3.2 (b) |
11.1 | 4.8.3 (a) | 28.1 | 4.8.6 (а) [4.8.3 (а)] |
11.2 | 4.8.3 (с) и 4.8.3 (l) | 28.2 | 4.5.3 |
11.3 | 4.8.3 (c) | 28.3 | 4.8.6 (a) [4.8.3 (b)] |
11.4 | 4.8.3 (k) | 28.4 | 3.4, 4.3.1 и 4.8.6 (с) |
11.5 | 4.8.3 (e) | 28.5 | 4.3.1 и 4.8.6 (с) |
11.6 | 4.8.3 (b) и 4.8.3 (d) | 28.6 | 4.7.3 |
11.7 | 4.8.3 (b) и 4.2.3 | 28.7 | 4.8.6 (m) |
11.8 | 4.3.1 | 28.8 | 4.8.6 (b) и 4.8.6 (о) |
11.9 | 4.2.3 | 28.9 | 4.8.6 (c) |
11.10 | 4.8.3 (g) | 28.10 | 4.8.6 (d) |
12.1 | 4.2.3 | 28.11 | 4.8.6 (e) |
12.2 | 4.2.3 | 28.12 | 4.3.2 (с) и 4.8.6 (f) |
12.3 | 3.5 | 28.13 | 4.3.2 (c) |
13.1 | 4.8.5 | 28.14 | 4.3.2 (c) |
13.2 | 4.8.5 | 28.15 | 4.3.2 (c) |
13.3 | 4.7.3 | 28.16 | 4.8.6 (а) [3.8.3 (с)] |
13.4 | 4.4.1.2 | 28.17 | 4.8.6 (g) и 4.8.6 (h) |
14.1 | 4.2.1, 4.2.3, 4.2.4 и 4.2.5 | 28.18 | 4.8.6 (а) [4.8.3 (f)] |
14.2 | 4.1.2 и 4.2.5 | 28.19 | 4.8.6 (k) |
14.3 | 4.1.1 (а), 4.1.1 (b) и 4.1.2 | 28.20 | 4.8.6 (k) |
14.4 | 4.1.4 | 28.21 | 4.8.6 (a) [4.8.3 (h)] |
15.1 | 4.3.2 (a) | 28.22 | 4.8.6 (I) |
15.2 | 4.3.2 (a) | 28.23 | 3.4 |
16.1 | 4.7.6.1 | 28.24 | 4.8.6 (n) |
Приложение СС
(справочное)
Примечания к ЕС 45502-2-3 (основание для настоящего стандарта)
СС.1 Общие положения
Настоящий стандарт является основой для развития EН 45502 и попыткой установить необходимые количественные требования Директивы 90/385/EEC. В настоящем стандарте данная задача решается путем детализации конкретных аспектов необходимых требований и определения процедур оценки или испытаний.
Для некоторых рисков в настоящем стандарте приведены специальные требования вместе с мерами оценки соответствия (например, оценки уровня тока утечки), которые в случае их удовлетворения будут уточнять конкретный аспект необходимых требований Директивы. Для прочих опасностей настоящий стандарт определяет риски, требующие идентификации и оценки в соответствии с ИСО 14971. Соответствие проверяется путем обзора файла менеджмента риска, предоставленного изготовителем.
В некоторых случаях никакое лабораторное испытание ограниченной продолжительности не может обеспечить достаточную уверенность в характеристиках конкретного изделия или подтвердить рабочие характеристики изделия спустя несколько лет после имплантации. В таких случаях настоящий стандарт требует от изготовителя изделия предоставить документально зафиксированные исследования, необходимые для проведения экспертной оценки изделия.
СС.2 Замечания по специальным разделам и подразделам
Изложенные ниже замечания к некоторым положениям настоящего стандарта призваны облегчить его понимание. Настоящее приложение предназначено для лиц, знакомых с конструкцией или с использованием активных имплантируемых медицинских изделий, но не принимавших участия в составлении настоящего стандарта. Содержание настоящего приложения относится не ко всем разделам настоящего стандарта; именно поэтому нумерация параграфов в приложении не является последовательной.
За исключением разделов 5, 7, и 8 настоящего стандарта, оставшиеся разделы организованы таким образом, что они могут быть адресованы ко всей последовательности действий, начиная от проверки маркировок на упаковке, процесса создания упаковки, испытания изделия, заканчивая проверкой сопровождающей документации.
3.3.2, 3.3.3, 3.3.4, 3.3.5 Большинство НЕИМПЛАНТИРУЕМЫХ в настоящее время ЧАСТЕЙ могут стать имплантируемыми в будущем.
3.20.4 СРОК ГОДНОСТИ применяется к пунктам 9.7 и 11.5.
5.1 При взаимодействии с НОСИМЫМИ НА ТЕЛЕ частями изделия (например, со сменными батарейками) пациент расценивается как оператор, а не как пациент в контексте МЭК 60601-1, и в связи с чем требования к уровням тока утечки не применяются.
5.2 ЕН 45502-1:1997 (подраздел 5.1) определяет требования, применимые к НЕИМПЛАНТИРУЕМЫМ ЧАСТЯМ. Они охватывают аспекты электрической безопасности, требования по электромагнитной совместимости и т.д., перечисленные в МЭК 60601-1-2. Современные технологии СИСТЕМ ИМПЛАНТАЦИИ используют передачу энергии через индуктивные радиочастотные связи между НЕИМПЛАНТИРУЕМОЙ ЧАСТЬЮ и имплантируемой частью (радиочастотный трансформатор).
13.3 Настоящий подпункт затрагивает фундаментальный принцип, отраженный в Директиве: любое используемое изделие должно быть идентифицировано без проведения хирургической операции и без необходимости в специализированном оборудовании, характерном для изготовителя или модели изделия. На практике нанесение маркировки на ИМПЛАНТИРУЕМУЮ СИСТЕМУ может оказаться невозможным. Сегодня высоким уровнем развития технологий считается идентификация изготовителя и модели изделия с помощью рентген-контрастных символов-меток. Для ИМПЛАНТИРУЕМЫХ СИСТЕМ, не имеющих внутреннего источника питания, идентификация года изготовления не считается необходимой. Будущие технические усовершенствования могут сделать возможной телеметрическую идентификацию серийного номера или даты производства изделия. Считывание рентген-контрастного символа позволяет выбрать подходящее телеметрическое устройство.
14.2 Кроме соответствия имплантата специфическим требованиям стерильности также недопустимо отделение от имплантата твердых частиц ("стерильной пыли"). Метод оценки соответствия построен таким образом, что значимые количественные ограничения могут быть установлены для оценки соответствия результатов испытания. Изготовитель может выбрать другую признанную технику измерений с использованием аппаратуры, находящейся в свободном обращении. Частицы, намеренно добавленные (например, фармацевтические агенты) к имплантату для обеспечения терапевтического действия, покрытие имплантатов или элюции имплантата, не являются объектом данного испытания.
Число частиц связано с поверхностью изделия, а не с его объемом. Например, пустой мешок (большая поверхность, но незначительный объем) может выделить чрезмерное количество частиц, когда его замачивают в ванне. Та же заполненная емкость может пройти испытание даже при том, что общее количество частиц является тем же. То же самое справедливо для изделий, относящихся к настоящему стандарту, особенно к тем, которые имеют большую площадь поверхности, но небольшой объем. Для ИМПЛАНТИРУЕМЫХ СИСТЕМ этот подход определяет характеристики ванны, которые имеют тот же порядок величины, что и в ИСО 14708-1.
Испытательные пределы основываются на стандартах испытания корпускулярного загрязнения для большого объема парентеральных инъекций, приведенных в Европейской Фармакопее.
15.2 ИСО/ТС 150/SC6 признает необходимость проведения соответствующих испытаний в целях подтверждения того, что физические характеристики имплантируемых частей не вызывают чрезмерную раздражительную (воспалительную) реакцию. Например, изготовитель должен предоставлять информацию об испытаниях на животных или другую подтверждающую информацию.
16.2 Длительные небольшие постоянные токи (DC) от имплантируемых электродов могут вызывать повреждение тканей или коррозию электродов. Безопасный предел был снижен до 0,1 мкА в соответствии с мнением, изложенным в современной литературе. Метод испытаний должен быть применим к изделию даже при стимуляции с использованием уровней, представляющих нормальную клиническую практику. Настройки изделия, включая аргументацию для их выбора, должны быть задокументированы вместе с результатами испытаний. Должны быть предприняты необходимые действия, для того чтобы гарантировать, что любая чрескожная связь не влияет на измерение. Используется вольтметр постоянного тока, питаемый через фильтр низких частот с временной константой не менее 1 с. Например, это может быть осуществлено посредством применения четырехэлементного резистивно-емкостного фильтра низких частот с элементами, состоящими из 1 МОм резисторов и металлизированных полиэстеровых конденсаторов емкостью 1 мкФ. В таком случае входное сопротивление вольтметра постоянного тока должно быть 400 МОм.
19.2 Желательно, чтобы снижение энергоснабжения ИМПЛАНТИРУЕМОЙ СИСТЕМЫ не вызывало прекращения его функционирования без предварительного предупреждения. Используемый механизм предупреждения не должен признаваться недействительным различными методиками стимуляции, истощающими источник питания на различных уровнях. Индикатор может быть как внутренним, так и внешним.
19.5 ИСО/ТС 150/SC6 признает, что жизненный цикл ИМПЛАНТИРУЕМЫХ СИСТЕМ, доступных на сегодняшний день, может быть короче, чем ожидаемая продолжительность жизни пациентов, особенно при имплантации маленьким детям. Основываясь на текущем положении дел и с этической точки зрения, должна быть возможна замена ИМПЛАНТИРУЕМЫХ СИСТЕМ. Во время процесса разработки изготовитель должен предусмотреть следующие аспекты, которые могут негативно повлиять на замену изделия: совместимость размеров и формы, механическая устойчивость и биологические эффекты.
19.6 ИСО/ТС 150/SC6 признает целесообразность обеспечения изготовителями и экспертами уверенности в том, что изоляция СТИМУЛЯТОРА обеспечит защиту от любого контакта его компонентов с жидкостями организма. В противном случае такая ошибка может вызвать дисфункцию прибора и/или непреднамеренные стимуляции вблизи изделия и/или непреднамеренные биологические эффекты, вызванные внутренними биологически несовместимыми частями (то есть электронными компонентами) при контакте с жидкостями организма.
Процедуры испытаний и критерии отказа установлены стандартами электронной отрасли. Испытание было создано методом STD 883 MIL 1014. В ЕН 13185 и ЕН 1593 предложены различные методы, которые могут выбрать изготовители.
20.2 Дефибрилляторы обычно пропускают через торс напряжением порядка 5000 В, но современные имплантируемые системы не имеют частей, имплантируемых в торсе, в результате чего напряжение в области имплантата имплантируемой системы недостаточно высоко, чтобы быть источником опасности. Необходимо учитывать, что существует вероятность того, что части будущих изделий смогут быть имплантированы в области торса, например, батарея или индукционная катушка. В этом случае испытание, определенное ИСО 14708-1, будет необходимым. В случае контакта внешних частей и электродов дефибриллятора возникновение повреждения не считается вероятным в силу того, что требования ESD, описанные в разделе 24, являются достаточными.
21.1 Испытание проверяет помехоустойчивость в условиях высокочастотного электрического тока, возникающего от хирургической диатермии. Испытательная частота 500 кГц была выбрана как типичная для большинства электрохирургического оборудования. Выбранная амплитуда 20 В для испытания защиты изделия была заимствована из основного стандарта ЕН 45502-2-1. Нагрузочный резистор 4,7 кОм был выбран для моделирования сопротивления поверхности нервных тканей в улитке. Во время испытания ИМПЛАНТИРУЕМАЯ СИСТЕМА должна быть выключена. Требования не обеспечивают полной защиты, так как увеличение напряжения во время воздействия хирургической диатермии очень зависимо от расстояния между диатермическими электродами и любой проводящей частью ИМПЛАНТИРУЕМОЙ СИСТЕМЫ или ее электродного массива, и хирург может быть не осведомлен о местоположении таких частей.
22.1 Следует обратить внимание на то, что данное требование адресовано только к случаю проведения ультразвуковой диагностики. Настоящий стандарт требует размещения предупредительной надписи о возможных последствиях в случае, если ИМПЛАНТИРУЕМАЯ СИСТЕМА будет подвержена терапевтическим уровням ультразвука (см. 28.20).
22.2 ИСО/ТС 150/SC6 признает целесообразность обеспечения изготовителями уверенности в том, что пациенты с ИМПЛАНТИРУЕМЫМИ СИСТЕМАМИ могут проходить МРТ-обследования, не подвергая себя риску. В силу того, что на данный момент на рынке представлена широкая линейка аппаратуры МРТ и в связи с различными характеристиками чрескожных соединений, используемых различными ИМПЛАНТИРУЕМЫМИ СИСТЕМАМИ, было определено, что одновременно с указанием уровня безопасности МРТ в сопровождающей документации (см. раздел 28) изготовитель заявляет напряженность поля МРТ аппаратуры, в пределах которых заявляются требования безопасности. Независимо от уровня испытания, любое решение по назначению МРТ исследования остается медицинским решением, сопоставляющим риск возможного повреждения и пользу от информации, полученной в результате применения МРТ сканирования. Испытание на безопасность МРТ предполагает, что имплантат был размещен в соответствии с хирургическими инструкциями изготовителя и имплантат должным образом стабилизирован. Максимальная напряженность поля была определена в соответствии с [24] и [25]. Сила воздействия на имплантат или магнит (если магнит не содержится в твердой структуре) является последствием крутящего момента и соответствующих размеров изделия, в связи с чем считается, что испытание на напряженность поля считается достаточным. Для измерения напряженности поля был предложен теоретический подход, так как последующее размагничивание может недооценивать фактическую силу. Также могут использоваться методы, определенные в стандартах ASTM (ASTM F2052, ASTM F2213), для измерения напряженности поля и крутящего момента отдельно.
ИСО/ТС 150/SC6 признает, что нагревание проводника может вызвать серьезную проблему. Возможно, что во время написания стандарта будет разработан более точный метод измерения. Стандарт ASTM F2182 был использован для испытания тепловыделения в проводниках таких активных имплантируемых медицинских изделий, как кардиостимуляторы и кохлеарные имплантаты, и поэтому рассматривается как соответствующий. Имплантат помещается под височной мышцей, имеющей хорошее кровоснабжение. В связи с этим повышение температуры на 2°С является обоснованным.
Во время испытания на тепловыделение, для избежания незапланированных скачков энергии и повреждений имплантата, имплантируемая часть должна быть закреплена подходящим приспособлением во избежание смещения имплантата в аппарате МРТ. Имплантируемая часть может вызвать искажение МРТ изображения, но это не считается угрозой безопасности. Также возможное размагничивание внутреннего магнита в результате МРТ сканирования не считается угрозой безопасности. Маркировка должна содержать соответствующую информацию о потенциальных угрозах размагничивания (см. 28.12).
22.3 ИСО/ТС 150/SC6 признает, что настоящие и будущие конструкции кохлеарных имплантатов, вероятно, будут оставаться склонными к ухудшению своих функций или к неправильному функционированию в результате применения терапевтического ионизирующего излучения. Группа также признала необходимость того, чтобы пациенты, имеющие кохлеарные имплантаты, не получали вреда в случае необходимости проведения лучевого лечения. Несмотря на то, что лучевое лечение может проводиться через (или вблизи) имплантируемую часть, было отмечено, что большинство видов лечения осуществляется при других расположениях. В этой ситуации излучения, вероятно, будут основной причиной беспокойства. В соответствии с надлежащей клинической практикой активные имплантаты должны быть экранированы во время лучевого лечения, минимизировав таким образом чувствительность к вредному излучению.
Данные литературы об испытаниях облучением некоторых кохлеарных имплантатов ([14] и [30]) указывают, что несмотря на то, что современные изделия имеют ограниченный уровень "устойчивости" к эффектам ионизирующего излучения, никакое изделие не может быть разработано и произведено, чтобы стать полностью защищенным. Группа идентифицировала потребность в информации от изготовителя, демонстрирующей уровень помехоустойчивости, но согласилась, что минимальный уровень "устойчивости" к излучению приведет к несправедливым ограничениям. Решение, принятое ИСО/ТС 150/SC6, состояло в том, чтобы согласовать определенный метод испытаний облучения на основе общих принципов лучевой терапии (дробление накопленной дозы). Изготовитель объявляет максимальный уровень накопленной дозы, после которой изделие будет продолжать функционировать в обычном режиме. Маркировка на основе этого анализа позволяет клиническим врачам судить, окажет ли намеченная структура лучевого лечения постоянное влияние на функциональность имплантируемой части.
23.1 Карманные программируемые и переносимые анализирующие устройства могут являться объектами тяжелых механических повреждений при использовании неопытным пользователем. Если такое воздействие наносит постепенный ущерб не только пользователю, поврежденное устройство должно быть отделено от имплантата, так как может давать ошибочные данные, которые могут впоследствии выдавать ненужную информацию.
23.2 Это испытание предназначено для установления минимальных требований к прочности имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ по отношению к механическим воздействиям.
Вывод по испытаниям, первоначально озвученный в ЕН 45502-1, установил необходимость проведения нового испытания.
Замененный текст основан на новой части ЕН 60068-2-64:2008.
Жесткость испытаний определяется условиями испытаний по перечислениям a)-d). Диапазон испытательных частот основывается на опыте применения метода испытаний с синусоидальным сигналом, широко используемым в течение многих лет в производстве кардиостимуляторов.
Значение для ускорения спектральной плотности было также получено посредством синусоидального метода испытаний в 8.1.1 из ЕН 50061:1988. Это испытание определяет пиковое ускорение 25 м/с, что эквивалентно среднеквадратичному значению 1,77 г. Ускорение спектральной плотности 0,7 (м/с
)
/Гц эквивалентно среднеквадратичному значению 1,86 г. Последнее вычисление является приближением, которое может варьироваться в зависимости от оборудования, используемого для генерации случайной вибрации. Заметим, что уровень испытательного воздействия на ИМПЛАНТИРУЕМУЮ СИСТЕМУ сопоставим с уровнем, приведенном в методе в ЕН 50061.
В целом испытание короткой продолжительности приведет к низкому доверительному уровню результатов. Значение продолжительности для этого испытания является средним из рекомендуемых значений в МЭК 60068-2-64:2008, подраздел 5.5. Это должно обеспечить обоснованную уверенность в воспроизводимости результатов испытаний с оптимальным полным временем проведения испытаний.
Защита изделия во время поставки и хранения обеспечена подходящей конструкцией, которая оценена относительно вибрации в 10.1.
23.3 Данные литературы указывают на рост костей черепа 12 мм (допустимое отклонение 5 мм) с момента рождения к взрослому состоянию ([24]). Это приводит к тому, что часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ должна быть разработана для устойчивости к растяжению, которое может быть во время периода роста черепа. В случае если проводник не допускает удлинения 15 мм, изготовитель должен избежать натяжения электродного массива от улитки. ИСО/ТС 150/SC6 полагает, что сила 1 Н была репрезентативной для силы удлинения, действующей во время роста кости и во время имплантации. Метод испытаний, описанный в 23.3 для ИМПЛАНТИРУЕМЫХ СИСТЕМ, учитывает отличающиеся геометрии проводников. Несмотря на то, что выбор адекватного метода крепления проводника оставляют на усмотрение изготовителя, требуется, чтобы проводник, подвергающийся удлинению при росте черепа, был подвергнут стандартному испытанию.
23.5 При составлении настоящего стандарта было отмечено, что в соответствии с клинической практикой рекомендовано имплантировать имплантируемую часть ИМПЛАНТИРУЕМОЙ СИСТЕМЫ в костное ложе. На текущий момент считается, что это обеспечивает максимальную стабильность имплантируемой части и связанного с ней электродного массива. Испытания 1 и 2 предназначены для установления минимальных требований для длительности изгибания имплантируемых ПРОВОДНИКОВ. Испытание 1 разработано для моделирования любых неблагоприятных условий обработки, которые могут возникнуть во время извлечения имплантата из СТЕРИЛЬНОЙ УПАКОВКИ и в период до имплантации. При испытании 2 признается, что изменения в методе имплантации могут вызывать микродвижения проводника после имплантации, особенно в области височной мышцы. Также признано, что с рекомендуемым методом имплантации микродвижения наконечника ПРОВОДНИКА могут быть значительно уменьшены.
Несмотря на то, что точные условия не могут быть определены, считается, что процессы растяжения и изгибания вызывают схожие нагрузки для испытуемых изделий in vivo. К испытательному сегменту присоединен груз массой 3 г, чтобы вынудить испытательный образец соответствовать требуемому угловому смещению без обеспечения значительной растягивающей нагрузки. Изгиб испытательного образца на ±15° для 100000 циклов создает больше существенных деформаций в электроде, чем ожидается in vivo.
23.6 EH 45502 оставляет определение метода обеспечения безопасного соединения за изготовителем. Таким образом, изготовитель обязан определить совместимые части соединителей (см. 9.9 и 28.9) так, чтобы указанные части могли быть отобраны для испытаний, гарантировав, что имплантируемые пары соединителей надежны, когда подвергаются растягивающему усилию.
23.8 ИСО/ТС 150/SC6 признал потребность в испытании на удар имплантируемой части для уменьшения риска отказов из-за травмы. В то время как взрослые редко испытывают падения, которые могли бы закончиться повреждением ИМПЛАНТИРУЕМОЙ СИСТЕМЫ, дети особенно уязвимы из-за подвижности, роста, недостаточной координации нижних конечностей. Испытание было разработано для обеспечения гарантии, что воздействия во время обычной ежедневной активности не поставят под угрозу имплантируемую часть. Такие воздействия включают в себя удары головы и падения во время ходьбы, бега или езды на велосипеде, которые не требуют медицинской или скорой помощи. Было проведено исследование по заказу немецкого Компетентного органа (BFARM), чтобы проанализировать виды отказа и предложить адекватные методы испытаний. Результат опубликован в [15]. На основе результатов того проекта комитет выбрал энергию 2,5 Дж как критичную. Эта энергия превосходит удар головы (в расположении имплантата) твердым объектом массой 1 кг и скоростью приблизительно 2,25 м/с. На основании имеющихся сведений опыт эксплуатации нескольких тысяч изделий, использующих модели устройства, идентичные тем, что проходят испытание Хольткамп при энергии 1-1,5 Дж, был сочтен достаточным для обеспечения приемлемой устойчивости к воздействиям окружающей среды. В ИСО/ТС 150/SC6 сделан вывод, что на момент введения стандарта для испытаний должна применяться энергия 1,5 Дж, а спустя три года после того, как стандарт становится обязательным, должна использоваться энергия 2,5 Дж, обеспечивая дополнительный запас прочности. В соответствии с Директивой активные имплантируемые медицинские изделия изготовитель может продемонстрировать соответствие существенным требованиям, не используя настоящий стандарт. В этом случае изготовитель должен продемонстрировать сопоставимый уровень безопасности, как описано в стандарте. Если ИМПЛАНТИРУЕМАЯ СИСТЕМА снабжена имплантируемым микрофоном или другим датчиком, они могут выйти из строя после испытания на удар. Это приемлемо, если существует избыточность в системе, не требующая замены имплантируемой части ИМПЛАНТИРУЕМОЙ СИСТЕМЫ.
24.1 Испытательная комплектация была выбрана для моделирования in vivo ситуации ношения имплантируемого предмета с изношенным элементом (например, речевой процессор с катушкой, но без дополнительного модуля FM), испытание применяется к НЕИМПЛАНТИРУЕМОЙ ЧАСТИ. Любой выброс, оказывающий влияние на носимую на теле часть, также повлияет на имплантируемую часть. Испытательные напряжения были выбраны из ЕН 45502-1:1997, подраздел 24.1. Более высокие испытательные напряжения не были бы адекватными для очень маленьких внешних частей (речевой процессор позади уха), используемых с ИМПЛАНТИРУЕМОЙ СИСТЕМОЙ.
25.2 Это испытание имитирует частичное повышение давления, которое может возникнуть во время таких профессиональных и развлекательных занятий, как дайвинг. Это было включено в результате увеличения потребностей пользователей.
27 В настоящее время это требование охватывает все обозримые электромагнитные среды, с которыми носитель ИМПЛАНТИРУЕМОЙ СИСТЕМЫ мог бы столкнуться, даже те, с которыми почти никогда не сталкиваются в местах общего доступа. Требование разделено на два подпункта: один направлен на защиту от вреда, повреждения ткани или изделия и возникновения у пациента боли в общественных местах при обычных ситуациях, даже тех, с которыми сталкиваются редко во время обычной ежедневной активности (см. 27.1). Другой гарантирует, что функции изделия не находятся под значительным влиянием воздействий, с которыми обычно сталкиваются во время нормальной жизни (см. 27.2).
Раздел 27 только содержит требования с точки зрения уровней, в рамках которых изделие может подвергаться воздействию других устройств (см. 27.3 и 27.4). Это требуется изготовителю для выбора адекватных средних значений для подтверждения соответствия, теоретического моделирования или прямых измерений электромагнитных помех.
Приложение DD приводит пример, как подтвердить соответствие посредством теоретического моделирования. Приложение ЕЕ
приводит пример, как продемонстрировать соответствие измерениям электромагнитных помех.
_______________
В оригинале ISO 14708-7:2013 ошибочно указано приложение ВВ.
В оригинале ISO 14708-7:2013 ошибочно указано приложение СС.
27.1 Это требование гарантирует, что изделие не будет повреждено, а лицу, которому оно имплантировано, не будет нанесен вред при электромагнитном воздействии. Это требование соответствует требованию 8 третьего абзаца Директивы 90/385/ЕЕС.
Соответствующие уровни для 27.1 (верхние уровни требований в 27.3 и 27.4) получены из основных ограничений Рекомендаций 1999/519/ЕС для широкого применения, которые охватывают пик и факторы локализации. Теоретически еще более высокие пиковые амплитуды и локальные пятна, как предполагается, не представляют риск также для людей без имплантата, но никакая известная область в широком применении действительно не использует такие параметры в настоящее время. ИСО/ТС 150/SC6 решил не координировать такие нереалистичные значения.
Требуемые уровни воздействия определены в 27.3 и 27.4. Эти подпункты обеспечивают два взаимосвязанных уровня: требование для функций, не оказывающих влияние (нижний уровень), и требование защиты (верхний уровень). Подраздел 27.1 применяется только на верхнем уровне и не предъявляет требований к функциям, не оказывающим влияние на ИМПЛАНТИРУЕМУЮ СИСТЕМУ во время воздействия.
Настройки были выбраны для отражения максимально возможных выходных сигналов. Любое значительное увеличение сигналов стимуляции выше максимального выходного уровня могло бы нанести ущерб носителю. Поэтому соответствие может быть продемонстрировано ограничением увеличения уровня выходного сигнала во время воздействия. Дополнительно должно быть подтверждено функционирование ИМПЛАНТИРУЕМОЙ СИСТЕМЫ после воздействия.
27.2 Требования соответствуют тем уровням, с которыми обычно сталкиваются пациенты в электромагнитной среде в местах общего доступа, и требованиям нормального функционирования во время воздействия. Требования для 27.2 (более низкие уровни требований в 27.3 и 27.4) получены из исходных уровней Рекомендаций 1999/519/ЕС без учета пиковых значений или фактора расположения источника. Тем не менее, эти более низкие уровни покрывают почти все известные области, так как эти значения будут превышены в очень редких ситуациях, которые ограничены по времени воздействия.
Требуемые уровни воздействия определены в 27.3 и 27.4. Эти подпункты устанавливают требования к двум взаимосвязанным уровням, для нормального функционирования (нижний уровень) и для защиты (верхний уровень). Подпункт 27.2 применяется только на более низком уровне и требует правильного функционирования ИМПЛАНТИРУЕМОЙ СИСТЕМЫ во время воздействия.
Требования были выбраны исходя из обычных соотношений, используемых в испытаниях выходных сигналов. Соответствие может быть продемонстрировано ограничением изменений амплитуды и фазы выходного сигнала во время воздействия. Формулировка "правильное функционирование" не означает, что не должно быть никаких обнаруживаемых изменений амплитуды и фазы выходного сигнала имплантируемой системы, но ее работа не должна создавать неудобства пользователю.
27.3 и 27.4 Число испытательных частот ограничено приблизительно двумя частотами на декаду с целью уменьшить время вычисления для имитации воздействий и теоретического моделирования или измерения времени воздействия электромагнитных помех. Частоты фиксированы. Такие большие шаги по частоте не охватывают отдельные узкие резонансные частоты в ИМПЛАНТИРУЕМОЙ СИСТЕМЕ, но позволяют обеспечить уверенность в хорошей общей эффективности испытаний. Между частотами 50 Гц и 1,66 кГц не представлена ни одна испытательная частота, потому что не известны никакие мощные воздействующие поля в этом диапазоне.
27.3 Диапазон частот от 16,6 Гц до 10 МГц. Требования ограничены только магнитными полями, потому что отношение допустимых уровней для электрических и магнитных полей ниже Z= 377 В/А. Кроме того, на низкой частоте экранирование ткани вокруг имплантируемой части намного более сильное для электрического поля, чем для магнитного.
Напряженность поля для обеспечения защиты (верхний уровень) определены в диапазоне частот 16,6 и 50 Гц, которые были выбраны исходя из самых мощных источников излучения, используемых в этом диапазоне: железнодорожные системы и источники питания (60 Гц также охватываются испытанием на 50 Гц). На низких частотах (выше 50 Гц и ниже 1,66 кГц) высокие напряженности поля приемлемы для людей без имплантатов, но такие напряженности не встречаются в повседневной жизни. Для частот между 1,66 и 100 кГц требование было установлено на основе ограничений, приведенных в Рекомендациях 1999/519/EC (которые соответствуют ANSI/IEEE C95.1) из предположения, что локальный фактор немного больше 20 в комбинации с пиково-среднеквадратичным значением отношения для непрерывной волны 1,4. Это означает, что максимальная пиковая напряженность поля неимпульсных помех может быть приблизительно в 28 раз больше исходного уровня (который является среднеквадратичным значением для воздействия на все тело). Пиковый фактор равен 1 для частот ниже 100 кГц. Это означает, что для импульсных полей никакие превышения пиковых амплитудных значений не принимаются. Выше 100 кГц локальный фактор уменьшается непрерывно до 5 на частоте 10 МГц, и пиковый фактор увеличивается непрерывно до 32 на частоте 10 МГц, приблизительно. Это означает, что пиковая напряженность поля в большей степени зависит от локального фактора на низких частотах и от пикового фактора на высоких частотах. 1999/519/EC определяет пик напряженности поля для импульсных областей выше 10 МГц и позволяет ему отличаться в 32 раза от исходных уровней (23·1,4=32). Максимальные уровни 27,3 покрывают оба фактора, локальные и пиковые, какой бы не был выше. В настоящее время известные области не используют пиковые факторы больше, чем приблизительно 5. Поэтому пиковые факторы до приблизительно 5 охвачены только верхними уровнями, приведенными в настоящем стандарте.
Напряженность поля (более низкий уровень) отражает исходный уровень, согласно Рекомендациям 1999/519/EC. Это означает, что локальное увеличение или пульсации напряженности поля охвачены частично. Более низкие уровни характерны для организма в целом в обычных условиях, но они не охватывают некоторые точечные источники помех, например, некоторые устройства электронных систем наблюдения (EAS) могут все же влиять на функцию имплантата. Требование для нормального функционирования на частотах между 1,66 и 100 кГц в 20 раз превышает требование защиты. Так как пиковый фактор, устанавливаемый в этих требованиях, увеличивается умеренно от 1 на частоте 100 кГц к 5 на частотах выше 10 МГц, различие между верхними и более низкими уровнями медленно уменьшается в верхней части диапазона частот. Рекомендация 1999/519/EC допускает функционирование на 5 МГц и на более высоких магнитных полях, чем определенные в таблице 101. При этом данная частота используется для телерадиовещания. В некоторых ситуациях (Е/Н = 377 В/А) компонент электронной области ограничивает уровень электромагнитного поля.
Примечание 1 - "Пиковая напряженность магнитного поля" описывает максимальную амплитуду вектора магнитного поля, а не максимальное кратковременное среднеквадратичное значение во время выделения энергии.
Теоретическое моделирование, а также измерения должны продемонстрировать, что требования соблюдаются в любом направлении вектора поля.
27.4 Диапазон частот от 10 МГц до 3 ГГц. На частотах выше 10 МГц для испытаний актуальны обе составляющие: электрическая и магнитная. Так как большинство воздействий может быть имитировано ситуациями в поле дальней области, то достаточно определять только напряженность электрического поля.
Теоретически локальный фактор 5 и дополнительный пиковый фактор 32 относительно исходных уровней, приведенных в Рекомендациях 1999/519/EC, приемлемы для людей без имплантатов. Но в реальной жизни не известны никакие общедоступные источники излучения с такими характеристиками. Для известных далеких источников излучения был выбран полный фактор 5 для частот до 450 МГц с уменьшением к 2,5 на частоте 2450 МГц. Это означает, что вероятное импульсное излучение мобильных устройств действует в тех точках, которые расположены непосредственно у места имплантации.
Напряженности поля для выполнения требований нормального функционирования (более низкий уровень) не отражают исходные уровни, приведенные в Рекомендации 1999/519/EC, которые не учитывают ни локальный фактор, ни пиковый фактор. Это обычно соответствует характеристикам далеких областей, которые характерны для открытого доступа.
Примечание 2 - "Пиковая напряженность электрического поля" описывает максимальную амплитуду вектора электрического поля, а не максимальное кратковременное среднеквадратичное значение во время выделения энергии.
Частоты выше 800 МГц, особенно в ситуации близкого поля, важны из-за широкого использования карманных сотовых телефонов. Тем не менее, представляется достаточным задание только напряженности электрического поля.
Теоретическое моделирование, а также измерения должны подтвердить, что в результате достигнуто соблюдение требований при любом направлении вектора поля.
27.5 Модуляция/форма импульса должны отразить два обстоятельства. Они должны иметь потенциал влияния на функцию имплантируемой системы, но в случае измерений они не должны давать потенциалы, которые можно не отличить от сигнала стимуляции в испытательном оборудовании. Амплитуда сигнала помехи определена в 27.3-27.4.
На частоте 16,6 Гц (некоторые европейские железнодорожные системы) и на 50 Гц (источники питания переменного тока) поля обычно имеют форму синусоидальной волны. На всех других частотах существуют всевозможные технические устройства, которые используют модулированные импульсные поля. Импульсные сигналы несущей частоты используются, чтобы смоделировать максимально возможное влияние на имплантаты.
28.4 На момент написания стандарта не существует ИМПЛАНТИРУЕМЫХ СИСТЕМ, использующих имплантируемые соединители. Цель данного подпункта состоит в том, чтобы гарантировать предоставление изготовителем необходимой информации о соответствующих соединителях и порядке сборки.
28.12 В то время как испытания были разработаны для подтверждения длительной безопасной работы ИМПЛАНТИРУЕМОЙ СИСТЕМЫ во время или после применения определенных диагностических процедур или методов лечения, для клинической практики все же требуются предупреждения о возможном влиянии на обеспечение нормального функционирования ИМПЛАНТИРУЕМОЙ СИСТЕМЫ. Нормальное функционирование не означает отсутствия рисков или их несущественность для пациентов во время диагностических процедур или при применении различных методов лечения.
28.15 ИМПЛАНТИРУЕМЫЕ СИСТЕМЫ могут быть устойчивы к определенным уровням лечебной ионизирующей радиации. Согласно 28.12 изготовитель должен предоставить информацию о максимальной допустимой дозе согласно испытанию, описанному в 22.3.
28.19 Во время написания настоящего стандарта известные ИМПЛАНТИРУЕМЫЕ СИСТЕМЫ не имеют имплантируемого источника энергии. ИМПЛАНТИРУЕМЫЕ СИСТЕМЫ, разработанные в будущем, вероятно, могут иметь перезаряжающуюся имплантируемую батарею. При оценке срока годности источника энергии для нормальных клинических и наихудших условий изготовитель должен принять во внимание следующие параметры: время работы батареи от одного заряда, общее число циклов перезарядки и время зарядки.
Приложение DD
(справочное)
Примечания к теоретическому моделированию для определения соответствия разделу 27
DD.1 Общие положения
В ИСО/ТК 150/SC6 определены и описаны следующие преимущества и недостатки теоретического моделирования:
a) Преимущества:
- для лаборатории не требуется дорогое измерительное оборудование;
- не требуются специалисты для проведения радиочастотных измерений;
- после разработки первой модели моделирование по аналогии проводится очень быстро;
- отсутствуют помехи, которые необходимо отфильтровывать.
b) Недостатки:
- используется не реальное воздействие, поэтому будут выявлены не все эффекты;
- требуется сложное разделение внутренних и внешних воздействий на имплантат;
- наряду с наличием опыта работы с полями и распределениями плотности тока в человеческой голове необходимо детальное знание внутреннего устройства имплантата;
- в случае внешних или внутренних изменений изделия может возникнуть необходимость повторного моделирования;
- необходим эксперт в моделировании и проверке моделей.
Все рекомендации, приведенные в настоящем приложении, являются только примерами. Для подтверждения соответствия требованиям возможно использование других моделей и методов.
Возможна замена некоторых вычислений измерениями и наоборот. Например, это может быть применено для вычисления "наихудших" токов и напряжений, действующих на имплантируемые проводники, и измерение устойчивости имплантата к сигналам, поступающим через его проводники-соединители.
Можно уменьшить сложность вычислений, исключив некоторые части изделия после проведения отдельных измерений, демонстрирующих достаточную помехоустойчивость этих частей. Например, можно пренебречь индуцированными сигналами между отделенными частями системы, если их соединение блокируется соответствующей фильтрацией.
Теоретическое моделирование должно содержать:
- численную модель человеческой головы, достаточно детализированную в области уха и ушной улитки, и численный метод вычисления напряженности полей и плотности индуцированного в голове тока (см. DD.2);
- расширенную численную модель имплантируемого изделия и численный метод для вычисления индуцированных токов в имплантируемых проводниках (см. DD.3);
- численную модель всех частей изделия и численный метод для вычисления индуцированных напряжений и токов во внутренних точках схемы при воздействии электромагнитных полей (см. DD.4). Должны быть учтены напряжения и токи, индуцированные между отдельными соединенными частями изделия;
- численное моделирование схемы демонстрации нарушения функционирования изделия расчетными индуцированными напряжениями и токами (включая токи в проводниках) (см. DD.5).
DD.2 Численная модель головы (без имплантата)
Существуют численные модели человеческой головы (см. [31]). Большинство других доступных моделей человеческой головы недостаточно подробны для детального представления уха и ушной улитки, поэтому следует принять меры для использования модели человеческой головы, оцифрованной должным образом. Модели и числовые методы воздействия первоначально были оптимизированы для вычисления SAR (удельный коэффициент поглощения равен нагреву ткани). Для расчета плотности индуцированного тока в тканях головы данные модели должны быть расширены. Доступен базовый опыт работы с такими расширениями (токи в тканях) (см. DD.3).
DD.3 Расчет токов в проводниках
Численные модели головы и численные методы расчета воздействия электромагнитных полей оптимизированы для непрерывной диэлектрической ткани. Для представления имплантируемых металлических проводящих структур и вычисления напряжений и токов, индуцированных в имплантируемых проводах, данные модели должны быть расширены. Базовый опыт работы с такими расширениями доступен из опубликованных исследований имплантируемых электрокардиостимуляторов [28].
DD.4 Расчет индуцированных токов и напряжений как внутри имплантируемых, так и внутри неимплантируемых частей и между соединенными частями
Подпункт DD.3 позволяет определить "внешние" сигналы помех, поступающие через проводники. Напряженность поля, воздействующего на НЕИМПЛАНТИРУЕМЫЕ ЧАСТИ, указана в таблицах 102 и 103. Подпункт DD.2 позволяет определить результирующее воздействие напряженности поля на части, имплантируемые в ткани головы. Для определения "внутренних" индуцированных помех может быть использовано численное моделирование.
DD.5 Описание подавления сигналов помех
После выполнения действий по DD.2-DD.4 все помехи будут известны. Инструменты моделирования схемы или анализа конструкции будут необходимы для определения нарушения функционирования.
Приложение ЕЕ
(справочное)
Примечания к измерениям электромагнитных помех для определения соответствия разделу 27
ЕЕ.1 Общие положения
В ИСО/ТК 150/SC6 определены и описаны следующие преимущества и недостатки прямого измерения электромагнитных помех:
a) Преимущества:
- используется реальное воздействие, поэтому будут выявлены все эффекты;
- не требуется сложное разделение внутренних и внешних воздействий на имплантат;
- может использоваться одно и то же измерительное оборудование, даже в случае замены изделия или изменения его формы;
- для проведения измерений не требуется детального знания внутреннего устройства имплантата.
b) Недостатки:
- необходима высокая напряженность поля, намного выше чем для обычного испытания электромагнитной совместимости, и в связи с этим может потребоваться разработка специального оборудования;
- необходима сложная фильтрация выходных сигналов изделия.
Все рекомендации, приведенные в настоящем приложении, являются только примерами. Для подтверждения соответствия требованиям возможно использование другого оборудования и методов.
Испытание должно быть проведено на имплантате, размещенном в модели головы, которая аппроксимирует свойства тканей человека и обеспечивает образование токов, подобных индуцированным в тканях человека. В ЕЕ.2 приведен пример подходящей модели головы.
На частотах ниже 10 МГц модель головы должна быть подвергнута воздействию магнитного поля. Это поле может быть создано планарной катушкой или катушкой Гельмгольца. В ЕЕ.3 приведены сведения по проведению данных измерений.
На частотах выше 10 МГц модель головы должна быть подвергнута воздействию электромагнитного поля. Необходимое поле дальней зоны может быть получено в камере поперечной электромагнитной волны (ТЕМ) или в гигагерцовой камере поперечной электромагнитной волны (GTEM). В ЕЕ.4 приведены сведения по проведению данных измерений.
На частотах выше 450 МГц модель головы альтернативно может быть подвергнута воздействию электромагнитного поля в ближней зоне. В ЕЕ.5 приведены сведения по проведению данных измерений.
Некоторые общие сведения применяются ко всем измерениям:
Из-за акустического шума, который может присутствовать в испытательной среде, порты микрофона должны быть акустически заблокированы, например, каплей эпоксидной смолы. Для оценки помехоустойчивости на начальном этапе необходимо использовать типичные клинические настройки (включая коэффициент усиления микрофона и частотный диапазон на входе). Для гарантии выходной сигнал должен составлять 50% (уровень комфорта) и 25% (пороговое значение) от максимального выходного сигнала (только для 27.2).
Для измерения в целях проверки требований защиты (верхние уровни по 27.1) необходимо настроить систему на максимально возможный выходной сигнал. Для стабилизации изображения на экране осциллографа пороговые уровни и уровни комфорта устанавливают на максимальные значения, и используются только два электрода. Для увеличения отношения сигнал/шум выходной сигнал можно измерить в нерабочее время т между импульсами. Поскольку реакция изделия на помехи сохраняется и во время отключения помех, то будут обнаружены все искажения выходного сигнала. Для тестовых сигналов непрерывной волны (CW) используется фильтр высоких частот, при проведении испытаний тестовый сигнал не должен искажать выходной сигнал. Для подавления помех, возникающих в результате индуктивной связи имплантируемого изделия и процессора, должен использоваться низкочастотный фильтр с частотой среза 100 кГц, схожий с приведенным в 22.2.
ЕЕ.2 Модель головы
Модель головы разработана на основе ИСО 14117, в котором описывается платформа модели. Модель головы, показанная ниже, приведена только в качестве примера. Размеры модели: ширина 15 см, длина 15 см и высота 12 см, отражают размеры головы. Испытания следует проводить при комнатной температуре.
Модель должна быть заполнена физиологическим раствором на высоту 10 см, для обеспечения соответствующей проводимости согласно ИСО 14117.
Не допускается поворот модели головы. Имплантируемая система может быть повернута на 90°. Должны быть проверены две перпендикулярные ориентации изделия, например, с вектором поля Е, перпендикулярным к выходному проводнику имплантата, и с вектором поля Е, параллельным выходному проводнику имплантата (ориентация 2). Для испытаний поля дальней зоны на частотах выше 10 МГц при обеих ориентациях, показанных на рисунке ЕЕ.101, вектор поля E должен быть вертикальным, и направление распространения волны должно быть нормальным к поверхности процессора и датчика.
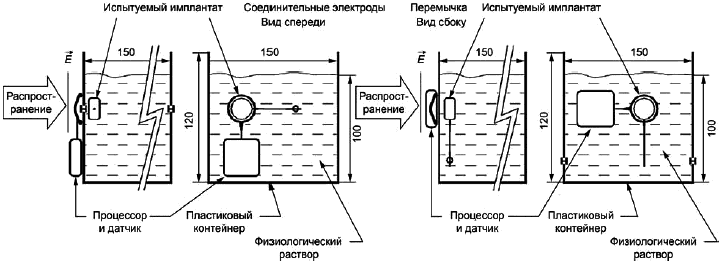
Примечание - Поле и вектор распространения показывают ситуацию в ТЕМ-камере.
Рисунок ЕЕ.101 - Модель головы для испытаний на электромагнитные помехи
Для испытания ближней зоны магнитного поля на частотах ниже 10 МГц с обеими ориентациями, показанными на рисунке ЕЕ.101, вектор поля Н должен быть горизонтальным и нормальным к вектору поля E и к направлению распространения. Дополнительная третья ориентация с вектором Н, нормальным к поверхности процессора и датчика, должна использоваться с одной из показанных ориентаций. Кроме того, должен быть определен и испытан худший случай воздействия.
Для дополнительных испытаний на частотах выше 450 МГц в поле ближней зоны испытательный диполь должен быть параллелен вектору Е, как показано на рисунке ЕЕ.101.
ЕЕ.3 Воздействие на изделие магнитного поля на частотах ниже 10 МГц
Во время испытания изделие включается и помещается в модель головы (ЕЕ.2). Модель головы помещается в катушку (планарную, Гельмгольца или аналогичную катушку диаметром по крайней мере 30 см) таким образом, чтобы имплантируемая система находилась в центре катушки. Испытание выполняется на дискретных частотах с амплитудными значениями напряженности поля в соответствии с таблицей 101. Испытание на каждой частоте должно проводиться в течение 10 с, не менее. Каждое испытание должно быть повторено для трех ортогональных ориентаций вектора магнитного поля относительно выходного проводника имплантата. В качестве альтернативы должны быть определены и проверены худшие случаи воздействия. Для достижения требуемой напряженности магнитного поля с необходимым током питания или выходной мощностью усилителя может потребоваться использование отдельных катушек с различным числом обмоток. На частотах выше 165 кГц для предотвращения резонанса катушки должна использоваться одна обмотка из очень толстой проволоки или из медной трубки диаметром около 1 см. Для уменьшения тока питания можно настроить катушки с параллельными конденсаторами на всех частотах. В связи с этим напряжение на катушке должно быть настроено в соответствии с напряженностью поля, а не тока питания.
Примечание - "Пиковая напряженность магнитного поля" описывает максимальную амплитуду вектора магнитного поля. На частотах выше 1 кГц при питании катушки генератор не должен обеспечивать постоянную выходную мощность. Например, на 5 МГц во время импульса достаточно мощности 1/200 от пикового значения (коэффициент амплитуды 23 дБ).
Калибровка катушки. На всех испытательных частотах отношение напряжения питания катушки к напряженности магнитного поля в центре катушки должно быть определено при отсутствии макета головы. Калибровка должна быть выполнена с использованием магнитного датчика диаметром менее 10 см и точностью не хуже ±10% и вольтметра с точностью не хуже ±5%. Испытания изделия, помещенного в модель головы, должны быть выполнены с применением калиброванного напряжения на катушке. Отличительной чертой этого вида калибровки от обычно используемого является измерение тока питания катушки. Калибровка по напряжению на катушке имитирует определение напряженности поля в отсутствие макета головы и испытуемого изделия. Калибровка по току будет моделировать напряженность поля в присутствии модели головы и испытуемого изделия. Принято считать, что поле одной планарной катушки в области модели головы не является однородным и что в модели головы поле немного смещено.
ЕЕ.4 Воздействие электромагнитного поля дальней зоны на частотах от 10 до 2450 МГц
Во время испытания изделие включается и помещается в модель головы (см. СС.2). Модель головы помещается в модуль ТЕМ (для частот ниже 1 ГГц) или в модуль GTEM с измерительной областью в диаметре 30 см, не менее. Испытание выполняется на дискретных частотах с амплитудными значениями напряженности поля в соответствии с таблицей 102. Направление распространения волны должно быть нормальным к поверхности процессора и датчика.
Калибровка. На всех испытательных частотах отношение напряжения питания камеры ТЕМ или GTEM к напряженности поля в центре измерительной области должно быть определено при отсутствии макета головы и испытуемого изделия. Калибровка должна быть выполнена с точностью, не хуже +30% и -25%. Испытания изделия, помещенного в модель головы, должны быть выполнены с применением калибровки напряжения или мощности питания камеры ТЕМ или GTEM. Принято считать, что модель головы и испытуемое изделие немного искажают поле.
В случае применения камеры ТЕМ или GTEM с расстоянием по высоте между пластинами, например, 50 см, амплитуда максимального напряжения составляет 100 В, что соответствует 71 В среднеквадратичного значения для верхнего уровня. Мощность генератора временного импульса камеры ТЕМ или GTEM с полным сопротивлением 50 Ом составила бы 100 Вт среднеквадратичного значения для верхнего уровня. Но обычно для испытательного сигнала на верхнем испытательном уровне среднеквадратичное значение мощности, интегрированное в течение рабочего цикла (время воздействия плюс время отсутствия сигнала), приблизительно в 25 раз ниже. Поэтому достаточно усилителя со среднеквадратичным значением средней мощности 4 Вт и среднеквадратичным значением пиковой мощности 100 Вт (коэффициент амплитуды 14 дБ).
Примечание - "Пиковая напряженность электрического поля" описывает максимальную амплитуду вектора электрического поля, а "пик мощности во время импульса" описывает среднеквадратичное значение мощности в течение короткого времени импульса.
ЕЕ.5 Воздействие электромагнитного поля ближней зоны на частотах выше 450 МГц
Настоящее испытание является альтернативой испытанию, описанному в ЕЕ.4, которое позволяет избежать большой напряженности поля в камерах ТЕМ или GTEM на частотах выше 450 МГц. Оно проще для выполнения, чем испытание СС.4, но с другой стороны камеры ТЕМ или GTEM по прежнему будут необходимы для испытаний на частотах ниже 450 МГц. Это испытание в поле ближней зоны особенно хорошо моделирует помехи от мобильных телефонов, которые представляются основными источниками помех в этом диапазоне частот.
Во время испытания изделие включается и помещается в модель головы (см. ЕЕ.2). Для каждой испытательной частоты должен быть использован отдельный испытательный диполь, согласно ИСО 14117. Испытание выполняется на дискретных частотах с пиковой мощностью диполя согласно указанной в таблице ЕЕ.101, приведенной ниже. Каждая испытательная частота должна быть применена для двух ортогональных ориентаций диполя относительно выходного проводника имплантата. Существует два варианта: либо используют обе типичные ориентации, приведенные на рисунке ЕЕ.101, с диполем, ориентированным вертикально, или используют только одну типичную ориентацию и поворачивают испытательный диполь в горизонтальную ориентацию. В любом случае расстояние от центра диполя до поверхности пластикового контейнера, в котором размещены процессор и датчик, должно составить 1 см.
Таблица ЕЕ.101 - Пиковое значение мощности диполя
Частота, МГц | Пиковое значение мощности диполя | |||
Нижние | Время импульса, мс | Максимальные | Время импульса, мс | |
900 | 40 | 10 | 2 | 1,4 |
1800 | 40 | 10 | 2 | 1,4 |
2450 | 40 | 10 | 2 | 1,4 |
| ||||
Погрешность мощности диполя для любой тестовой частоты должна быть не более ±15%.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным и действующим в этом качестве межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального, межгосударственного стандарта |
ISO 10993-1 | IDT | ГОСТ ISO 10993-1-2011 "Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования" |
ISO 11607-1 | - | * |
ISO 14155 | IDT | ГОСТ Р ИСО 14155-2014 "Клинические исследования. Надлежащая клиническая практика" |
ISO 14971 | IDT | ГОСТ ISO 14971-2011 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
ISO 14117 | - | * |
ISO 14708-1 | IDT | ГОСТ Р ИСО 14708-1-2012 "Имплантаты хирургические. Активные имплантируемые медицинские изделия. Часть 1. Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем" |
IEC 60068-2-27 | - | * |
IEC 60068-2-31 | - | * |
IEC 60068-2-47 | - | * |
IEC 60068-2-64 | IDT | ГОСТ 30630.1.9-2002 (МЭК 60068-2-64:1993) "Методы испытаний на стойкость к механическим внешним воздействующим факторам машин, приборов и других технических изделий. Испытания на воздействие случайной широкополосной вибрации с использованием цифровой системы управления испытаниями" |
IEC 60068-2-75:1997 | - | * |
IEC 60118-6 | - | * |
IEC 60601-1:2006 | IDT | ГОСТ Р МЭК 60601-1-6-2014 "Изделия медицинские электрические. Часть 1-6. Общие требования безопасности с учетом основных функциональных характеристик. Дополнительный стандарт. Эксплуатационная пригодность" |
IEC 60601-1-2 | IDT | ГОСТ Р МЭК 60601-1-2-2014 "Изделия медицинские электрические. Часть 1-2. Общие требования безопасности с учетом основных функциональных характеристик. Параллельный стандарт. Электромагнитная совместимость. Требования и испытания" |
IEC 61000-4-2 | - | * |
IEC 62304 | IDT | ГОСТ Р МЭК 62304-2013 "Изделия медицинские. Программное обеспечение. Процессы жизненного цикла" |
EN 1593 | - | * |
EN 13185 | - | * |
EN 50061:1998 | - | * |
EN 60068-2-64:2008 | - | * |
EN 45502-2-1 | - | * |
* Соответствующий национальный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного международного стандарта. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - IDT - идентичные стандарты. | ||
Библиография
[1] | ISO 5841-2, Implants for surgery - Cardiac pacemakers - Part 2: Reporting of clinical performance of populations of pulse generators or leads |
[2] | ISO 11137-1, Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices |
[3] | ISO 13485, Medical devices - Quality management systems - Requirements for regulatory purposes |
[4] | ISO 14117, Active implantable medical devices - Electromagnetic compatibility - EMC test protocols for implantable cardiac pacemakers and implantable cardioverter defibrillators |
[5] | ISO 14708-2, Implantsforsurgery - Activeimplantable medical devices - Part 2: Cardiac pacemakers |
[6] | ISO 17665-1, Sterilization of health care products - Moist heat - Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices |
[7] | IEC 60068-2-14, Environmental testing - Part 2-14: Tests - Test N: Change of temperature |
[8] | IEC 60068-2-17, Basic environmental testing procedures - Part 2-17: Tests - Test Q: Sealing |
[9] | EN 1041, Information supplied by the manufacturer of medical devices |
[10] | ANSI/IEEE C95.1, IEEE Standard for Safety Levels with Respect to Human Exposure to Radio Frequency Electromagnetic Fields, 3 kHz to 300 GHz |
[11] | ASTM F2052, Standard Test Method for Measurement of Magnetically Induced Displacement Force on Passive Implants in the Magnetic Resonance Environment |
[12] | ASTM F2213, Standard Test Method for Measurement of Magnetically Induced Torque on Medical Devices in the Magnetic Resonance Environment |
[13] | ASTM F2182, Standard Test Method for Measurement of Radio Frequency Induced Heating Near Passive Implants During Magnetic Resonance Imaging |
[14] | Baumann R., Lesinski Schiedat A., Goldring J.E., Gnadeberg D., Rittmann K.L., Battmer R.D. et al. The influence of ionizing radiation on the CLARION 1.2 cochlear implant during radiation therapy. Am. J. Otol. 1999, 20 (1) pp.50-52 |
[15] | Cochlea-lmplantate unter Stossbelastung - Auswertung von Unfallszenarien, Ermittlung von Beanspruchungsgrenzen und Entwicklung eines standardisierten |
[16] | Council Directive 65/65/EEC of 26 January 1965 on the approximation of provisions laid down by Law, Regulation or Administrative Action relating to proprietary medicinal products, OJ 22, 9.2.1965, pp.369-373 |
[17] | Council Directive 75/318/EEC of 20 May 1975 on the approximation of the laws of Member States relating to analytical, pharmaco-toxicological and clinical standards and protocols in respect of the testing of proprietary medicinal products, OJ L 147, 9.6.1975, pp.1-12 |
[18] | Council Directive 80/836/Euratom of 15 July 1980 amending the Directives laying down the basic safety standards for the health protection of the general public and workers against the dangers of ionizing radiation, OJ L 246, 17.9.1980, pp.1-72 |
[19] | Council Directive 84/466/Euratom of 3 September 1984 laying down basic measures for the radiation protection of persons undergoing medical examination or treatment, OJ L 265, 5.10.1984, pp.1-3 |
[20] | Council Directive 84/467/Euratom of 3 September 1984 amending Directive 80/836/Euratom as regards the basic safety standards for the health protection of the general public and workers against the dangers of ionizing radiation, OJ L 265, 5.10.1984, pp.4-156 |
[21] | Council Directive 89/341/EEC of 3 May 1989 amending Directives 65/65/EEC, 75/318/EEC and 75/319/EEC on the approximation of provisions laid down by law, regulation or administrative action relating to proprietary medicinal products, OJ L 142, 25.5.1989, pp.11-13 |
[22] | Council Recommendation 1999/519/EC of 12 July 1999 on the limitation of the exposure of the general public to electromagnetic fields (0 Hz to 300 GHz), OJ L 199, 30.07.1999, pp.59-79 |
[23] | Dahm M, Shepherd RK, Clark GM, Archives of Otolaryngology Stockholm, 1993 |
[24] | Gubbels S.P, & McMenomey S.O. Safetystudyofthe Cochlear Nucleus 24 device with internal magnet in the 1.5 Tesla Magnetic Resonance Imaging Scanner. Laryngoscope. 2006, 116 (June), pp.865-871 |
[25] | Hochmair E.S. Invited editorial: MRI safety of Med-EI C40/C40+ cochlear implants. Cochlear Implants Int. 2001, 2 (2), pp.98-114 |
[26] | ISBN 0 11 321543 6, British Pharmacopoeia 1993 Volume II |
[27] | European Pharmacopoeia. Fourth Edition, 2002 |
[28] | Landstorfer F.M., Geisbusch L., Jakobus U., Maier M., |
[29] | MIL-STD-883, Test method standard - Microcircuits |
[30] | Ralston A., Stevens G., Mahomudally E., Ibrahim I., Leckie E. Cochlear implants: response to therapeutic irradiation. Int. J. Radial Oncol. Biol. Phys. 1999, 44 (1), pp.227-231 |
[31] | Schutz von Personen mit Cochleaimplantaten in elektromagnetischen Feldern, Final report of project 37/02, German Federal Ministry of Economics and Work (BMWA), Dec. 2004 |
[32] | EN 45502-1, Active implantable medical devices - Part 1: General requirements for safety, marking and information to be provided by the manufacturer |
[33] | EN 45502-2-3, Particular requirements for cochlear and auditory brainstem implant systems |
[34] | CISPR 11 Industrial, scientific and medical equipment - Radio-frequency disturbance characteristics - Limits and methods of measurement |
УДК 616.28.76:006.354 | ОКС 11.040.40 | Р29 | ОКП 944480 |
| |||
Электронный текст документа
и сверен по:
, 2016

{kind=link}