ГОСТ Р 54882-2011
(GHTF/SG4/N30R20:2008)
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
РУКОВОДСТВО ПО АУДИТУ СИСТЕМ МЕНЕДЖМЕНТА КАЧЕСТВА ИЗГОТОВИТЕЛЕЙ МЕДИЦИНСКИХ ИЗДЕЛИЙ НА СООТВЕТСТВИЕ РЕГУЛИРУЮЩИМ ТРЕБОВАНИЯМ
Часть 2
Стратегия аудита
Guidelines for regulatory auditing of quality management systems of medical device manufacturers. Part 2. Regulatory auditing strategy
ОКС 11.040.01
ОКП 94 000
Дата введения 2013-01-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "МЕДИТЕСТ" на основе собственного аутентичного перевода на русский язык документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 13 декабря 2011 г. N 1189-ст
4 Настоящий стандарт является модифицированным по отношению к документу Целевой группы по глобальной гармонизации (Global Harmonization Task Force - GHTF) "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 2. Стратегия аудита"* (GHTF/SG4/N30R20:2008 "Guidelines for regulatory auditing of quality management systems of medical device manufacturers - Part 2: Regulatory auditing strategy") путем изменения его структуры, а также путем изменения содержания отдельных структурных элементов (терминологических статей 3.1; 3.3; 3.7), которые выделены вертикальной линией, расположенной слева от текста. При этом исключен раздел 4 "Обоснование" примененного оригинального документа, который нецелесообразно применять в национальном стандарте в связи с требованиями к структуре национального стандарта Российской Федерации, установленными в ГОСТ Р 1.0-2004, а также в связи с тем, что содержание данного раздела дублируется содержанием введения и раздела 1 "Область применения". Также изменено обозначение приложений настоящего стандарта в связи с требованиями, установленными в ГОСТ Р 1.0-2004 (в оригинальном документе приложения обозначены арабскими цифрами). Сравнение структуры настоящего стандарта со структурой указанного оригинального документа приведено в дополнительном приложении ДА.
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Оригинальный текст аутентичного перевода измененных структурных элементов примененного документа GHTF и объяснение причин внесения технических отклонений приведены в дополнительном приложении ДБ.
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных (региональных) стандартов соответствующие им национальные стандарты Российской Федерации, сведения о которых приведены в дополнительном приложении ДВ.
В связи с выпуском в 2010 г., в период публичного обсуждения окончательной редакции настоящего стандарта, новой редакции примененного оригинального документа GHTF, отличающейся наличием приложения Е "Аудит программного обеспечения", настоящее приложение добавлено к тексту настоящего стандарта и выделено двойной вертикальной линией, расположенной справа от текста
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
Введение
Документ "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 2. Стратегия аудита" разработан Целевой группой по глобальной гармонизации (GHTF) - добровольным сообществом, состоящим из представителей регулирующих органов медицинской промышленности.
Настоящий стандарт является руководством для регулирующих органов и организаций, проводящих аудиты систем менеджмента качества изготовителей медицинских изделий на основе процессного подхода к требованиям системы менеджмента качества.
Примечание - В контексте настоящего стандарта "аудит" означает "аудит на соответствие регулирующим требованиям".
Применение настоящего стандарта обеспечивает следующие преимущества регулирующим органам и организациям, проводящим аудиты:
- улучшение качества проведения аудитов, ведущее к улучшению систем менеджмента качества и улучшению качества продукции;
- достижение большей согласованности при проведении аудита как между аудиторами внутри одной организации, проводящей аудит, так и между аудиторами разных организаций, проводящих аудит;
- достижение более продуктивного сотрудничества между регулирующими органами в отношении аудитов;
- увеличение доверия к аудитам, осуществляемым организацией, проводящей аудиты, и признание результатов данных аудитов другими регулирующими органами;
- более эффективное использование ресурсов организации, проводящей аудиты;
- предоставление руководящих указаний для стран, намеревающихся разработать стратегию проведения аудитов систем менеджмента качества на соответствие регулирующим требованиям.
Применение настоящего стандарта обеспечивает следующие преимущества изготовителям медицинских изделий:
- улучшение качества проведения аудитов, ведущее к улучшению систем менеджмента качества и улучшению качества продукции;
- большую согласованность в процедурах проведения аудитов и в осуществлении обратной связи с изготовителями в отношении их систем менеджмента качества, что экономит ресурсы, облегчая подготовку к аудитам;
- сокращение числа аудитов, проводимых разными регулирующими органами у одного изготовителя;
- увеличение доверия к аудитам, осуществляемым организациями, проводящими аудиты, и признание результатов данных аудитов разными регулирующими органами.
К лицам, получающим выгоду, можно также отнести пациентов и пользователей медицинских изделий, испытывающих большую степень доверия к безопасности и клинической эффективности медицинских изделий, размещенных на рынке.
1 Область применения
Настоящий стандарт предназначен для регулирующих органов и организаций, проводящих аудиты систем менеджмента качества изготовителей медицинских изделий на основе процессного подхода к требованиям к системам менеджмента качества (см. ГОСТ Р ИСО 13485). Если деятельность организаций, проводящих аудиты, регламентируется регулирующими требованиями или требованиями к аккредитации, то стратегию аудита, представленную в настоящем стандарте, рекомендуется рассматривать как дополнение к регулирующим требованиям или требованиям к аккредитации, если целесообразно. Несмотря на то что аудит изготовителя медицинских изделий может включать в себя аудит на соответствие регулирующим требованиям, не связанным непосредственно с менеджментом качества, область применения настоящего стандарта ограничена требованиями к системам менеджмента качества. При наличии дополнительных регулирующих требований, являющихся частью области аудита, аудитор должен рассмотреть, идентифицировать и документировать данные требования, интегрировав их в цели и критерии аудита.
Настоящий стандарт применим к первоначальным и подтверждающим аудитам, но может также применяться к другим видам аудитов, описанным в ГОСТ Р 54421, включая все дополнения, разработанные GHTF SG4 в качестве руководства для организаций, проводящих аудиты. Цели других аудитов определяются элементами подсистем менеджмента качества, выбранными для проведения аудита.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ Р 40.003-2008 Система сертификации ГОСТ Р. Регистр систем качества. Порядок сертификации систем менеджмента качества на соответствие ГОСТ Р ИСО 9001-2008 (ИСО 9001:2008)
ГОСТ Р 53918-2010 Изделия медицинские. Руководство по интеграции принципов менеджмента риска в систему менеджмента качества (GHTF/SG3/N15R8:2005, IDT)
ГОСТ Р 54421-2011 Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 1. Общие требования (GHTF/SG4/N28R4.2008, MOD)
ГОСТ Р ИСО 9000-2008 Системы менеджмента качества. Основные положения и словарь (ИСО 9000:2008, IDT)
ГОСТ Р ИСО 13485-2004 Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования (ИСО 13485:2003, IDT)
ГОСТ Р ИСО/ТО 14969-2007 Изделия медицинские. Системы менеджмента качества. Руководство по применению ИСО 13485:2003 (ИСО/ТО 14969:2004, IDT)
ГОСТ Р ИСО 14971-2010* Изделия медицинские. Применение менеджмента риска к медицинским изделиям (ИСО 14971:2007, IDT)
________________
* Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 14971-2009, здесь и далее по тексту. - .
ГОСТ Р ИСО/МЭК 17021-2008 Оценка соответствия. Требования к органам, проводящим аудит и сертификацию систем менеджмента (ИСО/МЭК 17021:2006, IDT)
ГОСТ Р ИСО 19011-2003 Руководящие указания по аудиту систем менеджмента качества и/или систем экологического менеджмента (ИСО 19011:2002, IDT)
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодно издаваемому информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по соответствующим ежемесячно издаваемым информационным указателям, опубликованным в текущем году. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 аудит (проверка) (audit): Систематический, независимый и документированный процесс получения свидетельств аудита и объективного их оценивания с целью установления степени выполнения согласованных критериев аудита (заимствовано из ГОСТ Р ИСО 9000; пункт 3.9.1). |
3.2 аудит на соответствие регулирующим требованиям (regulatory audit): Аудит системы менеджмента качества с целью демонстрации ее соответствия регулирующим требованиям.
Примечание - В настоящем стандарте "аудит" означает "аудит на соответствие регулирующим требованиям".
3.3 критерии аудита (audit criteria): Совокупность политики, процедур или требований, которые применяют в виде ссылок (заимствовано из ГОСТ Р ИСО 9000; пункт 3.9.3). |
3.4 свидетельство аудита (audit evidence): Записи, изложение фактов или другая информация, которые связаны с критериями аудита и могут быть проверены (заимствовано из ГОСТ Р ИСО 9000, пункт 3.9.4).
Примечание - Свидетельство аудита может быть качественным и/или количественным и используется для доказательства наблюдений аудита.
3.5 организация, проводящая аудиты (auditing organization): Орган, предназначенный для проведения аудитов на базе конкретных регулирующих документов и в соответствии с поставленными задачами.
3.6 устанавливать (establish): Определять (учреждать), документировать (в письменной или электронной форме) и внедрять.
Примечание - Настоящее определение отличается от используемого в ГОСТ Р ИСО 13485 ("определять, разрабатывать"), где не приведено официальное определение данного термина и его применение носит менее директивный характер.
3.7 медицинское изделие (medical device): Определение данного термина приведено в соответствующей нормативной документации. |
3.8 процесс (process): Совокупность взаимосвязанных или взаимодействующих видов деятельности, преобразующая входы в выходы (заимствовано из ГОСТ Р ИСО 9000, пункт 3.4.1).
3.9 остаточный риск (residual risk): Риск, остающийся после выполнения мер по управлению риском (заимствовано из ГОСТ Р ИСО 14971, пункт 2.15).
3.10 менеджмент риска (risk management): Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания, управления и мониторинга рисков (заимствовано из ГОСТ Р ИСО 14971, пункт 2.22).
3.11 документация на продукцию (product documentation): Документы, представляющие собой выходные данные процесса проектирования и разработки конкретного изделия независимо от того, включен ли данный процесс в область применения системы менеджмента качества организации.
Примечание - В различных юрисдикциях используются разные термины и определения.
4 Общие замечания по стратегии аудита на соответствие регулирующим требованиям
При проведении аудита изготовителя медицинских изделий необходимо оценить его систему менеджмента качества на соответствие регулирующим требованиям к системам менеджмента качества и процедурам, установленным изготовителем. Система менеджмента качества может быть основана на применимых стандартах в отношении систем менеджмента качества (см. ГОСТ Р ИСО 13485) или на других регулирующих документах.
При проведении аудита рекомендуется использовать процессный подход, при этом желательно следовать блок-схеме процессов изготовителя медицинских изделий.
Проведение аудита должно быть основано на менеджменте риска и сфокусировано на основных процессах системы менеджмента качества, необходимых для изготовления медицинских изделий, охватываемых областью аудита. Аудитору следует сосредоточить свое внимание на факторах, в наибольшей степени воздействующих на безопасность медицинских изделий, гарантируя в то же время надлежащую полноту охвата всех классов медицинских изделий, подпадающих под область аудита.
4.1 Цели
Аудит рекомендуется планировать и проводить так, чтобы достичь следующих целей:
- оценивания результативности системы менеджмента качества изготовителя, включая систематическое и результативное оценивание выполнения регулирующих требований в разумно необходимый период времени;
- согласованности результатов аудита независимо от того, какая организация или какие индивидуальные аудиторы проводят аудит. Конечной целью являются гармонизация и взаимное признание результатов аудита;
- определения с помощью аудита того, как идентифицируются и решаются проблемы, связанные с медицинским изделием или системой менеджмента качества;
- прозрачности аудита для проверяемой организации.
4.2 Проверяемая система менеджмента качества
При проведении аудита рекомендуется сосредоточить внимание не столько на выполнении конкретных требований, сколько на результативности системы менеджмента качества изготовителя в целом. Для того чтобы разбить аудит на более управляемые части, необходимо идентифицировать подсистемы, приведенные в таблице 1.
Таблица 1 - Подсистемы или виды деятельности и связанные с ними разделы
Подсистемы | Связь с разделами ГОСТ Р ИСО 13485 |
1 Высшее руководство | 4, 5, 6, 7, 8 |
2 Проектирование и разработка | 7 |
3 Документация на продукцию | 4, 7 |
4 Управление продукцией и процессами (включая стерилизацию, где применимо) | 4, 6, 7, 8 |
5 Корректирующие и предупреждающие действия | 4, 5, 6, 7, 8 |
6 Управление закупками | 7 |
7 Документация и записи | 4 |
8 Процессы, связанные с потребителем | 7 |
Более подробные ссылки на разделы и подразделы ГОСТ Р ИСО 13485 приведены в разделе 5 настоящего стандарта. Основные подсистемы идентифицированы в таблице 1 под номерами 1-5. Данным подсистемам рекомендуется уделять первостепенное внимание в процессе аудита. В каких-то конкретных ситуациях может понадобиться рассмотреть в качестве основных подсистем другие подсистемы. Например, управление закупками рекомендуется рассматривать как основную подсистему в следующих случаях:
- если изготовитель закупает готовые медицинские изделия;
- если изготовитель передает стороннему исполнителю выполнение таких основных процессов или услуг, как проектирование и разработка, процессы жизненного цикла продукции, стерилизация и др.;
- если изготовитель закупает основные компоненты и узлы.
4.3 Подходы к аудиту
Существуют разные подходы к проведению аудита, например, "сверху вниз", "снизу вверх", комбинированный подход и аудит конкретного изделия.
Надлежащий подход следует выбрать в зависимости от цели и причины проведения аудита. Если в процессе аудита не рассматриваются какие-либо инциденты, то предпочтительнее выбрать подход "сверху вниз". Такой подход обычно практикуют при проведении первоначального аудита. Если аудит включает в себя рассмотрение возможной существенной проблемы безопасности, то обычно практикуют подход "снизу вверх". Для проведения подтверждающего аудита можно использовать комбинированный подход. Аудит конкретного изделия позволяет оценить взаимодействие между подсистемами.
Подход "сверху вниз", используемый для проведения аудита, начинается с оценивания структуры системы менеджмента качества изготовителя и ее подсистем: высшего руководства, проектирования и разработки, документации на продукцию, управления продукцией и процессами, корректирующих и предупреждающих действий. Выбранные подсистемы анализируются для того чтобы установить, как изготовитель выполняет основные требования посредством определения, документирования и внедрения соответствующих процедур. Важно отслеживать применение процессного подхода как к системе менеджмента качества в целом, так и к каждой ее подсистеме, например, используя цикл PDCA (планирование - осуществление - проверка - действие) (см. 4.4). Применяя подход "сверху вниз" аудитор должен подтвердить, что изготовитель разработал надлежащие процедуры и политику. Для этого аудитор должен проанализировать свидетельства аудита, включая записи, в целях верификации результативности внедрения изготовителем разработанных политики и процедур и соответствия системы менеджмента качества регулирующим требованиям.
Описанный подход является единым для всех сторон, участвующих в систематическом и прозрачном процессе аудита: регулирующих органов, организаций, проводящих аудиты, и изготовителей. Однако данный подход не позволяет сфокусировать внимание на конкретном изделии.
Подход "снизу вверх", используемый для проведения аудита, может иметь в качестве отправной точки проблему в области качества, например сообщение о неблагоприятном событии, произошедшем с медицинским изделием, или о несоответствующей продукции. Таким образом, аудитор начинает работать "снизу" и проходит весь путь "вверх" через систему менеджмента качества изготовителя вплоть до ответственности высшего руководства. Данный подход позволяет быстро получить представление о результативности выбранных для аудита подсистем и процессов, на функционирование которых повлияла какая-то конкретная проблема в области качества, и о причине(ах) возникновения проблемы в области качества. При использовании данного подхода труднее определить результативность системы менеджмента качества в целом.
Третий подход представляет собой комбинацию первых двух. Аудитор начинает с анализа верхнего слоя системы менеджмента качества (сверху вниз), затем проводит аудит некоторых элементов системы (например, процесса производства) и в итоге верифицирует соответствие проведенных процедур (снизу вверх). Применение комбинированного подхода часто более эффективно, чем подходов "сверху вниз" или "снизу вверх" по отдельности. Данный подход также часто является более гибким при исследовании конкретных проблем, связанных с оцениванием результативности системы менеджмента качества изготовителя.
При аудите конкретного изделия аудитор выбирает одно медицинское изделие, партию или серию и изучает историю данного образца (данной выборки) на примере разных процессов системы менеджмента качества (планирования, проектирования и разработки, закупок, производства, упаковывания, распределения и т.д.). Это необходимо делать либо начиная со стадии планирования и продвигаясь вперед, либо продвигаясь назад от стадии распределения. Кроме того, выбирая образец с известной проблемой, аудитор может также включить в аудиторское заключение подсистему корректирующих и предупреждающих действий.
4.4 Процессный подход к аудиту
Результативная система менеджмента качества является механизмом управления, способным предупреждать и выявлять отклонения и идентифицировать причины этих отклонений. Далее результативной системе менеджмента качества следует обеспечить идентификацию, осуществление и результативность корректирующих и предупреждающих действий. Аудитору рекомендуется оценить, структурированы ли применяемые подсистемы и процессы системы менеджмента качества как саморегулируемые, управляемые процессы и являются ли они результативными. ГОСТ Р ИСО 13485 помогает сформулировать следующие основополагающие вопросы, ответы на которые могут быть получены в процессе аудита.
4.4.1 Планирование
Разработал ли изготовитель в соответствии с регулирующими требованиями цели и процессы, необходимые для достижения его системой менеджмента качества требуемых результатов?
4.4.2 Осуществление
Руководствуется ли изготовитель в своей деятельности системой менеджмента качества?
4.4.3 Проверка
Регулярно ли оценивает изготовитель процессы системы менеджмента качества и результаты измерений в соответствии с разработанными целями и регулирующими требованиями? Оценивает ли изготовитель результативность системы менеджмента качества через запланированные периоды времени посредством проведения внутренних аудитов, анализа со стороны руководства и т.д.?
4.4.4 Действие
Осуществляет ли изготовитель результативные корректирующие и предупреждающие действия в целях обеспечения высокого качества медицинских изделий и их соответствия применимым законодательным и регулирующим документам?
4.5 Выборка
Аудиторы могут осуществлять выборку, основываясь на факторах, с наибольшей вероятностью воздействующих на безопасность применения медицинского изделия. При планировании аудитов систем менеджмента качества (см. также 4.6) аудиторам необходимо рассматривать многие факторы (например, область аудита, классификацию медицинского изделия, сложность медицинского изделия, предусмотренное применение, применимые регулирующие требования, результаты предыдущих аудитов и т.д.). Может возникнуть необходимость в проведении выборки внутри каждой подсистемы, для того чтобы оценить результативность внедрения данной подсистемы (и связанных с ней подсистем). Для определения соответствующих размеров статистической выборки можно использовать таблицы А.1 и/или А.2, приведенные в приложении А.
4.6 Планирование аудита
В дополнение к требованиям, приведенным в ГОСТ Р 54421, рекомендуется рассмотреть следующие факторы:
- информацию, полученную от изготовителя;
- продолжительность аудита, его периодичность и примерный расчет времени проведения аудита на месте.
Остальные факторы, которые необходимо рассмотреть, приведены в разделе 5 настоящего стандарта.
4.6.1 Информация, которую необходимо получить от изготовителя
На стадии планирования рекомендуется запросить следующую информацию от изготовителя, для того чтобы определить продолжительность аудита и разработать план аудита в соответствии с ГОСТ Р 54421, пункт 7.1.2:
- официальное наименование организации-изготовителя и все ее прочие наименования, ее адрес, а также структуру организации;
- наименование представителя, контактный телефон, факс и адрес электронной почты;
- общее число сотрудников (во всех сменах), попадающих под область аудита;
- номенклатуру и класс безопасности изготавливаемых медицинских изделий (класс безопасности медицинского изделия может отличаться у разных регулирующих органов);
- виды проданных и/или планируемых к продаже в страны и/или регионы медицинских изделий, регулирующие требования которых должны быть оценены, включая полный список авторизации (например, лицензий), выданных на рассматриваемые медицинские изделия (где применимо);
- местоположение и функции каждой производственной площадки, включенной в аудит;
- список видов деятельности, осуществляемых на каждой площадке;
- специальные производственные процессы, например разработка программного обеспечения, стерилизация и т.д.;
- перечень материалов и услуг, поставляемых наиболее значимыми поставщиками, и их местонахождение, а также способ управления процессами, переданными сторонней организации;
- все имеющиеся результаты аудитов, проведенных другими организациями, при наличии соответствующего разрешения;
- возможность монтажа или обслуживания медицинских изделий;
- сведения обо всех изменениях, произошедших со времени проведения последнего аудита, если таковые имели место.
4.6.2 Определение продолжительности аудита, его периодичности и примерный расчет времени проведения аудита на месте
4.6.2.1 Периодичность аудита
Периодичность аудита зависит от факторов, приведенных в ГОСТ Р 54421, подраздел 7.2, а также от регулирующих требований и предыстории изготовителя.
4.6.2.2 Продолжительность аудита
Продолжительность аудита оказывает существенное воздействие на регулирующие органы и промышленные структуры в зависимости от используемых ресурсов, а также от полноты охвата аудитом и тщательности его проведения.
Продолжительность аудита зависит от таких факторов, как область аудита, цели и конкретные регулирующие требования, соответствие которым необходимо оценить, а также от номенклатуры, класса безопасности, сложности медицинских изделий, размеров и сложности структуры организации-изготовителя. Если специально не оговорено, то перечисленные факторы относятся к первоначальным и подтверждающим аудитам.
4.6.2.3 Взаимосвязь между периодичностью и продолжительностью аудита
Продолжительность аудита зависит от его периодичности. Например, базовым уровнем является ежегодно проводимый аудит (см. ГОСТ Р ИСО/МЭК 17021).
4.6.2.4 Метод определения продолжительности аудита
Если организация, проводящая аудиты, планирует проведение аудита, то аудиторской группе следует предоставить достаточно времени для того, чтобы определить соответствие системы менеджмента качества изготовителя применимым регулирующим требованиям. Если необходимо дополнительное время для того, чтобы оценить соответствие национальным или региональным регулирующим требованиям, то эта необходимость должна быть обоснована.
Например, для определения базовой продолжительности первоначальных аудитов на соответствие стандартам серии ГОСТ Р ИСО 9000, измеряемой в человеко-днях, можно использовать таблицу из ГОСТ Р 40.003. Поскольку данная таблица не предназначена непосредственно для нужд аудиторов медицинских изделий, рекомендуется добавить дополнительное время с учетом требований ГОСТ Р ИСО 13485 и регулирующих требований. Данный стандарт можно также рассматривать как руководство для определения продолжительности других видов аудитов, например подтверждающих аудитов.
Увеличенное базовое время включает в себя время для подготовки к аудиту, предварительного анализа документации системы менеджмента качества и составления отчета. В него не входит время, необходимое для анализа пакета проектной документации, изучения типа, проверки одобрения выпуска изделия в продажу и других подобных видов деятельности, но входит время, необходимое для оценивания документов на продукцию на базе образца, выбранного для аудита. Увеличенное базовое время для первоначального аудита рекомендуется корректировать с учетом других видов аудитов и факторов, приведенных в приложении В, которые могут увеличивать или уменьшать установленную продолжительность аудита, но только если требование учета данных факторов содержится в применимых регулирующих документах.
4.6.2.5 Примерный расчет времени проведения аудита на месте (в процентах)
Примерно рассчитать время проведения аудита на месте (в процентах) для разных подсистем можно с помощью таблицы 2.
Таблица 2 - Примерный расчет времени проведения аудита на месте (в процентах)
Подсистема | Примерное время, рассчитанное на месте, % | Примечание |
Высшее руководство | 5-10 | - |
Проектирование и разработка | 0-20 | Зависит от регулирующих требований |
Документация на продукцию | 5-20 | - |
Управление продукцией и процессами | 20-30 | - |
Корректирующие и предупреждающие действия | 10-30 | - |
Управление закупками | 5-20 | Зависит от процентного соотношения и важности видов деятельности внешнего изготовителя, с которым заключен контракт |
Документация и записи | 5 | - |
Процессы, связанные с потребителем | 5 | - |
Примерное время проведения аудита на месте, рассчитанное и выраженное в процентах, будет различаться в зависимости от следующих факторов:
- области аудита;
- изменений графика;
- необходимости получения информации на удаленных площадках.
4.7 Руководство по материально-техническому обеспечению аудита
Для оказания помощи аудиторам в надлежащем проведении аудита рекомендуется рассмотреть следующие моменты:
- изменения, произведенные изготовителем, если они отличаются от ранее заявленных (например, изменения в структуре организации, в системе менеджмента качества, в технических средствах, процессах, изделиях), представленных на предварительном совещании;
- согласование графика проведения и продолжительности аудита между исполнительным руководством проверяемой организации и ответственным представителем организации, проводящей аудит;
- безотлагательный контроль работы над несоответствиями, выявленными при проведении последнего аудита, для определения результативности проведения изготовителем корректирующих действий;
- аудит склада готовой продукции на первоначальном этапе проведения аудита для выборки образцов с целью осуществления дальнейшей прослеживаемости (например, несоответствующих материалов, записей, относящихся к партии изделий, и т.д.);
- осуществление прослеживаемости на первоначальном этапе проведения аудита, позволяющее выбрать направление прослеживаемости - вперед или назад - и обеспечивающее изготовителю достаточно времени для оценивания поступающей информации;
- подтверждающие аудиты могут быть сфокусированы либо на проектировании, либо на производстве и связанных с ними видах деятельности, они должны также учитывать такие факторы, как номенклатура изделий и/или область сертификации;
- при проведении каждого аудита следует рассматривать внутренние аудиты, жалобы, корректирующие и предупреждающие действия и анализ со стороны руководства;
- документация по результатам аудита и тренинг по окончании аудита позволяют лучше понять ситуацию на примерах, рассматриваемых в ходе аудита;
- оценивание системы внутреннего аудита к концу аудита позволяет избежать предубеждения со стороны аудиторов;
- ситуация на месте может оказать влияние на последовательность действий при проведении аудита, ее следует учитывать во избежание потери времени.
Перечисленные выше факторы рекомендуется учитывать, но аудиторская группа может проводить аудит подсистем системы менеджмента качества изготовителя в любой последовательности.
4.8 Связи
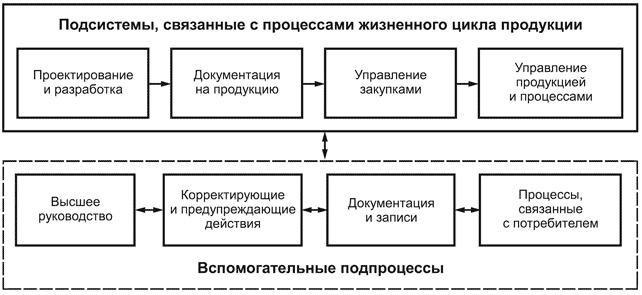
Рисунок 1 - Примеры связей между подсистемами
Примечание - На рисунке 1 представлены основные связи между подсистемами. Существует много других связей (например, обратная связь с процессами жизненного цикла продукции и связи между вспомогательными процессами).
Несмотря на то что большая часть времени аудиторов уходит на исследование процессов внутри подсистем, важно помнить о связях между подсистемами и связях между различными процессами.
Примеры
1 Корректирующие и предупреждающие действия и высшее руководство: доведение до сведения высшего руководства информации о корректирующих и предупреждающих действиях для анализа со стороны высшего руководства.
2 Управление проектированием и разработкой и управление закупками: использование выходных данных проекта для оценивания возможных поставщиков материалов и комплектующих и доведение до сведения поставщиков конкретных требований к закупкам.
3 Внутри каждого процесса его этапы обычно связаны между собой, так как выход одного процесса образует вход следующего процесса.
4 Имеются также некоторые очевидные связи между процессами: выход процесса проектирования будет входом процесса производства; эти связи необходимо проверять на обоих этапах аудита (в данном случае - проектирования и производства) с целью подтверждения наличия данной связи и функционирования системы менеджмента качества как единого целого.
5 Другие связи могут быть менее очевидными, но их также необходимо подвергать аудиту: например, при обнаружении несоответствующей продукции среди готовой проблема несоответствия может заключаться в хранении, производстве, окончательной проверке или проектировании.
6 Существуют также связи между подсистемами: например, если дефектные комплектующие поступают в производственные помещения, будет ли причиной этому поставщик, контроль при приемке, ошибочные данные, предоставленные поставщику, или проектирование? В таком случае требуется ли от системы менеджмента качества изготовителя обязательный отчет о корректирующих и предупреждающих действиях?
5 Проверяемые подсистемы
Проведение аудита каждой подсистемы преследует конкретную цель. План аудита каждой подсистемы рекомендуется разрабатывать на основе процессного подхода (см. 4.4) так, чтобы он удовлетворял поставленным целям. В данный план следует включать подтверждение соответствия требованиям, предъявляемым к каждой подсистеме.
При проведении аудита на соответствие регулирующим требованиям на протяжении процессов жизненного цикла медицинского изделия применяют принципы менеджмента риска, которые следует использовать для идентификации и решения проблем безопасности. Аудит видов деятельности по менеджменту риска рекомендуется проводить параллельно с аудитом соответствующих подсистем.
Целью аудита процесса менеджмента риска является обеспечение уверенности в разработке и поддержании надлежащего и результативного менеджмента риска на протяжении процессов жизненного цикла медицинского изделия.
Приложение Е настоящего стандарта содержит дополнительную информацию по аудиту программного обеспечения. Эта информация может быть применима к любой из подсистем системы менеджмента качества. |
Примечания
1 В некоторых национальных и региональных регулирующих документах требования менеджмента риска применимы ко всем стадиям жизненного цикла медицинского изделия.
2 Номера, приведенные ниже в скобках, относятся к соответствующим разделам ГОСТ Р ИСО 13485.
3 Подсистемы, обозначенные ниже "звездочкой" *, являются главными подсистемами, которым следует уделять основное внимание при аудите, если таково регулирующее требование (см. также 4.2).
5.1 Подсистема высшего руководства*
5.1.1 Цель
Целью аудита подсистемы высшего руководства является подтверждение того, что высшее руководство обеспечивает разработку и поддержание в рабочем состоянии надлежащей и результативной системы менеджмента качества.
5.1.2 Основные этапы
При проведении аудита подсистемы высшего руководства можно выделить следующие основные этапы:
a) подтверждение того, что руководство по качеству, процедуры анализа со стороны высшего руководства и процедуры внутреннего аудита качества, план в области качества и процедуры и инструкции системы менеджмента качества надлежащим образом разработаны и документированы (см. ГОСТ Р ИСО 13485, подразделы 4.1, 4.2);
b) подтверждение того, что политика и цели в области качества надлежащим образом разработаны и документированы и предприняты действия к их достижению (см. ГОСТ Р ИСО 13485, подразделы 5.3, 5.4);
c) подтверждение того, что процессы жизненного цикла продукции включают в себя планирование менеджмента риска и последующий анализ результативности деятельности по менеджменту риска, гарантируя разработку политики, процедур и практических действий с целью анализа, оценивания и управления риском (см. ГОСТ Р ИСО 13485, подраздел 7.1);
d) анализ организационной структуры изготовителя и относящихся к ней документов с целью подтверждения того, что в них включены положения в отношении распределения ответственности, полномочий (например, представителя высшего руководства), ресурсов, компетентности и подготовки (см. ГОСТ Р ИСО 13485, подразделы 5.1, 5.5.1, 5.5.2, 6.1, 6.2);
e) подтверждение неоднократного проведения анализа со стороны высшего руководства, включающего в себя анализ пригодности и результативности системы менеджмента качества (см. ГОСТ Р ИСО 13485, подраздел 5.6);
f) подтверждение проведения внутренних аудитов системы менеджмента качества, включающих в себя верификацию корректирующих и предупреждающих действий (см. ГОСТ Р ИСО 13485, пункт 8.2.2);
g) аудит должен начинаться и заканчиваться подсистемой высшего руководства, однако между началом и концом аудита подсистемы высшего руководства необходимо провести аудит других подсистем.
В заключение аудита следует принять решение о том, были ли предприняты высшим руководством необходимые действия для обеспечения постоянной пригодности надлежащей и результативной системы менеджмента качества.
5.2 Подсистема проектирования и разработки*
5.2.1 Цель
Целью аудита подсистемы проектирования и разработки является подтверждение того, что процесс проектирования и разработки является управляемым и обеспечивает соответствие медицинского изделия требованиям потребителя, предусмотренному применению и конкретным требованиям к изделию.
Примечание - В некоторых юрисдикциях для отдельных изделий, вследствие особенностей их классификации, не проводят аудит процесса управления проектированием. При аудите подсистемы проектирования и разработки аудит процесса управления проектированием проводится при необходимости.
5.2.2 Основные этапы
При проведении аудита подсистемы проектирования и разработки можно выделить следующие основные этапы:
a) подтверждение того, что медицинские изделия являются объектом регулирующих требований, предъявляемых к процедурам проектирования и разработки, включая процедуры менеджмента риска (например, идентификацию опасностей, оценивание риска и управление риском) (см. ГОСТ Р ИСО 13485, подразделы 7.1, 7.3);
b) анализ документов, описывающих процесс проектирования, и выбор достаточного количества записей, охватывающих всю номенклатуру выпускаемых изготовителем изделий. Особое внимание следует уделять отдельным изделиям, а не группам (сериям) изделий.
Критерии выбора:
1) риск применения изделия;
2) жалобы или известные проблемы;
3) период существования конструктивного решения (предпочтительнее новейшие разработки);
c) анализ плана проектирования выбранного(ых) изделия(ий) для лучшего понимания деятельности по проектированию и разработке, включая взаимодействие и распределение ответственности (см. ГОСТ Р ИСО 13485, пункт 7.3.1);
d) подтверждение на основе выбранных записей по проектированию изделий того, что процедуры проектирования и разработки были установлены и применены (см. ГОСТ Р ИСО 13485, пункт 7.3.1);
e) подтверждение того, что входные данные проектирования были разработаны с учетом требований потребителя к функционированию, эксплуатационным свойствам и безопасности, к предусмотренному применению изделия(ий), а также с учетом регулирующих требований и других требований, существенных для проектирования и разработки (см. ГОСТ Р ИСО 13485, пункты 7.2.1, 7.3.2);
f) анализ спецификаций на медицинские изделия для подтверждения того, что выходные данные проекта и разработки соответствуют входным требованиям; подтверждение того, что были определены выходные данные проекта, существенные для надлежащего функционирования медицинского изделия (см. ГОСТ Р ИСО 13485, пункт 7.3.3);
g) подтверждение того, что деятельность по менеджменту риска определена и осуществлена, критерии допустимости риска установлены и являются соответствующими на протяжении всего процесса проектирования и разработки медицинского изделия, любой остаточный риск оценен и, если необходимо и целесообразно, доведен до сведения потребителя (например, с помощью маркировок, инструкций по эксплуатации, пояснительных уведомлений и т.д.) (см. ГОСТ Р ИСО 13485, подраздел 7.1, пункт 7.3.2).
Примечание - Может возникнуть необходимость в проведении аудита других подсистем для подтверждения того, что критерии допустимости риска удовлетворены и информация об остаточном риске доведена, если необходимо и целесообразно, до сведения потребителя;
h) подтверждение того, что данные валидации проекта свидетельствуют о соответствии одобренного проекта требованиям к специальному или предусмотренному применению (см. ГОСТ Р ИСО 13485, пункт 7.3.6);
i) подтверждение того, что клинические исследования были проведены, а безопасность и функционирование изделия оценены в соответствии с требованиями национальных или региональных нормативных документов (см. ГОСТ Р ИСО 13485, пункт 7.3.6);
j) если медицинское изделие включает в себя программное обеспечение, то подтверждение того, что программное обеспечение является частью валидации процесса проектирования и разработки медицинского изделия (см. ГОСТ Р ИСО 13485, пункты 7.3.1, 7.3.6);
k) подтверждение того, что изменения проекта были управляемыми и верифицируемыми или, при необходимости, валидируемыми, а также были приведены в соответствие с регулирующими требованиями (см. ГОСТ Р ИСО 13485, подраздел 7.1, пункты 7.3.5, 7.3.7);
l) подтверждение того, что был проведен анализ проекта (см. ГОСТ Р ИСО 13485, пункты 7.3.1, 7.3.4);
m) подтверждение того, что изменения проекта были проанализированы с учетом их возможного влияния на ранее изготовленные и поставленные изделия и что записи о результатах анализа поддерживаются в рабочем состоянии (см. ГОСТ Р ИСО 13485, пункт 7.3.7);
n) подтверждение того, что проект был надлежащим образом передан в производство (см. ГОСТ Р ИСО 13485, пункт 7.3.1).
Соответствие подсистемы проектирования и разработки оценивают на основании данных исследований.
5.3 Подсистема документации на продукцию*
5.3.1 Цель
Целью аудита подсистемы документации на продукцию является подтверждение того, что документация изготовителя гарантирует соответствие изделий требованиям потребителя и регулирующим требованиям.
5.3.2 Основные этапы
При проведении аудита подсистемы документации на продукцию можно выделить следующие основные этапы:
a) подтверждение наличия документов, необходимых для того, чтобы организация могла обеспечить планирование, осуществление и управление процессами, разработанными в организации [см. ГОСТ Р ИСО 13485, пункт 4.2.1, перечисление d)];
b) выбор документов на продукцию для такого числа изделий, которого достаточно для того, чтобы охватить всю номенклатуру выпускаемых изготовителем изделий (см. ГОСТ Р ИСО 13485, подразделы 7.1, 7.2, пункт 7.3.3).
Критерии выбора:
1) риск, связанный с изделиями;
2) жалобы или известные проблемы;
3) период существования конструктивных решений (предпочтительнее новейшие разработки);
c) для выбранного(ых) изделия(ий) подтверждение того, что документы включают в себя (если таковы требования национальных или региональных нормативных документов):
1) свидетельство соответствия требованиям, в том числе требованиям применимых стандартов;
2) описание медицинского(их) изделия(ий), включая инструкции по эксплуатации, материалы и спецификацию;
3) сводную документацию по верификации и валидации проекта(ов), включая данные клинических исследований;
4) маркировку;
5) документы по менеджменту риска;
6) информацию по изготовлению, включая основных поставщиков.
Примечание - Выполнение данного перечня не избавляет аудиторов от необходимости оценивать дополнительные документы.
Соответствие подсистемы документации на продукцию оценивают на основании данных исследований.
5.4 Подсистема управления продукцией и процессами*
5.4.1 Цель
Целью аудита подсистемы управления продукцией и процессами (включая испытания, инфраструктуру, обслуживание и оборудование) является подтверждение того, что средства, с помощью которых изготовитель управляет продукцией и процессами, могут обеспечить соответствие упомянутой продукции техническим требованиям.
5.4.2 Основные этапы
При проведении аудита подсистемы управления продукцией и процессами можно выделить следующие основные этапы:
a) подтверждение того, что процессы жизненного цикла продукции, в том числе любые применяемые средства управления и управляемые условия, являются запланированными (см. ГОСТ Р ИСО 13485, подраздел 7.1, пункт 7.5.1);
b) подтверждение того, что планирование процессов жизненного цикла продукции взаимосвязано с требованиями, предъявляемыми к другим процессам системы менеджмента качества (см. ГОСТ Р ИСО 13485, подраздел 7.1);
c) анализ производственных процессов с учетом приведенных ниже критериев, выбор одного или нескольких процессов для аудита:
1) свидетельства проведения корректирующих и предупреждающих действий по проблемам, связанным с процессами;
2) использование производственных процессов для изделий высокой степени потенциального риска применения;
3) использование новых производственных процессов или технологий;
4) использование производственных процессов для изготовления серийной продукции;
5) процессы, не охваченные предыдущими аудитами.
Примечание - Рекомендации по аудиту процессов стерилизации приведены в приложении D;
d) если выходные данные процессов не могут быть верифицированы, то подтверждение валидации этих процессов, демонстрирующей способность упомянутых процессов достигать запланированных результатов (см. ГОСТ Р ИСО 13485, пункт 7.5.2);
e) подтверждение того, что оборудование, используемое при управлении производственными процессами, отрегулировано, откалибровано и содержится в исправности (надлежащим образом обслуживается) (см. ГОСТ Р ИСО 13485, подразделы 7.5, 7.6);
f) подтверждение того, что процессы являются управляемыми и контролируемыми и функционируют в установленных пределах, а также подтверждение того, что меры по управлению риском, идентифицированные изготовителем для производственных процессов, являются управляемыми, контролируемыми и оценены надлежащим образом (см. ГОСТ Р ИСО 13485, подразделы 7.1, 7.5);
g) подтверждение того, что меры по управлению риском применяются при поставке, монтаже (установке) и обслуживании (см. ГОСТ Р ИСО 13485, подпункты 7.5.1.1, 7.5.1.2.2, 7.5.1.2.3);
h) определение взаимосвязей с другими процессами (см. ГОСТ Р ИСО 13485, подразделы 4.1, 4.2);
i) подтверждение того, что персонал имеет надлежащую квалификацию и/или подготовку для осуществления/поддержания процессов в рабочем состоянии (см. ГОСТ Р ИСО 13485, пункт 6.2.2);
j) подтверждение того, что инфраструктура и производственная среда соответствуют регулирующим требованиям (см. ГОСТ Р ИСО 13485, подразделы 6.3, 6.4);
k) подтверждение идентификации и прослеживаемости процессов и продукции и их соответствия регулирующим требованиям (см. ГОСТ Р ИСО 13485, пункт 7.5.3);
I) если процесс управляется посредством программного обеспечения, то подтверждение валидации данного программного обеспечения с учетом его предусмотренного применения (см. ГОСТ Р ИСО 13485, подпункт 7.5.2.1);
m) подтверждение того, что управление устройствами для мониторинга и измерений соответствует регулирующим требованиям (см. ГОСТ Р ИСО 13485, подраздел 7.6);
n) подтверждение того, что система для мониторинга и измерений продукции соответствует регулирующим требованиям; обеспечение выполнения всех идентифицированных мер по управлению риском (см. ГОСТ Р ИСО 13485, подраздел 7.6, пункт 8.2.4);
о) подтверждение того, что деятельность по приемке гарантирует соответствие требованиям и документирована (см. ГОСТ Р ИСО 13485, пункт 8.2.4, подпункты 8.2.4.1, 8.2.4.2);
р) подтверждение надлежащего управления несоответствующей продукцией (см. ГОСТ Р ИСО 13485, подраздел 8.3).
Соответствие подсистемы управления продукцией и процессами оценивают на основании данных исследований.
5.5 Подсистема корректирующих и предупреждающих действий*
5.5.1 Цель
Целью аудита подсистемы корректирующих и предупреждающих действий (включая записи/прослеживание) является подтверждение того, что процессы, разработанные изготовителем, обеспечивают сбор и анализ информации; идентификацию проблем, связанных с процессами, готовой продукцией и полуфабрикатами; изучение данных проблем и выполнение адекватных и результативных корректирующих и предупреждающих действий.
5.5.2 Основные этапы
При проведении аудита подсистемы корректирующих и предупреждающих действий можно выделить следующие основные этапы:
a) подтверждение того, что разработаны процедуры подсистемы корректирующих и предупреждающих действий, отвечающие требованиям системы менеджмента качества (см. ГОСТ Р ИСО 13485, подразделы 4.1, 4.2, 8.5);
b) подтверждение того, что входные данные корректирующих и предупреждающих действий проанализированы и достоверны и что корректирующие и предупреждающие действия оказались результативными (см. ГОСТ Р ИСО 13485, подразделы 8.4, 8.5);
c) если корректирующие и предупреждающие действия приводят к изменениям проекта, то подтверждение того, что в рамках процесса менеджмента риска оценены все возникающие опасности и любые новые риски (см. ГОСТ Р ИСО 13485, подраздел 7.1);
d) подтверждение идентификации и мониторинга всех имеющихся источников данных для корректирующих и предупреждающих действий и необходимости осуществления конкретных действий. Подтверждение того, что данные, полученные из вышеуказанных источников, проанализированы с помощью применимых статистических методов с целью идентификации у выпускаемой продукции проблем в области качества, требующих проведения корректирующих действий (см. ГОСТ Р ИСО 13485, подраздел 8.1, пункт 8.2.3, подраздел 8.4);
e) определение того, были ли изучены причины отказов с целью идентификации, по возможности, причин несоответствий (см. ГОСТ Р ИСО 13485, пункт 8.5.2);
f) подтверждение того, что изготовитель имеет средства управления для предотвращения непреднамеренного использования несоответствующей продукции (см. ГОСТ Р ИСО 13485, подраздел 8.3);
g) подтверждение проведения результативных корректирующих и предупреждающих действий, которые были документированы и не оказали неблагоприятного воздействия на готовую продукцию (см. ГОСТ Р ИСО 13485, пункты 8.2.3, 8.5.2, 8.5.3);
h) определение того, была ли доведена до сведения высшего руководства информация в отношении несоответствующей продукции, проблем в области качества, корректирующих и предупреждающих действий, требующая анализа со стороны руководства (см. ГОСТ Р ИСО 13485, пункт 5.6.3);
i) подтверждение того, что рассматриваемое медицинское изделие изготовлено в соответствии с применимыми регулирующими требованиями (см. ГОСТ Р ИСО 13485, пункт 8.5.1);
j) подтверждение того, что изготовитель принимает результативные меры для накопления опыта посредством изучения постпроизводственной информации, работы с жалобами потребителей (см. ГОСТ Р ИСО 13485, пункт 7.8.3) и исследования причин несоответствий, имеющих отношение к рассылке пояснительных уведомлений/отзыву изделий, с целью осуществления обратной связи с подсистемой корректирующих и предупреждающих действий (см. ГОСТ Р ИСО 13485, пункты 7.2.3, 8.2.1);
k) подтверждение того, что изготовитель принял результативные меры для выпуска и рассылки пояснительных уведомлений, а также отзыва несоответствующей продукции (см. ГОСТ Р ИСО 13485, пункт 8.5.1).
Соответствие подсистемы корректирующих и предупреждающих действий оценивают на основании данных исследований.
5.6 Подсистема управления закупками
Подсистему управления закупками следует рассматривать как основную для тех изготовителей, которые передают на аутсорсинг выполнение работ по таким существенно важным видам деятельности, как проектирование и разработка и/или выпуск продукции, одному или более субподрядчикам.
5.6.1 Цель
Целью аудита подсистемы управления закупками является подтверждение того, что процессы, разработанные изготовителем, обеспечивают соответствие поставляемых изделий, комплектующих, материалов и услуг (включая исполнителей работ и консультантов) регулирующим требованиям. Это особенно важно, если готовая продукция и услуги не могут быть верифицированы путем внешнего осмотра (например, услуги по стерилизации).
5.6.2 Основные этапы
При проведении аудита подсистемы управления закупками можно выделить следующие основные этапы:
а) подтверждение того, что разработаны процедуры оценивания и выбора поставщиков (см. ГОСТ Р ИСО 13485, пункт 7.4.1);
b) подтверждение того, что изготовитель оценивает и поддерживает в рабочем состоянии результативные средства управления поставщиками, удовлетворяющие регулирующим требованиям (см. ГОСТ Р ИСО 13485, пункт 7.4.1);
c) подтверждение того, что изготовитель обеспечивает надлежащие спецификации на изделия и услуги, поставляемые поставщиками, распределяет ответственность за проведение менеджмента риска и принимает все необходимые меры по управлению риском (см. ГОСТ Р ИСО 13485, пункт 7.4.2);
d) подтверждение того, что записи по оцениванию поставщиков поддерживаются в рабочем состоянии (см. ГОСТ Р ИСО 13485, пункт 7.4.1);
e) определение того, что верификация закупленной продукции и услуг выполнена надлежащим образом (см. ГОСТ Р ИСО 13485, пункт 7.4.3).
Соответствие подсистемы управления закупками оценивают на основании данных исследований.
5.7 Подсистема документации и записей
5.7.1 Цель
Целью аудита подсистемы документации и записей является подтверждение того, что процессы документирования, разработанные изготовителем, обеспечивают надлежащее управление необходимыми документами и доступ к необходимым записям.
5.7.2 Основные этапы
При проведении аудита подсистемы документации и записей можно выделить следующие основные этапы:
a) подтверждение того, что разработаны процедуры идентификации, хранения, защиты, поиска, максимального срока хранения и удаления документов и записей (включая управление изменениями) (см. ГОСТ Р ИСО 13485, пункты 4.2.3, 4.2.4);
b) подтверждение того, что документы и изменения в них одобрены до их применения (см. ГОСТ Р ИСО 13485, пункт 4.2.3);
c) подтверждение того, что имеется доступ к действующим документам в местах их применения и что устаревшие документы больше не применяются (см. ГОСТ Р ИСО 13485, пункт 4.2.3);
d) подтверждение того, что все необходимые документы и записи хранятся в течение необходимого времени (см. ГОСТ Р ИСО 13485, пункты 4.2.1, 4.2.4).
Соответствие подсистемы документации и записей оценивают на основании данных исследований.
5.8 Подсистема процессов, связанных с потребителем
5.8.1 Цель
Целью аудита подсистемы процессов, связанных с потребителем, является подтверждение того, что система менеджмента качества обеспечивает соответствие требованиям, в том числе регулирующим, установленным к процессам, связанным с потребителем.
5.8.2 Основные этапы
При проведении аудита подсистемы процессов, связанных с потребителем, можно выделить следующие основные этапы:
a) анализ требований к продукции с целью подтверждения того, что она соответствует предусмотренному применению, а также регулирующим требованиям и требованиям потребителя (см. ГОСТ Р ИСО 13485, пункты 7.2.1, 7.2.2);
b) подтверждение того, что все поступающие заказы и связанная с ними информация анализируются с тем, чтобы гарантировать решение всех конфликтных вопросов и обеспечить выполнение изготовителем требований потребителя (см. ГОСТ Р ИСО 13485, пункт 7.2.2);
c) подтверждение того, что изготовитель принял результативные меры для установления связи с потребителем, включая документирование обратной связи с потребителем, с целью идентификации проблем в области качества и выполнения необходимых корректирующих и предупреждающих действий (см. ГОСТ Р ИСО 13485, пункты 7.2.3, 8.2.1);
d) подтверждение того, что обратная связь с потребителем анализируется в ходе процессов жизненного цикла продукции и используется для повторного оценивания риска и, при необходимости, для актуализации деятельности по менеджменту риска (см. ГОСТ Р ИСО 13485, подраздел 7.1, пункт 7.2.3).
Соответствие подсистемы процессов, связанных с потребителем, оценивают на основании данных исследований.
Приложение А
(справочное)
Схемы биномиального выбора
________________
Заимствовано из QSIT - Quality System Inspection Technique, метод контроля системы качества, 1999 г.
Таблица А.1 - Схемы биномиального выбора. Доверительный уровень в случае биномиального распределения
Граница доверительного интервала | 0 из | 1 из | 2 из | |
А | 0,30 ucl* | 11 | 17 | 22 |
В | 0,25 ucl* | 13 | 20 | 27 |
С | 0,20 ucl* | 17 | 26 | 34 |
D | 0,15 ucl* | 23 | 35 | 46 |
Е | 0,10 ucl* | 35 | 52 | 72 |
F | 0,05 ucl* | 72 | 115 | 157 |
* Верхний доверительный уровень. | ||||
Таблица А.2 - Схемы биномиального выбора. Доверительный уровень в случае биномиального распределения
Граница доверительного интервала | 0 из | 1 из | 2 из | |
А | 0,30 ucl* | 15 | 22 | 27 |
В | 0,25 ucl* | 19 | 27 | 34 |
С | 0,20 ucl* | 24 | 34 | 43 |
D | 0,15 ucl* | 35 | 47 | 59 |
Е | 0,10 ucl* | 51 | 73 | 90 |
F | 0,05 ucl* | 107 | 161 | 190 |
* Верхний доверительный уровень. | ||||
Биномиальный выбор может быть использован, если необходимо принять решение об ожидаемом результате, который может иметь только два возможных исхода (например, запись является соответствующей или несоответствующей).
Факторы, рассматриваемые при выборе необходимой таблицы и объема выборки, могут включать в себя риск применения медицинского изделия или процесса, выбранные записи и время, выделенное аудитором для проведения данной части аудита. Для анализа записей, относящихся к медицинским изделиям с низкой степенью потенциального риска применения, рекомендуется использовать таблицу А.1 (граница доверительного интервала 95%), а для анализа записей, относящихся к медицинским изделиям с высокой степенью потенциального риска применения, рекомендуется использовать таблицу А.2 (граница доверительного интервала 99%). Приведем два примера.
Примеры
1 Анализируя записи по стерилизации, аудитор должен определить, является ли процесс стерилизации контролируемым и управляемым. Процесс стерилизации представляет собой процесс высокой степени риска, поэтому аудитор использует таблицу А.2, приведенную в приложении А. Для анализа аудитор проводит произвольную выборку и отбирает серию из 24 записей по стерилизации. Все 24 записи демонстрируют, что процесс стерилизации был контролируемым и управляемым и его проводили при утвержденных эксплуатационных параметрах. На основе таблицы А.2 аудитор может быть на 99% уверен в том, что не более 20% записей по стерилизации могут продемонстрировать несоответствие процесса стерилизации утвержденным эксплуатационным параметрам.
2 Анализируя записи по подготовке, аудитор должен определить, проведена ли подготовка персонала в связи с последними пересмотрами процедур по рассмотрению жалоб потребителей. Например, изготовитель производит компьютерные томографы. Используя таблицу А.1, аудитор проводит произвольную выборку, состоящую из записей по подготовке 17 сотрудников. Аудитор обнаруживает, что один сотрудник не прошел подготовку по пересмотренным процедурам. На основе таблицы А.1 аудитор может быть на 95% уверен в том, что не более 30% сотрудников не получили подготовку по пересмотренным процедурам.
Приложение В
(справочное)
Факторы, применяемые для определения продолжительности аудита
a) Факторы, увеличивающие продолжительность аудита:
1) субподрядчики, задействованные в каких-либо процессах или поставке комплектующих, критично влияющих на функционирование медицинского изделия и/или безопасность пользователя или готовой продукции, включая продукцию с фирменной маркировкой изготовителя. Если изготовитель не может обеспечить достаточного свидетельства соответствия критериям аудита, то может понадобиться дополнительное время для аудита каждого субподрядчика.
Примечание - В США, например, поставщики комплектующих не подпадают под действие Руководства по системам качества FDA и освобождены от обычных проверок FDA;
2) монтаж изделия изготовителем на территории потребителя.
Примечание - Может потребоваться время для посещения объектов потребителя или анализа записей по монтажу;
3) проведение аудитов на иностранном языке (см. ГОСТ Р 54421, пункт 7.4.6);
4) требование изготовителя по проведению многоцелевого аудита;
5) недостаточное соответствие изготовителя регулирующим требованиям.
b) Факторы, сокращающие продолжительность аудита:
1) медицинские изделия с низкой и средней степенями потенциального риска применения;
2) наличие свидетельств об удовлетворительном проведении аудитов у любой третьей стороны или у организаций, проводящих аудиты у поставщиков;
3) если результаты предыдущих аудитов, проведенных той же организацией, демонстрировали соответствие изготовителя регулирующим требованиям;
4) сокращение номенклатуры изделий, производимых изготовителем, со времени проведения последнего аудита;
5) сокращение процесса проектирования или производственных процессов со времени проведения последнего аудита.
c) Изготовители, имеющие много производственных площадок
Если у изготовителя имеется много производственных площадок, то ему следует определить, какие виды деятельности будут осуществлены на каждой площадке.
Если на данных производственных площадках применяются разные системы менеджмента качества, то для определения продолжительности аудита каждую площадку следует рассматривать как отдельное целое.
Если изготовитель, имеющий одну или более производственных площадок, производит аналогичные изделия или услуги на разных площадках, на которых, однако, применяется единая система менеджмента качества, то продолжительность аудита можно оценить в три этапа:
1) оценить продолжительность аудита для каждой площадки отдельно, а затем совокупную продолжительность в человеко-днях;
2) определить общее количество персонала на всех площадках, а затем, если применимо, использовать рекомендации IAF (Inter-American Foundation) для определения основного значения;
3) вывести среднее значение на основании двух полученных результатов.
d) Аудиты других видов
Существуют аудиты различных видов, продолжительность которых меньше, чем это необходимо для полного первоначального аудита (см. ГОСТ Р 54421, раздел 7).
Для оценивания продолжительности других видов аудита следует рассмотреть факторы, приведенные в настоящем приложении.
Для частичных аудитов продолжительность можно рассчитать на основании количества исследуемых подсистем системы менеджмента качества. Такой способ применяют, например, при проведении повторных аудитов с целью верификации корректирующих действий, предпринятых по результатам первоначального аудита, или в тех случаях, когда проведение частичного аудита является требованием нормативных документов, например, проведение частичного аудита средств измерений класса 1.
В тех случаях, когда изготовителю необходимо произвести существенные изменения (см. ГОСТ Р 54421, подраздел 7.3), для проведения аудита может потребоваться дополнительное время.
Приложение С
(справочное)
Соответствие структурных элементов ИСО 13485:2003 и 21 CFR, часть 820
Таблица С.1
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.1 Подсистема высшего руководства | ||
4, 5, 6, 7, 8 | 820.5, 820.20, 820.22, 820.25, 820.30 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 4.1, 4.2 | 820.20(с), 820.20(d), 820.20(e), 820.22 |
2 | 5.3, 5.4 | 820.20(а) |
3 | 7.1 | 820.30(g), 820.30(i) |
4 | 5.1, 5.5.1, 5.5.2, 6.1, 6.2 | 820.20(b), 820.20(b)(1), 820.20(b)(2), 820.20(b)(3)(i) и (ii), 820.25 |
5 | 5.6 | 820.5, 820.20(c) |
6 | 8.2.2 | 820.22 |
Таблица С.2
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.2 Подсистема проектирования и разработки | ||
7 | 820.30, 820.70 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 7.1, 7.3 | 820.30 |
3 | 7.1 | 820.30(a) |
4 | 7.3.1 | 820.30(b) |
5 | 7.3.1 | 820.30(a), 820.30(b) - (j) |
6 | 7.2.1, 7.3.2 | 820.30(c) |
7 | 7.3.3 | 820.30(f), 820.30(d) |
8 | 7.1, 7.3.2 | 820.30(g) |
9 | 7.3.6 | 820.30(g) |
10 | 7.3.6 | 820.70(i), 820.30(g) |
11 | 7.3.1, 7.3.6 | 820.30(g) |
12 | 7.1, 7.3.5, 7.3.7 | 820.30(i), 820.70(b), 820.30(g) |
13 | 7.3.1, 7.3.4 | 820.30(e) |
14 | 7.3.7 | 820.30(i), 820.70(b) |
Таблица С.3
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.3 Подсистема документации на продукцию | ||
4, 7 | 820.30, 820.181, 820.50, 820.180, 820.184, 820.186, 820.75 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 4.2.1, перечисление d) | 820.180, 820.181, 820.184, 820.186 |
3 | 7.1, 7.2, 7.3.3 | 820.30(d), 820.30(g), 820.30(f), 820.181, 820.50, 820.75 |
Таблица С.4
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.4 Подсистема управления продукцией и процессами | ||
4, 6, 7, 8 | 820.50, 820.60, 820.65, 820.70, 820.72, 820.75, 820.80, 820.90, 820.20, 820.25, 820.30, 820.40, 820.100, 820.180, 820.140, 820.150, 820.184, 820.181, 820.86 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 7.1, 7.5.1 | 820.70, 820.70(с) |
2 | 7.1 | 820.30, 820.40, 820.50, 820.80, 820.181 |
3 | - | - |
4 | 7.5.2 | 820.75 |
5 | 7.5.1, 7.6 | 820.70(g)(3), 820.72(а), 820.70(g)(1) |
6 | 7.1, 7.5 | 820.70(а), 820.70(с), 820.70(е), 820.70(f), 820.70(g), 820.70(h), 820.72, 820.75(b), 820.80 |
7 | 7.5.1.1, 7.5.1.2.2, 7.5.1.2.3 | - |
8 | 4.1, 4.2 | 820.20, 820.25, 820.30, 820.40, 820.72, 820.90, 820.100, 820.180 |
9 | 6.2.2 | 820.20(b)(2), 820.25, 820.70, 820.70(d), 820.75(b)(1) |
10 | 6.3, 6.4 | 820.70(c), 820.70(g), 820.70(f) |
11 | 7.5.3, 7.5.3.1, 7.5.3.2, 7.5.3.3 | 820.60, 820.65 |
12 | 7.5.2.1 | 820.70(i) |
13 | 7.6 | 820.72 |
14 | 7.6, 8.2.4 | 820.72, 820.80(c), 820.80(d) |
15 | 8.2.4, 8.2.4.1, 8.2.4.2 | 820.80, 820.86, 820.184(d) |
16 | 8.3 | 820.90 |
Таблица С.5
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.5 Подсистема корректирующих и предупреждающих действий | ||
4, 5, 6, 7, 8 | 820.90, 820.100, 820.198, 820.250, 820.30 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 4.1, 4.2, 8.5 | 820.100(а), 820.100(b) |
2 | 8.4, 8.5 | 820.100(а)(1) |
3 | 7.1 | 820.30(i) |
4 | 8.1, 8.2.3, 8.4 | 820.100(а), 820.100(а)(1), 820.250 |
5 | 8.5.2 | 820.100(а)(2) |
6 | 8.3 | 820.90(b) |
7 | 8.2.3, 8.5.2, 8.5.3 | 820.100(а)(3), 820.100(а)(4), 820.100(а)(5), 820.100(b) |
8 | 5.6.3 | 820.100(а)(7) |
9 | 8.5.1 | 820.198(d) |
10 | 7.2.3, 8.2.1 | 820.100, 820.198 |
11 | 8.5.1 | - |
Таблица С.6
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.6 Подсистема управления закупками | ||
7 | 820.40, 820.50, 820.80 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 7.4.1 | 820.40, 820.50 |
2 | 7.4.1 | 820.50(а)(1), 820.50(а)(2) |
3 | 7.4.2 | 820.50(b) |
4 | 7.4.1 | 820.50(а)(3) |
5 | 7.4.3 | 820.50(а)(2), 820.80(а), 820.80(b) |
Таблица С.7
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.7 Подсистема документации и записей | ||
4 | 820.40, 820.65, 820.180, 820.100, 820.181, 820.184, 820.186, 820.198, 820.200 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 4.2.3, 4.2.4 | 820.180, 820.180(b) |
2 | 4.2.3 | 820.40(а), 820.40(b) |
3 | 4.2.3 | 820.40(а) |
4 | 4.2.1, 4.2.4 | 820.100(b), 820.180(b), 820.181, 820.184, 820.186, 820.198(a), 820.200(d) |
Таблица С.8
Разделы и подразделы ИСО 13485:2003 | Разделы 21 CFR, часть 820 | |
7.8 Подсистема процессов, связанных с потребителем | ||
7 | 820.30, 820.100, 820.198, 820.50, 820.160 | |
Этап | ИСО 13485:2003 | 21 CFR, часть 820 |
1 | 7.2.1, 7.2.2 | 820.30(с), 820.30(d), 820.30(f), 820.30(g) |
2 | 7.2.2 | 820.50, 820.160 |
3 | 7.2.3, 8.2.1 | 820.198, 820.100(а)(1) |
4 | 7.1, 7.2.3 | - |
Приложение D
(справочное)
Процессы стерилизации
D.1 Цель
Целью аудита процессов стерилизации (включая испытания, инфраструктуру, производственные помещения и оборудование) является подтверждение того, что данные процессы предназначены для выпуска стерильных изделий.
D.2 Основные этапы
При проведении аудита процессов стерилизации в подсистеме управления продукцией и процессами можно выделить следующие основные этапы:
a) определение того, что процессы стерилизации, включая управляемые условия, запланированы (см. ГОСТ Р ИСО 13485, подраздел 7.1, подпункт 7.5.1.3);
b) определение того, что планирование процессов стерилизации соответствует требованиям, предъявляемым к другим процессам системы менеджмента качества (см. ГОСТ Р ИСО 13485, подраздел 7.1, подпункт 7.5.1.3);
c) определение того, что записи параметров процесса стерилизации для каждой стерилизуемой партии изделий поддерживаются в рабочем состоянии и прослеживаются (см. ГОСТ Р ИСО 13485, подпункт 7.5.1.3);
d) выбор процесса(ов) стерилизации для анализа; при применении нескольких процессов стерилизации необходимо использовать следующие критерии:
1) степень сложности стерилизации медицинского изделия;
2) процесс, применяемый для стерилизации наибольшего числа медицинских изделий;
3) процесс, которым труднее всего управлять;
e) определение того, что процесс стерилизации валидирован и проанализирован на адекватность (валидация включает в себя квалификацию стерилизационной установки), подтверждение актуальности процесса валидации (см. ГОСТ Р ИСО 13485, подпункт 7.5.2.1);
f) определение того, что биологические индикаторы надлежащим образом валидированы и управляемы (см. ГОСТ Р ИСО 13485, пункт 8.2.3);
g) определение того, что процесс стерилизации является управляемым и контролируемым, в том числе для изделий с биологическим наполнителем. Подтверждение того, что содержимое изделий соответствует валидированному содержимому (см. ГОСТ Р ИСО 13485, подпункт 7.5.1.3);
h) определение того, что процесс протекает в соответствии с установленными параметрами (см. ГОСТ Р ИСО 13485, подпункт 7.5.1.3);
i) если данные показывают, что процесс не всегда протекает в соответствии с установленными параметрами, то определение того, что несоответствия исследуют и с ними проводят надлежащую работу, а также что в связи с данными несоответствиями осуществляют необходимые коррекции и корректирующие действия (см. ГОСТ Р ИСО 13485, подраздел 8.1, пункт 8.2.3, подразделы 8.3, 8.4, пункт 8.5.2);
j) если процессом стерилизации управляют с помощью программного обеспечения, то определение того, что программное обеспечение является валидированным (см. ГОСТ Р ИСО 13485, пункт 7.5.2.1);
k) определение того, что использованное оборудование отрегулировано, откалибровано и поддерживается в рабочем состоянии (см. ГОСТ Р ИСО 13485, подразделы 7.5, 7.6);
l) определение того, что персонал имеет надлежащие квалификацию и подготовку для валидации, выполнения и поддержания в рабочем состоянии процесса стерилизации (см. ГОСТ Р ИСО 13485, подраздел 6.2).
Соответствие процесса стерилизации оценивают в рамках оценивания подсистемы управления продукцией и процессами. Соответствие подсистемы управления продукцией и процессами оценивают на основании данных исследований.
Приложение Е
(справочное)
Аудит программного обеспечения
Е.1 Цель Целью аудита видов деятельности системы менеджмента качества, связанных с разработкой, закупкой и/или использованием программного обеспечения, является подтверждение того, что программное обеспечение удовлетворяет потребностям пользователя, предусмотренному применению и специальным требованиям. При проведении аудита программного обеспечения можно выделить этапы, перечисленные ниже. Не все идентифицированные этапы могут быть применимы ко всем видам деятельности по управлению программным обеспечением, и могут потребоваться дополнительные возможности управления (например, управление закупками).
Примечание - Ссылки на структурные элементы [1], приведенные ниже, имеют отношение к разработке программного обеспечения медицинских изделий. a) определение того, что разработка процессов жизненного цикла программного обеспечения запланирована (см. ГОСТ Р ИСО 13485, пункт 7.3.1; подпункт 7.5.1.1; [1], подразделы 5.1; 6.1); b) определение того, что рассмотрение программного обеспечения, способного привести к созданию опасной ситуации, является частью анализа риска (см. [1], подраздел 7.1); c) определение того, что запланированные процессы жизненного цикла программного обеспечения учитывают выходные данные по безопасности, полученные в процессе менеджмента риска медицинского изделия (см. ГОСТ Р ИСО 13485, подраздел 7.1; [1], подраздел 4.3); d) определение того, что требования, установленные к программному обеспечению, соответствуют целям программного обеспечения и включают в себя меры по управлению риском, встроенные в данное программное обеспечение (см. [1], подраздел 5.2); e) определение того, что каждый используемый многоцелевой компонент программного обеспечения удовлетворяет конкретным требованиям к функциональным и эксплуатационным характеристикам, необходимые для его предусмотренного применения, включая спецификацию аппаратного и программного обеспечения, необходимую для поддержания его надлежащего функционирования (см. [1], пункты 5.3.3; 5.3.4); f) подтверждение того, что изменения программного обеспечения были проанализированы с точки зрения возможности создания ими дополнительных причин для возникновения опасных ситуаций или препятствий для имеющихся мер по управлению риском, встроенных в программное обеспечение (см. [1], подраздел 7.4); g) подтверждение того, что проблемы, связанные с программным обеспечением, решены с помощью разработанного процесса решения проблем программного обеспечения, включающего в себя определение причины возникновения проблем и оценивание данных проблем с точки зрения безопасности (см. [1], раздел 9). |
Приложение ДА
(обязательное)
Сравнение структуры настоящего стандарта со структурой документа GHTF/SG4/N30R20:2006 "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 2. Стратегия аудита"
Таблица ДА.1
Номер раздела настоящего стандарта | Номер документа GHTF |
0 Введение | 1.0 Введение |
1 Область применения | 2.0 Область применения |
- | 3.0 Обоснование |
2 Нормативные ссылки | 4.0 Нормативные ссылки |
3 Термины и определения | 5.0 Определения |
4 Общие замечания по стратегии аудита на соответствие регулирующим требованиям | 6.0 Общие замечания по стратегии аудита на соответствие регулирующим требованиям |
5 Проверяемые подсистемы | 7.0 Проверяемые подсистемы |
Приложение А "Схемы биномиального выбора" | Приложение 1 "Схемы биномиального выбора" |
Приложение В "Факторы, применяемые для определения продолжительности аудита" | Приложение 2 "Факторы, применяемые для определения продолжительности аудита" |
Приложение С "Соответствие структурных элементов ИСО 13485:2003 и 21 CFR, часть 820" | Приложение 3 "Соответствие структурных элементов ИСО 13485:2003 и 21 CFR, часть 820" |
Приложение D "Процессы стерилизации" | Приложение 4 "Процессы стерилизации" |
Приложение Е "Аудит программного обеспечения" | Приложение 5 "Аудит программного обеспечения" |
Приложение ДБ
(обязательное)
Текст измененных структурных элементов настоящего стандарта и документа GHTF/SG4/N30R20:2006 "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 2. Стратегия аудита"
1) Текст раздела 3.0, исключенного из документа GHTF/SG4/N30R20:2006 "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 2. Стратегия аудита", так как нецелесообразно включать данный раздел в национальный стандарт в связи с требованиями к структуре национального стандарта Российской Федерации, установленными в ГОСТ Р 1.0-2004, а также в связи с тем, что содержание данного раздела дублируется содержанием введения и раздела 1 "Область применения" настоящего стандарта.
3.0 Обоснование
Настоящий документ предназначен для обеспечения регулирующих органов, организаций, проводящих аудиты, и аудиторов основной информацией о стратегии проведения аудита систем менеджмента качества на соответствие регулирующим требованиям.
Настоящий документ помогает достичь согласованности в проведении аудитов систем менеджмента качества изготовителей медицинских изделий, что необходимо для гармонизации и взаимного признания результатов аудитов.
2) Оригинальный текст измененных терминологических статей ГОСТ Р 54421.
Тексты терминологических статей 3.1 и 3.3 приведены в соответствие с текстом ГОСТ Р ИСО 9000, где данные термины стандартизованы. Соответственно ссылки на терминологический стандарт изменены с ИСО 19011 на ГОСТ Р ИСО 9000.
3.1 аудит (audit): Систематический, независимый, документированный процесс получения записей, изложения фактов (объективных свидетельств) или другой соответствующей информации и объективного их оценивания с целью определения степени выполнения установленных требований (заимствовано из ИСО 19011).
3.3 критерии аудита (audit criteria): Совокупность политики, процедур или требований (заимствовано из ИСО 19011).
3.7 медицинское изделие (medical device): Любой инструмент, аппарат, прибор, устройство, оборудование, имплантат, in vitro реагент или калибратор, программное обеспечение, материал или иные подобные или связанные с ними изделия, предназначенные изготовителем для применения к человеку по отдельности или в сочетании друг с другом в целях:
- диагностики, профилактики, мониторинга, лечения или облегчения заболеваний;
- диагностики, мониторинга, лечения, облегчения или компенсации последствий травмы;
- исследования, замещения или изменения анатомического строения или физиологических процессов;
- поддержания или сохранения жизни;
- управления зачатием;
- дезинфекции медицинских изделий;
- получения информации для медицинских целей посредством исследования in vitro проб, взятых из тела человека, при условии, что их функциональное воздействие на человеческий организм не реализуется за счет фармакологических, иммунологических или метаболических средств, но может поддерживаться такими средствами.
Приложение ДВ
(справочное)
Сведения о соответствии ссылочных национальных стандартов Российской Федерации международным стандартам, использованным в качестве ссылочных в примененном документе
Таблица ДВ.1
Обозначение ссылочного национального стандарта Российской Федерации | Степень соответствия | Обозначение и наименование ссылочного международного стандарта |
ГОСТ Р ИСО 9000-2008 | IDT | ИСО 9000:2008 "Системы менеджмента качества. Основные положения и словарь" |
ГОСТ Р ИСО 13485-2004 | IDT | ИСО 13485:2003 "Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования" |
ГОСТ Р ИСО/ТО 14969-2007 | IDT | ИСО/ТО 14969:2004 "Изделия медицинские. Системы менеджмента качества. Руководство по применению ИСО 13485:2003" |
ГОСТ Р ИСО 14971-2010 | IDT | ИСО 14971:2007 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
ГОСТ Р ИСО/МЭК 17021-2008 | IDT | ИСО/МЭК 17021:2006 "Оценка соответствия. Требования к органам, проводящим аудит и сертификацию систем менеджмента" |
ГОСТ Р ИСО 19011-2003 | IDT | ИСО 19011:2002 "Руководящие указания по аудиту систем менеджмента качества и/или систем экологического менеджмента" |
ГОСТ Р 54421-2011 | MOD | GHTF/SG4/N28R4:2008 "Руководство по аудиту систем менеджмента качества изготовителей медицинских изделий на соответствие регулирующим требованиям. Часть 1. Общие требования" |
ГОСТ Р 53918-2010 | IDT | GHTF/SG3/N15R8:2005 "Изделия медицинские. Руководство по интеграции принципов менеджмента риска в систему менеджмента качества" |
Примечание - В настоящей таблице использованы следующие условные обозначения степени соответствия стандартов: | ||
Библиография
[1] IEC 62304:2006 Medical device software - Software life cycle processes
Электронный текст документа
и сверен по:
, 2012

{kind=link}