ГОСТ Р 54329-2011
(GHTF/SG1/N011:2008)
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
СВОДНЫЙ КОМПЛЕКТ ТЕХНИЧЕСКОЙ ДОКУМЕНТАЦИИ ДЛЯ ДЕМОНСТРАЦИИ СООТВЕТСТВИЯ СУЩЕСТВЕННЫМ ПРИНЦИПАМ ОБЕСПЕЧЕНИЯ БЕЗОПАСНОСТИ И ОСНОВНЫХ ФУНКЦИОНАЛЬНЫХ ХАРАКТЕРИСТИК МЕДИЦИНСКИХ ИЗДЕЛИЙ
Summary technical documentation for demonstrating conformity to the essential principles of safety and performance of medical devices
ОКС 11.040.01
ОКП 94 4000
Дата введения 2012-03-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "МЕДИТЕСТ" на основе аутентичного перевода на русский язык документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 15 июня 2011 г. N 127-ст
4 Настоящий стандарт является модифицированным по отношению к международному документу Целевой группы по глобальной гармонизации (Global Harmonization Task Force - GHTF) GHTF/SG1/N011:2008* "Сводный комплект технической документации для демонстрации соответствия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий" (GHTF/SG1/N011:2008 "Summary technical documentation for demonstrating conformity to the essential principles of safety and performance of medical devices") путем изменения обозначений классов риска медицинских изделий, которые выделены в тексте курсивом.
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
Введение
Документ "Сводный комплект технической документации для демонстрации соответствия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий" разработан Целевой группой по глобальной гармонизации (GHTF) - добровольным сообществом, состоящим из представителей регулирующих органов медицинской промышленности.
Настоящий стандарт разработан для сближения различных регулирующих систем в разных странах. Настоящий стандарт предназначен для регулирующих органов и органов по оценке соответствия и призван обеспечить экономически целесообразный и результативный подход к управлению медицинскими изделиями. Настоящий стандарт содержит рекомендации по составу и содержанию сводного комплекта технической документации, помогающего изготовителю представить в регулирующий орган или орган по оценке соответствия документированные свидетельства того, что медицинское изделие соответствует существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий. Использование сводного комплекта технической документации позволяет изготовителю и контролирующему органу сокращать финансовые затраты, устранять торговые барьеры и облегчать доступ к медицинским изделиям в международном масштабе.
Международный документ подготовлен исследовательской группой 1 (SG1) Целевой группы по глобальной гармонизации (GHTF). Комментарии или вопросы о применении настоящего документа следует направлять на кафедру SG1, подробности изложены на веб-сайте www.ghtf.org.
1 Область применения
1.1 Обоснование
Изготовитель медицинских изделий должен подготовить техническую документацию на свои изделия и поддерживать ее в рабочем состоянии, а также обеспечивать своевременный доступ к данной технической документации, демонстрирующей, как было разработано, спроектировано и изготовлено медицинское изделие. Данная техническая документация, обычно управляемая системой менеджмента качества изготовителя, часто является обширной и делится на части, которые могут находиться, например, в разных структурных подразделениях или на разных производственных площадках.
Следует отметить, что как для регулирующего органа или органа по оценке соответствия, так и для регулируемых отраслей промышленности удобно, если для выполнения установленных действий по оценке соответствия вместо полной технической документации используется сводный комплект технической документации (далее - сводный комплект, СК) на рассматриваемое изделие. Сводный комплект должен быть подготовлен в установленной регулирующими требованиями суммарной или сокращенной форме, со степенью детализации, достаточной для регулирующего органа или органа по оценке соответствия. Документы, содержащиеся в данном комплекте, должны быть частью полной технической документации, хранящейся у изготовителя и позволяющей ему демонстрировать, что медицинское изделие, к которому относится данная техническая документация, соответствует существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (ГОСТ Р ИСО/ТО 16142).
Наличие сводного комплекта помогает уменьшить различия в требованиях к документации, имеющиеся в разных юрисдикциях, сокращая таким образом финансовые затраты на достижение соответствия регулирующим требованиям и помогая пациентам получить доступ к новым технологиям и методикам лечения.
1.2 Цель
Настоящий стандарт предназначен для обеспечения руководства информационным наполнением сводного комплекта, который необходимо собрать и предоставить в регулирующий орган или орган по оценке соответствия для предпродажного анализа и использования на послепродажной стадии с тем, чтобы оценить соответствие рассматриваемого медицинского изделия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (далее - существенные принципы, СП).
1.3 Область применения
Настоящий стандарт применим ко всем изделиям, подпадающим под определение "медицинское изделие", установленное в нормативно-правовых документах (например, в ГОСТ Р ИСО 14155-1), за исключением медицинских изделий для диагностических исследований in vitro проб, полученных из тела человека.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ Р ИСО 14155-1-2008 Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования
ГОСТ Р ИСО 15223-2002* Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации
________________
* На территории Российской Федерации документ не действует. Действует ГОСТ Р ИСО 15223-1-2010, здесь и далее по тексту. - .
ГОСТ Р ИСО/ТО 16142-2008 Изделия медицинские. Руководство по выбору стандартов, поддерживающих важнейшие принципы обеспечения безопасности и эксплуатационных характеристик медицинских изделий
ГОСТ Р ИСО/МЭК 17050-1-2009 Оценка соответствия. Декларация поставщика о соответствии. Часть 1. Общие требования
ГОСТ Р 51609-2000 Изделия медицинские. Классификация в зависимости от потенциального риска применения. Общие требования
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодно издаваемому информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по соответствующим ежемесячно издаваемым информационным указателям, опубликованным в текущем году. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 стандарт, поддерживающий существенные принципы (recognised standard): Стандарт, обеспечивающий презумпцию соответствия конкретным существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (ГОСТ Р ИСО/ТО 16142).
3.2 техническая документация (technical documentation): Документированные свидетельства, обычно являющиеся выходом системы менеджмента качества, демонстрирующие соответствие изделия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (ГОСТ Р ИСО/ТО 16142).
4 Подготовка и использование сводного комплекта
4.1 Подготовка
Изготовители медицинских изделий должны демонстрировать соответствие данных изделий существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий посредством подготовки и хранения технической документации, показывающей, как каждое медицинское изделие было разработано, спроектировано и изготовлено, и содержащей описания и объяснения, необходимые для понимания предусмотренного изготовителем применения данного медицинского изделия, призванного обеспечивать указанное соответствие. Данную техническую документацию обновляют по мере необходимости для того, чтобы отразить текущий статус, спецификации и конфигурацию медицинского изделия.
В целях оценки соответствия изготовитель формирует сводный комплект технической документации из имеющейся полной технической документации на медицинское изделие, обеспечивая тем самым для регулирующего органа/органа по оценке соответствия свидетельства того, что рассматриваемое медицинское изделие соответствует существенным принципам. Сводный комплект отражает статус медицинского изделия в конкретный момент времени (например, на предпродажной стадии при рассмотрении документации или, по требованию регулирующего органа, для послепродажных целей); его подготавливают для оценивания соответствия регулирующим требованиям. На рисунках 1 и 2 показан информационный поток, начиная с полной технической документации и заканчивая сводным комплектом.
Сводный комплект рекомендуется подготавливать на языке, требуемом регулирующим органом/органом по оценке соответствия.
Глубина и степень детализации информации, содержащейся в сводном комплекте, зависят от:
- классификации рассматриваемого медицинского изделия;
- сложности рассматриваемого медицинского изделия;
- имеет ли данное медицинское изделие новую технологию;
- отличается ли применение готового медицинского изделия от первоначального применения, предусмотренного изготовителем;
- является ли медицинское изделие новым для изготовителя;
- ассоциируется ли данный тип медицинского изделия со значительным числом неблагоприятных (нежелательных) событий, включая ошибки применения;
- включает ли медицинское изделие новые или потенциально опасные материалы;
- затрагивает ли данный тип медицинского изделия конкретные проблемы в области здравоохранения.
Сводный комплект должен содержать суммарную информацию по выбранным темам, подробную информацию по некоторым специальным темам (приведенным ниже) и таблицу соответствия медицинского изделия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий (далее - таблица соответствия существенным принципам). Данная информация может включать в себя, например, краткое описание документов, итоговую информацию, проанализированную высшим руководством, или, когда целесообразно, имеющиеся в наличии управляемые документы, достаточные для передачи основной информации и позволяющие проверяющему лицу понять суть вопроса. Таблица соответствия существенным принципам разработана как часть технической документации изготовителя и должна быть документом, управляемым системой менеджмента качества изготовителя. Это позволяет обеспечить краткий обзор существенных принципов и идентифицировать те из них, которые применимы к рассматриваемому медицинскому изделию, выбрать метод демонстрации соответствия медицинского изделия каждому относящемуся к нему существенному принципу и обеспечить ссылку на документы, поддерживающие конкретный существенный принцип. Несмотря на то, что на поддерживающие документы рекомендуется иметь ссылки в таблице соответствия существенным принципам, только некоторые из этих документов следует включать в сводный комплект. Упомянутые ссылки на поддерживающие документы облегчают выполнение запросов от регулирующего органа/органа по оценке соответствия, обеспечивая получение дополнительной информации.
4.2 Использование сводного комплекта на предпродажной стадии
На предпродажной стадии сводный комплект подготавливают и передают в регулирующий орган/орган по оценке соответствия для медицинских изделий классов 2б и 3. Для медицинских изделий классов 1 и 2а сводный комплект подготавливают и передают только по требованию регулирующего органа/органа по оценке соответствия (см. рисунок 1).
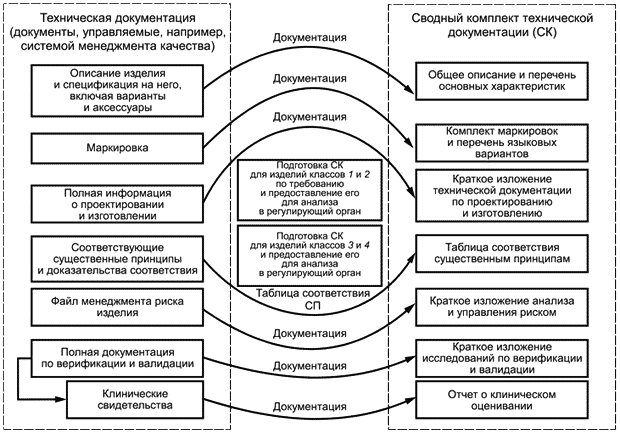
Рисунок 1 - Использование сводного комплекта на предпродажной стадии
Примечания
1 Для медицинских изделий классов 1 и 2а, когда сводный комплект подготавливают по требованию, изготовитель должен быть готов собрать и передать сводный комплект в период времени, указанный регулирующим органом/органом по оценке соответствия. Данный период может быть очень коротким.
2 Копии переданного сводного комплекта должны храниться изготовителем для будущих ссылок.
4.3 Использование сводного комплекта на послепродажной стадии
На послепродажной стадии регулирующий орган/орган по оценке соответствия может затребовать сводный комплект на рассматриваемое медицинское изделие либо для исследования соответствия медицинского изделия класса 1 или 2а, либо для исследования продолжающегося соответствия медицинского изделия класса 2б или 3 (см. рисунок 2). Сводный комплект обычно не используют в целях послепродажного исследования неблагоприятных (нежелательных) событий либо для сообщения информации, содержащейся в послепродажных регистрационных записях, либо для сообщения данных исследований, в которых использованы различные виды информации.
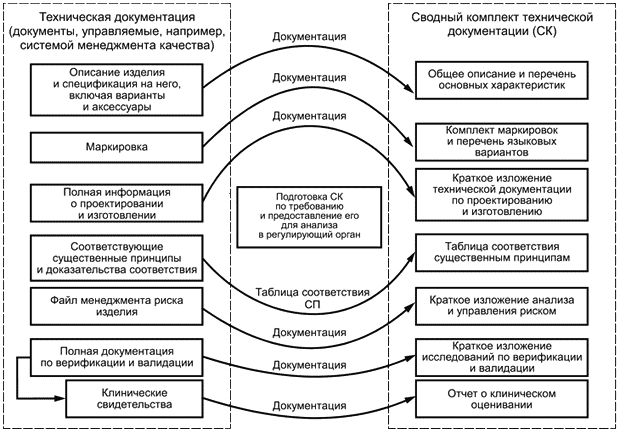
Рисунок 2 - Использование сводного комплекта на послепродажной стадии
Примечания
1 Изготовитель должен быть готов собрать и передать сводный комплект в период времени, указанный регулирующим органом/органом по оценке соответствия. Данный период может быть очень коротким.
2 Копии переданного сводного комплекта должны храниться изготовителем для будущих ссылок.
4.4 Использование сводного комплекта для сообщения об изменениях в регулирующий орган/орган по оценке соответствия
Если для внесения изменения в медицинское изделие необходимо предварительное одобрение регулирующего органа, то для его получения можно использовать сводный комплект. Для этих целей будет подготовлен отдельный руководящий документ.
5 Описание медицинского изделия и его технических характеристик, включая варианты и принадлежности
5.1 Описание медицинского изделия
Сводный комплект должен содержать следующую информацию, описывающую медицинское изделие:
a) общее описание, включая предусмотренное применение медицинского изделия;
b) предусмотренных пользователей и заболевания, которые можно диагностировать и лечить с помощью рассматриваемого медицинского изделия, а также другие факторы, например, критерии отбора пациентов;
c) принципы действия медицинского изделия;
d) класс риска и применимые классификационные правила согласно установленным в ГОСТ Р 51609 принципам классификации медицинских изделий;
e) объяснение новых свойств и характеристик медицинского изделия;
f) описание принадлежностей, других медицинских изделий и изделий, не являющихся медицинскими, но предусмотренных для использования в комбинации с рассматриваемым медицинским изделием;
g) описание или полный перечень различных возможных конфигураций/вариантов рассматриваемого медицинского изделия;
h) общее описание основных функциональных элементов, например, частей и компонентов (включая программное обеспечение при его наличии), их формулировки, состав, функциональные возможности. При необходимости может присутствовать иллюстративный материал (например, диаграммы, фотографии и рисунки), четко демонстрирующий основные части/компоненты медицинского изделия и включающий в себя пояснительные надписи, достаточные для понимания рисунков и диаграмм;
i) описание материалов, входящих в состав основных функциональных элементов, а также материалов, вступающих в непосредственный или опосредованный контакт с телом человека, например, при экстракорпоральной циркуляции биологических жидкостей.
5.2 Техническое описание медицинского изделия
Сводный комплект должен содержать перечень основных характеристик, размеров и эксплуатационных свойств медицинского изделия, его вариантов и принадлежностей (входящих в область применения сводного комплекта), который, как правило, имеется в техническом описании медицинского изделия, доступном конечному пользователю, например, в брошюрах, каталогах и т.п.
5.3 Ссылка на подобные и предыдущие варианты медицинского изделия
В случае необходимости демонстрации соответствия существенным принципам и представления общих сведений сводный комплект должен содержать краткое описание:
a) предыдущих поколений рассматриваемого медицинского изделия, если таковые существовали;
b) подобных медицинских изделий, имеющихся на региональных и международных рынках.
6 Маркировка
Как правило, сводный комплект должен содержать набор всех маркировок, относящихся к рассматриваемому медицинскому изделию, и перечень языковых вариантов для стран, в которых будет реализовано данное медицинское изделие (ГОСТ Р ИСО 15223). Информация по маркированию должна включать в себя:
- маркировки на медицинском изделии и его упаковке;
- инструкции по применению медицинского изделия;
- рекламные материалы.
Набор маркировок должен быть на языке, требуемом регулирующим органом или органом по оценке соответствия.
7 Информация по проектированию и производству
7.1 Проектирование медицинского изделия
Сводный комплект должен содержать информацию, позволяющую контролирующему лицу получать общее представление об основных стадиях проектирования рассматриваемого медицинского изделия. При этом данную информацию нельзя использовать вместо более подробной информации, которая требуется для аудита системы менеджмента качества или для другой деятельности по оценке соответствия. Упомянутая информация может быть представлена в виде блок-схемы.
7.2 Производственные процессы
Сводный комплект должен содержать информацию, позволяющую контролирующему лицу получать общее представление о производственных процессах. При этом данную информацию нельзя использовать вместо более подробной информации, которая требуется для аудита системы менеджмента качества или для другой деятельности по оценке соответствия. Упомянутая информация может быть представлена в виде блок-схемы процессов, дающей, например, общее представление о производстве, сборке, всех заключительных испытаниях медицинского изделия и об окончательной упаковке готового медицинского изделия.
7.3 Производственные площадки
Для видов деятельности, описанных в 7.1 и 7.2, сводный комплект должен идентифицировать производственные площадки, на которых эти виды деятельности были выполнены. Если для данных площадок имеются сертификаты системы менеджмента качества или равноценные документы, то они должны быть приложены к сводному комплекту.
8 Таблица соответствия существенным принципам
Сводный комплект должен включать в себя таблицу соответствия существенным принципам, определяющую:
a) существенные принципы;
b) применим ли каждый существенный принцип к рассматриваемому медицинскому изделию и если нет, то почему;
c) метод(ы), используемый(ые) для демонстрации соответствия рассматриваемого медицинского изделия каждому применимому существенному принципу;
d) ссылку на применяемый(ые) метод(ы) (например, стандарт);
e) точное наименование документа(ов), обеспечивающего(их) свидетельство соответствия каждому применяемому методу.
Для демонстрации соответствия можно использовать один или несколько следующих методов:
a) соответствие методам, поддерживающим существенные принципы, или другим стандартам;
b) соответствие общепринятым в промышленности методам испытаний;
c) соответствие методам испытаний, принятым в данной организации;
d) оценивание доклинических и клинических данных (свидетельств);
e) сравнение с подобным медицинским изделием, уже выпущенным в продажу.
Таблица соответствия существенным принципам должна включать в себя перекрестные ссылки на каждое свидетельство соответствия как в полной технической документации, находящейся у изготовителя, так и в сводном комплекте.
Образец таблицы соответствия существенным принципам приведен в приложении А.
9 Основные результаты анализа и управления риском
Сводный комплект должен содержать краткий перечень рисков, идентифицированных в процессе анализа риска, и описание способов управления данными рисками в целях снижения их до допустимого уровня. Предпочтительно, чтобы данный анализ рисков был основан на стандартах, поддерживающих существенные принципы, и являлся частью плана менеджмента риска изготовителя.
10 Деятельность по верификации и валидации
10.1 Общие положения
Сводный комплект должен содержать документы по верификации и валидации. При этом степень детализации может быть различной (см. 4.1).
Как правило, в сводном комплекте должны быть кратко изложены результаты верификации и валидации, предпринятых для демонстрации соответствия применимым существенным принципам, содержащие:
a) результаты технических испытаний;
b) результаты лабораторных испытаний;
c) результаты испытаний в условиях, имитирующих эксплуатационные;
d) результаты испытаний на животных для демонстрации технической применимости и доказательства правильности концепции готового медицинского изделия;
e) все опубликованные источники касательно рассматриваемого медицинского изделия или подобных медицинских изделий.
Информация о верификации и валидации может также включать в себя:
a) декларацию/свидетельство соответствия стандарту(ам), поддерживающему(им) существенные принципы, и краткое изложение данных, если в упомянутом(ых) стандарте(ах) не установлены критерии приемки;
b) декларацию/свидетельство соответствия опубликованному(ым) стандарту(ам), который не был официально принят, с обоснованием его (их) применения, и краткое изложение данных, если в упомянутом(ых) стандарте(ах) не установлены критерии приемки;
c) декларацию/свидетельство соответствия профессиональному(ым) руководству(ам), производственному(ым) методу(ам) или методу(ам), используемым в организации, с обоснованием их применения, описанием данных методов и краткой информацией со степенью детализации, позволяющей оценить ее достаточность;
d) обзор опубликованных источников касательно рассматриваемого медицинского изделия или подобных медицинских изделий.
Кроме того, если применимо к рассматриваемому медицинскому изделию, сводный комплект должен содержать подробную информацию о:
a) биологической совместимости;
b) лекарственных средствах, входящих в состав рассматриваемого медицинского изделия, включая информацию о совместимости данного медицинского изделия с данными лекарственными средствами;
c) биологической безопасности медицинских изделий, включающих в себя клетки, ткани или их производные, взятые у человека или животных;
d) методах стерилизации;
e) верификации и валидации программного обеспечения;
f) исследованиях на животных, обеспечивающих прямые свидетельства безопасности, и основных функциональных характеристиках рассматриваемого медицинского изделия, особенно если не проводились клинические испытания данного медицинского изделия;
g) клинических данных.
Сводный комплект должен содержать подробную информацию о плане испытаний, протоколы испытаний или исследований в полном объеме, методы анализа полученных данных в дополнение к резюмирующим и к результатам испытаний. Если не были проведены новые испытания, то сводный комплект должен включать в себя обоснование решения о проведении или не проведении испытаний (например, испытание биологической совместимости идентичных материалов было проведено при включении данных материалов в предыдущую, введенную в обращение версию рассматриваемого медицинского изделия). Данное обоснование может быть включено в таблицу соответствия существенным принципам.
10.2 Биологическая совместимость
Сводный комплект должен содержать перечень всех материалов, находящихся в непосредственном или опосредованном контакте с пациентом или пользователем. Если для того, чтобы охарактеризовать физические, химические, токсикологические и биологические характеристики материала, необходимо провести испытания биологической совместимости, то в сводный комплект следует включить подробную информацию о проведенных испытаниях, примененных стандартах, протоколах испытаний, анализ данных и краткое изложение результатов испытаний. Испытания следует проводить на образцах готового, простерилизованного (если поставляется стерильным) медицинского изделия.
10.3 Лекарственные средства
Если медицинское изделие включает в себя лекарственное(ые) средство(а), то сводный комплект должен содержать подробную информацию о применяемом(ых) лекарственном(ых) средстве(ах), его (их) идентичности и источнике(ах), причине его (их) включения в медицинское изделие, безопасности применения и механизме воздействия при предусмотренном применении.
10.4 Биологическая безопасность
Сводный комплект должен содержать перечень всех материалов животного или человеческого происхождения, использованных в рассматриваемом медицинском изделии. Сводный комплект должен содержать подробную информацию об этих материалах, относящуюся к выбору источников/доноров, взятию проб, обработке, хранению, исследованию и обращению с тканями, клетками и веществами животного или человеческого происхождения.
В сводный комплект следует включать результаты валидации процесса, подтверждающие наличие производственных процедур, минимизирующих биологические риски, в частности в отношении вирусов и других возбудителей болезней.
Также следует включать полное описание системы хранения записей, позволяющей осуществлять прослеживаемость от источников материалов до готового медицинского изделия.
10.5 Стерилизация
В тех случаях, когда рассматриваемое медицинское изделие поставляется стерильным, сводный комплект должен содержать подробную информацию о первоначальной валидации процесса стерилизации, включая испытания на биологическую нагрузку, наличие пирогенных веществ, наличие остаточного количества стерилизующего вещества (если применимо) и валидацию процесса упаковывания.
Как правило, подробная информация о валидации должна включать в себя примененный метод, достигнутый уровень обеспечения стерильности, примененные стандарты, протокол стерилизации, разработанный в соответствии с данными стандартами, и краткое изложение полученных результатов.
Необходимо также обеспечить свидетельство продолжающегося статуса валидации процесса стерилизации. Как правило, при этом следует принимать меры или получать свидетельство повторной валидации процессов упаковывания и стерилизации.
10.6 Верификация и валидация программного обеспечения
Сводный комплект должен содержать информацию о процессе проектирования и разработки программного обеспечения и свидетельство валидации программного обеспечения, используемого в готовом медицинском изделии. Данная информация обычно включает в себя краткое изложение результатов всей деятельности по верификации, валидации и результатов испытаний, выполненных как в организации-изготовителе, так и в моделируемой или реальной окружающей пользователя среде до заключительного выпуска медицинского изделия. Данная информация должна также включать в себя все имеющиеся конфигурации аппаратных средств и, когда применимо, операционные системы, идентифицированные в маркировке.
10.7 Исследования на животных
Если для обеспечения соответствия существенным принципам были предприняты исследования на животных, то сводный комплект должен содержать подробную информацию об этих исследованиях.
В сводном комплекте должны быть описаны цели данных исследований, методология, результаты, анализ и заключения, а также соответствие принципам надлежащей лабораторной практики. Следует также описать обоснование (и ограничения) выбора конкретного экспериментального животного.
10.8 Клиническое свидетельство
Сводный комплект должен содержать клиническое свидетельство, демонстрирующее соответствие рассматриваемого медицинского изделия применимым существенным принципам. Данное клиническое свидетельство должно быть оформлено так, как описано в отчете о клинических испытаниях/исследованиях (ГОСТ Р ИСО 14155-1).
11 Формат сводного комплекта
Несмотря на то, что настоящий стандарт не содержит конкретных рекомендаций по формату сводного комплекта, изготовителям и контролирующим органам рекомендуется включать в сводный комплект элементы, описанные в настоящем стандарте (описание медицинского изделия, спецификацию на медицинское изделие и т.д).
12 Декларация соответствия
Декларация соответствия не является составной частью сводного комплекта. Однако она может быть присоединена к сводному комплекту в качестве приложения после оценивания соответствия. Содержание декларации соответствия приведено в ГОСТ Р ИСО/МЭК 17050-1.
Приложение А
(справочное)
Таблица соответствия существенным принципам
Таблица соответствия существенным принципам может быть использована регулирующими органами, органами по оценке соответствия и изготовителями; она позволяет быстро понять, как изготовитель демонстрирует соответствие конкретного медицинского изделия существенным принципам обеспечения безопасности и основных функциональных характеристик медицинских изделий. Таблица соответствия существенным принципам позволяет также легко идентифицировать документы и данные, необходимые в целях оценки соответствия.
Содержание таблицы соответствия существенным принципам меняется в зависимости от конкретного медицинского изделия. Для сложных медицинских изделий таблица будет содержать большое число ссылок на стандарты, протоколы испытаний и другие документы. В таких случаях таблица соответствия существенным принципам может быть очень длинной. Очень простые медицинские изделия будут иметь менее обширные таблицы соответствия существенным принципам, т.к. многие существенные принципы не будут востребованы. В этих случаях в таблицы соответствия может быть включено минимальное число поддерживающих ссылок.
Ниже приведен рекомендуемый формат таблицы соответствия существенным принципам. Подготовка таблицы с использованием данного формата полезна для анализа соответствия медицинского изделия существенным принципам. Последовательное применение данного формата поддерживает гармонизацию между различными юрисдикциями.
Таблицу соответствия существенным принципам заполняют следующим образом.
a) Идентификация медицинского изделия
Изготовитель должен идентифицировать медицинское изделие и, если применимо, различные конфигурации/варианты, охватываемые таблицей соответствия существенным принципам.
b) Применимость внесенного в таблицу соответствия существенного метода к медицинскому изделию
Применим ли внесенный в таблицу существенный принцип к рассматриваемому медицинскому изделию. Ответ может быть либо положительным, либо отрицательным. Если ответ отрицательный, то в столбце, озаглавленном "Метод, применяемый для демонстрации соответствия", должно быть изложено краткое обоснование.
Пример - Для медицинского изделия, не включающего в себя биологические вещества, ответ на существенный принцип 5.8.2 будет отрицательным, а в столбце, озаглавленном "Метод, применяемый для демонстрации соответствия", будет изложено следующее обоснование: "Медицинское изделие не включает в себя биологические вещества".
c) Метод, применяемый для демонстрации соответствия
В данном столбце изготовитель должен установить метод(ы), выбранный(ые) для демонстрации соответствия: стандарт(ы), поддерживающий(ие) существенные принципы; метод(ы) испытаний, применяемый(ые) в промышленности или в данной организации; сравнительное(ые) исследование(ия) или другой(ие) примененный(ые) метод(ы).
d) Ссылка на метод
Установив метод в предыдущем столбце, в данном столбце изготовитель должен привести наименование и ссылку на стандарт(ы), поддерживающий(ие) существенные принципы, метод(ы) испытаний, применяемый(ые) в промышленности или в данной организации, сравнительное(ые) исследование(ия) или другой метод, примененный для демонстрации соответствия. Для стандартов следует привести дату и, при необходимости, раздел(ы) и пункт(ы), демонстрирующие соответствие конкретному существенному принципу.
e) Ссылка на подтверждающие документы
Данный столбец должен содержать ссылку на фактическую техническую документацию, демонстрирующую соответствие существенным принципам, т.е. сертификаты, протоколы испытаний, протоколы валидации, аналитические отчеты или другие документы, появившиеся в результате применения конкретного метода демонстрации соответствия, а также на местонахождение данных документов в сводном комплекте.
Примечание - Приведенная ниже таблица предназначена только для справочных целей. Существенные принципы, перечисленные в первом столбце, взяты из ГОСТ Р ИСО/ТО 16142.
Таблица А.1 - Таблица соответствия существенным принципам
Медицинское изделие: | ||||
Существенный принцип | Применим ли к меди- | Метод, применяемый для демонстрации соответствия | Ссылка на метод | Ссылка на подтверж- |
1 Общие требования | ||||
1.1 Медицинские изделия (далее - изделия) должны быть спроектированы и изготовлены так, чтобы при использовании их по назначению в надлежащих условиях и (при необходимости) с применением технических знаний, опыта, образования или подготовки потенциальных пользователей они не подвергали риску клинические условия или безопасность пациентов либо безопасность и здоровье пользователей или других лиц; любые риски, связанные с их применением, должны быть допустимыми и меньшими, чем приносимая польза пациенту, а также соответствовать высокому уровню защиты здоровья и обеспечения безопасности | ||||
1.2 Решения, принятые изготовителем при проектировании и разработке изделий, должны соответствовать принципам безопасности с учетом современного состояния науки и техники. Чтобы выбрать наиболее правильные решения, изготовитель в следующем порядке должен: - идентифицировать опасности и сопутствующие им риски, связанные с предусмотренным применением или прогнозируемым неправильным использованием медицинского изделия; - исключать или уменьшать, насколько это возможно, риски (обеспечение безопасности изделия на этапах проектирования и разработки); - принимать, по возможности, соответствующие защитные меры, включая, где необходимо, срабатывание тревожной сигнализации в случае невозможности исключения рисков; - информировать пользователей об остаточных рисках, возникающих при наличии недостатков в предпринятых способах защиты | ||||
1.3 Изделия должны функционировать в соответствии со своим назначением и быть спроектированы, изготовлены и упакованы так, чтобы выполнять одну или несколько функций из официально утвержденной области применения конкретного медицинского изделия | ||||
1.4 Технические и эксплуатационные характеристики медицинских изделий, описанные в 1.1-1.3 настоящей таблицы, не должны оказывать вредное воздействие настолько, чтобы подвергать риску клинические условия, а также безопасность пациентов и других лиц в течение срока службы изделия, указанного изготовителем, если изделие находится в нормальных условиях эксплуатации и поддерживается в рабочем состоянии надлежащим образом и в соответствии с инструкциями изготовителя | ||||
1.5 Изделия должны быть спроектированы, изготовлены и упакованы так, чтобы их технические и эксплуатационные характеристики при использовании по назначению не испытывали вредного воздействия при транспортировании и хранении, осуществляемыми в соответствии с инструкциями и с учетом информации изготовителя | ||||
1.6 При функционировании в соответствии с назначением польза от применения медицинского изделия должна превышать вред от нежелательных побочных эффектов | ||||
2 Требования к проектированию и разработке | ||||
2.1 Химические, физические и биологические свойства | ||||
2.1.1 Изделия должны быть спроектированы и изготовлены так, чтобы обеспечивать технические и эксплуатационные характеристики в соответствии с разделом 1 настоящей таблицы. Особое внимание следует уделять: - выбору используемых материалов, особенно их токсичности и, при необходимости, воспламеняемости; - совместимости используемых материалов с биологическими тканями, клетками и физиологическими жидкостями организма с учетом предусмотренного применения изделия; - выбору используемых материалов с учетом, при необходимости, их твердости, износа и усталостной прочности | ||||
2.1.2 Изделия должны быть спроектированы, изготовлены и упакованы так, чтобы свести к минимуму риск, который может быть причинен загрязняющими веществами и прочими отходами производства лицам, занятым транспортированием, хранением и эксплуатацией изделий в процессе лечения с учетом их назначения. Особое внимание следует уделять биологическим тканям, которые подвергаются какому-либо воздействию, а также длительности и частоте этого воздействия | ||||
И т.д. | ||||
Приложение ДА
(справочное)
Сведения о соответствии ссылочных национальных стандартов международным стандартам, использованным в качестве ссылочных в примененном международном документе
Таблица ДА.1
Обозначение ссылочного национального стандарта | Степень соответствия | Обозначение и наименование ссылочного международного стандарта |
ГОСТ Р ИСО 14155-1-2008 | IDT | ИСО 14155-1:2003 "Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования" |
ГОСТ Р ИСО 15223-2002 | IDT | ИСО 15223:2000 "Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации" GHTF/SG1/N43:2005 "Маркировка медицинских изделий" |
ГОСТ Р ИСО/ТО 16142-2008 | IDT | ИСО/ТО 16142:2006 "Изделия медицинские. Руководство по выбору стандартов, поддерживающих важнейшие принципы обеспечения безопасности и эксплуатационных характеристик медицинских изделий" GHTF/SG1/N41:2005 "Существенные принципы обеспечения безопасности и эксплуатационных характеристик медицинских изделий" |
ГОСТ Р ИСО/МЭК 17050-1-2009 | IDT | ИСО/МЭК 17050-1:2004 "Оценка соответствия. Декларация поставщика о соответствии. Часть 1. Общие требования" GHTF/SG1/N40:2006 "Принципы оценки соответствия медицинских изделий" |
ГОСТ Р 51609-2000 | NEQ | Директива 93/42/ЕЕС от 14 июня 1993 г. "О медицинских изделиях", приложение 9 "Критерии классификации" GHTF/SG1/N15:2006 "Принципы классификации медицинских изделий" |
Примечание - В настоящей таблице использованы следующие условные обозначения степени соответствия стандартов: - IDT - идентичные стандарты; - NEQ - неэквивалентные стандарты. | ||
Электронный текст документа
и сверен по:
, 2011

{kind=link}