ГОСТ Р 57449-2017/ISO/TR 24971:2013
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИЗДЕЛИЯ МЕДИЦИНСКИЕ
Руководство по применению ИСО 14971
Medical devices. Guidance on the application of ISO 14971
ОКС 03.120.10
11.040.01
Дата введения 2018-03-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "МЕДИТЕСТ" (ООО "МЕДИТЕСТ") на основе собственного перевода на русский язык англоязычной версии документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 21 апреля 2017 г. N 295-ст
4 Настоящий стандарт идентичен международному документу ISO/TR 24971:2013* "Изделия медицинские. Руководство по применению ИСО 14971" (ISO/TR 24971:2013 "Medical devices - Guidance on the application of ISO 14971", IDT)
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
5 ВВЕДЕН ВПЕРВЫЕ
6 ПЕРЕИЗДАНИЕ. Июнь 2020 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
ИСО/ТО 24971 подготовлен совместно Техническим комитетом ISO/TC 210 "Менеджмент качества и соответствующие общие аспекты медицинских изделий" и Техническим комитетом IEC/SC 62А "Общие аспекты электрооборудования, используемого в медицинской практике". Проект был направлен на голосование в национальные органы как ИСО, так и МЭК.
Опыт показывает, что изготовители испытывают сложности при практическом применении некоторых разделов ИСО 14971. Настоящий стандарт представляет собой руководство, помогающее при разработке, применении и поддержании процесса менеджмента риска, применяемого к медицинским изделиям, целью которого является выполнение требований ИСО 14971. Также настоящий стандарт включает его специальные аспекты для широкого диапазона медицинских изделий, таких как активные, неактивные, имплантируемые и неимплантируемые медицинские изделия и медицинские изделия для in vitro диагностики.
Настоящий стандарт не является полным руководством по применению ИСО 14971 для организаций. Он дополняет руководства, содержащиеся в информативных приложениях ИСО 14971, касающиеся следующих областей:
- руководство по роли международных стандартов по безопасности продукции и стандартов на процессы в менеджменте риска;
- руководство по разработке политики установления критериев допустимости риска;
- руководство по использованию информации, полученной по обратной связи на стадиях производства и постпроизводства;
- руководство по дифференциации информации по безопасности как меры по управлению риском и сообщаемых сведений об остаточном риске;
- руководство по оцениванию совокупного остаточного риска.
В настоящем стандарте приведено несколько подходов, которые может использовать организация для применения и поддержания некоторых аспектов системы менеджмента риска, соответствующей ИСО 14971. Могут использоваться альтернативные подходы, если они удовлетворяют требованиям ИСО 14971.
При оценке применимости руководящих указаний настоящего стандарта необходимо рассматривать природу медицинского(их) изделия(ий), к которому(ым) они применяются, риски, связанные с использованием данного медицинского изделия и применимые регулирующие требования.
1 Область применения
Настоящий стандарт представляет собой руководство, касающееся определенных областей ИСО 14971 при осуществлении деятельности по менеджменту риска.
Стандарт предназначен для оказания помощи изготовителям и другим пользователям стандарта в области:
- понимания роли международных стандартов по безопасности продукции и стандартов на процессы в менеджменте риска;
- разработки политики установления критериев допустимости риска;
- включения производственной и постпроизводственной информации, полученной с помощью обратной связи, в процесс менеджмента риска;
- дифференциации между "информацией по безопасности" и "сообщаемыми сведениями об остаточном риске";
- оценивания полного остаточного риска.
2 Роль международных стандартов по безопасности продукции и стандартов на процессы в менеджменте риска
2.1 Обзор
Международные стандарты по безопасности продукции и стандарты на процессы играют существенную роль в менеджменте риска, как описано в ИСО 14971. В принципе эти стандарты разрабатывают, уже используя концепцию менеджмента риска, которая может включать идентификацию опасностей и опасных ситуаций, определение рисков, оценивание рисков и определение мер по управлению риском.
Более подробную информацию по процессу разработки стандартов на медицинские изделия с использованием концепции менеджмента риска можно найти в таких документах, как ИСО/МЭК Руководство 51 и ИСО/МЭК Руководство 63.
Международные стандарты по безопасности продукции и стандарты на процессы разрабатываются экспертами в данной области и отражают общепризнанный уровень развития науки (см. D.4 ИСО 14971:2007).
Данные стандарты играют важную роль в менеджменте риска. При осуществлении менеджмента риска изготовитель, в первую очередь, должен рассмотреть проект разрабатываемого медицинского изделия, его предполагаемое использование и связанные с ним опасности/опасные ситуации. Изготовитель при выборе применимых стандартов может отдать предпочтение тем, которые уже содержат конкретные требования, помогающие управлять рисками, связанными с конкретными опасностями/опасными ситуациями.
Для медицинских изделий, которые соответствуют требованиям и удовлетворяют критериям данных стандартов, остаточные риски для конкретных опасностей/опасных ситуаций можно счесть допустимыми, если не представлены объективные данные, свидетельствующие об обратном. Некоторыми возможными источниками таких объективных свидетельств могут являться отчеты о неблагоприятных событиях, изъятии продукции из обращения и претензии. Требования международных стандартов, в числе которых инженерные и аналитические процессы, конкретные предельные значения выходных характеристик изделия, предупреждения или проектные спецификации, могут считаться мерами по управлению риском, установленными разработчиками стандартов и предназначенными для реагирования на риски от конкретных опасных ситуаций, которые идентифицированы и оценены как нуждающиеся в управлении.
Во многих случаях разработчики стандартов взяли на себя, выполнили элементы менеджмента риска и предоставили изготовителям готовые решения в виде требований к конструкции и методам испытаний для установления соответствия.
При осуществлении деятельности по менеджменту риска изготовители могут использовать результаты работы разработчиков стандартов и не повторять анализ, ведущий к требованиям стандарта. Следовательно, международные стандарты представляют собой ценную информацию по допустимости рисков, которая валидирована в рамках всемирного процесса оценивания, включая многократные этапы анализа, комментирования и голосования.
2.2 Использование международных стандартов по безопасности продукции в менеджменте риска
Международный стандарт по безопасности продукции может устанавливать требования, которые, при их выполнении, приводят к допустимому риску для конкретных опасных ситуаций (например, ограничения для обеспечения безопасности). Изготовитель может использовать эти требования в менеджменте риска следующим образом.
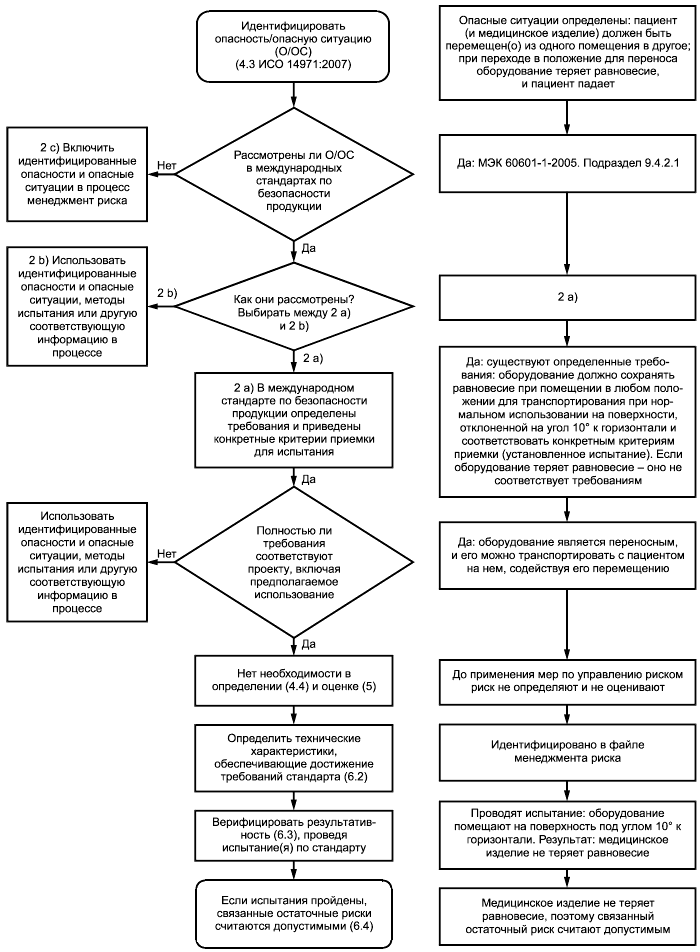
а) Если в международных стандартах по безопасности продукции установлены конкретные технические требования, касающиеся определенных опасностей и опасных ситуаций, а также конкретные критерии приемки, то соответствие этим требованиям считается достаточным для установления того, что остаточные риски снижены до допустимых уровней, если не представлено объективных свидетельств обратного. Например, в МЭК 60601-1 должны контролироваться токи утечки для достижения допустимого уровня риска. В МЭК 60601-1 приведены ограничения в отношении токов утечки, которые, как считается, приводят к допустимым уровням риска при измерении в соответствии с условиями, установленными в 8.7 МЭК 60601-1:2005. Для данного примера дальнейший менеджмент риска не является необходимым. В этом случае необходимо выполнить следующие шаги:
1) выполнить требования пунктов 4.2 и 4.3 ИСО 14971:2007 для идентификации характеристик, касающихся безопасности, и идентификации опасностей и опасных ситуаций, связанных с изделием, настолько полно, насколько это возможно;
2) идентифицировать опасности и опасные ситуации, связанные с рассматриваемым медицинским изделием, которые полностью охватываются международным стандартом по безопасности продукции;
3) для тех идентифицированных опасностей и опасных ситуаций, которые полностью охватываются международными стандартами по безопасности продукции, изготовитель может счесть возможным не определять (п.4.4 ИСО 14971:2007) или оценивать (п.5 ИСО 14971:2007) эти риски. В этом случае для демонстрации завершенности определения рисков и оценивания рисков следует опираться на требования, содержащиеся в международном стандарте;
4) по возможности, изготовитель должен идентифицировать технические характеристики конструкции, которые удовлетворяют требованиям, содержащимся в стандарте, и использовать их как меры по управлению риском (6.2 ИСО 14971:2007).
Примечание - Для некоторых международных стандартов по безопасности продукции возможность идентификации всех конкретных мер по управлению риском ограничена. Одним из примеров является испытание на электромагнитную совместимость в МЭК 60601-1-2 для сложных медицинских изделий;
5) верификация выполнения мер по управлению риском в отношении этих опасных ситуаций может быть проведена посредством анализа проектно-конструкторской документации. Верификация результативности мер по управлению риском может быть проведена посредством испытаний и анализа их результатов, которые должны демонстрировать, что изделие удовлетворяет соответствующим требованиям международного стандарта по безопасности продукции;
6) если применимые требования выполняют, соответствующие остаточные риски считаются допустимыми.
b) Если в международном стандарте по безопасности продукции технические требования и связанные с ними испытания, а также критерии приемки результатов испытаний установлены не в полном объеме, то ситуация является более сложной. В некоторых случаях стандарты предписывают изготовителю проводить конкретные испытания, связанные с известными опасностями или опасными ситуациями, но не приводят критерии приемки (например, МЭК 60601-2-16, к оборудованию для гемодиализа, гемодиафильтрации и гемофильтрации). В других случаях стандарты просто указывают изготовителю на необходимость исследовать конкретные опасности или опасные ситуации в рамках анализа риска (например, 10.2 МЭК 60601-1:2005). Диапазон альтернативных вариантов слишком велик, чтобы предоставить конкретные указания по использованию таких стандартов в процессе менеджмента риска. Тем не менее изготовителям рекомендуется использовать содержание таких стандартов в их менеджменте риска конкретных медицинских изделий.
c) Для опасностей или опасных ситуаций, которые идентифицированы для конкретного медицинского изделия, но специально не рассмотрены ни в одном из стандартов, изготовитель должен учесть эти опасности или опасные ситуации в процессе менеджмента риска. Изготовитель должен определять, оценивать риски и, при необходимости, управлять этими рисками (см. 4.4, 5 и 6 ИСО 14971:2007).
На рисунке 1 представлена блок-схема и пример использования международных стандартов по безопасности продукции.
2.3 Международные стандарты на процессы и ИСО 14971
Международные стандарты на процессы, как показано в примере ниже, часто могут использоваться совместно с ИСО 14971. Это реализуется одним из двух путей:
- в международном стандарте на процесс требуется применение ИСО 14971 как части использования международного стандарта на процесс, например МЭК 62304 на процессы жизненного цикла программного обеспечения; или
- международный стандарт на процессы предназначен для использования в менеджменте риска, например, МЭК 62366 по проектированию с учетом эксплуатационной пригодности и серия ИСО 10993 по биологической оценке.
В любом случае для корректного применения международного стандарта к процессу и с целью достижения допустимых уровней риска в отношении медицинского изделия требуется уделить особое внимание его взаимосвязи с ИСО 14971. Эти два стандарта должны работать совместно так, чтобы их входные данные, выходные данные и согласованность были оптимизированы. Ниже приведены три примера, демонстрирующие такую идеальную ситуацию.
а) МЭК 62304
Взаимосвязь между МЭК 62304 и ИСО 14971 хорошо описана во введении к МЭК 62304.
За основу берется, что программное обеспечение медицинских изделий разрабатывают и обслуживают в рамках системы менеджмента качества (см. 4.1 МЭК 62304:2006) и процесса менеджмента риска (см. 4.2 МЭК 62304:2006). Процесс менеджмента риска уже хорошо рассмотрен в ИСО 14971. Таким образом, МЭК 62304 задает необходимую взаимосвязь напрямую при нормативной ссылке на ИСО 14971. Для программного обеспечения менеджмент риска должен выдвигать некоторые незначительные дополнительные требования, особенно в области идентификации факторов программного обеспечения, связанных с опасностями. Эти требования объединены и включены в п.7 МЭК 62304:2006 как процесс менеджмента риска программного обеспечения.
Приводит ли программное обеспечение к дополнительным опасностям, определяют во время деятельности по идентификации опасностей в процессе менеджмента риска. При определении, приводит ли программное обеспечение к дополнительным опасностям, необходимо учитывать опасности, которые могут быть вызваны программным обеспечением не напрямую (например, предоставление ложной информации, которая может привести к неадекватному лечению). Решение об использовании программного обеспечения для управления риском принимают во время деятельности по управлению риском в процессе менеджмента риска. Процесс менеджмента риска программного обеспечения, требуемый настоящим стандартом, включен в процесс менеджмента риска изделия в соответствии с ИСО 14971.
|
 .
.
Рисунок 1 - Использование международных стандартов по безопасности продукции и пример такого стандарта, в котором определены требования и приведены конкретные критерии приемки
В МЭК 62304 дана нормативная ссылка на ИСО 14971 и установлены специальные требования:
- планирование разработки программного обеспечения (см. 5.1 МЭК 62304:2006), что согласуется с планом менеджмента риска, необходимым согласно ИСО 14971; и
- процесс менеджмента риска программного обеспечения (см. 7 МЭК 62304:2006), основанный на ИСО 14971.
b) МЭК 62366
Блок-схема на рисунке А.1 МЭК 62366:2007 показывает взаимосвязь и взаимодействие двух параллельных и взаимосвязанных процессов. В дополнение к нормативным ссылкам на ИСО 14971, в МЭК 62366:2007 определены три специальных пункта, в которых процесс проектирования с учетом эксплуатационной пригодности может дополнять и взаимодействовать с менеджментом риска, как описано в ИСО 14971:
- п.5.3.1 МЭК 62366:2007 требует: "Идентификация характеристик, связанных с безопасностью (часть анализа риска), которая сфокусирована на эксплуатационной пригодности, должна быть проведена в соответствии с ИСО 14971:2007, 4.2.";
- п.5.3.2 МЭК 62366:2007 требует: "изготовитель должен идентифицировать известные или предполагаемые опасности (часть анализа риска), связанные с эксплуатационной пригодностью, в соответствии с ИСО 14971:2007, 4.3.";
- п.5.9 МЭК 62366:2007 по валидации эксплуатационной пригодности содержит несколько ссылок на деятельность, проводимую как часть менеджмента риска.
c) МЭК 10993 (все части), Биологическая оценка медицинских изделий
Во введении к ИСО 10993-1 установлено, что он предназначен для применения в качестве руководящего документа по биологической оценке медицинских изделий в рамках менеджмента риска как части общей оценки и разработки конкретного изделия.
В приложении ИСО 10993-1:2009 указан ИСО 14971 как стандарт, обеспечивающий руководство по подходу, с точки зрения менеджмента риска, к идентификации биологических опасностей, связанных с медицинскими изделиями, определению и оцениванию рисков, управлению рисками, а также мониторингу результативности мер по управлению риском.
Данный подход объединяет как анализ и оценку существующих данных из всех источников, так и выбор и применение дополнительных испытаний (при необходимости), тем самым обеспечивая проведение совокупного оценивания биологической реакции на каждое медицинское изделие, связанное с безопасностью его применения.
ИСО 10993-1 точно встраивается в менеджмент риска, определенный в ИСО 14971.
Биологическое оценивание должно быть проведено способом, подобным тому, который используют для оценивания других рисков продукции и включать:
- анализ риска (Каковы опасности и связанные риски?);
- оценивание риска (Является ли он допустимым?);
- управление риском (Как он будет управляться?);
- оценивание соотношения совокупный остаточный риск/польза.
Как установлено в ИСО 14971, если в ходе оценивания совокупного остаточного риска, основанного на существующих данных, установлено, что идентифицированные риски являются допустимыми, то дальнейшее управление риском не требуется. В противном случае необходимо принять соответствующие меры для дальнейшего оценивания или уменьшения рисков.
Результатом такого оценивания является отчет о биологической оценке.
Применение
Состояния, идентифицированные в ИСО 10993-1 как опасности, включают:
- острую токсичность;
- хроническую токсичность;
- раздражение (кожи, глаз, слизистых оболочек);
- гиперчувствительность;
- генотоксичность;
- концерогенность*.
________________
* Текст документа соответствует оригиналу. - .
Вызывают ли материалы, предполагаемые для изготовления конкретного медицинского изделия, такие состояния?
Методы, которые используют для определения возможности материалов конкретного медицинского изделия вызывать такие состояния, включают:
- определение и оценку химических параметров;
- литературный обзор;
- испытания (in vitro/in vivo, не клинические);
- практический опыт.
Допустимы ли уровни воздействия?
В соответствии с ИСО 10993-1 лицо, проводящее экспертную оценку, должно определить, достаточно ли доступной информации или данных для определения того, является ли допустимым связанный с биологическими опасностями совокупный остаточный риск. Данный вывод фиксируют в отчете о биологической оценке, который становится составной частью файла менеджмента риска.
3 Разработка политики установления критериев допустимости риска
В соответствии с 3.2 ИСО 14971 высшее руководство должно разрабатывать и документировать политику установления критериев допустимости риска. Эта политика должна обеспечивать, чтобы критерии:
a) основывались на применимых национальных или региональных нормативных документах;
b) основывались на соответствующих международных стандартах;
c) учитывали доступную информацию, такую как общепринятый уровень развития науки и потребности заинтересованных сторон.
Примечание - Также может быть включена другая подходящая информация.
Данная политика может охватывать либо весь диапазон медицинских изделий изготовителя, либо может иметь различные формы в зависимости от разнообразия выпускаемых в обращение групп медицинских изделий.
При разработке или актуализации политики необходимо учитывать следующее:
- применимые региональные регулирующие требования, которые действуют там, где продается медицинское изделие;
- соответствующие международные стандарты на конкретные медицинские изделия или предполагаемое применение медицинских изделий, которые могут помочь в определении принципов установления критериев допустимости риска (см. 2.2);
- информацию о современном уровне научно-технического развития, которая может быть получена на основании обзора литературы и другой информации по аналогичным медицинским изделиям, включая изделия конкурентов;
- одобренные и всеобъемлющие запросы от основных заинтересованных лиц. Некоторые возможные источники информации по пациентам и клиническим перспективам могут включать новостные сообщения, общественное мнение, форумы пациентов, а также входные данные от внутренних подразделений с экспертным знанием проблем заинтересованных лиц, таких как клиническое подразделение.
Изготовитель должен предоставить руководящие указания по разработке актуальных критериев допустимости риска, которые будут использовать в плане менеджмента риска в отношении рассматриваемых конкретных медицинских изделий (см. 3.4 ИСО 14971:2007).
Анализ пригодности процесса менеджмента риска через запланированные интервалы времени, как требуется в 3.2 ИСО 14971:2007, может продемонстрировать обоснованность ранее используемых критериев допустимости риска или привести к изменениям в политике. Такие изменения могут также привести к пересмотру правильности ранее принятых решений о допустимости риска.
4 Производственная и постпроизводственная обратная связь
4.1 Обзор
Как правило, первоначальная оценка риска основана на опыте работы с аналогичными медицинскими изделиями, или принадлежностями на рынке, или на предположениях, когда новые медицинские изделия выпускают на рынок. Информация, полученная после выпуска изделия в обращение, является ценной для подтверждения или коррекции оценки и допущений (как завышенных, так и заниженных), или идентификации упущений, сделанных в ходе этапов анализа и управления риском. Раздел 9 ИСО 14971:2007 требует, чтобы в организации изготовителя была установлена петля обратной связи для сбора и оценивания такой информации, которая может иметь отношение к безопасности медицинского изделия (см. рисунок 2). Петля обратной связи должна состоять из следующих этапов:
|
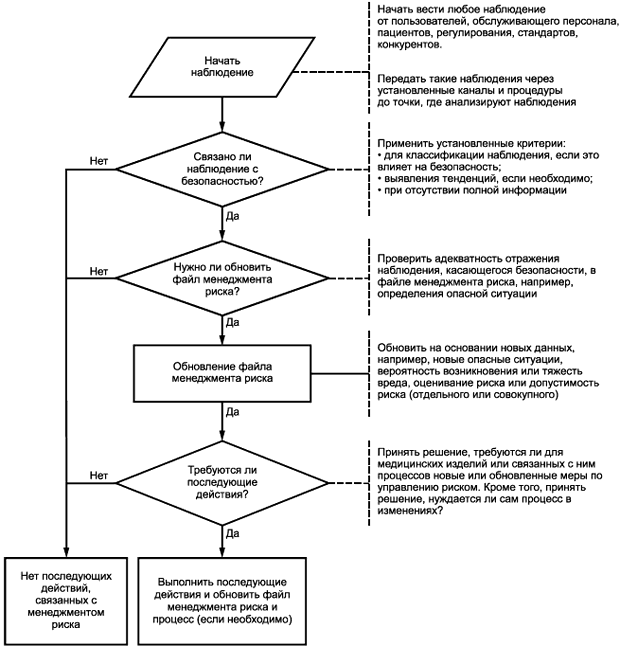
Рисунок 2 - Производственная и постпроизводственная обратная связь
- наблюдение и передача;
- оценка;
- действия
Чтобы обратная связь была эффективной, необходимо, чтобы было определено лицо, ответственное за поддержание файла менеджмента риска.
4.2 Наблюдение и передача
Наблюдение обеспечивает информацией о медицинских изделиях или опытом работы с ними, которые необходимо сравнить с текущим файлом менеджмента риска. Входными данными процесса наблюдения могут служить ряд различных источников, каждый из которых может влиять на безопасность медицинских изделий, например:
- информация по производственной или научно-исследовательской и опытно-конструкторской (R&D) деятельности, выполняемой изготовителем или по его заказу;
- информация по монтажу, обслуживанию и/или обучению персонала, выполняемых изготовителем или по его заказу;
- информация от пользователя(ей) медицинских изделий (например, претензии потребителей, опрос пользователей);
- информация на основе опыта применения медицинских изделий конкурентов, полученная из отчетов об инцидентах (например, из баз данных, которые региональные регулирующие органы используют для сбора и анализа опыта применения изделий);
- клиническая информация (например, клинические наблюдения на стадии применения медицинского изделия изготовителя или другая опубликованная клиническая литература по аналогичным медицинским изделиям или изделиям конкурентов);
- информация о новых или измененных стандартах и регулирующих документах;
- для изделий, содержащих лекарственное средство, информация, касающаяся такого лекарственного средства.
Информацией, имеющей отношение к медицинскому изделию изготовителя, может служить и информация, не связанная напрямую с их собственным изделием или продукцией конкурента. Информация, касающаяся аналогичных медицинских изделий с аналогичными характеристиками предусмотренного назначения/применения или аналогичными принципами работы, также может дать полезную информацию с рынка об актуальности рисков медицинского изделия изготовителя.
При разработке способов получения или обнаружения информации с рынка изготовители должны быть осторожными, чтобы не сделать процесс предвзятым. Средства получения информации или способы обратной связи должны быть нейтральными в отношении получения отрицательной или положительной обратной связи. Кроме того, обратная связь должна включать в себя события, которые произошли (включая корректирующие действия), а также события, которые могут произойти (включая предупреждающие действия).
Чтобы информация с рынка была полезной, она должна быть доведена до сведения лиц или подразделения в пределах организации, которые несут ответственность и имеют полномочия проводить сравнения с текущим файлом менеджмента риска и вносить при необходимости в него изменения.
Средства передачи данной информации зависят от источника информации. Один вид информации запрашивают (инициирует изготовитель), а другой вид информации передают (инициируют такие источники, как потребители, регулирующие органы или пациент). В любом случае организация должна обеспечить планирование и создание эффективных каналов связи для своевременного и точного получения информации. Частота, с которой изготовитель запрашивает информацию от различных источников (включая пользователей), зависит от зрелости медицинского изделия, его технологии и конкретного рынка.
Различные подразделения в рамках организации изготовителя могут получать и обрабатывать различные виды информации, например:
- претензии потребителей и отчеты о неблагоприятных событиях;
- отчеты об обслуживании и монтаже;
- новые или пересмотренные регулирующие требования, стандарты или руководства;
- отчеты о выявленных несоответствиях на производстве.
Важно, чтобы вся соответствующая информация по всем видам информации рассматривалась и передавалась в ту часть организации изготовителя, которая несет ответственность и уполномочена проводить оценку риска (см. 4.3).
Если вероятность некоторых событий (например, отказа компонентов) является важным фактором, способствующим оцениванию риска, необходимо учитывать статистический анализ тенденций таких событий.
4.3 Оценка
Любой пересмотр оценки риска, основанный на новых наблюдениях, должен подвергаться тому же уровню управления и анализа, что и первоначальная оценка риска. Если потребуется, он должен включать идентификацию любых последующих мер по управлению риском. Это управление должно включать анализ и утверждение лицами, имеющими те же функции или из тех же подразделений, как и те, которые подписали изначально. Новые наблюдения в отношении безопасности необходимо оценивать с использованием текущих критериев допустимости риска.
Новые наблюдения в отношении безопасности необходимо сравнивать с установленными в файле менеджмента риска с целью проверки обоснованности любых сделанных допущений. Некоторые вопросы предложены ниже:
a) Остается ли обоснованным предусмотренное применение/назначение?
b) Есть ли тенденция увеличения случаев использования не по назначению?
c) Случается ли обоснованно прогнозируемое неправильное применение, не рассмотренное в первоначальном процессе менеджмента риска?
d) Есть ли свидетельства присутствия новых опасностей или опасных ситуаций, не идентифицированных изначально в процессе идентификации опасностей?
e) Остаются ли обоснованными определения тяжести и вероятности возникновения конкретных рисков?
f) Есть ли какие-либо свидетельства, что критерии допустимости риска должны быть скорректированы?
g) Адекватно ли доказана результативность мер по управлению риском?
h) Точно ли отражает анализ соотношения риск/польза реальный опыт применения изделия?
Если по результатам анализа данных предполагается исправление или корректировка текущего файла менеджмента риска, то остаточные риски должны быть оценены на основании новых данных. Кроме того, должен быть пересмотрен совокупный остаточный риск.
4.4 Действия
Если в результате анализа новых данных остаточный риск становится недопустимым и анализ соотношения риск/польза показывает, что польза не перевешивает риск, то дальнейшее управление риском необходимо проводить в двух областях:
a) Медицинские изделия, уже установленные и используемые на рынке, должны быть подвергнуты корректировке.
b) Конструкция медицинских изделий, изготавливаемых с этого момента времени, или связанные процессы должны быть пересмотрены, а изменения реализованы.
Для уже установленных и используемых на рынке медицинских изделий меры по управлению риском могут отличаться от тех, которые применяют к изделиям в текущем производстве.
В отношении уже установленных и используемых на рынке медицинских изделий экстренная информация (например, письмо клиента) незамедлительно может быть представлена пользователям еще до разработки и верификации мер по управлению риском на предмет их результативности.
В том случае, если необходима модификация или замена медицинских изделий, скорость проведения данных мероприятий способствует результативности действий по уменьшению риска.
Примечание - Данная информация известна как пояснительное уведомление в ИСО 13485, а также как уведомление, касающееся обеспечения безопасности в общеевропейском документе MEDDEV 2.12-1.
Результат оценки постпроизводственной информации может служить в качестве входных данных для анализа пригодности процесса менеджмента риска в запланированные интервалы времени для обеспечения постоянной результативности процесса менеджмента риска (см. 3.2 ИСО 14971:2007).
5 Дифференциация информации для обеспечения безопасности и раскрытия остаточного риска
5.1 Различие между "информацией о безопасности" и "сообщаемыми сведениями об остаточном риске"
Разница между "информацией по безопасности" и "сообщаемыми сведениями об остаточном риске" объяснена в приложении J ИСО 14971:2007. Тем не менее накопленный опыт изготовителей показывает, что существует путаница между этими двумя понятиями. Настоящий стандарт предназначен для разъяснения этих различий.
Информация по безопасности считается мерой по управлению риском. Эта мера может предписывать или предупреждать. В ИСО 14971 требуется, чтобы была верифицирована результативность таких мер. Она может быть представлена в виде предупреждения или мер предосторожности.
Остаточный риск определен в ИСО 14971:2007 как риск, остающийся после принятия всех мер по управлению риском (которые могут включать информацию по безопасности).
В ИСО 14971 требуется, чтобы вся информация по безопасности прослеживалась в файле менеджмента риска. Решение изготовителя, касающееся информирования в отношении сообщаемых сведений об остаточном риске, может быть зарегистрировано в файле менеджмента риска.
5.2 Информация по безопасности
Хотя информацию по безопасности рассматривают как меру по управлению риском в ИСО 14971:2007 (п.6.2с), это наименее предпочтительный вариант после попытки создания безопасной конструкции и разработки мер защиты. Это означает, что информация по безопасности должна быть использована только после того, как изготовитель установил, что дальнейшие способы уменьшения риска, делающие медицинское изделие более безопасным, и принятие защитных мер являются практически невозможными. Содержание информации по безопасности может быть предписано локальными регулирующими документами.
Верификация результативности информации по безопасности может проводиться в рамках процесса проектирования с учетом эксплуатационной пригодности (МЭК 62366).
Информация по безопасности должна давать пользователю четкие инструкции, какие действия следует предпринимать или каких избегать для предотвращения опасных ситуаций или вреда от них. Она обычно представляется в виде предупреждений или мер предосторожности (см. J.2 ИСО 14971:2007).
Информация по безопасности может быть дана в форме этикеток с предупреждениями, прикрепленными к медицинскому изделию, или в виде предупреждений в инструкции по эксплуатации/применению.
Некоторые примеры приведены ниже:
- предупреждение: Не наступайте на поверхность;
- предупреждение: Не удаляйте покрытие, риск электрического удара;
- предупреждение: Используйте с осторожностью. Образцы плазмы, содержащие более 60 мг/дл гемоглобина, влияют на принцип исследования, тем самым ограничивая диагностический результат.
5.3 Сообщаемые сведения об остаточном риске
Раскрытие информации в отношении остаточного риска является описательным и может содержать справочные сведения об остаточных рисках, связанных с использованием медицинского изделия. Целью является указание в сопроводительной документации информации, позволяющей пользователю и потенциальному пациенту принять взвешенное решение с учетом остаточных рисков и пользы от применения медицинского изделия (см. J.3 ИСО 14971:2007).
Изготовитель должен рассмотреть вопрос о способе и носителях информации для раскрытия остаточного риска. Данная информация может быть существенной в процессе принятия клинического решения. В рамках использования по назначению, оператор или пользователь может решить, какие клинические параметры изделия могут быть использованы для достижения определенной пользы для пациента. Информирование в отношении остаточного риска может быть также полезным для оператора, пользователя или лечебного учреждения для подготовки пациента к возможным побочным эффектам или опасностям, которые могут возникнуть во время или после использования медицинского изделия. Следует отметить, что оператор, пользователь и пациент могут быть одним лицом, например для медицинского изделия, предназначенного для использования в домашних условиях.
Ниже приведены некоторые примеры для иллюстрации остаточных рисков, связанных с использованием медицинских изделий, а также побочных эффектов, которые обычно подлежат раскрытию:
- Для лечения опухолей могут использовать линейные ускорители. Остаточные риски облучения опухолей могут включать вероятность эритемы или выпадения волос.
- Когда пациенты подвергаются исследованиям на аппарате магниторезонансной томографии (МРТ), они иногда испытывают тревогу из-за нахождения в замкнутом пространстве, громкого шума, создаваемого оборудованием, и необходимости оставаться неподвижным во время получения изображения.
6 Оценивание совокупного остаточного риска
6.1 Обзор
После оценивания каждой идентифицированной отдельной опасной ситуации изготовитель затем должен рассмотреть совокупное влияние индивидуальных остаточных рисков и принять решение о допустимости совокупного остаточного риска на основе критериев для остаточного риска, установленных в плане менеджмента риска. Данный этап особенно важен для сложных медицинских систем и для медицинских изделий с большим количеством отдельных рисков. Такое оценивание следует использовать для принятия окончательного решения о безопасности продукции.
В разделе 7 ИСО 14971:2007 требуется, чтобы совокупный остаточный риск оценивался по критериям, установленным в плане менеджмента риска. Тем не менее определение совокупного остаточного риска является сложной задачей, которая не может быть решена простым численным сложением всех отдельных рисков. Неизвестно, можно ли в принципе складывать риски, т.к. каждой вероятности возникновения вреда соответствует разная тяжесть этого вреда. Такие сложности могут возникнуть по следующим причинам:
- Даже на поздних этапах разработки медицинских изделий достоверность оценки вероятности может существенно различаться. Некоторые вероятности известны достаточно точно либо на основании исторических данных со схожими медицинскими изделиями, либо на основании испытаний. Другие вероятности можно оценить только приблизительно или невозможно оценить вообще, например вероятность отказа программного обеспечения. Также обычно не представляется возможным объединить тяжести вреда отдельных рисков в широкие категории, используемые для анализа риска.
- В ИСО 14971 не установлено, что критерии допустимости риска для отдельных рисков должны быть теми же самыми, что и критерии допустимости совокупного риска. Критерии, используемые для оценивания отдельных рисков, обычно основаны на вероятности возникновения конкретной тяжести вреда.
В D.4 и D.7 ИСО 14971:2007 перечислены некоторые возможные общие методы или методики оценивания совокупного остаточного риска и некоторые полезные соображения, влияющие на их выбор. В отношении критериев ИСО 14971 в целом указывает, что они должны основываться на политике установления критериев допустимости риска, а соответствующее руководство можно найти в разделе 3. Как критерий, так и связанный с ним метод должны быть определены в плане менеджмента риска. Настоящий стандарт предназначен для помощи в установлении этих критериев и методов.
6.2 Входные данные и другие подходы по оцениванию совокупного остаточного риска
Совокупный остаточный риск может быть оценен только после реализации и верификации всех мер по управлению риском. Этот означает, что все идентифицированные опасные ситуации были оценены и что все риски были снижены до допустимых уровней или были приняты на основании анализа соотношения риск/польза. Некоторые примеры входных данных и их использования приведены ниже. Они могут использоваться как входные данные по оцениванию совокупного остаточного риска и как соображения, которые необходимо использовать при определении, является ли совокупный остаточный риск допустимым.
a) Изготовитель может сравнить рассматриваемое медицинское изделие с аналогичным медицинским изделием, представленным на рынке (см. D.7.7 ИСО 14971:2007). Для того чтобы изготовитель сделал обоснованное заключение о совокупном остаточном риске с учетом медицинской пользы от рассматриваемого медицинского изделия, необходимо рассмотреть текущую актуальную информацию о предусмотренном применении/назначении и связанных с ним неблагоприятных событиях в отношении аналогичных медицинских изделий, представленных на рынке, а также информацию из научной литературы, включая информацию о клиническом опыте. Ключевым вопросом является вопрос о том, имеет ли рассматриваемое медицинское изделие такой же или лучший уровень безопасности, что и медицинское изделие, которое уже можно считать имеющим допустимый совокупный остаточный риск.
b) Для оценивания совокупного остаточного риска с учетом медицинской пользы от рассматриваемого медицинского изделия изготовитель может также использовать мнение экспертов, не входящих в организацию изготовителя (см. D.7.8 ИСО 14971:2007). Эти эксперты могут быть отобраны из различных дисциплин и включать тех, кто имеет клинический опыт, и тех, кто продает аналогичные медицинские изделия. Такие специалисты могут помочь изготовителю учитывать интересы заинтересованных лиц. Следует обратить внимание на требования к профессиональной подготовке и опыту, установленные в 3.2 и 3.3, 3.4 b) и c), и A.2.3.3 ИСО 14971:2007.
c) Даже если все индивидуальные риски были идентифицированы и сочтены допустимыми, для некоторых из них, как часть оценивания совокупного остаточного риска, может потребоваться дальнейший анализ.
Одним из примеров такой ситуации может быть то, что многие отдельные риски близки к недопустимым уровням. Следовательно, допустимость совокупного остаточного риска может быть подвергнута сомнению и может быть принято обоснованное решение о дальнейшем исследовании медицинского изделия и связанного с ним файла менеджмента риска. Другим примером может быть то, что существуют риски, которые являются взаимозависимыми как от вызвавших их причин, так и от принятых мер по управлению риском. Меры по управлению риском должны быть верифицированы на результативность не только по отдельности, но также и совместно с другими мерами по управлению риском. Это относится и к мерам по управлению риском, которые разработаны для уменьшения нескольких рисков одновременно. Дерево отказов или анализ дерева событий могут быть полезными инструментами для демонстрации таких взаимосвязей между рисками и используемыми мерами по управлению риском.
d) Другие подходы по оцениванию совокупного остаточного риска:
1) Полезную информацию можно получить, используя результаты оценивания эксплуатационной пригодности или клинического опыта, полученного при валидации проекта.
2) Может быть полезно визуальное представление остаточных рисков. Каждый отдельный остаточный риск может быть показан в матрице рисков, как на рисунках D.4 и D.5 ИСО 14971:2007, давая графическое представление о распределении рисков. Если многие риски находятся в верхней области тяжести или в верхней области вероятности матрицы рисков либо кластеры рисков пограничны, то распределение рисков может показывать, что совокупный остаточный риск является недопустимым, хотя каждый индивидуальный риск был обоснованно сочтен допустимым.
3) При оценивании совокупного риска необходимо учитывать все отдельно проведенные анализы соотношения риск/польза.
4) Если при анализе риска имели место компромиссы между рисками, это может указывать на то, что совокупный остаточный риск необходимо анализировать более тщательно. Это могут быть случаи, когда один из рисков может быть несколько увеличен с тем, чтобы другой риск мог быть уменьшен. Например, риск для одного лица (пользователя) допускается увеличить, тогда как риск для другого лица (пациента) может быть уменьшен. Это называется параллаксом риска. Оценивание может носить характер маневрирования между основными рисками с обязательным описанием того, почему баланс компромиссных решений практичен и почему уровень комбинированного риска при принятии компромиссных решений в отношении рисков является допустимым.
В конечном итоге оценивание совокупного остаточного риска основано на клинической оценке. Результаты оценивания совокупного остаточного риска образуют части файла менеджмента риска. Крайне полезно документировать обоснованность допустимости совокупного остаточного риска.
УДК 006.83:006.354 | ОКС 03.120.10 |
11.040.01 | |
Ключевые слова: система менеджмента качества, изделия медицинские, риск, менеджмент риска, управление риском, информация по безопасности, остаточный риск, опасные ситуации, вред, изготовители медицинских изделий | |
Электронный текст документа
и сверен по:
, 2020

{kind=link}