ГОСТ IEC 62321-5-2016
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ОПРЕДЕЛЕНИЕ РЕГЛАМЕНТИРОВАННЫХ ВЕЩЕСТВ В ЭЛЕКТРОТЕХНИЧЕСКИХ ИЗДЕЛИЯХ
Часть 5
Определение кадмия, свинца и хрома в полимерах и электронных частях систем, а также кадмия и свинца в металлах методами AAS, AFS, ICP-OES и ICP-MS
Determination of certain substances in electrotechnical products. Part 5. Cadmium, lead and chromium in polymers and electronics and cadmium and lead in metals by AAS, AFS, ICP-OES and ICP-MS
МКС 43.040.10
Дата введения 2022-03-01
Предисловие
Цели, основные принципы и общие правила проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0 "Межгосударственная система стандартизации. Основные положения" и ГОСТ 1.2 "Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены"
Сведения о стандарте
1 ПОДГОТОВЛЕН Научно-производственным республиканским унитарным предприятием "Белорусский государственный институт стандартизации и сертификации" (БелГИСС) на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 5
2 ВНЕСЕН Государственным комитетом по стандартизации Республики Беларусь
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 28 июня 2016 г. N 49)
За принятие стандарта проголосовали:
Краткое наименование страны по МК (ИСО 3166) 004-97 | Код страны по МК (ИСО 3166) 004-97 | Сокращенное наименование национального органа по стандартизации |
Армения | AM | ЗАО "Национальный орган по стандартизации и метрологии" Республики Армения |
Беларусь | BY | Госстандарт Республики Беларусь |
Казахстан | KZ | Госстандарт Республики Казахстан |
Киргизия | KG | Кыргызстандарт |
Россия | RU | Росстандарт |
Таджикистан | TJ | Таджикстандарт |
Узбекистан | UZ | Узстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 26 августа 2021 г. N 838-ст межгосударственный стандарт ГОСТ IEC 62321-5-2016 введен в действие в качестве национального стандарта Российской Федерации с 1 марта 2022 г.
5 Настоящий стандарт идентичен международному стандарту IEC 62321-5:2013* "Определение регламентированных веществ в электротехнических изделиях. Часть 5. Определение кадмия, свинца и хрома в полимерах и электронных частях систем, а также кадмия и свинца в металлах методами AAS, AFS, ICP-OES и ICP-MS" ("Determination of certain substances in electrotechnical products - Part 5: Cadmium, lead and chromium in polymers and electronics and cadmium and lead in metals by AAS, AFS, ICP-OES and ICP-MS", IDT).
Международный стандарт разработан Техническим комитетом по стандартизации IEC TC 111 "Стандартизация в области окружающей среды относительно электрических и электронных товаров и систем" Международной электротехнической комиссии (IEC).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
6 ВВЕДЕН ВПЕРВЫЕ
Информация о введении в действие (прекращении действия) настоящего стандарта и изменений к нему на территории указанных выше государств публикуется в указателях национальных стандартов, издаваемых в этих государствах, а также в сети Интернет на сайтах соответствующих национальных органов по стандартизации.
В случае пересмотра, изменения или отмены настоящего стандарта соответствующая информация также будет опубликована на официальном интернет-сайте Межгосударственного совета по стандартизации, метрологии и сертификации в каталоге "Межгосударственные стандарты"
Введение
Широкое использование изделий электротехнического назначения повысило внимание к их воздействию на окружающую среду. Во многих странах мира были приняты технические документы, предусматривающие определенный порядок работы с отходами, веществами и затраченной энергией при использовании электротехнических изделий.
Использование таких веществ, как свинец (Pb), ртуть (Hg), кадмий (Cd), шестивалентный хром (Cr(VI)), содержащийся в неорганических и органических соединениях, а также два типа бромированных огнестойких ингибиторов, включая полибромбифенилы (РВВ) и полибромированные дифениловые эфиры (PBDE), в электротехнических изделиях регламентируется национальным законодательством.
Целью стандартов серии IEC 62321 является установление методов контроля, которые позволят определить уровень регламентированных веществ в электротехнических изделиях.
1 Область применения
В настоящем стандарте установлены методы определения содержания свинца, кадмия и хрома в полимерах, металлах и электронике методами AAS, AFS, ICP-OES and ICP-MS.
В настоящем стандарте установлены методы определения уровней кадмия (Cd), свинца (Pb) и хрома (Cr) в электротехнических изделиях. Настоящий стандарт охватывает три типа материалов: полимеры/полимерные детали, металлы и сплавы, электронику.
В настоящем стандарте образец рассматривается, как объект, подлежащий обработке и измерениям. Что представляет собой образец или как его получить определяется субъектом, проводящим испытания. Дальнейшие руководство по получению типовых образцов из готовых электронных изделий, которые будут проверены на содержание регламентированных веществ, установлено в IEC 62321-2. Следует отметить, что выбор и/или определение образца может повлиять на интерпретацию результатов испытаний. В настоящем стандарте приводится описание четырех методов, а именно: AAS (атомно-абсорбционная спектрометрия), AFS (атомно-флуоресцентная спектрометрия), ICP-OES (оптическая эмиссионная спектрометрия с индуктивно связанной плазмой) и ICP-MS (масс-спектрометрия с индуктивно связанной плазмой), а также рассматривается несколько процедур подготовки раствора образца, на основании которого эксперты могут выбрать наиболее подходящий метод анализа.
Как показал анализ шестивалентного хрома, его иногда трудно определить в полимерах и электронике, поэтому в настоящем стандарте приведены методы скрининга для хрома в полимерах и электроники, за исключением AFS. Анализ хрома предоставляет информацию о присутствии шестивалентного хрома в материалах. Тем не менее элементный анализ не может выборочно обнаруживать шестивалентный хром; он определяет количество хрома во всех состояниях окисления в образцах. Если количество хрома превышает предел шестивалентного хрома, должны быть выполнены испытания шестивалентного хрома.
Методы испытаний, рассматриваемые в настоящем стандарте, должны обеспечить максимальную точность и достоверность для концентрации Pb, Cd и Cr, которая может составлять от 10 мг/кг для Pb, Cd и Cr при использовании ICP-OES и AAS и от 0,1 мг/кг для Pb и Cd при использовании ICP-MS, от 10 мг/кг для Pb, Cd и от 1,5 мг/кг для Cr при использовании AFS. Данные методы испытаний могут использоваться и для более высоких концентраций.
Настоящий стандарт не распространяется на материалы, содержащие полифторированные полимеры из-за их стабильности. Если серную кислоту используют в аналитической процедуре, существует риск потери Pb, в результате чего получают ошибочно низкие значения для этого аналита. Кроме того, серная и плавиковая кислоты не пригодны для определения Cd при использовании AFS, потому что влияют на его восстановление.
На стадии растворения образца могут иметь место определенные ограничения и риски, т.к. может произойти осаждение целевых или других элементов; в этом случае остатки необходимо проверить отдельно или растворить с помощью другого метода, а затем объединить их с испытательным раствором образца.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты [для датированных ссылок применяют только указанное издание ссылочного стандарта, для недатированных - последнее издание (включая все изменения)]:
IEC 62321-1, Determination of certain substances in electrotechnical products - Part 1: Introduction and overview (Определение регламентированных веществ в электротехнических изделиях. Часть 1. Введение и обзор)
IEC 62321-2, Determination of levels of certain substances in electrotechnical products - Part 2: Disassembly, disjointment and mechanical sample preparation (Определение регламентированных веществ в электротехнических изделиях. Часть 2. Разборка, отсоединение и механическая подготовка образца)
IEC 62321-3-1, Determination of certain substances in electrotechnical products - Part 3-1: Screening - Lead, mercury, cadmium, total chromium and total bromine using X-ray fluorescence spectrometry (Определение регламентированных веществ в электротехнических изделиях. Часть 3-1. Скрининг. Анализ свинца, ртути, кадмия, общего хрома и общего брома методом рентгенофлуоресцентной спектрометрии)
ISO 3696, Water for analytical laboratory use - Specification and test methods (Вода для лабораторного анализа. Технические требования и методы испытаний)
ISO 5961, Water quality - Determination of cadmium by atomic absorption spectrometry (Качество воды. Определение содержания кадмия методом атомной абсорбционной спектрометрии)
3 Термины, определения и сокращения
3.1 Термины и определения
В настоящем стандарте применены термины по IEC 62321-1, а также следующие термины с соответствующими определениями:
3.1.1 достоверность (accuracy): Точность соответствия между результатами испытаний и принятым эталонным значением.
3.1.2 калибровочный образец (calibration standard): Вещество в твердом или жидком состоянии с известной и стабильной концентрацией аналита(ов), используемое для определения характеристики прибора (калибровочной кривой) по отношению к концентрации аналита(ов).
3.1.3 калибровочный раствор (calibration solution): раствор, используемый при калибровке прибора, подготовленный как из исходного раствора так и из сертифицированного стандартного образца.
3.1.4 сертифицированный стандартный образец (certified reference material): Эталонный материал, сопровождаемый сертификатом, одно или более значений параметров которого сертифицированы по процедуре, которая устанавливает его неопределенность и прослеживаемость.
3.1.5 лабораторный контрольный образец (laboratory control sample): проба матрицы с известным составом типового представителя целевого аналита, используемая для представления документации лаборатории [1]
3.1.6 холостой раствор реагентов (reagent blank solution): раствор, подготовленный путем добавления в растворитель реагентов в том же количестве в котором они были добавлены в раствор анализируемого образца (с тем же конечным объемом).
3.1.7 раствор анализируемого образца (test sample solution): раствор, подготовленный на основе пробы анализируемого образца в соответствии с установленными требованиями таким образом, чтобы он мог использоваться для последующих измерений.
3.2 Сокращения
В настоящем стандарте использованы следующие сокращения:
CCV - непрерывная проверка калибровки;
LCS - лабораторный контрольный образец.
4 Реактивы
4.1 Общая информация
Для определения элементов на уровне следов должны использоваться реактивы соответствующей чистоты. Концентрация аналита или мешающих веществ в реактивах и воде должна быть ничтожно малой по сравнению с наименьшей измеряемой концентрацией.
Для измерений методами ICP-OES и ICP-MS, эффект памяти возникает в тех случаях, когда вводятся элементы с высокими концентрациями. Разбавление раствора образца требуется для высоких уровней каждого элемента. Если эффект памяти не уменьшается при разбавлении, требуется тщательное мытье оборудования.
4.2 Реактивы
Должны использоваться следующие реактивы:
a) Для приготовления и разбавления всех растворов образцов должна использоваться вода класса 1 по ISO 3696.
b) Серная кислота:
2) Разбавленный раствор серной кислоты, (1:2): разбавить 1 объем раствора концентрированной серной кислоты (4.2, перечисление b, пункт 1) двумя (2) объемами воды (4.2, перечисление a).
c) Азотная кислота:
2) 10%-ный (по массе) раствор азотной кислоты, пригодный для анализа следовых количеств металлов.
3) Раствор азотной кислоты с молярной концентрацией 0,5 моль/л, пригодный для анализа следовых количеств металлов.
4) Разбавленный раствор азотной кислоты 1:2: разбавить 1 объем раствора концентрированной азотной кислоты (4.2, перечисление c, пункт 1) двумя объемами воды (4.2, перечисление a).
d) Соляная кислота:
2) Раствор соляной кислоты, разбавленный 1:2 (одна часть раствора соляной кислоты (4.2, перечисление d1), разбавленный 2 частями воды (4.4, перечисление а)), пригодный для анализа следовых количеств металлов.
3) 5%-ный (по массе) раствор соляной кислоты, пригодный для анализа следовых количеств металлов.
4) 10%-ный (по массе) раствор соляной кислоты, пригодный для анализа следовых количеств металлов.
I) Смешанная кислота:
1) Смешанная кислота 1 (две части раствора соляной кислоты (4.2, перечисление d, пункт 1), одна часть раствора азотной кислоты (4.2, перечисление c1) и две части воды (4.2, перечисление a);
2) Смешанная кислота 2 (одна часть раствора азотной кислоты (4.2, перечисление c, пункт 1) и три части раствора фтористоводородной кислоты (4.2, перечисление e);
3) Смешанная кислота 3 (три части раствора соляной кислоты (4.2, перечисление d, пункт 1) и одна часть раствора азотной кислоты (4.2, перечисление с, пункт 1).
m) Гидроксид калия (KOH), пригодный для анализа следовых количеств металлов.
Добавить примерно 800 мл воды (4.2, перечисление a) в 1000 мл мерную колбу (5.2, перечисление e, пункт 3), затем добавить 2 г гидроксида калия (4.2, перечисление m). Добавить 15,0 г борогидрида калия (4.2, перечисление n) и 10 г феррицианида калия (4.2, перечисление o), размешать до растворения. Добавить воды (4.2, перечисление a) до метки. Такой раствор приготавливается ежедневно.
q) Восстановители:
Добавить примерно 800 мл воды (4.2, перечисление a) в 1000 мл мерную колбу (5.2, перечисление e, пункт 3), затем добавить 2 г гидроксида калия (4.2, перечисление m). Добавить 30 г борогидрида калия (4.2, перечисление n), размешать до растворения. Добавить воды (4.2, перечисление a) до метки. Такой раствор приготавливается ежедневно.
Добавить примерно 800 мл воды (4.2, перечисление a) в 1000 мл мерную колбу (5.2, перечисление e, пункт 3) затем добавить 8 г гидроксида калия (4.2, перечисление m). Добавить 40 г борогидрида калия (4.2, перечисление n), размешать до растворения. Добавить воды (4.2, перечисление а) до метки. Такой раствор приготавливается ежедневно.
r) Несущий поток:
1) Несущий поток 1: HCI с массовой долей 1,5%.
2) Несущий поток 2: HCI с массовой долей 1,0%.
t) Маскирующий агент:
Добавить 10 г щавелевой кислоты, 10 г сульфационата калия и 1 г о-фенантролина к 200 мл воды (4.2, перечисление a). Нагреть при низкой температуре и размешать до растворения, не доводя раствор до кипения. Раствор необходимо использовать до выкристаллизовывания твердых частиц. Не использовать раствор после потемнения, необходимо приготовить новый.
2) Маскирующий агент 2: тиокарбамид с массовой долей 10% и раствор аскорбиновой кислоты с массовой долей 10%.
Растворить 10 г тиокарбамида и 10 г аскорбиновой кислоты в 100 мл воды. Приготавливается ежедневно.
u) Раствор кобальта концентрации 50 мг/л.
v) Стандартный раствор:
1) Стандартный раствор свинца (Pb) концентрации 1000 мг/кг.
2) Стандартный раствор кадмия (Cd) концентрации 1000 мг/кг.
3) Стандартный раствор хрома (Cr) концентрации 1000 мг/кг.
4) Стандартный раствор железа (Fe) концентрации 1000 мг/кг.
5) Стандартный раствор меди (Cu) концентрации 1000 мг/кг.
w) Стандартный раствор внутреннего стандарта.
1) Должны использоваться элементы внутреннего стандарта, которые не создают помех для целевого элемента. При этом присутствие данных элементов внутреннего стандарта в растворе образца должно находиться на ничтожно низком уровне. В качестве элементов внутреннего стандарта для данной специфической спектрометрии могут использоваться Sc, In, Tb, Lu, Re, Rh, Bi и Y.
2) Для метода ICP-OES рекомендуется использовать Sc или Y. Рекомендуемая концентрация 1000 мг/л.
3) Для метода ICP-MS рекомендуется использовать Rh. Рекомендуемая концентрация 1000 мкг/л.
Токсичность реактивов, используемых в данном методе, не имеет точного определения, поэтому каждое химическое соединение должно рассматриваться как потенциально опасное вещество. С этой точки зрения контактирование с данными веществами должно быть сведено к минимуму с использованием всех возможных средств защиты.
Способы приготовления растворов предусматривают использование сильных кислот, которые являются коррозионно-активными веществами, способными вызвать ожоги.
При работе с такими кислотами следует использовать лабораторные костюмы, перчатки и защитные очки.
Азотная кислота выделяет токсичные пары. Поэтому разложение должно всегда проводиться в вытяжном шкафу. Это требование распространяется также и на случаи добавления кислоты в образцы, так как при этом возможно выделение токсичных газов.
Отходящие газы плазмы должны удаляться с использованием эффективной вытяжной системы.
Особые меры предосторожности должны соблюдаться при работе с фтористоводородной кислотой, в местной аптечке должен быть гелевый HF-антидот (2,5%-ного глюконата кальция в водорастворимом геле) для оказания первой помощи в случае ожога кожи фтористоводородной кислотой (HF).
В качестве альтернативы могут использоваться химически чистые реагенты, за исключением проведения методов ICP-MS.
5 Оборудование
5.1 Общая информация
Сбор и хранение стеклянной лабораторной посуды представляют собой важную часть анализа элементов на уровне следов независимо от типа испытательного образца. Ввиду чувствительности рассматриваемых методов анализа на содержание Pb, Cd и Cr выполнение каждой отдельной стадии отбора образцов требует большой осторожности и внимания. Все оборудование для отбора образцов, их обработки и хранения не должны содержать металлов. Всю стеклянную посуду необходимо погрузить в 10%-ный раствор азотной кислоты (4.2, перечисление с, пункт 2) на 24 ч при комнатной температуре, а затем тщательно промыть ее водой (4.2, перечисление а).
5.2 Оборудование
Должно использоваться следующее оборудование.
a) Аналитические весы с точностью измерения 0,0001 г.
b) HF-стойкая система введения образца: система, в которой секция ввода образца и горелка обработаны с целью повышения их устойчивости к воздействию фтористоводородной кислоты (HF).
c) Газообразный аргон: газ, степень чистоты которого превышает 99,99% (по объему).
d) Газообразный ацетилен: газ, степень чистоты которого превышает 99,99% (по объему).
e) Стеклянная посуда: перед использованием вся стеклянная лабораторная посуда должна быть очищена 10%-ной азотной кислотой (4.2, перечисление c2):
1) колба Кьельдаля: 100 мл;
2) химические стаканы: 100, 200 мл и т.д.;
3) мерные колбы: 50, 100, 200, 500, 1000 мл и т.д. Если позволяют условия, вместо мерных колб могут использоваться другие типы объемных мерных приспособлений с приемлемой точностью и достоверностью;
4) пипетки: 1, 5, 10, 20 мл и т.д.;
5) смотровое стекло.
f) Платиновые тигели: 50, 150 мл и т.д.
g) Фарфоровые тигели: 50, 150 мл и т.д.
h) Устройства из политетрафторэтилена/перфторалкоксилалкановой смолы (PTFE/PFA): перед использованием все устройства промываются 10%-ой азотной кислотой (4.2, перечисление c, пункт 2).
1) химические стаканы: 100, 200, 500 мл и т.д.;
2) крышки для химических стаканов;
3) мерные колбы: 100, 200, 500 мл и т.д.
i) Микропипетки: 10, 100, 200, 500, 1000 мкл и т.д.
j) Контейнеры: для хранения стандартного раствора и калибранта.
Контейнеры должны быть изготовлены из полиэтилена высокой плотности (PE-HD) или из перфторалкоксилалкановой смолы (PFA).
k) Для определения ультраследового уровня должны использоваться контейнеры из перфторалкоксилалкановой смолы (PFA) или перфтор (этилен-пропиленового) пластика (FEP). В любом случае пользователь должен проверить пригодность выбранного контейнера.
I) Электроплитка или нагретая песочная ванна.
m) Муфельная печь: способная поддерживать температуру на уровне (550±25)°С.
n) Бунзеновская горелка или газовая горелка аналогичного типа.
o) Гидролитическое разложение в царской водке: аппарат разложения с микроконтроллером времени и температуры, термостат нагревательного блока, набор сосудов, каждый из которых оснащен дефлегматорами и абсорбционными емкостями.
p) Микроволновая система разложения образцов, оснащенная держателем образца, сосудами из политетрафторэтилена/модифицированного тетрафторэтилена высокого давления (PTFE/TFM) или перфторалкоксилалкановой смолы/модифицированного тетрафторэтилена (PFA/TFM) или другими сосудами из фторуглеродистых материалов.
Имеется много рекомендаций по технике безопасности и правилам эксплуатации микроволновых устройств в лабораториях в зависимости от их модели и фирмы-изготовителя. Перед тем как приступить к работе персонал лаборатории должен ознакомиться с соответствующими инструкциями и технической литературой, а также проконсультироваться с изготовителем микроволнового оборудования и сосудов по поводу их правильной и безопасной эксплуатации.
q) Термоизоляционная плита.
r) Стекловолоконный фильтр (боросиликатное стекло) с размером ячеек 0,45 мкм и подходящей чашкой фильтра.
s) Оптический эмиссионный спектрометр с индуктивно связанной плазмой (ICP-OES).
t) Масс-спектрометр с индуктивно связанной плазмой (ICP-MS).
u) Атомно-абсорбционный спектрометр (AAS).
v) Атомно-флуоресцентный спектрометр (AFS).
6 Подготовка образцов и проб
6.1 Общая информация
Для разных аналитических методов, которые могут использоваться согласно настоящему стандарту в качестве альтернативных вариантов, требуется разное количество образца для получения результатов необходимого качества. Обычно рекомендуется начинать с самого большого количества образца, подходящего для выбранного метода.
В случае если образец отбирается из электроники, то его необходимо механически разрушить соответствующим способом (например, измельчить, размолоть или отрезать) до начала кислотного разложения.
Чтобы обеспечить репрезентативность образца на данной стадии, необходимо обеспечить определенный размер частиц как функции начального количества образца (см. IEC 62321-2).
Рекомендуется проводить анализ водного раствора образца непосредственно после подготовки образца. Если такая возможность отсутствует, необходимо подходящим способом стабилизировать раствор и хранить его не более 180 дней при комнатной температуре.
6.2 Пробы образца
6.2.1 Полимеры
Для кислотного разложения необходимо измельчить, размолоть или отрезать 400 мг образца и отмерить с точностью до 0,1 мг. Для метода сухого озоления или микроволнового разложения необходимо измельчить, размолоть или отрезать 200 мг образца и отмерить с точностью до 0,1 мг.
6.2.2 Металлы
Один грамм образца измеряется с точностью до 0,1 мг и помещается в стеклянный лабораторный химический стакан (5.2, перечисление h, пункт 1) или химический стакан из PTFE/PFA (4.2, перечисление e) при использовании фтористоводородной кислоты HF (4.2, перечисление e). Для применения метода AFS измеряется 0,2 г образца.
6.2.3 Электроника
Для гидролитического разложения в царской водке необходимо взвесить примерно 2 г измельченного образца (максимальный размер частиц: 250 мкм) с точностью до 0,1 мг. Для микроволнового разложения необходимо взвесить примерно 200 мг измельченного образца (максимальный размер частиц: 250 мкм) с точностью до 0,1 мг.
7 Процедура испытаний
7.1 Полимеры
7.1.1 Общая информация
Образцы вырезаются и/или измельчаются до соответствующего размера в соответствии с выбранным методом испытаний согласно процедуре, описание которой приводится в разделе 6. В зависимости от способа приготовления испытательного раствора количество образца может меняться, как будет описано ниже.
Испытательный раствор можно приготовить посредством сухого озоления или посредством разложения образца с помощью азотной или серной кислоты. Кислотное разложение может осуществляться в закрытой системе с использованием системы микроволнового разложения. В зависимости от присутствия определенных элементов методика разложения может претерпевать некоторые изменения. Информацию о присутствии таких элементов можно получить из предыдущих исследовательских экспериментов (см. IEC 62321-3-1). Для определения содержания свинца, кадмия и хрома в растворе разложения могут использоваться такие способы измерения, как оптическая эмиссионная спектрометрия с индуктивно связанной плазмой (ICP-OES), масс-спектрометрия с индуктивно связанной плазмой (ICP-MS) или атомная абсорбционная спектрометрия (AAS). В случае если применяется AFS, перед определением содержания Pb и Cd раствор для разложения должен быть дополнительно обработан.
7.1.2 Метод сухого озоления
Если образец не содержит галогеносодержащих соединений (возможно потребуется информация предыдущих отсеивающих экспериментов), необходимо выполнить следующие операции.
a) Отмерить необходимое количество образца в тигель (5.2, перечисление g), установленный в отверстии жаропрочной термоизоляционный* плиты (5.2, перечисление q).
b) Плавно нагреть тигель (5.2, перечисление g) с помощью горелки (5.2, перечисление n) в хорошо проветриваемом вытяжном колпаке, обращая внимание на то, чтобы образец не воспламенился.
c) После разложения образца до обугленного состояния нагревание постепенно повышается до удаления летучих продуктов разложения, после чего в тигле остается сухой углеродистый остаток.
d) Перенести тигель и его содержимое в муфельную печь (5.2, перечисление m) при температуре (550±25)°С, при этом дверца должна оставаться немного приоткрытой для того, чтобы обеспечить достаточный приток воздуха, необходимого для окисления углерода.
e) Нагревание продолжается до тех пор, пока углерод не будет полностью окислен и не останется только зола.
f) Достать тигель (5.2, перечисление g) и его содержимое из печи (5.2, перечисление m) и дать ему возможность остыть до температуры окружающей среды (для AFS см. 7.1.2, перечисление h).
g) Добавить 5 мл азотной кислоты (4.2, перечисление c, пункт 1), перелить полученный раствор в 50 мл мерную колбу (5.2, перечисление h, пункт 3) и заполнить водой (4.2, перечисление a) до метки. Это концентрированный раствор образца.
Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт (4.2, перечисление w), он должен добавляться перед заполнением водой. Перед заполнением до окончательного объема 50 мл необходимо добавить 500 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000).
h) Переместить полученный раствор в 100 мл мерную колбу (5.2, перечисление е, пункт 3) и заполнить водой (4.2, перечисление а) до метки. С помощью пипетки поместить 2,50 мл раствора в 100 мл химический стакан (5.2, перечисление е, пункт 2).
Поместить стакан на электроплитку (5.2, перечисление I). Нагреть при низкой температуре до полного высыхания раствора. Протереть внутренние стенки стакана водой (4.2, перечисление a) и добавить либо 1,0 мл (для определения Cd) или 1,5 мл (для определения Pb) соляной кислоты (4.2, перечисление d, пункт 2). Немного нагреть для растворения соли в стакане. Охладить раствор при комнатной температуре и переместить его в мерную колбу (5.2, перечисление e, пункт 3). Раствор в 50 мл колбе обрабатывается в следующей последовательности соответственно.
- Для определения Pb заполнить водой (4.2, перечисление a) до метки и тщательно перемешать.
- Для определения Cd необходимо получить образец, не содержащий примесей меди, железа, цинка или никеля и т.д., добавить 1,0 мл раствора кобальта (4.2, перечисление u) и 5,0 мл раствора тиокарбамида (4.2, перечисление s) в мерную колбу. Если образец содержит перечисленные чужеродные примеси, необходимо заменить раствор тиокарбамида (4.2, перечисление s) 10,0 мл маскирующего агента 2 (4.2, перечисление t, пункт 2). Заполнить водой (4.2, перечисление a) до метки и тщательно перемешать.
Если образец содержит существенное количество галогенных соединений (возможно, потребуется информация предыдущих отсеивающих экспериментов), необходимо выполнить следующие операции.
i) Отмерить необходимое количество образца в тигель (5.2, перечисление g).
j) Добавить от 5 мл до 15 мл серной кислоты (4.2, перечисление b) и медленно нагреть тигель (5.2, перечисление g) и его содержимое на горячей плитке или в песочной ванне (5.2, перечисление I) так, чтобы пластик расплавился и почернел.
k) После охлаждения добавить 5 мл азотной кислоты (4.2, перечисление c, пункт 1) и продолжить нагревание до полного разложения пластика и появления белых паров.
I) После охлаждения поставить тигель (5.2, перечисление g) в муфельную печь (5.2, перечисление m) и при температуре (550±25)°С подождать, пока образец испарится, высохнет и озолится так, чтобы углерод оказался полностью сожженным.
m) После озоления добавить 5 мл азотной кислоты (4.2, перечисление c, пункт*), перелить полученный раствор в 50 мл мерную колбу (5.2, перечисление e, пункт 3) и заполнить водой (4.2, перечисление a) до метки, получив концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением водой. Для окончательного объема 50 мл необходимо добавить 500 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000) перед заполнением.
n) Все остатки образца необходимо отделить с помощью центрифуги или фильтра. Остатки необходимо проверить посредством соответствующих измерений (например, с помощью рентгенофлуоресцентной спектроскопии) и убедиться в отсутствии в них целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
Примечание - Данный метод не распространяется на фтороуглерод.
7.1.3 Метод кислотного разложения
a) Отмерить необходимое количество образца в колбу (5.2, перечисление e, пункт 1). Добавить 5 мл серной кислоты (4.2, перечисление b, пункт 1), 1 мл азотной кислоты (4.2, перечисление c, пункт 1) и нагреть колбу так, чтобы произошло озоление образца и выделение белых паров. После прерывания нагревания, добавить азотную кислоту (4.2, перечисление c, пункт 1) мелкими порциями (примерно по 0,5 мл) и продолжить нагревания до появления белых паров. Нагревание и разложение азотной кислотой (4.2, перечисление c, пункт 1) повторяется до тех пор, пока разложенный раствор не станет бледно-желтого цвета.
b) Подождать несколько минут, чтобы образец остыл. Добавить перекись водорода (4.2, перечисление k) мелкими порциями (по несколько миллилитров за один раз) и нагреть образец до появления белых паров. После охлаждения перелить полученный раствор в 100 мл мерную колбу (5.2, перечисление e, пункт 3) и заполнить водой (4.2, перечисление a) до метки. Это концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением. Для окончательного объема 100 мл, необходимо добавить 1000 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000) перед заполнением.
c) Если общее разложение проведено неправильно или если образец содержит значительное количество Si, Zr, Hf, Ti, Ta, Nb, W (возможно потребуется информация предыдущих отсеивающих экспериментов), необходимо выполнить следующие операции:
- Отмерить необходимое количество образца в колбу. Добавить 5 мл серной кислоты, 1 мл азотной кислоты и нагреть колбу так, чтобы произошло озоление образца и выделение белых паров. Прервать нагревание, добавить азотную кислоту (4.2, перечисление c, пункт 1) мелкими порциями (примерно по 0,5 мл) и продолжить нагревания до появления белых паров. Нагревание и разложение азотной кислотой (4.2, перечисление c, пункт 1) повторяется до тех пор, пока разложенный раствор не станет бледно-желтого цвета.
- Подождать несколько минут, чтобы образец остыл. Добавить перекись водорода мелкими порциями (по несколько миллилитров за один раз) и нагреть образец до появления белых паров. После охлаждения перелить полученный раствор в сосуд из фторполимеров (5.2, перечисление h, пункт 1). Добавить 5 мл фтористоводородной кислоты (4.2, перечисление e) и нагреть сосуд до появления белых паров. По желанию добавить борную кислоту (4.2, перечисление j) для образования фторидных соединений с целью защиты кварцевой плазменной горелки (если нет кислотостойкой системы введения образца). После остывания перелить раствор в 100 мл мерную колбу PTFE/PFA (5.2, перечисление h, пункт 3) и залить водой до метки. Полученный раствор - это концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением. Для окончательного объема 100 мл необходимо добавить 1000 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000) перед заполнением.
d) Все остатки образца необходимо отделить с помощью центрифуги или фильтра. Остатки необходимо проверить посредством соответствующих измерений (например, с помощью РФ-спектроскопии) и убедиться в отсутствии в них целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
Примечание - Данный метод не подходит для проведения AFS.
7.1.4 Микроволновое разложение
a) Отмерить необходимое количество образца и поместить в сосуд микроволнового разложения, добавить 5 мл азотной кислоты (4.2, перечисление c, пункт 1). Добавить перекись водорода (4.2, перечисление k) небольшими или каталитическими порциями (по 0,1-1 мл) для поддержания полного окисления органического вещества. Закрыть сосуд крышкой и поместить в аппарат микроволнового разложения (5.2, перечисление p). Разложение образца в микроволновой печи должно производиться согласно предварительно разработанной рабочей программе. После остывания образца перелить раствор в 50 мл мерную колбу (5.2, перечисление e, пункт 3) и залить водой (4.2, перечисление a) до метки. Полученный раствор - это концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением. Для окончательного объема 50 мл необходимо добавить 500 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000) перед заполнением.
Перекись водорода может добавляться только тогда, когда известны активные компоненты образца. Перекись водорода может быстро и агрессивно вступать в реакцию с легко окисляемыми материалами; поэтому она не должна добавляться, если образец содержит большие количества легко окисляемых органических составляющих.
b) Если общее разложение протекает неправильно или если образец содержит значительное количество Si, Zr, Hf, Ti, Ta, Nb, W (возможно потребуется информация предыдущих отсеивающих экспериментов), необходимо выполнить следующие операции:
- Отмерить необходимое количество образца в сосуд микроволнового разложения. Добавить 5 мл азотной кислоты (4.2, перечисление c, пункт 1) и 1 мл фтористоводородной (4.2, перечисление e). Добавить каталитическое количество перекиси водорода (4.2, перечисление k) (по 0,1-1 мл) для достижения полного окисления органического вещества. Закрыть сосуд крышкой и поместить в аппарат микроволнового разложения (5.2, перечисление p). Разложение образца в микроволновой печи должно производиться согласно предварительно разработанной рабочей программе. По своему усмотрению добавить борную кислоту (4.2, перечисление j) для образования фторидных соединений с целью защиты кварцевой плазменной горелки (если нет, кислотостойкой системы введения образца). После остывания перелить раствор в 50 мл мерную колбу PTFE/PFA (5.2, перечисление h, пункт 3) и залить (4.2, перечисление a) до метки. Полученный раствор - это концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением. Для окончательного объема 50 мл необходимо добавить 500 мкл внутреннего стандарта (4.2, перечисление w) для ICP-OES и ICP-MS (после разбавления 1:1000) перед заполнением.
Перекись водорода может добавляться, только если известны активные компоненты образца. Перекись водорода может быстро и агрессивно вступать в реакцию с легко окисляемыми материалами; поэтому она не должна добавляться, если образец содержит большие количества легко окисляемых органических составляющих.
Примечание - Данный метод не подходит для проведения AFS.
c) Остатки образца необходимо отделить с помощью центрифуги или фильтра. Содержание остатков необходимо проверить (например, с помощью РФ-измерений) и убедиться в отсутствии в них целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
7.2 Металлы
7.2.1 Общая информация
Процедура приготовления раствора испытательного образца, описание которой приводится в настоящем стандарте, не обязательно охватывает все металлы и их соединения. Обычно для приготовления раствора рекомендуется использовать соляную кислоту, азотную кислоту или их смесь. Для образцов, которые с трудом растворяются под воздействием этих кислот, по мере необходимости может добавляться перхлорная кислота, серная кислота и т.д. При этом следует помнить о риске использования серной кислоты для определения содержания свинца, так как это может привести к потере некоторой части целевого элемента. В условиях высоких температур образцы должны растворяться полностью без остатка. Для растворения образцов может использоваться также фосфорная кислота.
При растворении металлов или их смесей сильными кислотами всегда существует риск осаждения (например, растворение Pb и Ba серной кислотой и Ag соляной кислотой. AI может образовать оксиды/гидраты оксидов и их аналоги). Даже если на данные элементы не распространяются нормативные положения, существует риск потери целевого элемента в результате осаждения. В рамках данного раздела необходимо убедиться в отсутствии потерь целевых элементов в растворе испытательного образца. Для проверки всех остатков и определения в них содержания или отсутствия целевых элементов должен использоваться другой метод измерения. Альтернативно после кислотного разложения остатки необходимо подвергнуть полному дополнительному разложению с помощью других методов (например, посредством щелочного плавления или использования герметичного сосуда под давлением). Обработанные таким образом остатки затем объединяются с кислотно-растворенным раствором и измеряются.
Остатки образца необходимо отделить с помощью центрифуги или фильтра. Содержание остатков необходимо проверить (например, с помощью РФ-измерений) и убедиться в отсутствии в них целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
Если в присутствующем серебре содержится большое количество олова, т.е. припой, не содержащий Pb, в качестве растворяющей кислоты должна использоваться соляная кислота с добавлением 10 мл перекиси водорода до тех пор, пока разложение не будет завершено.
7.2.2 Общие методы разложения образца
а) Стеклянный химический стакан (5.2, перечисление e, пункт 2), в котором находится образец, закрыть смотровым стеклом (5.2, перечисление e, пункт 5). Добавить 20 мл смешанной кислоты 1 (4.2, перечисление I, пункт 1) и нагреть стакан до растворения образца. После того как стакан остынет до комнатной температуры, промыть его внутренние стенки и нижнюю часть смотрового стекла водой (4.2, перечисление a). Перелить раствор в 100 мл мерную колбу (5.2, перечисление e, пункт 3) и заполнить водой (4.2, перечисление a) до метки. Полученный раствор представляет собой концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до необходимой концентрации для каждого измерительного прибора. Если необходимо, добавить раствор внутреннего стандарта (4.2, перечисление w) с содержанием, например, Rh, до заполнения колбы (5.2, перечисление e, пункт 3) водой (4.2, перечисление a). Тип элемента и его количество зависят от выбранного аналитического метода. При расчете результатов необходимо учитывать соответствующие способы разбавления. И разбавление, и добавление внутреннего стандарта должно документироваться в протоколе испытаний.
b) При применении метода AFS вначале необходимо до разложения концентрированного раствора с помощью пипетки поместить 2,50 мл раствора в 100 мл химический стакан (5.2, перечисление е, пункт 2).
Поместить стакан на электроплитку (5.2, перечисление I). Нагреть при низкой температуре до полного высыхания раствора. Протереть внутренние стенки стакана водой (4.2, перечисление a) и добавить либо 1,0 мл (для определения Cd) или 1,5 мл (для определения Pb) соляной кислоты (4.2, перечисление d, пункт 2). Немного нагреть для растворения соли в стакане. Охладить раствор при комнатной температуре и переместить его в мерную колбу (5.2, перечисление e, пункт 3). Раствор в 50 мл колбе обрабатывается в следующей последовательности соответственно.
- Для определения Pb добавить 4,0 мл маскирующего агента 1 (4.2, перечисление t, пункт 1) в мерную колбу и заполнить водой (4.2, перечисление a) до метки. После смешивания оставить оседать примерно на 30 мин, а затем пропустить через медленнодействующую фильтровальную бумагу. Фильтраты необходимо оставить для испытаний.
- Для определения Cd добавить 1,0 мл раствора кобальта (4.2, перечисление u) и 5,0 мл маскирующего агента 2 (4.2, перечисление t, пункт 2) в мерную колбу и заполнить водой (4.2, перечисление a) до метки. Оставить оседать примерно на 30 мин. Раствор необходимо оставить для испытаний.
7.2.3 Образцы, содержащие Zr, Hf, Ti, Ta, Nb или W
Закрыть химический стакан PTFE/PFA (5.2, перечисление h, пункт 1), в котором находится образец, крышкой (5.2, перечисление h, пункт 2). Добавить 20 мл смешанной кислоты 2 (4.2, перечисление I, пункт 2) и нагреть стакан до растворения образца.
После того как стакан остынет до комнатной температуры, промыть его внутренние стенки (5.2, перечисление h, пункт 1) и нижнюю часть крышки (5.2, перечисление h, пункт 2) водой (4.2, перечисление a). Снять крышку (5.2, перечисление h, пункт 2). Перелить раствор в 100 мл мерную колбу (5.2, перечисление h, пункт 3) и заполнить водой (4.2, перечисление a) до метки.
Полученный раствор представляет собой концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление а) до необходимой концентрации для каждого измерительного прибора. Если необходимо, добавить раствор внутреннего стандарта (4.2, перечисление w) с содержанием, например Rh до заполнения колбы (5.2, перечисление h, пункт 3) водой (4.2, перечисление a). Так как здесь используется фтористоводородная кислота (4.2, перечисление e), раствор внутреннего стандарта (4.2, перечисление w) не должен содержать редкоземельных элементов. Тип элемента и его количество зависят от выбранного аналитического метода. При расчете результатов необходимо учитывать соответствующие способы разбавления. И разбавление, и добавление внутреннего стандарта должно документироваться в протоколе испытаний.
Примечание - Данный метод не подходит для проведения AFS.
7.2.4 Образцы, содержащие Sn
Повторить данный процесс три раза. После охлаждения до комнатной температуры добавить 10 мл азотной кислоты (4.2, перечисление c, пункт 1) для растворения солей. Перелить раствор в 100 мл мерную колбу (5.2, перечисление е, пункт 3) и заполнить водой (4.2, перечисление a) до метки. Полученный раствор представляет собой концентрированный раствор образца. Разбавить данный раствор водой (4.2, перечисление a) до необходимой концентрации для каждого измерительного прибора. Если необходимо, добавить раствор внутреннего стандарта (4.2, перечисление w) с содержанием, например Rh до заполнения колбы (4.1, перечисление e, пункт 3) водой (4.2, перечисление a). Тип элемента и его количество зависят от выбранного аналитического метода. При расчете результатов необходимо учитывать соответствующие способы разбавления. И разбавление, и добавление внутреннего стандарта (4.2, перечисление w) должно документироваться в протоколе испытаний.
Альтернативно 1 г образца растворяется посредством добавления 40 мл воды (4.2, перечисление а), 12 мл азотной кислоты (4.2, перечисление c, пункт 1) и 6 мл свежеприготовленной фтороборной кислоты (4.2, перечисление f) (200 мл 40%-ной (по массе) фтористоводородной кислоты (4.2, перечисление e) с 75 г борной кислоты (4.2, перечисление j). При этом должен использоваться химический стакан PTFE/PFA (5.2, перечисление e, пункт 3) и мерная колба из полиэтилена высокой плотности или PTFE/PFA (5.2, перечисление e, пункт 3).
Примечание - Данный метод не подходит для проведения AFS.
7.3 Электронные компоненты
7.3.1 Общая информация
Процедура приготовления раствора испытательного образца, описание которой приводится в настоящем стандарте, не обязательно охватывает всю электронику. После разложения в растворе могут присутствовать твердые остатки. Следует убедиться (например, с помощью РФ-спектрометрии), что в данных остатках нет значительных количеств целевых элементов. Если же они будут обнаружены, данные остатки необходимо растворить другими химическими методами и объединить с раствором испытательного образца.
7.3.2 Гидролитическое разложение в царской водке
Полученный раствор представляет собой концентрированный раствор образца. Данный раствор можно разбавить 5%-ной HCI (4.2, перечисление d, пункт 3) до определенного уровня концентрации для каждого измерительного прибора. Если будет использоваться внутренний стандарт, он должен добавляться перед заполнением. Для окончательного объема 100 мл необходимо добавить 1000 мкл внутреннего стандарта для ICP-OES и ICP-MS (после разбавления 1:1000).
b) При применении метода AFS вначале необходимо до разложения концентрированного раствора с помощью пипетки поместить 2,50 мл раствора в 100 мл химический стакан (5.2, перечисление e, пункт 2).
Поместить стакан на электроплитку (5.2, перечисление I). Нагреть при низкой температуре до полного высыхания раствора. Протереть внутренние стенки стакана водой (4.2, перечисление a) и добавить либо 1,0 мл (для определения Cd) или 1,5 мл (для определения Pb) соляной кислоты (4.2, перечисление d, пункт 2). Немного нагреть для растворения соли в стакане. Охладить раствор при комнатной температуре и переместить его в мерную колбу (5.2, перечисление e, пункт 3). Раствор в 50 мл колбе обрабатывается в следующей последовательности соответственно.
- Для определения Pb добавить 4,0 мл маскирующего агента 1 (4.2, перечисление t, пункт 1) в мерную колбу и заполнить водой (4.2, перечисление a) до метки. После смешивания, оставить оседать примерно на 30 мин, а затем пропустить через 0,45 мкм стекловолоконный микрофильтр (5.2, перечисление r). Фильтраты необходимо оставить для испытаний.
- Для определения Cd, добавить 1,0 мл раствора кобальта (4.2, перечисление u) и 5,0 мл маскирующего агента 2 (4.2, перечисление t, пункт 2) в мерную колбу и заполнить водой (4.2, перечисление a) до метки. Оставить оседать примерно на 30 мин. Раствор необходимо оставить для испытаний.
Если на фильтре имеются остатки образца, их необходимо проверить с помощью соответствующих измерений (например, посредством рентгеновской флуоресцентной спектрометрии) для подтверждения отсутствия целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
Если в лаборатории нет оборудования, указанного выше, она может использовать более простой подход, если пользователь сможет подтвердить его пригодность. Отклонения от процедур, описание которых приводится выше, необходимо оценить и задокументировать в протоколе испытаний. Такой упрощенный подход можно реализовать следующим образом: стеклянный химический стакан (5.2, перечисление e, пункт 2) с образцом закрыть смотровым стеклом (5.2, перечисление e, пункт 5). Добавить смешанную кислоту (4.2, перечисление I, пункт 3) и нагревать химический стакан (5.2, перечисление е, пункт 2) в течение 2 ч при температуре 120°С, после чего оставить его на 12 ч при комнатной температуре. Промыть водой (4.2, перечисление а) нижнюю часть смотрового стекла (5.2, перечисление e, пункт 5) и внутренние стенки химического стакана (5.2, перечисление е, пункт 2). Снять смотровое стекло (5.2, перечисление e, пункт 5). После остывания образец пропускается через 0,45 мкм стекловолоконный микрофильтр (5.2, перечисление r). Твердые остатки промываются соляной кислотой 5%-ной (по массе) (4.2, перечисление d, пункт 3). Полученный раствор переливается в мерную колбу (5.2, перечисление e) и заполняется соляной кислотой 5%-ной (по массе) (4.2, перечисление d, пункт 3) до метки. Полученный раствор используется для проведения дальнейших измерений.
7.3.3 Микроволновое разложение
b) Сосуд открывается после охлаждения до комнатной температуры (примерно через 1 ч), после чего добавляется 4 мл HCI (4.2, перечисление d, пункт 1). После повторного закрытия сосуда происходит растворение других элементов в соляной кислоте (4.2, перечисление d, пункт 1): это является второй стадией (стадией В) микроволнового разложения с усилением. Пример подходящей микроволновой программы (стадии А и В) можно найти в таблице А.6.
c) После охлаждения до комнатной температуры (примерно через 1 ч) сосуд открывают и раствор пропускается через стекловолоконный микрофильтр (5.2, перечисление r) в 25-миллиметровую колбу (5.2, перечисление e, пункт 3), промывается и заполняется соляной кислотой 5%-ной (по массе) (4.2, перечисление d, пункт 3). Если на фильтре имеются остатки образца, их необходимо проверить с помощью соответствующих измерений (например, посредством рентгеновской флуоресцентной спектрометрии) для подтверждения отсутствия целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
Процедура, описание которой приведено выше, содержит минимальные требования, предъявляемые к системе микроволнового разложения. Рекомендуется проводить двухкратный или трехкратный анализ для каждого образца в течение одного цикла.
Если в ходе одного цикла проводится двухкратный или трехкратный анализ, необходимо взвешивать одно и то же количество образцов одинакового типа.
Если для получения репрезентативной части измеряемого материала требуется более 200 мг образца, необходимо использовать следующую процедуру. Разделить образец на порции примерно одинаковой массы. Взвесить каждую порцию, помещаемую в отдельный сосуд разложения, выполнить процедуру разложения в каждом сосуде и объединить полученные растворы разложения.
Пример - Для разложения печатной платы минимальное необходимое количество образца должно составлять 1,2 г. Поэтому в шести сосудах необходимо взвесить 6х200 мг измельченного образца. После охлаждения в конце стадии В микроволнового разложения сосуды открываются, а растворы объединяются, пропускаются через стекловолоконный микрофильтр 0,45 мкм (5.2, перечисление r) в 100 мл мерную колбу (5.2, перечисление e, пункт 3), промываются и заполняются соляной кислотой 5%-ной (по массе) (4.2, перечисление d, пункт 3) до метки.
d) Если на фильтре имеются остатки образца, их необходимо проверить с помощью соответствующих измерений (например, посредством рентгеновской флуоресцентной спектрометрии) для подтверждения отсутствия целевых элементов. Руководство по проведению РФА приведено в IEC 62321-3-1.
7.4 Подготовка реагентного холостого раствора
Данная процедура идентична процедуре подготовки образца; она выполняется параллельно, но без образца.
8 Калибровка
8.1 Общая информация
Допускается, что образец имеет неизвестный состав и поэтому рекомендуется использовать метод внутреннего стандарта (метод сравнения интенсивности). При необходимости может использоваться также метод добавления стандартного раствора. Если отсутствуют мешающие матричные элементы или если известен состав образца, может применяться метод калибровочной кривой.
8.2 Подготовка калибровочного раствора
После постепенного разбавления стандартного раствора каждого элемента разбавленные стандартные растворы, содержащие от 0 мкг до 100 мкг каждого элемента, переливаются в 100 мл мерную колбу (5.2, перечисление e, пункт 3). Затем необходимо добавить определенное количество каждого реактива и, если используется метод AFS или метод внутреннего стандарта, соответствующее количество раствора для раствора кобальта (4.2, перечисление u) и раствора тиокарбамида (4.2, перечисление s) или маскирующего агента (4.2, перечисление u) или раствора внутреннего стандарта (4.2, перечисление w) для достижения концентрации реактива, идентичной концентрации раствора образца.
Полученный раствор представляет собой смешанный раствор калибранта для ICP-OES, ICP-MS или AAS.
8.3 Построение калибровочной кривой
Спектрометры подготавливаются для квантификации. Часть раствора, полученного согласно 8.2, распыляется в аргоновую плазму или воздушно/ацетиленовое пламя в случае применения методов ICP-OES, ICP-MS или AAS. Если раствор образца содержит фтористоводородную кислоту (HF), должна использоваться HF-стойкая система подачи образца.
a) ICP-OES (Оптическая эмиссионная спектрометрия с индуктивно связанной плазмой):
- Показания определяются для интенсивности излучения целевых элементов (и, если необходимо, элементов внутреннего стандарта). Если используется метод калибровочной кривой, кривая, показывающая зависимость интенсивности излучения целевых элементов от их концентрации, выводится как калибровочная кривая. Если используется метод внутреннего стандарта, кривая, показывающая зависимость коэффициента интенсивности от концентрации целевых элементов по отношению к кривой элементов внутреннего стандарта, выводится как калибровочная кривая.
- Рекомендуемые значения длины волн и мешающих элементов указаны в таблице А.1 и А.2.
b) ICP-MS (Масс-спектрометрия с индуктивно связанной плазмой):
- Показания определяются для массы/заряда (m/z) целевых элементов (и, если необходимо, элементов внутреннего стандарта). Если используется метод калибровочной кривой, кривая, показывающая зависимость интенсивности массы/заряда целевых элементов от их концентрации, выводится как калибровочная кривая. Если используется метод внутреннего стандарта, кривая, показывающая зависимость коэффициента интенсивности от концентрации целевых элементов по отношению к кривой элементов внутреннего стандарта, выводится как калибровочная кривая.
- Коэффициент массы/заряда может определяться на основании данных, приведенных в таблице А.3.
c) AAS (Атомная абсорбционная спектрометрия):
- Показания определяются для поглощения целевых элементов. Если используется метод калибровки, кривая, показывающая зависимость поглощения целевых элементов от их концентрации, выводится как калибровочная кривая.
- Если используются стандартные добавки, стандартные растворы добавляются в раствор образца, а неизвестная концентрация определяется по экстраполяции кривой добавок относительно нулевого поглощения.
- Значения длины волн должны выбираться по отношению к типичным значениям длины волн измерения для элементов, указанных в таблице А.4. Если со стороны находящихся веществ наблюдаются помехи, должен использоваться метод стандартных добавок.
d) AFS (Атомная флуоресцентная спектрометрия):
- Для определения Pb должны быть использованы несущий поток 1 (4.2 перечисление r, пункт 1) и окислительно-восстановительный агент (4.2, перечисление p). Для определения Cd должны быть использованы несущий поток 1 (4.2 перечисление r, пункт 2) и восстановитель 1 (4.2, перечисление q, пункт 1). Показания определяются по интенсивности флуоресценции целевых элементов. В калибровочном методе, кривая, показывающая зависимость между интенсивностью флуоресценции целевых элементов и концентрацией разработана в качестве калибровочной кривой.
- В методе стандартных добавок стандарты добавляют в раствор образца и неизвестную концентрацию определяется путем экстраполяции дополнительных кривых к нулю плотности.
- Длины волн должны быть выбраны с учетом типичных измеренных длин волн элементов, приведенных в таблице А.5.
8.4 Измерение образца
После построения калибровочной кривой производится измерение холостой пробы лабораторного реактива и раствора образца. Если концентрация образца находится выше диапазона кривой концентрации, раствор должен разбавляться до области калибровочной кривой, обеспечивая должное подкисление калибрантов, с проведением повторных измерений.
Точность измерений проверяется с помощью стандартного вещества, калибровочного раствора и т.д. через регулярные интервалы (например, один раз для каждых 10 образцов). Если необходимо, калибровочная кривая создается по-новому.
Если результат калибрантов отличается от ожидаемого значения более чем на 20%, для калибровки и всех образцов в последовательности требуются повторные измерения.
Если образец разбавляется в пределах области калибровки, концентрация внутреннего стандарта в разбавленном растворе образца должна настраиваться на стандартный раствор.
9 Расчеты
Концентрация, измеренная в 8.4, - это концентрация каждого элемента в растворе образца. Концентрация каждого элемента в образце рассчитывается с помощью следующей формулы:
10 Прецизионность результатов
Таблица 1 - Повторяемость и воспроизводимость результатов
Тип материала | Межлабо- раторное сравни- тельное испытание | Метод | Элемент | Среднее значение, мг/кг | Предел повторяемости , мг/кг | Предел воспроизводимости , мг/кг |
Полимер | 2 | AAS | Pb | 480,0 | 21,1 | Недостаточно данных |
Полимер | 4А |
| Pb | 953,8 | 22,5 | 60,4 |
|
|
|
| 98,3 | 3,5 | 3,6 |
Металл | 2 |
| Pb | 26,2 | 4,7 | 10,6 |
|
|
|
| 188,0 | 11,9 | Недостаточно |
Электроника | 2 |
| Pb | 17050 | 990 | данных |
Полимер | 4А |
| Cd | 98,2 | 4,2 | 7,3 |
|
|
|
| 138,5 | 11,1 | 9,5 |
Электроника | 2 |
| Cd | 14,0 | 7,9 | Недостаточно данных |
Полимер | 4А |
| Cr | 15,2 | 3,4 | 7,8 |
|
|
|
| 98,1 | 9,7 | 9,9 |
Полимер | 2 | AFS | Pb | 109,0 | 10,1 | Недостаточно |
|
|
|
| 17,3 | 4,3 | данных |
|
|
|
| 15,6 | 2,0 | 2,6 |
|
|
|
| 902,1 | 36,2 | 143,4 |
|
|
|
| 15,6 | 2,00 | 2,6 |
|
|
|
| 902,1 | 36,2 | 143,4 |
|
|
|
| 1016,0 | 259,6 | Недостаточно |
Полимер | 2 |
| Cd | 131,3 | 26,0 | данных |
|
|
|
| 21,3 | 3,2 |
|
| 4А |
|
| 173,6 | 6,5 | 18,0 |
|
|
|
| 91,0 | 7,1 | 20,5 |
Полимер | 2 | ICP- | Pb | 444,0 | 25,9 | 119,4 |
|
| OES |
| 426,2 | 21,3 | 307,1 |
|
|
|
| 106,8 | 15,4 | 19,7 |
|
|
|
| 14,7 | 5,2 | 6,7 |
|
|
|
| 94,8 | 4,5 | 17,5 |
| 4А |
|
| 933 | 57,0 | 133,4 |
|
|
|
| 16,5 | 2,4 | 10,8 |
|
|
|
| 950,8 | 32,59 | 114,8 |
Металл | 2 |
| Pb | 206,0 | 7,4 | Недостаточно данных |
|
|
|
| 988,0 | 26,4 | 61,9 |
|
|
|
| 23,0 | 1,6 | 3,0 |
|
|
|
| 193,1 | 16,9 | 87,9 |
Электроника | 2 |
| Pb | 16790 | 739 | 2097 |
|
|
|
| 22450 | 1293 | 1153 |
|
|
|
| 207483 | 42942 | 74907 |
Полимер | 4А |
| Cd | 179,3 | 8,0 | 15,7 |
|
|
|
| 98,1 | 4,0 | 11,8 |
Электроника | 2 |
| Cd | 16,5 | 3,8 | 13,1 |
Полимер | 4А |
| Cr | 46,1 | 3,1 | 10,9 |
|
|
|
| 15,5 | 3,3 | 9,8 |
Полимер | 2 | ICP- | Pb | 481,2 | 35,5 | 124,6 |
|
| MS |
| 462,3 | 39,8 | 194,1 |
|
|
|
| 102,3 | 1,6 | Недостаточно данных |
|
|
|
| 16,2 | 8,1 | 15,4 |
|
|
|
| 103,8 | 5,1 | 7,3 |
|
|
|
| 1049 | 59,9 | 332,3 |
|
|
|
| 15,1 | 0,73 | 3,3 |
| 4А |
|
| 949,2 | 52,8 | 70,5 |
Металл | 2 |
| Pb | 26,5 | 2,1 | 15,1 |
|
|
|
| 156,1 | 10,8 | 15,5 |
|
|
|
| 922,4 | 74,5 | 327,0 |
|
|
|
| 36,7 | 4,3 | Недостаточно |
|
|
|
| 190,7 | 18,6 | данных |
Электроника | 2 |
| Pb | 23633 | 1060 |
|
|
|
|
| 22220 | 4167 | 6707 |
Полимер | 2 |
| Cd | 137,3 | 8,1 | Недостаточно |
|
|
|
| 22,0 | 0,0 | данных |
|
|
|
| 10,0 | 0,6 | 1,3 |
|
|
|
| 103,7 | 3,8 | 33,1 |
| 4А |
|
| 180,9 | 7,8 | 20,1 |
|
|
|
| 94,4 | 3,6 | 12,4 |
Электроника | 2 |
| Cd | 24,7 | 1,6 | Недостаточно данных |
Полимер | 4А |
| Cr | 39,7 | 8,7 | 20,0 |
| 4А |
|
| 14,2 | 2,1 | 3,7 |
Примечание - Повторяемость и воспроизводимость данных для некоторых методов и типов материалов недоступны из-за ограниченного количества участвующих лабораторий и соответствующих образцов для международных межлабораторных сравнительных испытаний. | ||||||
Дополнительная информация приведена в приложении В.
11 Обеспечение качества
11.1 Общая информация
Раздел об обеспечении качества и контроля в отдельных стандартах на методы контроля должен включать в себя требования к контрольному образцу относительно частоты тестирования и критерия приемки. Этот раздел также должен включать особенности методов контроля установленного качества в отношении определения предела обнаружения (ПО) и предела квантификации (ПК).
Содержание разделов ПО и ПК должно быть совместимо с описанием, приведенным в 11.2.
Примеры других особенностей методов контроля установленного качества включают в себя требования, касающиеся первоначальной проверки калибровки, холостого метода, контрольного лабораторного образца (LCS) и т.д., перечислены в таблице 2.
Таблица 2 - Критерии приемлемости объектов для контроля качества
Объект | Концентрация мг/кг в испытуемом образце | Критерии приемлемости |
Калибровочная кривая |
| 0,995 |
Первичная поверка калибровки | например, для Pb, Cd или Cr 1 мг/кг | Восстановление: (90-110)% |
Непрерывная проверка калибровки | например, для Pb, Cd или Cr 1 мг/кг | Восстановление: (90-110)% |
Холостой метод |
| <ПОМ |
Лабораторный контрольный образец (LCS) | Середина диапазона калибровки | Восстановление: (80-120)% |
Дубликат контрольного лабораторного образца | Середина диапазона калибровки | Относительное отклонение <20% |
a) Первичная поверка калибровки выполняется каждый раз, когда устанавливается калибровочная кривая, с использованием стандарта из источника, отличного от калибровочного стандарта.
b) На каждую партию должно быть проведено одно контрольное испытание. Холостая матрица, которая не содержит Pb, Cd или Cr, может быть использована в качестве метода холостой пробы.
c) В каждой партии один контрольный лабораторный образец (LCS) и дубликат лабораторного образца должны быть проанализированы с помощью добавления Pb, Cd или Cr в холостую матрицу. Кроме того, сертифицированный эталонный материал, содержащий Pb, Cd или Cr может быть испытан в двух дубликатах.
d) После прогона каждого десятого образца и в конце каждого набора образцов необходимо произвести анализ стандарта непрерывной проверки калибровки (CCV). Процентная величина восстановления для Pb, Cd или Cr должна составлять от 90% до 110%. Если процентная величина восстановления для Pb, Cd или Cr в стандарте CCV выходит за пределы данного диапазона, стандарт CCV должен анализироваться повторно в пределах 12 ч. Если величина восстановления остается за пределами диапазона и после этого анализ необходимо прервать и произвести техническое обслуживание системы, чтобы вернуть ее в оптимальное рабочее состояние. Все образцы, загруженные перед последним успешным стандартом CCV, могут указываться в протоколе испытаний, а все образцы, использованные после неудачного стандарта CCV, должны подвергаться повторному анализу с новой калибровкой.
11.2 Предел обнаружения (ПО) и предел квантификации (ПК)
Следующая экспериментальная процедура выполняется для определения предела обнаружения метода и предела квантификации Pb, Cd или Cr.
a) Произвести точное взвешивание соответствующего количества образца без содержания Pb, Cd или Cr (например, стандартного, сертифицированного образца) или какого-либо другого соединения, создающего помехи для анализа в соответствии с процедурой, установленной в разделе 7. Поместить образец в каждый лабораторный стакан. Данную операцию необходимо повторить не менее 5 раз.
b) Добавить в каждый химический стакан 10 мкг Pb, Cd или Cr с использованием матричного раствора (4.2, перечисление v).
c) Выполнить процедуру испытания согласно разделу 7 с применением разложения и спектроскопических измерений.
d) Произвести расчет концентрации каждого элемента (мкг/г) согласно разделу 9 и определить процентное восстановление добавки для каждого из образцов.
Процентная величина восстановления ртути должна находиться в диапазоне от 70% до 125% для каждого из образцов. Если значение восстановления выходит за данные пределы для любой из выборок, всю процедуру экстракции и анализа необходимо повторить.
Примечание - Все анализы, используемые для расчета ПОМ, должны быть последовательными.
Число образцов (выборок) | Значение критерия Стьюдента ( -критерия) (доверительный уровень 99%) |
6 | 3,36 |
7 | 3,14 |
8 | 3,00 |
9 | 2,90 |
10 | 2,82 |
f) ПК определяется посредством умножения ПОМ на коэффициент 5.
Разные лаборатории могут иметь разные значения ПОМ и ПК.
В общем ПОМ со значением 2 мкг/г (ПК 10 мкг/г) считается достижимым при использовании данного метода.
Приложение A
(справочное)
Практическое определение содержания Cd, Pb и Cr в полимерах и электронных устройствах и Cd и Pb в металлах посредством AAS, AFS, ICP-OES и ICP-MS
A.1 Блок-схема для анализа хрома
 |
A.2 ICP-OES
Таблица A.1 - Спектральная интерференция для длины волн кадмия и свинца
Элемент | Cd | Cd | Cd | Cd | Pb | Pb | Pb | Pb |
нм | 214,439 | 226,502 | 228,802 | 361,051 | 217,000 | 220,353 | 261,417 | 283,305 |
Ag | + | + | + | + | + | + | + | + |
As | ++ | + | +++ | + | + | + | + | + |
Au | + | + | ++ | + | + | + | + | +++ |
B | + | + | + | +++ | + | + | ++ | + |
Ca | + | + | + | + | + | + | + | + |
Co | + | ++ | +++ | +++ | ++ | +++ | +++ | ++ |
Cr | + | + | + | + | + | + | ++ | + |
Cu | + | + | + | + | + | + | + | ++ |
Eu | + | + | + | +++ | ++ | + | +++ | +++ |
Ga | + | + | + | + | + | + | + | + |
Ge | + | + | + | + | + | + | + | + |
In | + | + | + | + | + | + | + | + |
lr | ++ | ++ | ++ | ++ | +++ | +++ | +++ | +++ |
Mg | + | + | + | + | + | + | + | ++ |
Mn | + | + | + | +++ | + | ++ | +++ | + |
Mo | ++ | + | + | +++ | ++ | + | ++ | +++ |
Ni | + | + | ++ | +++ | +++ | ++ | + | + |
Pd | + | + | + | + | + | +++ | + | + |
Pt | +++ | + | ++ | + | + | + | + | + |
Re | ++ | ++ | + | +++ | ++ | +++ | ++ | +++ |
Ru | ++ | + | ++ | + | ++ | + | +++ | + |
Sb | ++ | + | + | + | ++ | + | + | + |
Sc | + | + | +++ | ++ | ++ | ++ | +++ | ++ |
Sn | + | + | + | + | ++ | + | + | ++ |
V | + | + | ++ | +++ | ++ | ++ | ++ | + |
W | ++ | ++ | ++ | ++ | +++ | + | +++ | ++ |
Zn | + | + | + | + | +++ | + | + | + |
Al | + | + | + | + | +++ | +++ | + | ++ |
Ti | + | + | + | ++ | + | +++ | + | ++ |
Fe | +++ | +++ | + | ++ | +++ | ++ | +++ | +++ |
Nb | + | + | + | - | - | + | - | +++ |
Hf | - | - | - | - | - | + | - | +++ |
Ta | - | - | - | - | - | + | - | ++ |
Pb | + | + | + | + | - | - | - | - |
Cd | - | - | - | - | + | + | + | + |
Примечание - В таблице A.1 указана сила интерференции для длины волн Cd и Pb во время ввода 1000 мг/кг соответствующих матричных элементов. | ||||||||
+ - отсутствует или небольшая интерференция (обычно менее 0,05 мг/кг). ++ - средняя интерференция (обычно от 0,05 мг/кг до 0,2 мг/кг). +++ - сильная интерференция (обычно более 0,2 мг/кг). | ||||||||
Таблица A.2 - Спектральная интерференция для длины волн хрома
Элемент | Cr | Cr | Cr | Cr | Cr | Cr |
нм | 205,560 | 206,158 | 267,716 | 283,563 | 284,325 | 357,896 |
Ag | - | - | - | - | + | - |
As | - | - | - | - | - | - |
Au | - | - | - | + | - | - |
B | - | - | - | - | - | - |
Ca | - | - | - | - | - | + |
Co | - | - | - | - | - | + |
Cr | - | + | - | + | + | - |
Cu | - | - | - | - | - | - |
Eu | - | - | - | - | - | - |
Ga | - | - | - | - | - | - |
Ge | - | - | - | - | - | - |
In | - | - | - | + | - | - |
lr | + | + | - | + | - | - |
Mg | - | - | - | - | - | - |
Mn | - | - | + | - | - | - |
Mo | + | - | + | ++ | ++ | + |
Ni | + | + | - | - | - | - |
Pd | - | - | - | - | - | - |
Pt | - | + | + | - | - | - |
Re | - | - | - | - | + | + |
Ru | - | - | + | + | + | - |
Sb | - | ++ | - | - | - | - |
Sc | - | - | - | - | - | - |
Sn | - | - | - | - | - | - |
V | + | + | + | + | + | + |
W | - | + | ++ | + | + | - |
Zn | - | + | - | - | - | - |
Al | - | + | - | - | - | - |
Ti | - | - | - | + | - | + |
Fe | - | - | + | ++ | ++ | - |
Nb | - | - | ++ | + | ++ | + |
Hf | - | + | ++ | + | - | - |
Ta | - | + | - | + | ++ | - |
Pb | - | + | - | - | - | - |
Cd | - | - | + | - | - | - |
Примечание - В таблице A.2 указана сила интерференции для длины волн Cr во время ввода 1000 мг/кг соответствующих матричных элементов. | ||||||
+ - отсутствует или небольшая интерференция (обычно менее 0,05 мг/кг). ++ - средняя интерференция (обычно от 0,05 мг/кг до 0,2 мг/кг). +++ - сильная интерференция (обычно более 0,2 мг/кг). | ||||||
A.3 ICP-MS
Если обнаружен стабильный изотоп, число масса/заряд (m/z) нескольких изотопов может измеряться для оценки уровня спектральной интерференции. Это показано в таблице A.3. Если образец содержит олово или молибден, необходимо обратить внимание на положительную интерференцию при измерении массы кадмия.
Таблица A.3 - Примеры соотношений масса/заряд (m/z)
Элемент | Изотоп | Изобара | Многоатомный ион |
Cd |
| MoO, MoOH, ZrOH | |
| Sn | MoO, MoOH | |
| In | MoO, MoOH, ZrOH, RuO | |
| Sn | MoO, MoOH, RuO | |
Pb |
| ||
|
| PtO | |
|
| IrO | |
|
| PtO | |
Cr |
| SO, CIO, HCIO, ArC, ArN, ArO | |
|
| HSO, CIO, HCIO, ArC |
A.4 AAS
Рекомендуемые измерения длины волн для AAS указаны в таблице A.4.
Таблица A.4 - Примеры длины волн для AAS
Элемент | Длина волны, нм | Ширина щели, нм |
Cd | 228,8 | 0,7 |
Pb | 261,4 | 0,7 |
| 217,0 | 0,7 |
| 283,3 | 0,7 |
Cr | 357,9 | 0,7 |
Источник света: безэлектродная разрядная лампа или лампа с полым катодом; тип газа: воздушно/ацетиленовая смесь.
A.5 AFS
Рекомендуемые измерения длины волн для AAS указаны в таблице A.5.
Таблица А.5 - Примеры длины волн для AFS
Элемент | Длина волны, нм |
Cd | 228,8 |
Pb | 283,3 |
Источник света: безэлектродная разрядная лампа или лампа с полым катодом; тип газа: аргон.
А.6 Коррекция фона
Если происходит изменение фона под воздействием главной матрицы раствора, которое влияет на интенсивность излучения (Iх), значение данной интенсивности можно получить посредством вычитания фоновой интенсивности (Iх’). На рисунке A.1 показан пример эффекта поправки на фон. На рисунке A.1а показан пример равномерного фона относительно длины волн. В этом случае фон может корректироваться с помощью положений A и B. На рисунке A.1b приведен пример изменяющегося фона относительно длины волны. В этом случае значения фоновой интенсивности могут корректироваться посредством вывода фоновой интенсивности (lx’), которая рассчитывается с помощью положения A и положения B интенсивности излучения.
 |
Рисунок A.1а - Равномерный фон относительно длины волны
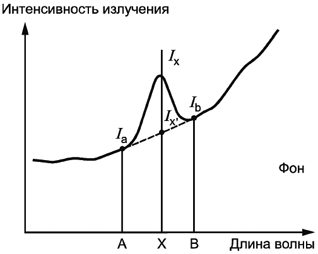 |
Рисунок A.1b - Изменяющийся фон относительно длины волны
Рисунок А.1 - Коррекция фона
Если используется метод стандартной добавки перед ее калибровкой, необходимо произвести вычитание фона с помощью метода коррекции фона (см. выше).
A.7 Программа микроволнового разложения
В таблице A.6 приведен пример программы микроволнового разложения образцов.
Операция | Время, мин | Выходная мощность, Вт | Давление ограничено до, МПа |
1A | 5 | 300 | 2,5 |
2A | 5 | 350 | 2,5 |
3A | 17 | 450 | 2,5 |
4A | 2 | 300 | 2,5 |
Стадия вентиляции A | 3 | 0 | 2,5 |
1B | 5 | 300 | 2,5 |
2B | 5 | 400 | 2,5 |
3B | 17 | 450 | 2,5 |
Стадия вентиляции B | 3 | 0 | 2,5 |
Выходная мощность для пяти сосудов. | |||
Приложение B
(справочное)
Результаты межлабораторных сравнительных испытаний Nos. 2 (IIS2) и 4А (IIS 4А)
Образец из полимера | Параметр | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | |
IIS 2-B09 | Pb | 480,0 | 380-640 | 3 | 7,5 | 21,1 | Недостаточно данных | |
IIS 4А-05 | Pb | 953,8 | 954,3 | 10 | 8,04 | 22,51 | 21,56 | 60,37 |
IIS 4А-08 | Pb | 98,3 | 98 | 10 | 1,26 | 3,54 | 1,29 | 3,60 |
IIS 4А-05 | Cd | 98,2 | 100 | 10 | 1,50 | 4,20 | 2,619 | 7,333 |
IIS 4A-08 | Cd | 138,5 | 137 | 10 | 3,98 | 11,14 | 3,39 | 9,49 |
IIS 4A-05 | Cd | 15,2 | 16 | 10 | 1,21 | 3,38 | 2,78 | 7,79 |
IIS 4A-08 | Cr | 98,1 | 100 | 9 | 3,45 | 9,67 | 3,54 | 9,92 |
Образец из металла |
|
|
|
|
|
|
|
|
IIS 2-D17 | Pb | 26,2 | 30 | 6 | 1,7 | 4,7 | 3,8 | 10,6 |
IIS 2-D15 | Pb | 118,0 | 190 | 2 | 4,2 | 11,9 | Недостаточно данных | |
Образец электроники |
|
|
|
|
|
|
|
|
IIS 2-F20 | Cd | 14,0 | 20 | 2 | 2,8 | 7,9 | Недостаточно данных | |
IIS 2-F20 | Pb | 17050 | 23000 | 2 | 354 | 990 | Недостаточно данных | |
Таблица B.2 - Статистические данные для AFS
Образец из полимера | Параметр | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | |
IIS2-C10 | Pb | 109,0 | 107,6 | 3 | 3,6 | 10,1 | Недостаточно | |
IIS2-C11 | Pb | 17,3 | 13,8 | 3 | 1,5 | 4,3 | данных | |
IIS 4А-04 | Pb | 15,6 | 15,7 | 18 | 0,71 | 1,98 | 0,94 | 2,63 |
IIS 4А-05 | Pb | 902,1 | 954 | 18 | 12,93 | 36,21 | 51,21 | 143,38 |
IIS2-C10 | Cd | 131,3 | 140,8 | 3 | 9,3 | 26,0 | Недостаточно | |
IIS2-C11 | Cd | 21,3 | 21,7 | 3 | 1,2 | 3,2 | данных | |
IIS 4А-04 | Cd | 173,6 | 183 | 18 | 2,33 | 6,53 | 6,44 | 18,02 |
IIS 4A-05 | Cd | 91,0 | 100 | 18 | 2,53 | 7,08 | 7,33 | 20,51 |
Образец из металла |
|
|
|
|
|
|
|
|
IIS 2-D16 | Pb | 1016,0 | 930 | 3 | 92,7 | 259,6 | Недостаточно данных | |
Таблица B.3 - Статистические данные для ICP-OES
Образец из полимера | Параметр | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | |
IIS2-B08 | Pb | 444,0 | 390-665 | 9 | 9,3 | 25,9 | 42,6 | 119,4 |
IIS2-B09 | Pb | 426,2 | 380-640 | 9 | 7,6 | 21,3 | 109,7 | 307,1 |
IIS2-C10 | Pb | 106,8 | 107,6 | 25 | 5,5 | 15,4 | 7,0 | 19,7 |
IIS2-C11 | Pb | 14,7 | 13,8 | 23 | 1,9 | 5,2 | 2,4 | 6,7 |
IIS2-C12 | Pb | 94,8 | 108,9 | 12 | 1,6 | 4,5 | 6,3 | 17,5 |
IIS2-C13 | Pb | 933 | 1084 | 12 | 20,4 | 57,0 | 47,6 | 133,4 |
IIS 4A-04 | Pb | 16,5 | 15,70 | 21 | 0,85 | 2,37 | 3,87 | 10,84 |
IIS 4A-05 | Pb | 950,8 | 954 | 24 | 11,64 | 32,59 | 40,99 | 114,78 |
IIS2-C10 | Cd | 142,4 | 140,8 | 23 | 2,6 | 7,3 | 3,9 | 11,0 |
IIS2-C11 | Cd | 20,9 | 21,7 | 28 | 1,5 | 4,3 | 2,6 | 7,3 |
IIS2-C12 | Cd | 9,7 | 10,8 | 12 | 0,6 | 1,5 | 0,7 | 2,0 |
IIS2-C13 | Cd | 95,8 | 106,9 | 12 | 2,7 | 7,5 | 6,6 | 18,5 |
IIS 4A-04 | Cd | 179,3 | 183 | 27 | 2,84 | 7,96 | 5,62 | 15,73 |
IIS 4A-05 | Cd | 98,1 | 100 | 24 | 1,43 | 4,00 | 4,22 | 11,82 |
IIS 4A-04 | Cr | 46,1 | 47 | 18 | 1,11 | 3,10 | 3,89 | 10,90 |
IIS 4A-05 | Cr | 15,5 | 16 | 21 | 1,18 | 3,29 | 3,52 | 9,84 |
Образец из металла |
|
|
|
|
|
|
|
|
IIS 2-D18 | Pb | 206,0 | 174 | 3 | 2,6 | 7,4 | Недостаточно данных | |
IIS 2-D16 | Pb | 988,0 | 930 | 6 | 9,4 | 26,4 | 22,1 | 61,9 |
IIS 2-D17 | Pb | 23,0 | 30 | 6 | 0,6 | 1,6 | 1,1 | 3,0 |
IIS 2-D15 | Pb | 193,1 | 190 | 11 | 6,0 | 16,9 | 31,4 | 87,9 |
Образец электроники |
|
|
|
|
|
|
|
|
IIS 2-F20 | Cd | 16,5 | 20 | 11 | 1,3 | 3,8 | 4,7 | 13,1 |
IIS 2-F20 | Pb | 16790 | 23000 | 11 | 264 | 739 | 749 | 2097 |
IIS 2-F21 | Pb | 22450 | 22000 | 6 | 462 | 1293 | 412 | 1153 |
IIS 2-F22 | Pb | 207483 | 222534 | 12 | 15336 | 42942 | 26752 | 74907 |
Таблица B.4 - Статистические данные для ICP-MS
Образец из полимера | Параметр | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | , мг/кг | |
IIS2-B08 | Pb | 481,2 | 390-665 | 12 | 12,7 | 35,5 | 44,5 | 124,6 |
IIS2-B09 | Pb | 462,3 | 380-640 | 6 | 14,2 | 39,8 | 69,3 | 194,1 |
IIS2-C10 | Pb | 102,3 | 107,6 | 3 | 0,6 | 1,6 | Недостаточно данных | |
IIS2-C11 | Pb | 16,2 | 13,8 | 6 | 2,9 | 8,1 | 5,5 | 15,4 |
IIS2-C12 | Pb | 103,8 | 108,9 | 6 | 1,8 | 5,1 | 2,6 | 7,3 |
IIS2-C13 | Pb | 1049 | 1084 | 6 | 21,4 | 59,9 | 118,7 | 332,3 |
IIS 4А-04 | Pb | 15,1 | 15,7 | 15 | 0,26 | 0,73 | 1,17 | 3,28 |
IIS 4А-05 | Pb | 949,2 | 954 | 15 | 18,84 | 52,75 | 25,19 | 70,52 |
IIS2-C10 | Cd | 137,3 | 140,8 | 3 | 2,9 | 8,1 | Недостаточно | |
IIS2-C11 | Cd | 22,0 | 21,7 | 3 | 0,0 | 0,0 | данных | |
IIS2-C12 | Cd | 10,0 | 10,8 | 6 | 0,2 | 0,6 | 0,5 | 1,3 |
IIS2-C13 | Cd | 103,7 | 106,9 | 6 | 1,4 | 3,8 | 11,8 | 33,1 |
IIS 4А-04 | Cd | 180,9 | 183 | 12 | 2,78 | 7,79 | 7,19 | 20,14 |
IIS 4A-05 | Cd | 94,4 | 100 | 15 | 1,27 | 3,55 | 4,44 | 12,43 |
IIS 4A-04 | Cr | 39,7 | 47 | 15 | 3,10 | 8,69 | 7,16 | 20,04 |
IIS 4A-05 | Cr | 14,2 | 16 | 15 | 0,75 | 2,09 | 1,32 | 3,69 |
Образец из металла |
|
|
|
|
|
|
|
|
IIS 2-D14 | Pb | 26,5 | 30 | 9 | 0,7 | 2,1 | 5,4 | 15,1 |
IIS 2-D18 | Pb | 156,1 | 174 | 9 | 3,9 | 10,8 | 5,5 | 15,5 |
IIS 2-D16 | Pb | 922,4 | 930 | 12 | 26,6 | 74,5 | 116,8 | 327,0 |
IIS 2-D17 | Pb | 36,7 | 30 | 3 | 1,5 | 4,3 | Недостаточно | |
IIS 2-D15 | Pb | 190,7 | 190 | 3 | 6,7 | 18,6 | данных | |
Образец электроники |
|
|
|
|
|
|
|
|
IIS 2-F20 | Cd | 24,7 | 20 | 3 | 0,6 | 1,6 | Недостаточно | |
IIS 2-F20 | Pb | 23633 | 23000 | 3 | 379 | 1060 | данных | |
IIS 2-F21 | Pb | 22220 | 22000 | 12 | 1488 | 4167 | 2395 | 6707 |
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего межгосударственного стандарта |
IEC 62321-1:2013 | IDT | ГОСТ IEC 62321-1-2016 "Определение регламентированных веществ в электротехнических изделиях. Часть 1. Введение и обзор" |
IEC 62321-2:2013 | IDT | ГОСТ IEC 62321-2-2016 "Определение регламентированных веществ в электротехнических изделиях. Часть 2. Разборка, отсоединение и механическая подготовка образца" |
IEC 62321-3-1:2013 | IDT | ГОСТ IEC 62321-3-1-2016 "Определение регламентированных веществ в электротехнических изделиях. Часть 3-1. Скрининг. Анализ свинца, ртути, кадмия, общего хрома и общего брома методом рентгенофлуоресцентной спектрометрии" |
ISO 3696:1987 | MOD | ГОСТ Р 52501-2005 "Вода для лабораторного анализа. Технические условия" |
ISO 5961 | - | * |
* Соответствующий межгосударственный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного стандарта. Примечание - В настоящей таблице использованы следующие условные обозначения степени соотетствия стандартов: - IDT - идентичные стандарты; - MOD - модифицированный стандарт. | ||
Библиография
[1] | ERNST, Т., РОРР, R., WOLF, М., VAN ELDIK, R., Analysis of eco-relevant elements and noble metals in printed wiring boards using AAS, ICP-OES and EDXRF, Anal. Bioanal. Chem., 2003, 375, p.805-814 | |
[2] | IEC 62321-7-2, | Determination of certain substances in electrotechnical products - Part 7-2: Hexavalent chromium (Cr(VI)) in polymers and electronics by the colorimetric method (Определение регламентированных веществ в электротехнических изделиях. Часть 7-2. Определение шестивалентного хрома (Cr(VI)) в полимерах и электронных частях колориметрическим методом) |
Дополнительные источники, не процитированные по тексту | ||
EN 1122, | Plastics - Determination of cadmium - Wet decomposition method (Пластмассы. Определение содержания кадмия методом влажного разложения) | |
EN 13346, | Characterization of sludges - Determination of trace elements and phosphorus - Aqua regia extraction methods (Характеристика шламов. Определение содержания микроэлементов и фосфора. Методы экстракции с применением царской водки) | |
EDGELL, K., US ЕРА Method Study 37 - SW-846 Method 3050 Acid Digestion of Sediments, Sludges, and Soils. EPA Contract No. 68-03-3254, November 1988 | ||
United States Environmental Protection Agency (EPA), EPA SW-846 Method 3052, Microwave-assisted acid digestion of siliceous and organically based matrices | ||
United States Environmental Protection Agency (EPA), EPA SW-846 Method 6010B, Inductively coupled plasma-atomic emission spectrometry | ||
United States Environmental Protection Agency (EPA), EPA SW-846 Method 7000, Series measurement methods for lead, cadmium, chromium and mercury | ||
RITTER, A. MICHEL, E. SCHMID, M. AFFOLTER, S., Interlaboratory test on polymers: determination of heavy metals in polymer matrices, Polymer testing 23 (2004), 467-474 | ||
ASTM D 4004-93, | Standard test methods for rubber determination of metal content by flame atomic absorption (AAS) analysis (Стандартный метод испытания для определения резины в содержании металла посредством пламенной атомно-абсорбционной спектрометрии (AAS) | |
ASTM D 3335-85A , | Standard test method of low concentration of lead, cadmium and cobalt in paint by atomic absorption spectroscopy (Стандартный метод испытаний низкой концентрации свинца, кадмия и кобальта в краске методом атомной абсорбционной спектроскопии) | |
ASTM D 1224-92, | Standard test methods for zinc and cadmium in paper (Стандартные методы испытаний для цинка и кадмия в бумаге) | |
ASTM D 3624-85A , | Standard test method for low concentration of mercury in paint by atomic absorption spectroscopy (Стандартный метод испытаний для низкой концентрации ртути в краске по атомно-абсорбционной спектроскопии) | |
ASTM C 146-94A, | Standard test methods for chemical analysis of glass sand (Стандартные методы испытаний для химического анализа стекольных песков) | |
ASTM С 169-92, | Standard test methods for chemical analysis of soda-lime and borosilicate glass (Стандартные методы испытаний для химического анализа натриево-кальциево* и боросиликатного стекла) | |
EN 10181, | Chemical analysis of ferrous materials determination of lead in steels flame atomic absorption spectrometric method (Химический анализ черных металлов определения свинца в кражах пламя* спектрометрического метода атомной абсорбции) | |
ASTM E 350-95, | Standard test methods for chemical analysis of carbon steel, low-alloy steel, silicon electrical steel, ingot iron, and wrought iron (Стандартные методы испытаний для химического анализа углеродистых сталей, низколегированных сталей, кремния электротехнической стали, слитков железа, и кованого железа) | |
ASTM E 353-93, | Standard test methods for chemical analysis of stainless, heat resisting, maraging, and other similar chromium-nickel-iron alloys (Стандартные методы испытаний для химического анализа из нержавеющей, жаропрочной, мартенситностареющих и других подобных хром-никель-железных сплавов) | |
ASTM E 363-93, | Standard methods for chemical analysis of chromium and ferrochromium (Стандартные методы химического анализа хрома и феррохрома) | |
ASTM E 361-99, | Standard test methods for the determination of arsenic and lead in ferromanganese (Стандартные методы испытаний для определения мышьяка и свинца в ферромарганце) | |
ASTM E 362-99, | Standard test methods for the determination of arsenic and lead in silicomanganese and ferrosilicon manganese (Стандартные методы испытаний для определения мышьяка и свинца в силикомарганце марганца и железобетона силикомарганца) | |
ASTM E 34-94, | Standard test methods for chemical analysis of aluminum and aluminum-base alloys (Стандартные методы испытаний для химического анализа из алюминия и алюминиевых сплавов) | |
ASTM E 478-03, | Standard test methods for chemical analysis of copper alloys (Стандартные методы испытаний для химического анализа медных сплавов) | |
ASTM E 1834-96, | Standard test method for determination of lead in nickel alloys by electrothermal atomic absorption spectrometric method (Стандартный метод испытаний для определения свинца в никелевых сплавов электротермическим атомно-абсорбционным методом спектрометрии) | |
ASTM E 536, | Standard test methods for chemical analysis of zinc and zic alloys (Стандартные методы испытаний для химического анализа цинка и ZIC сплавов) | |
EN 12441-3, | Zinc and zinc alloys - Chemical analysis - Part 3: Determination of lead, cadmium and copper - Flame atomic absorption spectrometric method (Цинк и цинковые сплавы. Химический анализ. Часть 3: определение свинца, кадмия и меди. Пламя методом атомной абсорбционной спектрометрии) | |
United States Environmental Protection Agency (EPA), EPA SW-846 Chapter 1-Quality Control, Revision 1: July 1992 | ||
УДК 621.3:543.632.495(083.74)(476) | МКС 43.040.10 | IDT |
Ключевые слова: изделия электротехнические, изделия электронные, образец, отбор образцов, регламентированные вещества | ||

{kind=link}