
ОГНЕУПОРЫ
И ОГНЕУПОРНЫЕ ИЗДЕЛИЯ


ГОСУДАРСТВЕННЫЕ СТАНДАРТЫ СОЮЗА ССР
ОГНЕУПОРЫ И ОГНЕУПОРНЫЕ ИЗДЕЛИЯ
Издание официальное
ИЗДАТЕЛЬСТВО СТАНДАРТОВ Москва — 1975
УДК 666.76(083.74)
ОТ ИЗДАТЕЛЬСТВА
Сборник «Огнеупоры н огнеупорные изделия* содержит стандарты, утвержденные до I декабря 1974 г.
В стандарты внесены все изменения, принятые до указанного срока. Около номера стандарта, в который внесено изменение, стоит знак •.
Текущая информация о вновь утвержденных и пересмотренных стандартах, а также о принятых к ним изменениях публикуется в выпускаемом ежемесячно «Информационном указателе стандартов*.
© Издательство стандартов, 1975
Группа И29
ГОСУДАРСТВЕННЫЙ стандарт СОЮЗА ССР
ЦИРКОНИЯ ДВУОКИСЬ (ТЕХНИЧЕСКАЯ), ЦИРКОНОВЫЙ КОНЦЕНТРАТ И ОГНЕУПОРЫ НА ИХ ОСНОВЕ
гост
13997-68
Методы анализа
Circonium dioxide (technical), circonium concentrate and circonium fireproof materials. Methods of analisis
Постановлением Комитета стандартов, мер и измерительных приборов при Совете Министров СССР от 4/Х 1968 г. № 22 срок введения установлен
с 1/V1I 1969 г.
Несоблюдение стандарта преследуется по закону
Настоящий стандарт распространяется на цирконовый концентрат, циркон обезжелезненный первого и второго сортов, двуокись циркония техническую, двуокись циркония стабилизированную окисью кальция или окисью магния и огнеупоры на их основе и устанавливает методы определения содержания двуокисей циркония, кремния, титана, гафния, окисей алюминия, железа, кальция, магния, калия, натрия и пятиокиси фосфора.
Применение методов предусматривается в стандартах и технических условиях, устанавливающих технические требования на продукцию.
1. ОБЩИЕ УКАЗАНИЯ
1.1. Для химического анализа отбирают среднюю пробу не менее 200 г размером частиц не более 2 мм. Пробу измельчают до прохождения через сетку № 05 по ГОСТ 6613—73, перемешивают, сокращают квартованием до 50—60 г и снова измельчают до прохождения через сетку № 02 по ГОСТ 6613—73. Если пробу измельчали в стальной ступке или в истирателе со стальными дисками, то частицы железа, попавшие в пробу при измельчении, должны быть удалены магнитом.
Издание официальное
Перепечатка воспрещена
После удаления железа и тщательного перемешивания методом квартования пробу сокращают до 10—15 г и снова измельчают в ступке из двуокиси циркония до прохождения через сетку № 0063 по ГОСТ 6613—73.
1.2. Проба материала перед взятием навески должна быть высушена при 105—110° С и тщательно перемешана.
1.3. Навеску испытуемой пробы и навеску материала, используемую для приготовления стандартных растворов, взвешивают с точностью ±0,0002 г.
1.4. Во всех случаях проведения анализа применяют реактивы квалификации не ниже ч. д. а.
1.5. Для приготовления водных растворов и проведения анализа применяют дистиллированную воду по ГОСТ 6709—72.
1.6. Под концентрацией растворов в процентах следует понимать количество вещества в граммах в 100 мл раствора.
1.7. В выражении «разбавленная 1 : 1, 1 : 2» и т. д. первые цифры означают объемные части кислоты или какого-либо раствора, вторые — объемные части воды.
1.8. Расчет титров производят до четвертой значащей цифры.
1.9. За результат анализа принимают среднее арифметическое двух параллельных определений.
Максимальное расхождение между результатами параллельных определений не должно превышать абсолютной величины допускаемых отклонений.
При получении результатов с расхождениями более допускаемых определение повторяют.
2. МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА ЦИРКОНОВОГО КОНЦЕНТРАТА И ОГНЕУПОРНЫХ ИЗДЕЛИЙ НА ЕГО ОСНОВЕ
2.1. Определение потери при прокаливании
2.1.1. Сущность метода
Потерю при прокаливании определяют весовым методом по разности между массой тигля с навеской до и после прокаливания.
2.1.2. Аппаратура
Муфельная печь с нагревом до 950—1000° С.
Тигли фарфоровые.
2.1.3. Проведение анализа
В прокаленный и доведенный до постоянной массы фарфоровый тигель помещают 1 г пробы, высушенной при 105—110° С, постепенно нагревают в электрической муфельной печи до 950—1000° С и выдерживают при этой температуре не менее 1 ч, после чего охлаждают в эксикаторе и взвешивают. Прокаливание повторяют (по 10 мин) до достижения постоянной массы.
2.1.4. Подсчет результатов анализа
Потерю при прокаливании (X) в процентах вычисляют по формуле:
у * G1 — ■ 100
Х =-----в-----•
где
Gj — масса тигля с навеской до прокаливания в г;
С2 — масса тигля с навеской после прокаливания в г;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений при содержании потери при прокаливании до 1,0% не должны превышать 0,10 абс.%.
2.2. Определение содержания двуокиси кремния
2.2.1. Сущность метода
Содержание двуокиси кремния определяют весовым методом. После разложения пробы щелочным плавлением кремниевую кислоту обезвоживают в азотнокислой среде, отфильтровывают, прокаливают и отгоняют в виде четырехфтористого кремния путем обработки фтористоводородной кислотой и прокаливания остатка.
2.2.2. Реактивы и растворы
Натрия гидрат окиси (натр едкий) по ГОСТ 4328—66.
Калий пиросернокислый по ГОСТ 7172—65.
Кислота соляная по ГОСТ 3118—67.
Кислота азотная по ГОСТ 4461—67 и разбавленная 1:1.
Кислота фтористоводородная (плавиковая кислота) по ГОСТ 10484—73, 40%-ный раствор.
Кислота серная по ГОСТ 4204—66.
Серебро азотнокислое по ГОСТ 1277—63, 1%-ный раствор.
Индикатор. Метиловый оранжевый (пара-диметиламиноазобен-золсульфокислый натрий) по ГОСТ 10816—64, 0,2%-ный раствор.
2.2.3. Проведение анализа
В никелевом, серебряном или железном тигле сплавляют при 400—450° С 4—5 г едкого натра до прекращения выделения пузырьков (спокойная поверхность расплавленной щелочи означает полное ее обезвоживание). На сплавленную и остывшую щелочь помещают навеску концентрата 0,5 г и сплавляют при 600—700° С в течение 20—30 мин.
В стакан из жаростойкого стекла вместимостью 400 мл предварительно наливают 20 мл разбавленной 1 : 1 азотной кислоты, куда выщелачивают сплав и тщательно отмывают тигель и крышку горячей водой из промывалки. После этого приливают 25—30 мл азотной кислоты. Раствор в стакане нагревают на электроплитке до прозрачного состояния, после чего количественно переносят в чашку и выпаривают содержимое чашки на водяной бане досуха. Образовавшиеся комочки осторожно растирают стеклянной палочкой. Остаток высушивают до полного удаления кислоты и выдерживают чашку на водяной бане еще 1 ч. После этого чашку снимают с бани, охлаждают, приливают 10 мл концентрированной азотной кислоты и снова выпаривают до полного удаления кислоты. Чашку с сухим остатком выдерживают в сушильном шкафу при 115—117° С в течение 1 ч, затем охлаждают, приливают 25 мл концентрированной соляной кислоты, выдерживают 10 мин, приливают 50—70 мл кипящей воды, перемешивают, дают раствору отстояться, после чего фильтруют через фильтр диаметром 9 см «белая лента».
Осадок на фильтре промывают горячей водой до удаления ионов хлора (реакция с 1%-ным раствором азотнокислого серебра). Фильтрат с промывными водами переводят в ту же фарфоровую чашку и вторично выпаривают на водяной бане досуха. Чашку с сухим остатком нагревают в сушильном шкафу при 115—117° С в течение 1 ч.
После охлаждения сухой остаток смачивают 20—25 мл соляной кислоты, выдерживают 10 мин, добавляют 30—40 мл горячей воды и отфильтровывают осадок кремниевой кислоты на другой фильтр диаметром 9 см.
Частички кремниевой кислоты, приставшие к чашке, переносят на фильтр при помощи слегка увлажненного кусочка фильтра.
Осадок на фильтре промывают горячей водой до удаления ионов хлора (реакция с1%-ным раствором азотнокислого серебра). Фильтры с осадками кремниевой кислоты помещают в платиновый тигель, осторожно высушивают, озоляют, не допуская воспламенения фильтров, затем прокаливают при 1100° С в течение 1 ч, охлаждают в эксикаторе и взвешивают. Прокаливание повторяют (по 10—15 мин) до достижения постоянной массы.
Осадок двуокиси кремния загрязнен примесями двуокисей титана, гафния, алюминия и железа. Для получения массы чистой двуокиси кремния прокаленный осадок смачивают несколькими каплями воды и приливают 0,5 мл серной и 6—10 мл фтористоводородной кислот. Содержимое тигля выпаривают досуха на закрытой электроплитке, не доводя до кипения. Для полного удаления серной кислоты тигель прокаливают 10 мин при 1100° С, затем охлаждают в эксикаторе и взвешивают.
Остаток в тигле после определения содержания двуокиси кремния сплавляют с 2 г пиросульфата калия. Остывший сплав растворяют в разбавленной соляной кислоте, присоединяют к фильтрату, полученному после отделения кремниевой кислоты, и разбавляют водой в мерной колбе вместимостью 250 мл (раствор А).
Раствор используют для определения содержания суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиоки-си фосфора.
В таких же условиях проводят контрольный опыт на содержание двуокиси кремния в применяемых реактивах. Необходимость в проведении контрольного опыта отпадает, если едкий натр, применяемый для сплавления концентрата, квалификации х. ч. или ч. д. а. и упакован не в стеклянную тару.
Если при выпаривании 6—10 мл фтористоводородной кислоты' с двумя каплями серной кислоты и прокаливании тигля при 1000° С остается нелетучий осадок, то массу последнего вычитают из веса тигля с остатком после обработки фтористоводородной кислотой.
2.2.4. Подсчет результатов анализа
Содержание двуокиси кремния (Л\) в процентах вычисляют по формуле:
(Gi~ Gab 100
G
где
G\ — масса тигля с осадком двуокиси кремния до обработки фтористоводородной кислотой в г;
С% — масса тигля с остатком после обработки фтористоводородной кислотой и прокаливания остатка в г;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений двуокиси кремния не должны превышать 0,40 абс.%.
2.3. Определение содержания суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора
2.3.1. Сущность метода
Метод основан на двукратном осаждении вышеуказнных элементов аммиаком с последующим получением их в виде окисей в результате прокаливания. Далее определение проводят весовым методом.
2.3.2. Реактивы и растворы
Аммиак водный по ГОСТ 3760—64, 25%-ный раствор.
Аммоний азотнокислый по ГОСТ 3761—72, 2%-ный раствор, к которому добавляют аммиак до слабощелочной реакции по метиловому красному.
Метиловый красный по ГОСТ 5853—51, 0,1%-ный спиртовой раствор; готовят следующим образом: 0,1 г метилового красного растворяют в 60 мл этилового спирта и разбавляют водой до 100 мл.
Аммоний хлористый по ГОСТ 3773—72, 20%-ный раствор.
Серебро азотнокислое по ГОСТ 1277—63, 1%-ный раствор.
Кислота соляная по ГОСТ 3118—67, разбавленная 1 : 1.
Кислота азотная по ГОСТ 4461—67, разбавленная 1 : 1.
2.3.3. Проведение анализа
100 мл раствора А помещают в стакан вместимостью 300 мл, добавляют бумажной массы (из беззольных фильтров) и 5 мл 20%-ного раствора хлористого аммония. Раствор нагревают до кипения и в присутствии нескольких капель раствора метилового красного осаждают аммиаком цирконий, гафний, алюминий, железо, титан и фосфор. Аммиак прибавляют до перехода окраски индикатора в желтый цвет и появления слабого запаха.
После осаждения гидроокисей раствор нагревают до кипения, дают осадку осесть и фильтруют через фильтр диаметром 11 см «красная» или «белая лента». Осадок промывают несколько раз горячим 2%-ным раствором азотнокислого аммония. Фильтр с осадком переносят в стакан, в котором производилось осаждение, приливают для растворения осадка 30 мл разбавленной соляной кислоты, растирают фильтр в кашицеобразную массу, разбавляют водой до 100—150 мл и вторично осаждают алюминий, железо, титан, цирконий, гафний и фосфор, как описано выше.
Раствор с осадком фильтруют на фильтр диаметром 11 см «красная» или «белая лента» и промывают осадок горячим 2%-ным раствором азотнокислого аммония до исчезновения реакции на ион хлора (реакция с 1%-ным раствором азотнокислого серебра на подкисленную азотной кислотой промывную воду).
Объединенный фильтрат после двукратного осаждения циркония, гафния, алюминия, железа, титана и фосфора используют для определения содержания окисей кальция и магния.
Фильтр с осадком помещают во взвешенный платиновый тигель, подсушивают, озоляют и прокаливают 1,5 ч при 1200° С. После охлаждения в эксикаторе тигель с осадком взвешивают. Прокаливание повторяют (по 20 мин) до достижения постоянной массы. Осадок суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора используют для определения содержания гафния спектральным методом.
2.3.4. Подсчет результатов анализа
Содержание суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора (Х2) в процетнах вычисляют по формуле: где
■ 250 - 100 G . 100 *
Gi — масса осадка суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора в г;
250— исходный объем испытуемого раствора в мл;
100 — аликвотная часть испытуемого раствора в мл;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,50 абс.%.
2.4. Фотоколориметрический метод определения содержания окиси железа (при содержании окиси железа от 0,01 до 0,50%)
2.4.1. Сущность метода
В основу фотоколориметрического метода определения окиси железа положена реакция образования в аммиачной среде окрашенного в желтый цвет трисульфосалицилата железа при условии связывания в комплексы мешающих элементов раствором винной кислоты.
2.4.2. Аппаратура, реактивы и растворы Фотоэлектроколориметр типа ФЭК-М или ФЭК-56. Аммиак водный по ГОСТ 3760—64, 25%-ный раствор.
Кислота серная по ГОСТ 4204—66, разбавленная 1 : 1, 1 : 2 и 1 : 9.
Аммоний виннокислый средний по ГОСТ 4951—71, 25%-ный раствор.
Кислота сульфосалициловая по ГОСТ 4478—68, 20%-ный раствор.
Железа окись по ГОСТ 4173—66, х. ч., стандартные растворы.
Раствор Б; готовят следующим образом: 0,1 г высушенной окиси железа помещают в коническую колбу вместимостью 500 мл, приливают 50 мл разбавленной 1 : 1 соляной кислоты и, накрыв колбу часовым стеклом или стеклянным шариком, нагревают на водяной бане до растворения окиси железа. Затем содержимое колбы охлаждают, переводят в мерную колбу вместимостью 1000 мл, доливают до метки водой и перемешивают.
1 мл раствора Б содержит 0,0001 г окиси железа.
Раствор В; готовят следующим образом: 10 мл раствора Б переносят пипеткой в мерную колбу вместимостью 100 мл, прибавляют 1 мл серной кислоты, разбавленной 1 : 9, и доводят водой до метки.
1 мл раствора В содержит 0,00001 г окиси железа.
2.4.3. Проведение анализа
25 мл раствора А (см. п. 2.2.3) помещают в мерную колбу вместимостью 50 мл, приливают 2 мл 25%-ного раствора виннокислого аммония, 10 мл 20%-него раствора сульфосалициловой кислоты, раствор аммиака до появления устойчивой желтой окраски и в избыток 5 мл. Раствор охлаждают, доводят до метки дистиллированной водой, перемешивают и через 15 мин колориметрируют на фотоэлектроколориметре ФЭК-М с синим светофильтром или ФЭК-56 со светофильтром № 4 при длине волны 440 нм в кюветах с толщиной слоя 30 или 50 мм.
В качестве раствора сравнения применяют раствор контрольного опыта, содержащий те же реактивы и в тех же количествах, кро-39* 611
ме исследуемого раствора. Количество окиси железа определяют по калибровочному графику.
Построение калибровочного графика
В мерные колбы вместимостью по 100 мл отбирают аликвотные части свежеприготовленного раствора В в количестве: 0,5; 1; 2,5; 5; 10; 15; 20; 25 мл.
В каждую колбу прибавляют по 2 мл 25%-ного раствора виннокислого аммония, сульфосалициловую кислоту, аммиак до перехода окраски раствора в желтую и в избыток 5 мл. Растворы в колбах доводят до метки водой, перемешивают и через 15 мин колориметрируют на фотоэлектроколориметре ФЭК-М с синим светофильтром или ФЭК-56 со светофильтром № 4 в кюветах с толщиной слоя 30 или 50 мм, используя в качестве раствора сравнения раствор контрольного опыта.
Калибровочный график строят в координатах: количество окиси железа в миллиграммах — оптическая плотность раствора.
2.4.4. Подсчет результатов анализа
Содержание окиси железа (Х3) в процентах вычисляют по формуле: где
Gi • 250 • 100
25 • G • 1000 ’
250 — исходный объем испытуемого раствора в мл;
25 — аликвотная часть испытуемого раствора в мл;
Gj — количество окиси железа, найденное по калибровочному графику, в мг;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать:
при содержании окиси железа от 0,01 до 0,10% — 0,01 абс.%;
при содержании окиси железа от 0,10 до 0,50% —0,03 абс.%.
Фотоколориметрический метод определения содержания окиси железа является арбитражным.
2.5. Объемный метод определения содержания окиси железа (при содержании окиси железа свыше 0,20%).
2.5.1. Реактивы и растворы
Реактивы и растворы указаны в разд. 6, ГОСТ 2642.1—71.
2.5.2. Проведение анализа
1,0 г пробы помещают в кварцевый или платиновый тигель, смешивают с 7—10 г пиросульфата калия и сплавляют при 800° С. Далее определение ведут как указано в разд. 6, ГОСТ 2642.1—71.
Объемный метод определения содержания окиси железа является арбитражным.
2.6. Фотоколориметрический метод определения содержания двуокиси титана
2.6.1. Сущность метода
Метод основан на измерении интенсивности окраски окрашенного в желтый цвет комплекса титана с перекисью водорода.
2.6.2. Аппаратура, реактивы и растворы Фотоэлектроколориметр ФЭК-М или ФЭК-56.
Кислота серная по ГОСТ 4204—66, 10%-ный и 5%-ный рас
творы.
Перекись водорода (пергидроль) по ГОСТ 177—71, 3%-ный раствор.
Калий пиросернокислый по ГОСТ 7172—65.
Титана двуокись.
Стандартный раствор соли титана; готовят следующим образом: навеску двуокиси титана 0,2 г сплавляют в кварцевом или платиновом тигле с 6—7 г пиросернокислого калия при800°С дополучения прозрачного расплава. После охлаждения сплав растворяют в 150 мл 10%-ного раствора серной кислоты при нагревании. Раствор переводят в мерную колбу вместимостью 1000 мл, доводят до метки 5%-ным раствором серной кислоты и перемешивают.
Титр стандартного раствора титана устанавливают весовым методом.
1 мл стандартного раствора соли титана содержит 0,0002 г двуокиси титана.
2.6.3. Проведение анализа
Из мерной колбы с фильтратом после выделения кремниевой кислоты отбирают 100 мл раствора А (см. п. 2.2.3) в стакан вместимостью 300 мл, осаждают гидроокиси циркония, гафния, титана, алюминия, железа аммиаком и отфильтровывают осадок на фильтр диаметром 11 см «красная» или «белая лента». Осадок промывают 6—7 раз горячей водой и смывают его струей воды из промывалки в стакан, в котором проводилось осаждение. Затем осадок растворяют при нагревании в 5%-ном растворе серной кислоты (раствор должен быть совершенно прозрачным), охлаждают, переводят в мерную колбу вместимостью 100 мл, прибавляют 3 мл перекиси водорода, доводят до метки 5%-ным раствором серной кислоты и перемешивают.
Оптическую плотность растворов определяют на фотоэлектроколориметре ФЭК-М с синим светофильтром или ФЭК-56 со светофильтром № 3, используя в качестве раствора сравнения раствор контрольного опыта.
Количество двуокиси титана определяют по калибровочному графику.
Построение калибровочного графика
В мерные колбы вместимостью по 100 мл отмеривают стандартный раствор титана (0,0002 г/мл TiO2) в количестве 1; 4; 8; 12; 16 и 20 мл. В каждую колбу добавляют по 3 мл 3%-ного раствора перекиси водорода, доливают до метки 5%-ным раствором серной кислоты и перемешивают.
Оптическую плотность всех полученных растворов определяют на ФЭК-М. с синим светофильтром в кювете с толщиной слоя 20 мм, используя в качестве раствора сравнения 5%-ный раствор серной кислоты. По полученным данным строят калибровочный график в координатах: количество двуокиси титана в миллиграммах — оптическая плотность раствора.
2.6.4. Подсчет результатов анализа
Содержание двуокиси титана (Х4) в процентах вычисляют по формуле:
Gi ■ 250 > 100 100 - G - 1000
где
— количество двуокиси титана, найденное по калибровочному графику, в мг;
250 — исходный объем испытуемого раствора в мл;
100 — аликвотная часть испытуемого раствора в мл;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать:
при содержании двуокиси титана до 2% — 0,05 абс.%; при содержании двуокиси титана свыше 2% — 0,10 абс.%.
2.7. Трилонометрический метод определения содержания окиси алюминия с индикатором «ПАН»
2.7.1. Сущность метода
Метод основан на обратном титровании избытка трилока Б раствором сернокислой меди в среде ацетатного буферного раствора при pH 4,8 с применением спиртового раствора индикатора 1,2-(пи-ридил-азо)-2-нафтола («ПАН»).
2.7.2. Реактивы и растворы
Натрия гидрат окиси (натр едкий) по ГОСТ 4328—66 и 50%-ный раствор; хранят в полиэтиленовом сосуде.
Кислота серная по ГОСТ 4204—66, плотностью 1,84 г/см3.
Кислота соляная по ГОСТ 3118—67, плотностью 1,19 г/см3.
Кислота уксусная по ГОСТ 61—69, 2 М раствор.
Аммиак водный по ГОСТ 3760—64, 25%-ный раствор.
Натрий уксуснокислый по ГОСТ 199—68, раствор, содержащий 540 г уксуснокислого натрия в 1000 мл раствора.
Медь сернокислая по ГОСТ 4165—68, 0,05 М раствор; готовят следующим образом: 12.5 г медного купороса (СиБО^бЫгО) растворяют в воде и приливают 2 мл серной кислоты на 1000 мл.
Индикатор 1,2- (пиридил-азо)-2-нафтол («ПАН»), 0,2%-ный спиртовой раствор.
Буферный раствор с pH 4,8; готовят следующим образом: 500 мл 2 М раствора уксусной кислоты смешивают с 500 мл раствора, содержащего 540 г уксуснокислого натрия в 1000 мл раствора.
Стандартный раствор хлористого алюминия; готовят следующим образом: 0,65—0,66 г точно взвешенного, «особо чистого» алюминия растворяют в 25 мл 50%-ного раствора едкого натра, нейтрализуют соляной кислотой и прибавляют избыток соляной кислоты 30 мл, затем доводят до 1 л водой.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73; 0,025 М раствор (9,3 г трилона Б на 1 л раствора).
Соотношение между растворами трилона Б и сернокислой меди устанавливают следующим образом: отмеривают бюреткой 10 мл 0.025 М раствора трилона Б, прибавляют 100 мл воды, 10 мл ацетатного буферного раствора с pH 4,8 и пять капель индикатора «ПАН», после чего титруют раствором сернокислой меди до перехода окраски из желто-зеленой в ярко-фиолетовую. Проводят не менее трех титрований и берут среднее арифметическое значение.
Соотношение между растворами трилона Б и сернокислой меди (К) рассчитывают с точностью до второго знака после запятой по формуле:
где
V — объем раствора трилона Б, взятый для установки соотношения, в мл;
Vi — объем раствора сернокислой меди, израсходованный на титрование, в мл.
Для определения титра 0,025 Л1 раствора трилона Б по окиси алюминия пипеткой отбирают 10 мл стандартного раствора хлористого алюминия в коническую колбу вместимостью 250—300 мл, прибавляют несколько капель соляной кислоты плотностью 1,19 г/см3, прибавляют 100 мл воды, приливают 25 мл 0,025 М раствора трилона Б, нагревают до 60—70° С, нейтрализуют раствором аммиака по бумаге «конго» до слабо-розового цвета, прибавляют 10 мл ацетатного буферного раствора с pH 4,8 и пять капель индикатора «ПАН». Нагретый раствор оттитровывают 0,05 М раствором сернокислой меди до перехода окраски из желто-зеленой в сине-фиолетовую.
Титр раствора трилона Б (7), выраженный в г/мл окиси алюми ния, вычисляют по формуле:
где
Г) — титр стандартного раствора хлористого алюминия, выраженный в г/мл окиси алюминия;
V — объем стандартного раствора хлористого алюминия, взятый для установки титра раствора трилона Б, в мл;
Vi — объем раствора трилона Б в мл;
V2 — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой
меди.
2.7.3. Проведение анализа
В никелевом или серебряном тигле сплавляют 4 г едкого натра до получения прозрачного расплава. На сплавленную и остывшую щелочь помещают навеску концентрата 0,2 г и сплавляют при 500—700° С в течение 20—30 мин. Плав выщелачивают горячей водой в стакан вместимостью 300 мл и тщательно отмывают тигель от сплава. Для определения содержания окиси алюминия может быть использовано 100 мл раствора А (см. п. 2.2.3), взятого в стакан вместимостью 300 мл и нейтрализованного 50%-ным раствором едкого натра с добавлением 7—10 мл избытка последнего. Раствор с осадком кипятят 5 мин, добавляют в него беззольной бумажной массы и фильтруют на фильтр диаметром 11 см «белая лента», собирая фильтрат в коническую колбу вместимостью 250—300 мл. Фильтр промывают горячей водой 8—10 раз.
Полученный раствор алюмината натрия нейтрализуют соляной кислотой до синего цвета бумаги «конго» и приливают в избыток 1 мл соляной кислоты. Затем к раствору приливают 15—20 мл 0,025 М раствора трилона Б, нагревают до 60—70° С, нейтрализуют по бумаге «конго» аммиаком до слабо-розового цвета, приливают 20 мл ацетатного буферного раствора с pH 4,8, пять капель индикатора «ПАН» и титруют избыток трилона Б 0,05 М раствором сернокислой меди до перехода окраски раствора из желто-зеленой з сине-фиолетовую.
2.7.4. Подсчет результатов анализа
Содержание окиси алюминия (Х5) в процентах вычисляют по формуле:
где
У — объем раствора трилона Б в мл;
Vt— объем раствора сернокислой меди, израсходованный на титрование, в мл;
К — соотношение между растворами трилона Б и сернокислой меди;
Т — титр раствора трилона Б, выраженный в г/мл окиси алюминия;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,15 абс.%-
Трилонометрический метод определения содержания окиси алюминия с индикатором «ПАН» является арбитражным.
2.8. Трилонометрический метод определения содержания окиси алюминия с индикатором ксиленоловым оранжевым
2.8.1. Сущность метода
Метод основан на трилонометрическом титровании избытка трилона Б раствором азотнокислого висмута в присутствии ксиленолового оранжевого в качестве индикатора. Навеску материала разлагают сплавлением с едким натром.
2.8.2. Реактивы и растворы
Натрий гидрат окиси (натр едкий) по ГОСТ 4328—66, 20%-ный и 4%-ный растворы.
Кислота азотная по ГОСТ 4461—67, разбавленная 1:1.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73. 0,025 М раствор; готовят следующим образом: 9,3 г трилона Б растворяют в воде при слабом нагревании, отфильтровывают раствор в мерную колбу вместимостью 1 л и разбавляют водой до метки.
Кислота уксусная по ГОСТ 61—69, 2 М раствор; готовят следующим образом: 120 мл уксусной кислоты разбавляют до метки водой в мерной колбе вместимостью 1 л.
Натрий уксуснокислый по ГОСТ 199—68, 2 М раствор, готовят следующим образом: 272 г уксуснокислого натрия растворяют в воде, фильтруют раствор в мерной колбе вместимостью 1 л и доводят объем раствора до метки дистиллированной водой.
Буферный раствор с pH 4,8; готовят следующим образом: смешивают два объема 2 Л1 раствора уксуснокислого натрия с одним объемом 2 М раствора уксусной кислоты.
Висмут по ГОСТ 10928—64.
Висмут азотнокислый по ГОСТ 4110—62, 0,025 М раствор; готовят следующим образом: 75 г азотнокислого висмута растворяют при нагревании в 60 мл азотной кислоты, разбавленной 1:1. Полученный раствор фильтруют, добавляют 60 мл азотной кислоты, разбавленной 1 : 1, и доливают водой до 1 л.
0,025 М раствор азотнокислого висмута может быть приготовлен также из металлического висмута. Для этого 5,22 г металлического висмута растворяют в 60 мл азотной кислоты плотностью 1,4 г/см3 и кипятят до удаления окислов азота. Полученный раствор переводят в колбу вместимостью 1 л и разбавляют водой до метки.
Аммиак водный по ГОСТ 3760—64, 25%-ный раствор и разбавленный 1:1.
Аммоний азотнокислый по ГОСТ 3761—72, 2%-ный раствор: к раствору добавляют аммиак до слабощелочной реакции по метиловому красному.
Метиловый красный по ГОСТ 5853—51, 1%-ный спиртовой раствор; готовят следующим образом: 0,1 г метилового красного растворяют в 60 мл спирта и разбавляют водой до 100 мл.
Ксиленоловый оранжевый, 0,5%-ный водный раствор.
Бумага индикаторная «конго».
Алюминий первичный по ГОСТ 11069—74.
Стандартный раствор азотнокислого алюминия, содержащий 0,003 г/мл окиси алюминия; готовят следующим образом: 1,5 г металлического алюминия растворяют при нагревании в 25 мл 20%-ного раствора едкого натра, разбавляют до 150—200 мл дистиллированной водой, фильтруют через фильтр «белая лента», предварительно обработанный три раза 4%-ным раствором едкого натра, в мерную колбу вместимостью 1 л, прибавляют при помешивании 50 мл азотной кислоты плотностью 1,4 г/см3 и доводят водой до 1 л.
Для установки титра стандартного раствора азотнокислого алюминия в стакан вместимостью 300 мл помещают 10 мл стандартного раствора, прибавляют 100 мл воды, нагревают до кипения, прибавляют 1—2 капли раствора метилового красного и аммиак, разбавленный 1 ; 1, до перехода окраски раствора из розовой в желтую.
Раствор с осадком кипятят 2 мин, затем горячий раствор фильтруют через беззольный фильтр. Осадок промывают на фильтре 5—6 раз горячим 2%-ным раствором азотнокислого аммония. Фильтр с осадком помещают во взвешенный платиновый тигель, озоляют и прокаливают при 1200° С в муфельной печи до постоянной массы.
Титр стандартного раствора алюминия (7\), выраженный в г/мл окиси алюминия, вычисляют по формуле:
где
G — масса прокаленного осадка в г;
V — объем стандартного раствора алюминия, взятый для установки титра, в мл.
Соотношение между растворами трилона Б и азотнокислого висмута устанавливают следующим образом: в коническую колбу вместимостью 250 мл помещают 3 мл раствора трилона Б, добавляют 50—60 мл воды, 15 мл буферного раствора, четыре капли индикатора ксиленолового оранжевого и титруют из микробюретки раствором азотнокислого висмута до перехода окраски из желтой в малиновую.
Соотношение между раствором трилона Б и азотнокислого висмута (X;) рассчитывают по формуле:
и — количество раствора трилона Б, взятое для титрования.
в мл;
— количество раствора азотнокислого висмута, израсходованное на титрование, в мл.
Для установки титра раствора трилона Б по стандартному раствору алюминия в коническую колбу вместимостью 250 мл помещают 1 мл стандартного раствора азотнокислого алюминия, 50 мл дистиллированной воды, 5 мл 20%-ного раствора едкого натра. 5 мл раствора трилона Б, нейтрализуют азотной кислотой, разбавленной 1:1, до перехода цвета бумаги «конго» в сине-фиолетовый и кипятят в течение 1—2 мин.
Затем раствор охлаждают, прибавляют 15 мл буферной смеси, четыре капли раствора индикатора ксиленолового оранжевого и титруют из микробюретки раствором азотнокислого висмута до перехода окраски раствора из желтой в малиновую.
Титр раствора трилона Б (T2)t выраженный в г/мл окиси алюминия, вычисляют по формуле:
где
Т[ — титр стандартного раствора алюминия, выраженный в г/мл окиси алюминия;
V — объем стандартного раствора алюминия, взятый для установки титра раствора трилона Б, в мл;
Vi — объем раствора трилона Б в мл;
1'г — объем раствора азотнокислого висмута, израсходованный на титрование, в мл;
Ki — соотношение между растворами трилона Б и азотнокислого висмута.
2.8.3. Проведение анализа
В никелевом тигле при температуре 400—450° С расплавляют
4 г едкого натра до прекращения выделения пузырьков (спокойная поверхность расплавленной щелочи означает полное ее обезвоживание). На расплавленную остывшую поверхность щелочи помещают 0,10—0,15 г концентрата и сплавляют при 600—700° С в течение 20—30 мин. Сплав выщелачивают теплой водой в стакан вместимостью 300 мл и тщательно отмывают тигель. Раствор с осадком кипятят 3—5 мин, после чего охлаждают и переводят в мерную колбу вместимостью 200 мл. Объем раствора доводят до метки дистиллированной водой, перемешивают и фильтруют через сухой фильтр «белая лента». Первые две порции фильтрата отбрасывают, а затем отбирают 100 мл фильтрата, помещают в коническую колбу вместимостью 250 мл, прибавляют из микробюретки
5 мл раствора трилона Б и нейтрализуют азотной кислотой, разбавленной 1 : 1, до перехода цвета бумаги «конго» в сине-фиолетовый. Раствор кипятят 1—2 мин, охлаждают, приливают 15 мл буферного раствора, четыре капли раствора индикатора ксиленолового оранжевого и титруют из микробюретки раствором азотнокислого висмута до перехода окраски раствора из желтой в малиновую.
2.8.4. Подсчет результатов анализа
Содержание окиси алюминия (Х6) в процентах вычисляют по формуле:
х = ■ Уг)г2 . 100. 200
6 (7-100
где
V — объем раствора трилона Б в мл;
Pi — объем раствора азотнокислого висмута, израсходованный на титрование, в мл;
7(1 — соотношение между растворами трилона Б и азотнокислого висмута;
?2 — титр раствора трилона Б, выраженный в г/мл окиси алюминия;
100 — аликвотная часть раствора в мл;
200 — исходный объем раствора в мл;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений при содержании окиси алюминия до 2% не должны превышать 0,30 абс. %.
2.9. Спектральный метод определения содержания двуокиси гафния (при содержании двуокиси гафния от 0,1 до 3%).
2.9.1. Сущность метода
Содержание двуокиси гафния в двуокиси циркония определяют методом эмиссионного спектрального анализа с применением фотографического фотометрирования. Пробу смеси с буфером испаряют из отверстия в электроде в угольной дуге постоянного тока.
При анализе цирконового концентрата используют выделенный химическим методом осадок (Х2), содержащий двуокиси циркония, гафния, титана, окиси алюминия, железа и пятиокись фосфора (п. 2.3.3).
2.9.2. Аппаратура, материалы и реактивы
Спектрограф средней дисперсии типа ИСП-28. Система освещения трехлинзовая с промежуточной диафрагмой.
Источник постоянного тока напряжением 220 В, силой тока 15 А.
Микрофотометр МФ-2 или другой любой системы.
Станок настольный токарный.
Весы аналитические.
Ступка из двуокиси циркония или агатовая.
Секундомер.
Кюветы для фоторабот.
Угольный порошок, спектрально-чистый.
Фотопластинки «спектрографические» или «микро» светочувствительностью 16—32 ед. по ГОСТ 10691—63.
Барий азотнокислый по ГОСТ 3777—69.
Проявитель:
вода дистиллированная по ГОСТ 6709—72 — 1 л;
метол (пара-метиламинофеносульфат) по ГОСТ 5.1177—71—• 1 г;
натрий сернистокислый (сульфит натрия) кристаллический по ГОСТ 429—66 — 52 г;
или натрий сернистокислый (сульфит натрия) безводный по ГОСТ 195—66 — 26 г;
гидрохинон (парадиоксибензол) по ГОСТ 19627—74 — 5 г; натрий углекислый кристаллический по ГОСТ 84—66 — 54 г; или натрий углекислый безводный по ГОСТ 83—63 — 22 г; калий бромистый по ГОСТ 4160—74— 1 г.
При приготовлении проявителя сохраняют указанную последовательность растворения составляющих. Для предохранения от окисления проявитель рекомендуется хранить в темном месте в колбах с узким горлом с притертой пробкой вместимостью примерно 200 мл. Проявитель должен полностью заполнять колбу почти до пробки. Может быть использозан также стандартный проявитель № 1 (ГОСТ 2817—50), который рекомендуется сохранять в виде двух растворов.
Фиксаж:
вода дистиллированная по ГОСТ 6709—72 — 1 л;
тиосульфат натрия кристаллический (гипосульфит натрия) по ГОСТ 244—68 — 400 г;
натрин сернистокислый (сульфит натрия) кристаллический по ГОСТ 429—66 — 50 г;
или натрий сернистокислый (сульфит натрия) безводный по ГОСТ 195—66 — 25 г;
кислота уксусная по ГОСТ 61—69 — 8 мл.
Буфер, состоящий из смеси угля и азотнокислого бария в соотношении 1:1, измельченный до прохождения через сетку № 0063. Смесь тщательно перемешивают до получения равномерного распределения составляющих.
Электроды; готовят из спектрально-чистых угольных стержней диаметром 6±0,2 мм. Верхние электроды затачивают на усеченный конус диаметром торца 3 мм. Длина электрода 40 мм. Расстояние от основания конуса до торца 10 мм. Нижние электроды с рабочей стороны по высоте 10 мм от торца стачивают до диаметра 4 мм; с центра торца вдоль оси сверлят отверстие диаметром 2 мм глубиной 5 мм. Длина электрода 40 мм (черт. 1).
Эталонные образцы двуокиси циркония со следующим содержанием двуокиси гафния приведены в таблице.
Первый эталон готовят введением 3% двуокиси гафния в очищенную от гафния двуокись циркония, содержащую обычные для анализируемых образцов концентрации окислов титана, алюминия, железа, фосфора и других элементов. Эталоны № 2—4 готовят последовательным разбавлением предыдущего эталона в три раза указанной двуокиси циркония.
Номер эталона | Содержание | двуокиси гафния в % |
1 | 3.0 | |
2 | 1,0 | |
3 | 0,33 | |
4 | 0,11 |
2.9,3. Проведение анализа
Составляют рабочие смеси, состоящие из 0,2 г пробы или эталонного образца и 0,4 г буфера. Отверстия в электродах плотно заполняют рабочей смесью. Кассету спектрографа заряжают спектрографической фотопластинкой типа II или «микро», укладывая ее в центральной части кассеты. Ширина щели спектрографа — 0,01 мм. Дутовой промежуток — 3 мм. Сила тока дуги — 15 А. Время экспозиции — 90 с. На фотопластинку снимают спектры проб и эталонных образцов № 3—4. Спектр каждой пробы и эталонного образца снимают по три раза, каждый раз на новом месте фотопластинки.
При работе по методу постоянной аналитической кривой на той же фотопластинке снимают спектр железных электродов через девятиступенчатый ослабитель со временем экспозиции 90 с.
о о
Фотометрируют линии гафния 2638,7 А и циркония 2665,18 А.
Угольные электроды
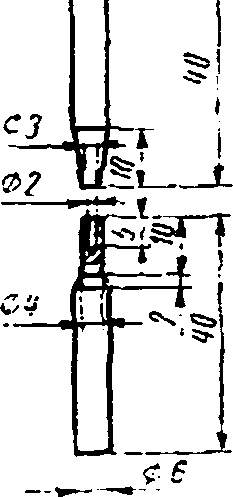
Черт. 1
При выполнении анализа методом трех эталонов на калибровочном графике по оси ординат откладывают ASzr-nt = Szr—Shi — разность почернений линий циркония (Szr) и гафния (Sm), по оси абсцисс — логарифм концентрации гафния в эталонных образцах (IgC). Определив для пробы значение ASzr-нь по калибровочной кривой находят IgC и затем С. (В случае построения калибровочного графика на полулогарифмической бумаге оперируют не с IgC, а непосредственно с С).
При применении метода постоянной аналитической кривой по оси ординат откладывают lg , где /иг и /zr — интенсивность линий гафния и циркония, по оси абсцисс — IgC. Переход от почернений к интенсивностям осуществляется с помощью спектра железа, снятого через девятиступенчатый ослабитель на той же фотопл астинке.
2.9.4. Подсчет результатов анализа
Содержание двуокиси гафния (Х7) в процентах вычисляют по формуле:
100
где
Х2 — содержание суммы двуокисей циркония, гафния, титана, окисей железа, алюминия и пятиокиси фосфора в % (п. 2.3.4);
С — содержание двуокиси гафния по данным спектрального анализа в %.
Допускаемые расхождения между результатами параллельных определений двуокиси гафния не должны превышать 0,40 абс. %.
Спектральный метод определения содержания двуокиси гафния является арбитражным.
2.10. Спектральный метод определения содержания двуокиси гафния
2.10.1. Сущность метода
Содержание двуокиси гафния определяют методом эмиссионного спектрального анализа с применением фотографического фо-тометрирования. Анализ выполняют возбуждением спектра в конденсированной искре при содержании двуокиси гафния 0,3—4,0% или в дуге переменного тока при содержании двуокиси гафния 0,19—2,6%.
2.10.2. Аппаратура, материалы и реактивы
Спектрограф ДФС-8 с трехлинзовой системой освещения.
Генератор искры ИГ-2 или ИГ-3.
Генератор активизированной дуги переменного тока.
Микрофотометр нерегистрирующий МФ-2.
Станок для заготовки электродов.
Гидравлический пресс (сила давления до 6000 кг).
Стальная прессформа для прессования таблеток (черт. 2).
Весы аналитические с разновесом.
Шкаф сушильный с нагревом до t= 150° С.
Кислота фтористоводородная (плавиковая кислота) по ГОСТ 10484—73.
Кислота азотная по ГОСТ 4461—67.
Кислота серная по ГОСТ 4204—66.
Кислота соляная по ГОСТ 3118—67, разбавленная 1 : 1.
Аммоний роданистый по ГОСТ 3768—64, 10-ный раствор.
Аммиак водный по ГОСТ 3760—64, 5%-ный раствор.
Весы торзионные.
Ступка яшмовая.
Чашка платиновая.
Графит металлургический.
Двуокись циркония.
Угольный порошок. Молибденовый ангидрид.
Кварц молотый (порошок).
Угольные электроды спектральные марки С-3.
Двуокись гафния.
Фотопластинки спектральные, чувствительностью 10—16 ед. по ГОСТ 10691—63.
Стандартные образцы — изоморфные смеси, свободные от гафния, двуокиси циркония и двуокиси гафния, охватывающие интервал концентраций 0,3—4,0% НЮ2. Первый эталон готовят введением в двуокись циркония 4 вес. % двуокиси гафния. Остальные эталоны готовят последовательным разбавлением предыдущего эталона двуокисью циркония (не более чем в три раза). При анализе цирконовых концентратов дуговым методом стандартные образцы предварительно смешивают с кварцем.
Очищенный графит, используемый для изготовления таблеток при анализе искровым методом; готовят следующим образом: порошок металлургического графита очищают от кремния, выпаривая его два раза в платиновой чашке с плавиковой кислотой с добавлением первый раз 2—3 мл азотной кислоты и второй раз — серной кислоты до появления густых паров серного ангидрида. После охлаждения содержимое чашки переводят в химический стакан и многократным кипячением с раствором соляной кислоты, разбавленной 1:1, очищают от железа и других примесей. Очистку ведут до отрицательной реакции на ион трехвалентного железа в промывных водах (проба с 10%-ным раствором роданистого аммония). После промывания графита водой порошок высушивают при 120—140° С.
Угольный порошок с внутренним стандартом для дугового анализа; готовят следующим образом: навеску молибденового ангидрида (из расчета 0,75% МоО3 в угольном порошке) растворяют в слабом растворе аммиака и кипятят до полного удаления аммиака. В фарфоровой чашке навеску угольного порошка (50—100 г) смачивают раствором молибденовокислого аммония таким образом, чтобы порошок был смочен полностью, и выпаривают на водяной бане при перемешивании почти досуха. Затем угольный порошок смачивают еще два—три раза водой, выпаривая после каждого смачивания. Последний раз выпаривают досуха. Угольный порошок, приставший к стенкам чашки, отделяют и прокаливают на электроплитке с закрытой спиралью при 300—400° С в течение 1 ч. После охлаждения полученную смесь растирают и перемешивают в ступке в течение 2—3 ч.
2.10.3. Проведение анализа с применением высоковольтной конденсированной искры (при содержании двуокиси гафния от 0,3 до 4,0%)
Стандартные и анализируемые образцы, измельченные до прохождения через сито 200 меш, смешивают с очищенным графитовым порошком в соотношении 1 : 3 (20 мг образца и 60 мг графита) в течение 5 мин.
Из полученных смесей готовят таблетки прессованием в стальной прессформе (черт. 2) ручным масляным прессом при давлении 6000 кг/см2. Таблетки устанавливают в углублении нижнего угольного электрода следующих размеров:
внешний диаметр — 6 мм;
диаметр отверстия — 4,1 мм;
глубина отверстия — 1,0 мм.
Верхним электродом служит угольный стержень диаметром 4 мм и высотой заточки 7 мм.
Спектры фотографируют при ширине щели спектрографа 0,015 мм и расстоянии между электродами 2 мм. В качестве источника возбуждения используют генератор искры ИГ-2 (ИГ-3), работающий по сложной схеме в следующем режиме: емкость 0,005 мкФ, самоиндукция 0,015 мГн, сила тока в первичной цепи 1,2 А. При фотографировании устанавливают круглый вырез промежуточной диафрагмы.
Отпрессованные таблетки предварительно обыскривают в течение 30 с. Таблетка пригодна для суммарного обыскривания в течение 20—30 мин.
Фотографируют по три раза спектры стандартных и анализируемых образцов на спектральные фотопластинки типа II, чувствительностью 10—16 ед.
Обработку фотопластинок ведут в одинаковых условиях. Фо-тометрируют почернения следующих аналитических линий:
гафния — 2641.4 А
о
циркония — 2669,5 А
о
гафния — 2641,4 А
О
циркония — 2667,8 А
п НЮ,
Соотношение---
ZrOt
определяют
по аналитическим графикам,
построенным в координатах:
AS—lg
CzrO. ’
где
AS — разность почернений аналитических линий гафния и циркония;
С — концентрация двуокисей гафния и циркония в процентах.
2.10.4. Подсчет результатов анализа
Содержание двуокиси гафния (Xg) в процентах вычисляют по формуле:
= С •
где
С — содержание суммы двуокисей циркония и гафния в (определяется химическим методом по п. 2.13);
„ v НЮ»
по
Л2 — соотношение концентрации -■■■ , установленное аналитическим графикам.
Прессформа
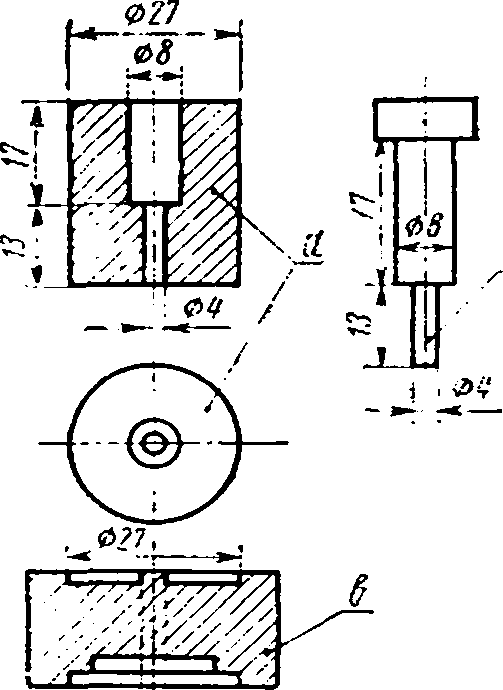
Г; 0#
а — матрица; б — пуансон; в — подставка.
Черт. 2
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,2 абс. %.
2.10.5. Проведение анализа с применением дуги переменного тока (при содержании двуокиси гафния от 0,19 до 2,60%).
Навеску материала 50 мг смешивают с 200 мг угольного порошка, содержащего внутренний стандарт, в течение 10 мин. Эталонные образцы в этом случае получают смешением в течение 15 мин изоморфной смеси окислов циркония и гафния (каждого стандартного образца) в количестве 65 мг с 35 мг молотого кварца и 400 мг угольного порошка, содержащего внутренний стандарт. Смеси плотно набивают в канал нижнего угольного электрода следующих размеров:
диаметр заточенной части — 2,6 мм; длина заточенной части — 10 мм;
диаметр канала — 1,3 мм;
глубина канала — 5,0 мм.
Верхний электрод той же формы и размеров, но без кратера. Спектры фотографируют при ширине щели спектрографа 0,015 мм. Дугу включают при сомкнутых электродах, затем устанавливают на расстоянии 2 мм и сжигают в течение 2 мин 15 с, причем первые 15 с горения дуги щель спектрографа должна быть закрыта. Сила тока 8,5 А.
На фотопластинках чувствительностью 10—16 ед. спектры эталонных и анализируемых образцов фотографируют по три раза. Обработку фотопластинок ведут в одинаковых условиях.
Измеряют почернение аналитических линий:
гафния — 2641,4 А молибдена — 2683,23 А
гафния — 2641,4 А молибдена — 2646,48 А.
Содержание гафния (в пересчете на двуокись) определяют непосредственно по аналитическим графикам, построенным в координатах: AS — lgC, где: AS — разность почернения аналитических линий гафния и молибдена; С — концентрация гафния в стандартном образце.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,4 абс. %.
2.11. Фотоколориметрический метод определения содержания пятнокиси фосфора (при содержании пятиокиси фосфора до 0,50%).
2.11.1. Сущность метода
Содержание пятиокиси фосфора определяют фотоколориметрически по желтой окраске фосфорномолибденованадиевого комплекса. Пробу вскрывают сплавлением с едким натром.
2.11.2. Аппаратура, реактивы и растворы
Фотоэлектроколориметр ФЭК-56.
Натрия гидрат окиси (натр едкий) по ГОСТ 4328—66.
Кислота серная по ГОСТ 4204—66, 6 и раствор; готовят следующим образом: 165 мл серной кислоты плоттюстью 1,84 г/см3 вливают тонкой струей в воду при постоянном помешивании и охлаждают. Объем раствора доводят до 1 л водой.
Кислота азотная по ГОСТ 4461—67, плотностью 1,4 и 1,15 г/см3; готовят разведением водой бесцветной азотной кислоты плотностью 1,4 г/см3 в соотношении 1:2с последующим корректированием по ареометру.
Аммоний молибденовокислый по ГОСТ 3765—72.
Аммоний ванадиевокислый мета по ГОСТ 9336—60.
Раствор комплексообразователя; готовят следующим образом: растворяют при нагревании 50 г молибденовокислого аммония в 400—500 мл воды. Этот раствор при перемешивании постепенно вливают в колбу, содержащую 135 мл бесцветной азотной кислоты плотностью 1,4 г/см3, закрывают пробкой и выдерживают двое суток в темном месте. После этого раствор фильтруют в мерную колбу вместимостью 1 л и добавляют в нее отфильтрованный раствор ванадиевокислого аммония, который готовят следующим образом: 2,4 г ванадиевокислого аммония растворяют в 100 мл воды с добавлением 160 мл 6 н раствора серной кислоты.
Раствор молибденованадиевокислого аммония доводят до метки водой, перемешивают и переводят в темную склянку с притертой пробкой. Раствор устойчив длительное время. При появлении мути допускается его применять после фильтрования.
Спирт этиловый ректификованный по ГОСТ 5962—67.
Фенолфталеин по ГОСТ 5850—72, 1%-ный раствор; готовят следующим образом: 1 г фенолфталеина растворяют в 70 мл этилового спирта и разбавляют дистиллированной водой до 100 мл.
Калий фосфорнокислый однозамещенный по ГОСТ 4198—65.
Раствор А; готовят следующим образом: 0,4394 г перекристал-лизированного однозамещенного фосфата калия растворяют в мерной колбе вместимостью 1 л, разбавляют до метки водой и перемешивают. 1 мл раствора А содержит 0,0001 г фосфора.
Раствор Б; готовят пятикратньш разбавлением водой раствора А. 1 мл раствора Б содержит 0,00002 г фосфора.
Натрий углекислый безводный по ГОСТ 83—63.
Кислота кремневая водная по ГОСТ 4214—70, стандартный раствор; готовят следующим образом: 1 г кремневой кислоты сплавляют в платиновой чашке с 15 г углекислого натрия при 900° С до получения однородного сплава. Сплав выщелачивают горячей водой, раствор фильтруют в мерную колбу вместимостью 500 мл, охлаждают, разбавляют водой до метки и перемешивают. Раствор хранят в полиэтиленовой посуде.
2.11.3. Проведение анализа
В никелевом тигле при температуре 400—450е С расплавляют 5 г едкого натра до прекращения выделения пузырьков (спокойная поверхность расплавленной щелочи означает полное ее обезвоживание). На остывшую поверхность щелочи помещают 0,2 г пробы и сплавляют при 600—700° С в течение 20—30 мин. Сплав выщелачивают теплой водой в стакан вместимостью 300 мл, кипятят 1—2 мин, охлаждают, переводят в мерную колбу вместимостью 100 мл. разбавляют до метки водой и перемешивают. Раствор фильтруют через сухой фильтр «белая лента», отбрасывая первые две порции фильтрата. 25 мл фильтрата переносят пипеткой в мерную колбу вместимостью 50 мл. Фильтрат в мерной колбе нейтрализуют по фенолфталеину 6 н раствором серной кислоты, затем прибавляют 1 мл 6 н раствора серной кислоты, 8 мл азотной кислоты плотностью 1,15 г/см3 и 5 мл раствора комп-лексообразователя. Через 3—5 мин объем раствора доводят до метки водой. Спустя 10 мин раствор фотометрируют на фотоэлектроколориметре ФЭК-56 со светофильтром № 3 в кюветах с толщиной слоя 50 мм. Раствором сравнения служит раствор контрольного опыта, проведенный через весь ход анализа. Измерения оптической плотности должны быть прозедены в течение 10 мин. Количество пятиокиси фосфора в граммах находят по калибровочному графику.
Построение калибровочного графика
В мерные колбы вместимостью по 50 мл помещают 0,2; 0,4; 0,6; 1,0; 1,4; 1,8; 2,0 и 2,5 мл стандартного раствора Б, 15 мл стандартного раствора кремния, обмывают стенки колб водой, нейтрализуют 6 н раствором серной кислоты по фенолфталеину и добавляют еще по 1 мл 6 н раствора серной кислоты, затем приливают по 8 мл азотной кислоты плотностью 1,15 г/см3 и по 5 мл раствора комплексообразователя. Через 3—5 мин растворы доводят водой до метки и перемешивают. Спустя 10 мин растворы фотометрируют на фотоэлектроколориметре ФЭК-56 со светофильтром № 3 в кюветах с толщиной слоя 50 мм. Раствором сравнения служит раствор, содержащий все реактивы, кроме стандартных растворов фосфора и кремния.
По полученным значениям оптических плотностей строят калибровочный график, в координатах: количество фосфора в граммах в 50 мл раствора — оптическая плотность раствора или составляют расчетную таблицу.
2.11.4. Подсчет результатов анализа
Содержание пятиокиси фосфора (X9) в процентах вычисляют по формуле:
О- v
где
G.— количество фосфора, найденное по калибровочному гра фику или таблице, в г;
v — аликвотная часть анализируемого раствора в мл;
100— исходный объем раствора в мл;
2,29 — коэффициент пересчета фосфора на пятиокись фосфора;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,08 абс. %.
Фотоколориметрический метод определения содержания пяти-окиси фосфора является арбитражным.
2.12. Объемный метод определения содержания пятиокиси фосфора (при содержании пятиокиси фосфора от 0,20% и выше)
2.12.1. Сущность метода
Метод основан на титровании избытка щелочи раствором азотной кислоты после предварительного осаждения фосфора в растворе молибденовой жидкостью и растворения осадка фос-формолибдата в растворе едкого натра.
2.12.2. Реактивы и растворы
Натрий углекислый безводный по ГОСТ 83—63 и 1%-ный раствор.
Кислота азотная по ГОСТ 4461—67, разбавленная 1:1, 1 : 19; 1 : 99 и 0,1 н раствор.
Железо азотнокислое по ГОСТ 4111—74, 10%-ный раствор.
Аммиак водный по ГОСТ 3760—64, разбавленный 1 : 9 и 1 : 99.
Кислота фтористоводородная (плавиковая кислота) по ГОСТ 10484—73.
Борфтористоводородная кислота, раствор; готовят следующим образом: к 5 г борной кислоты, помещенной в полиэтиленовый стакан, добавляют 80 мл воды, перемешивают и вводят 10 мл плавиковой кислоты. Раствор разбавляют водой до 100 мл и хранят в полиэтиленовой бутылке.
Аммоний молибденовокислый по ГОСТ 3765—72.
Аммоний азотнокислый по ГОСТ 3761—72.
Молибденовая жидкость; готовят следующим образом: 150 г молибденовокислого аммония растворяют в 1 л воды. Полученный раствор вливают тонкой струей при перемешивании в 1 л азотной кислоты, разбавленной 1:1. Раствор оставляют на двое суток. Если образуется осадок, его отфильтровывают и отбрасывают.
Калий азотнокислый по ГОСТ 4217—73, 1%-ный раствор.
Метиловый красный по ГОСТ 5853—51, 1%-ный спиртовой раствор.
Кислота соляная по ГОСТ 3118—67, 0,1 н раствор, приготовленный из фиксанала.
Индикатор. Метиловый оранжевый (пара-диметиламиноазобен-золсульфокислый натрий) по ГОСТ 10816—64.
Натрий гидрат окиси (натр едкий) по ГОСТ 4328—66, 0,1 н раствор.
Точную концентрацию (нормальность) раствора едкого натра устанавливают по 0,1 н раствору соляной кислоты, приготовленному из фиксанала, с индикатором метиловым оранжевым.
2.12.3. Проведение анализа
Навеску цирконового концентрата 0,5 г сплавляют в платиновом тигле с 4 г углекислого натрия. Плав выщелачивают горячей водой, осадок отфильтровывают, промывают несколько раз раствором углекислого натрия и отбрасывают. В фильтрат добавляют 5 мл раствора азотнокислого железа и в присутствии нескольких капель метилового красного осаждают аммиаком гидроокиси и фосфор, затем нагревают на водяной бане, отфильтровывают через фильтр «белая лента» диаметром 9 см и промывают горячим раствором аммиака, разбавленным 1 : 99.
Осадок растворяют 30—40 мл горячей азотной кислоты, разбавленной 1 : 19, содержащей 2 мл борфтористоводородной кислоты. К полученному раствору прибавляют 5 г азотнокислого аммония, нагревают до 50° С и осаждают фосфорную кислоту добавлением 15 мл раствора молибденовой жидкости при перемешивании. Раствор с осадком оставляют на 2 ч, после чего фильтруют через плотный фильтр «синяя лента» диаметром 9 см и промывают раствором азотнокислого калия до удаления кислоты (5 мл промывных вод при добавлении одной капли 0,1 н раствора едкого натра и одной капли раствора фенолфталеина должны окрашиваться в розовый цвет).
Фильтр с осадком переносят в коническую колбу вместимостью 100—200 мл, приливают 15 мл свежепрокипяченной и охлажденной воды, не содержащей двуокиси углерода, три капли фенолфталеина и из бюретки небольшой избыток 0,1 н раствора едкого натра (8—10 мл). Колбу закрывают пробкой и взбалтывают до тех пор, пока фильтр не распадется на мелкие кусочки. Если при этом розовая окраска раствора исчезает, то приливают еще несколько миллилитров 0,1 н раствора едкого натра. Стенки колбы ополаскивают водой, не содержащей двуокиси углерода, и избыток щелочи отти-тровывают 0,1 н раствором азотной кислоты до исчезновения розовой окраски.
Одновременно проводят контрольный опыт через все стадии анализа.
2.12.4. Подсчет результатов анализа
Содержание пятиокиси фосфора (Х10) в процентах вычисляют по формуле:
v (V —Ks-VjJT-IOO --5,
где
V — объем 0.1 н раствора едкого натра, прилитый с избытком, в мл;
Vi — объем 0,1 н раствора азотной кислоты, израсходованный на титрование избытка щелочи, в мл;
Кз — соотношение между 0,1 н раствором едкого натра и 0,1 н раствором азотной кислоты; устанавливают с индикатором фенолфталеином;
Т — титр раствора едкого натра, выраженный в г/мл пятиокиси фосфора;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,05 абс.%.
Объемный метод определения содержания пятиокиси фосфора является арбитражным.
2.13. Определение содержания двуокиси циркония
После определения содержания двуокисей титана, гафния, окисей железа и алюминия и пятиокиси фосфора, содержание двуокиси циркония (Хп) вычисляют по разности:
Хп = Х2— (Х3 4" Х4 4~ Хъ + Х7 4“ X9J или
Хц = Х2— (Х3 4- -^4 + Х6 + Xg т Х10).
При отсутствии спектральной установки содержание двуокиси циркония вычисляют в сумме с содержанием двуокиси гафния:
(XU 4" в -^2- (Х3 4" ^4 + + Хе) ИЛИ
(Хп 4* Х8) = Х2— (Х3 4- Х4 4- Хь 4~ Х10).
Допускается определение содержания циркония по разности от 100% при условии определения всех остальных компонентов пробы.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,50 абс.%.
2.14. Определение содержания окиси кальция
Содержание окиси кальция определяют в соответствии с разд. 9 ГОСТ 2642.1—71.
Для определения используют объединенный фильтрат, полученный посте двукратного осаждения циркония, гафния, титана, алюминия, железа и фосфора (см. п. 2.3.3).
2.15. Определение содержания окиси магния
2.15.1. Сущность метода
Метод основан на осаждении магния из расвора в виде магний — аммоний фосфата, последующем растворении его в соляной кислоте и обратном трилонометрическом титровании с индикатором 1,2—(пиридил-азо)-2-нафтолом («ПАН»).
2.15.2. Реактивы и растворы
Кислота серная по ГОСТ 4204—66.
Кислота соляная по ГОСТ 3118—67, разбавленная 1 : 1.
Аммоний хлористый по ГОСТ 3773—72.
Аммоний фосфорнокислый двузамещенный по ГОСТ 3772—74, 10%-ный раствор.
Аммиак водный по ГОСТ 3760—64, 25 и 2,5%-ный раствор.
Метиловый красный по ГОСТ 5853—51, 0,1%-ный раствор; готовят, как указано в п. 2.3.2.
Медь сернокислая поГОСТ4165—68, 0,1 н раствор; готовят следующим образом: 12,5 г медного купороса (CuSO4-5H2O) растворяют в воде и приливают 2 мл серной кислоты плотностью 1,8 г/см3 на 1000 мл раствора.
Индикатор 1,2- (пиридил-азо) -2-нафтол («ПАН»), 0,2% -ный
спиртовой раствор.
Бумага индикаторная «конго».
Буферный раствор с pH 10; для приготовления 1000 мл буферного раствора берут 67,5 г хлористого аммония и 570 мл 25%-ного водного раствора аммиака.
Стандартный раствор сернокислого магния, 0,02 и раствор; готовят из фиксанала. Титр стандартного раствора сернокислого магния по окиси магния равен 0,0004035 г/мл.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73, 0,025 Л4 раствор (9,3 г трилона Б на I л раствора).
Соотношение между растворами трилона Б и сернокислой меди устанавливают следующим образом: отмеряют бюреткой 10 мл 0,025 Л1 раствора трилона Б, прибавляют примерно 100 мл воды, 10 мл аммиачного буферного раствора с pH 10, пять капель индикатора «ПАН» и титруют раствором сернокислой меди до перехода окраски из желто-зеленой в ярко-фиолетовую. Для оценки соотношения проводят не менее трех титрований и берут среднее арифметическое объемов в миллилитрах раствора сернокислой меди, израсходованных на титрование.
Значение К рассчитывают, как указано в п. 2.7.2.
Для определения титра раствора трилона Б по окиси магния в коническую колбу вместимостью 300 мл отбирают 25 мл 0,02 н раствора сернокислого магния, прибавляют примерно 100 мл воды, приливают 0,025 М раствор трилона Б с избытком в 20 мл, 10 мл аммиачного буферного раствора с pH 10 и пять капель индикатора «ПАН». Избыток раствора трилона Б оттитровывают 0,05 М раствором сернокислой меди до перехода цвета раствора из желто-зеленого в фиолетовый. Для установки значения титра раствора трилона Б по окиси магния проводят не менее трех титрований.
Титр раствора трилона Б (Г3), выраженный в г/мл окиси магния, вычисляют по формуле:
Г — титр стандартного раствора сернокислого магния, выраженный в г/мл окиси магния;
V — объем стандартного раствора сернокислого магния, взятый для установки титра раствора трилона Б. в мл;
— объем раствора трилона Б в мл;
У2 — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди.
2.15.3. Проведение анализа
Фильтрат после выделения щавелевокислого кальция в соответствии с разд. 9 ГОСТ 2642.1—71 подкисляют соляной кислотой по метиловому красному, выпаривают примерно до 150 мл. приливают 10—15 мл 10%-ного раствора двузамещенного фосфорнокислого аммония и постепенно приливают аммиак при постоянном перемешивании до изменения окраски раствора. Затем добавляют избыток аммиака по 20 мл на каждые 100 мл анализируемого раствора, тщательно перемешивают, накрывают стакан стеклом и оставляют на 12—15 ч при комнатной температуре, после чего фильтруют через фильтр «синяя лента» диаметром 9 см и 4—5 раз промывают осадок 2,5%-ным раствором аммиака не переводя его количественно на фильтр.
В стакан, в котором проводилось осаждение магния, приливают 10 мл соляной кислоты, разбавленной 1 : 1, и растворяют осадок при нагревании. Этим же раствором растворяют осадок на фильтре. Раствор собирают в коническую колбу вместимостью 300 мл. Стакан и фильтр промывают горячей водой 7—8 раз.
К полученному раствору (объем около 120—130 мл) прибавляют 15 мл 0,025 М раствора трилона Б и нейтрализуют содержимое колбы аммиаком по бумаге «конго» до красного цвета, после чего прибавляют 10 мл аммиачного буферного раствора с pH 10 и пять капель индикатора «ПАН». Избыток раствора трилона Б оттитровывают 0,05 М раствором сернокислой меди до перехода окраски раствора из желто-зеленой в розово-фиолетовую.
2.15.4. Подсчет результатов анализа
Содержание окиси магния (X!2) в процентах вычисляют по формуле:
G
где
V — объем прилитого раствора трилона Б в мл;
Vi — объем раствора сернокислой меди, израсходованный на титрование, в мл;
К — соотношение между растворами трилона Б и сернокислой меди;
Т3— титр раствора трилона Б, выраженный в г/мл окиси магния;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,10 абс.%.
2.16. Определение содержания окисей натрия и калия
Содержание окисей натрия и калия определяют в соответствии с разд. II ГОСТ 2642.1—71, но в отличие от ГОСТ 2642.1—71 обработку пробы фтористоводородной кислотой проводят дважды: первый раз приливают 7—8 мл фтористоводородной кислоты и выпаривают до получения влажного остатка, затем добавляют еще 5 мл фтористоводородной кислоты и выпаривают досуха.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,10 абс.%.
3. УСКОРЕННЫЕ МЕТОДЫ АНАЛИЗА ЦИРКОНОВОГО КОНЦЕНТРАТА И ОГНЕУПОРНЫХ ИЗДЕЛИЙ НА НЕГО
3.1. Фотоколориметрический метод определения содержания двуокиси кремния
3.1.1. Сущность метода
Метод основан на измерении интенсивности окраски восстановленного аскорбиновой кислотой синего кремнемолибденового комплекса.
3.1.2. Аппаратура, реактивы и растворы Фотоэлектроколориметр типа ФЭК-М.
Натрий углекислый безводный по ГОСТ 83—63.
Натрий тетраборнокислый (бура) по ГОСТ 4199—66.
Смесь для сплавления, состоящая из двух весовых частей безводной соды и одной весовой части безводной буры.
Кислота уксусная по ГОСТ 61—69, разбавленная 1:1.
Кислота соляная по ГОСТ 3118—67, разбавленная 1 : 3.
Кислота серная по ГОСТ 4204—66, 0,5 н раствор.
Кислота аскорбиновая (витамин С) по ГОСТ 4815—54.
Кислота винная (виннокаменная кислота) по ГОСТ 5817—69.
Аммоний молибденовокислый по ГОСТ 3765—72, х. ч., 5%-ный раствор; готовят следующим образом: 50 г молибдата аммония растворяют в 500—600 мл дистиллированной воды при нагревании. Не доводя до кипения, полученный раствор фильтруют через плотный фильтр, прибавляют к раствору 100 мл уксусной кислоты и доводят до I л водой. Если после прибавления уксусной кислоты раствор остается мутным, его еще раз фильтруют. Раствор хранят в темной склянке; пригоден для применения в течение недели.
Восстановитель; готовят следующим образом: 15 г винной кислоты и 1 г аскорбиновой кислоты растворяют на холоду в 100 мл воды. Раствор пригоден в течение 4—5 дней.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73, 0,025 М раствор (9,3 г трилона Б на 1 л раствора).
Стандартный раствор двуокиси кремния; готовят, используя стандартный образец шамота № 55-а с содержанием двуокиси кремния 58,57%, следующим образом: навеску шамота 0,0427 г сплавляют в платиновом тигле с 2—3 г смеси соды и буры в течение 10 мин в муфельной печи при 900—1000° С. Сплав равномерно распределяют по стенкам тигля и охлаждают. Затем тигель со сплавом и крышку опускают в стакан с 100 мл соляной кислоты, разбавленной 1 : 3, и с 20 мл 0,025 М раствора трилона Б и осторожно перемешивают до полного растворения сплава. Растворение производят без нагревания. Стакан во время растворения накрывают часовым стеклом. После растворения сплава стекло тщательно ополаскивают водой. Прозрачный раствор переносят количественно в мерную колбу вместимостью 1000 мл, доливают до метки водой и хорошо перемешивают.
1 мл стандартного раствора содержит 0,000025 г двуокиси кремния. Во избежание полимеризации кремниевой кислоты переведение солянокислого раствора в мерную колбу производят сразу. Раствор следует хранить в полиэтиленовом сосуде.
3.1.3. Проведение анализа
Навеску материала 0,05 г сплавляют в платиновом тигле с 2—3 г смеси соды с бурой (2:1) при 900—1000° С в течение 10 мин. Сплав выщелачивают 100 мл соляной кислоты, разбавленной 1 : 3, и 20 мл 0,025 М раствора трилона Б таким образом, как при приготовлении стандартного раствора. Раствор переносят в мерную колбу вместимостью 500 мл, доводят до метки водой и тщательно перемешивают.
Для определения двуокиси кремния в мерную колбу вместимостью 100 мл отбирают аликвотную часть раствора 10 мл, приливают 25 мл воды, 25 мл 0,5 н раствора серной кислоты, 10 мл раствора молибдата аммония, через 20 мин прибавляют 5 мл восстанови-тельной смеси, перемешивают, доводят до метки водой, еще раз перемешивают и через 50—60 мин, фотоколориметрируют, используя в качестве раствора сравнения контрольную пробу.
Количество двуокиси кремния находят по калибровочному графику.
Во время измерения на фотоэлектроколориметре допускаются колебания напряжения сети переменного тока ± 10 В.
Построение калибровочного графика
В мерные колбы вместимостью по 100 мл отбирают 10; 11; 12; 13; 14; 15 и 16 мл стандартного раствора двуокиси кремния.
Во все колбы мерным цилиндром прибавляют по 25 мл воды, по 25 мл 0,5 н раствора серной кислоты, по 10 мл раствора молибдата аммония и оставляют на 15—20 мин. После этого прибавляют 5 мл восстановительной смеси. Через 50 мин раствор фотометриру-ют на фотоэлектроколориметре ФЭК-М с красным светофильтром з кюветах с толщиной слоя 20 мм. В качестве раствора сравнения используют раствор контрольного опыта.
Калибровочный график строят в координатах: оптическая плотность раствора — количество двуокиси кремния в граммах.
3.1.4. Подсчет результатов анализа
Содержание двуокиси кремния (Xi3) в процентах вычисляют по формуле:
где
G] — количество двуокиси кремния, найденное по калибровочному графику, в г;
500 — исходный объем раствора в мл;
v — аликвотная часть раствора в мл;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,30 абс.%.
3.2. Определение содержания двуокиси циркония методом обратного титрования раствором трилона Б с индикатором «ПАНэ
3.2.1. Сущность метода
Метод основан на обратном трилонометрическом титровании суммы циркония, железа, титана, алюминия и вычислении циркония по разности после определения железа, титана и алюминия. В качестве индикатора применяют «ПАН».
3.2.2. Реактивы и растворы
Натрий углекислый безводный по ГОСТ 83—63.
Натрий тетраборнокислый (бура) по ГОСТ 4199—66.
Смесь для сплавления, состоящая из двух весовых частей безводной соды и одной весовой части безводной буры.
Кислота серная по ГОСТ 4204—66, 5 и 10%-ные растворы.
Кислота соляная по ГОСТ 3118—67, разбавленная 1 : 1.
Кислота уксусная по ГОСТ 61—69, 2 М раствор.
Натрий уксуснокислый по ГОСТ 199—68, раствор, содержащий 540 г уксуснокислого натрия на 1 л раствора.
Буферный раствор с pH 4,8; готовят следующим образом: 1 л 2 М раствора уксусной кислоты смешивают с 1 л раствора, содержащего 540 с уксуснокислого натра.
Аммиак водный по ГОСТ 3760—64, 25%-ный раствор.
Медь сернокислая по ГОСТ 4165—68, 0,05 М раствор; готовят следующим образом: 12,5 г медного купороса (CuSO4-5H2O) растворяют в воде и приливают 2 мл серной кислоты плотностью 1,84 г/см3 на 1 л раствора.
Индикатор 1,2- (пиридил-азо) -2-нафтол («ПАН»), 0,2 % -ный
спиртовой раствор.
Калий пиросернокислый по ГОСТ 7172—65.
Титана двуокись, х. ч., стандартный раствор; готовят следующим образом: навеску двуокиси титана 0,2 г сплавляют с 6—7 г пиросернокислого калия при 800—850° С до получения прозрачного расплава. Остывший сплав растворяют в 150 мл 10%-ного раствора серной кислоты при нагревании, переводят раствор в мерную колбу вместимостью 250 мл и доводят до метки 5%-ным раствором серной кислоты.
Титр стандартного раствора титана, выраженный в г/мл двуокиси титана, равен 0,0008.
Железа окись, х. ч., стандартный раствор; готовят следующим образом: навеску окиси железа 1 г растворяют при нагревании на водяной бане в 50 мл соляной кислоты, разбавленной 1 : 1, в конической колбе вместимостью 500 мл, накрытой часовым стеклом. Затем раствор охлаждают, переводят в мерную колбу вместимостью 500 мл и доводят водой до метки.
1 мл стандартного раствора хлорного железа содержит 0,002 г окиси железа.
Стандартный раствор цирконового концентрата; готовят следующим образом: навеску стандартного образца цирконового концентрата 0,5 г помещают в платиновый тигель, тщательно перемешивают с 7—8 г смеси для сплавления, закрывают тигель платиновой крышкой и сплавляют в электрической муфельной печи при 1000—1100°С в течение 10—15 мин. Сплав выщелачивают 100 мл горячей соляной кислоты, разбавленной 1 : 1, в стакане вместимостью 300—400 мл, накрытом часовым стеклом. После охлаждения раствор переводят в мерную колбу вместимостью 250 мл и доводят водой до метки. Для правильного учета влияния содержания двуокиси гафния на значение титра раствора трилона Б по двуокиси циркония, стандартный образец цирконового концентрата изготовляется из сырья того же месторождения, которое используется для производства огнеупоров.
Содержание двуокиси циркония в цирконовом концентрате устанавливают весовым методом, усредняя не менее трех результатов.
На основании этих данных титр стандартного раствора цирконового концентрата, выраженный в г/мл двуокиси циркония, определяют расчетным путем.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксуснон кислоты) по ГОСТ 10652—73, 0,025 М раствор *9,3 г трилона Б на 1 л раствора).
Для установки титра раствора трилона Б по двуокиси циркония отбирают 50 мл стандартного раствора цирконового концентрата в коническую колбу вместимостью 300 мл, прибавляют 40—45 мл раствора трилона Б, нагревают и умеренно кипятят 3—5 мин, после чего нейтрализуют раствором аммиака по индикаторной бумаге «конго» до слаборозового цвета. Затем приливают 10 мл ацетатного буферного раствора с pH 4,8, 5—6 капель раствора индикатора «ПАН» и титруют избыток раствора трилона Б 0,05 М раствором сернокислой меди до перехода окраски раствора из желто-зеленой в сине-фиолетовую. Переход должен быть четким.
Титр раствора трилона Б (Г4), выраженный в г/мл двуокиси циркония, вычисляют по формуле: где
Т — титр стандартного раствора цирконового концентрата, выраженный в г/мл двуокиси циркония;
V — объем стандартного раствора цирконового концентрата, взятый для титрования, в мл;
Vj — объем раствора трилона Б, взятый с избытком, в мл;
у2 — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди (см. п. 2.7.2).
Для установки титра раствора трилона Б по окиси железа отбирают 10 мл стандартного раствора окиси железа, прибавляют 50—70 мл воды, 20—25 мл раствора трилона Б, нагревают до 60—70° С, нейтрализуют раствором аммиака по индикаторной бумаге «конго» до слаборозового цвета, далее поступают, как указано гри определении титра раствора трилона Б по двуокиси циркония.
Титр раствора трилона Б (Т5), выраженный в г/мл окиси железа, вычисляют по формуле:
VT-K. V2
Т — титр стандартного раствора окиси железа, выраженный в г/мл окиси железа;
V — объем стандартного раствора окиси железа, взятый для
титрования, в мл;
Vi — объем раствора трилона Б, взятый с избытком, в мл;
V2 — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди (см. п. 2.7.2).
Для установки титра раствора трилона Б по двуокиси титана отбирают 25 мл стандартного раствора двуокиси титана, прибавляют 50 мл воды, 20—25 мл раствора трилона Б, нагревают до 60—70° С. нейтрализуют раствором аммиака по индикаторной бумаге «конго» до слаборозового цвета и далее поступают, как указано при определении титра раствора трилона Б по двуокиси циркония.
Титр раствора трилона Б (Те), выраженный в г/мл двуокиси титана, вычисляют по формуле:
Т = V ' Т
6 Kj - KVi ’
где
Т — титр стандартного раствора двуокиси титана, выраженный в г/мл двуокиси титана;
V — объем стандартного раствора двуокиси титана, взятый для
титрования, в мл;
Vi —объем раствора трилона Б, взятый с избытком, в мл;
V2 — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди.
3.2.3. Проведение анализа
Навеску анализируемого материала 0,5 г помещают в платиновый тигель, тщательно смешивают с 7—8 г смеси для сплавления, закрывают тигель платиновой крышкой и сплавляют в электрической муфельной печи при 1100° С в течение 10—15 мин.
Сплав выщелачивают 100 мл горячей соляной кислоты, разбавленной 1 : 1, в стакане вместимостью 300—400 мл, накрытом часовым стеклом. Холодный прозрачный раствор переносят в мерную колбу вместимостью 250 мл, доводят раствор водой до метки и тщательно перемешивают (раствор Г);
а) для определения содержания двуокиси циркония отбирают аликвотную часть раствора Г 50 мл в коническую колбу вместимостью 300 мл, прибавляют 40—45 мл раствора трилона Б, нагревают и умеренно кипятят 3—5 мин, после чего нейтрализуют раствором аммиака по индикаторной бумаге «конго» до слаборозового цвета, затем приливают 10 мл ацетатного буферного раствора с pH 4,8, 5—6 капель 0,2%-ного спиртового раствора индикатора «ПАН» и титруют избыток раствора трилона Б 0,05 М раствором сернокислой меди до перехода желто-зеленой окраски в сине-фиолетовую. Переход должен быть четким.
Для подсчета результатов анализа необходимо определить содержание окиси железа, двуокиси титана и окиси алюминия;
б) для определения содержания окиси железа пипеткой отбирают 25 мл раствора Г в мерную колбу вместимостью 100 мл (или в мерную колбу вместимостью 50 мл при содержании окиси железа ниже 0,1 %).
Далее анализ проводят, как указано в разд. 2.4;
в) для определения содержания двуокиси титана навеску материала 0,2 г помещают в платиновый тигель, тщательно смешивают с 3—4 г смеси для сплавления и сплавляют в электрической муфельной печи при 1100° С в течение 10—15 мин. Сплав выщелачивают в стакан вместимостью 300 мл 5%-ным раствором серной кислоты при нагревании (раствор должен быть совершенно прозрачным). Далее анализ проводят, как указано в разд. 2.6;
г) содержание окиси алюминия определяют, как указано в разд. 2.7.
3.2.4. Подсчет результатов анализа
Содержание двуокиси циркония (Х14) в процентах вычисляют по формуле:
Хм=[ - (иг + V, + ^4)] • Л . 100,
где
V — объем раствора трилона Б в мл;
Vj — объем раствора сернокислой меди, израсходованный на титрование, в мл;
К — соотношение между растворами трилона Б и сернокислой меди, определенное по п. 2.7.2;
G — навеска пробы, взятая для титрования, в г;
V2, V3, V4— объемы раствора трилона Б, израсходованные на комплексообразование с железом, титаном и алюминием соответственно их содержанию в анализируемой пробе материала, в мл;
Т4 — титр раствора трилона Б, выраженный в г/мл двуокиси циркония.
Расход раствора трилона Б на комплексообразование с железом, титаном, алюминием рассчитывают на основании результатов анализа по этим элементам.
Объем раствора трилона Б (У2), связанного с железом, находящимся в 1 г пробы, в миллилитрах вычисляют по формуле:
V = —'^2—
2 100- т6 ’
где
Х3 — содержание окиси железа в %, найденное по п. 2.4,5;
Т5 — титр раствора трилона Б, выраженный в г/мл окиси железа, устанавливают, как указано в п. 3.2.2.
Объем раствора трилона Б (V3), связанного с титаном, находящимся в 1 г пробы, в миллилитрах вычисляют по формуле:
где
Х4 — содержание двуокиси титана в %, найденное по п. 2.6.5;
Г6 — титр раствора трилона Б, выраженный в г/мл двуокиси титана, устанавливают, как указано в п. 3.2.2.
Объем раствора трилона Б (У«), связанного с алюминием, находящимся в I г пробы, в миллилитрах вычисляют по формуле:
где
Х3 — содержание окиси алюминия в %, найденное по п. 2.7.4;
Т — титр раствора трилона Б, выраженный в г/мл окиси алюминия, устанавливают, как указано в п. 2.7.2.
Допускаемые расхождения между результатами параллельных определений содержания двуокиси циркония не должны превышать
0,50 абс.%.
3.3. Определение содержания двуокиси циркония методом об
ратного титрования раствора трилона Б с индикатором ксиленоловым оранжевым
3.3.1. Сущность метода
Цирконий определяют трилонометрическим методом обратного титрования избытка раствора трилона Б азотнокислым висмутом в 0,4 н сернокислой среде. Пробу разлагают сплавлеием со смесью соды и буры.
3.3.2. Реактивы и растворы
Натрий тетраборнокислый (бура) по ГОСТ 4199—66, прокаленный при 300—400° С.
Натрий хлористый по ГОСТ 4233—66.
Натрий углекислый безводный по ГОСТ 83—63.
Кислота соляная по ГОСТ 3118—67, разбавленная I : 1.
Кислота азотная по ГОСТ 4461—67, плотностью 1,4 г/см3.
Кислота серная по ГОСТ 4204—66, 4 н раствор и разбавленная 1:1 и 3 : 97.
Магний сернокислый по ГОСТ 4523—67.
Цинк металлический гранулированный по ГОСТ 989—62.
Цинк хлористый 0,05 М раствор; готовят следующим образом: навеску металлического цинка 3,2690 г растворяют при слабом нагревании в 20—30 мл соляной кислоты, разбавленной 1:1, раствор переводят в мерную колбу вместимостью 1 л и разбавляют водой до метки.
Аммиак водный по ГОСТ 3760—64, плотностью 0,9 г/см3.
Аммоний хлористый по ГОСТ 3773—72, х. ч.
Аммиачный буферный раствор; готовят следующим образом: к 10 мл аммиака плотностью 0,9 г/см3 прибавляют 20 г хлористого аммония марки х. ч. и доводят до 1 л дистиллированной водой.
Аскорбиновая кислота (витамин С) по ГОСТ 4815—54, кристаллическая или 5%-ный раствор.
Спирт этиловый ректификованный по ГОСТ 5962—67.
Индикатор бромфеноловый синий, 0,1%-ный раствор; готовят следующим образом: 0,1 г индикатора растворяют при слабом нагревании в 20 мл этилового спирта и разбавляют до 100 мл водой.
Индикатор ксиленоловый оранжевый, 0,5 %-ный водный раствор. Индикатор эриохромчерный Т (хромоген черный специальный ЕТ-00). Индикатор смешивают в ступке путем растирания с хлористым натрием в соотношении 1 : 50.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73, 0,05 М раствор; готовят следующим образом: 18,6 г трилона Б растворяют при слабом нагревании в 200—300 мл дистиллированной воды; раствор фильтруют и доводят до 1 л дистиллированной водой.
Для установки титра раствора трилона Б по двуокиси циркония в конусообразную колбу вместимостью 250 мл помещают 10 мл 0,05 М раствора сернокислого магния или хлористого цинка, прибавляют 10 мл аммиачного буферного раствора, объем раствора доводят дистиллированной водой до 100 мл и титруют раствором трилона Б в присутствии индикатора эриохромчерного Т до перехода окраски из виннокрасной в сине-зеленую. Параллельно проводят контрольный опыт на загрязнение реактивов.
Титр раствора трилона Б (Г?), выраженный в г/мл окиси циркония, вычисляют по формуле:
т = Ум ■ 123,22
7 (Vi — г2) - 1000’
где
V — объем раствора магния или цинка, взятый для титрования, в мл;
М — молярность раствора магния или цинка;
Vt — объем раствора трилона Б, израсходованный на титрование, в мл;
V2— объем раствора трилона Б, израсходованный на титрование раствора контрольного опыта, в мл;
123,22 — молекулярный вес двуокиси циркония.
Висмут по ГОСТ 10928—-64.
Висмут азотнокислый, 0,05 А1 раствор; готовят следующим образом: 20,9 г металлического висмута растворяют в 50 мл азотной кислоты плотностью 1,4 г/см3, удаляют при кипячении окислы азота и выпаривают до 30 мл. Раствор переносят в мерную колбу вместимостью 2 л, обмывают стенки стакана таким количеством азотной кислоты, чтобы общий объем раствора составлял 100 мл. Объем раствора доводят до метки водой.
Соотношение между растворами трилона Б и азотнокислого висмута устанавливают следующим образом: в коническую колбу вместимостью 250 мл помещают 10 мл раствора трилона Б, 10 мл серной кислоты, разбавленной 1:1, прибавляют 50 мл дистиллированной воды, три капли бромфенолового синего и нейтрализуют аммиаком до окрашивания раствора в синий цвет. После этого к раствору прибавляют 10 мл 4 н раствора серной кислоты, разбавляют дистиллированной водой до 100 мл, охлаждают, прибавляют пять капель ксиленолового оранжевого и титруют раствором азотнокислого висмута до перехода окраски раствора в красную.
Соотношение между растворами трилона Б и азотнокислого висмута (Xi) рассчитывают с точностью до второго знака после запятой по формуле:
где
V — объем раствора трилона Б, израсходованный на титрование, в мл;
Vi—объем раствора азотнокислого висмута, израсходованный на титрование, в мл.
3.3.3. Проведение анализа
Навеску концентрата 0,1 г помещают в платиновую чашку, куда заранее насыпают 3 г смеси соды и буры в отношении 1,5 : 1. Содержимое чашки сплавляют в муфельной печи при 800—900° С до получения прозрачного сплава. После охлаждения сплав выщелачивают горячей дистиллированной водой, собирая раствор в стакан вместимостью 300 мл, на котором заранее должна быть проставлена метка 100 мл. Раствор разбавляют дистиллированной водой до 100—150 мл и нагревают до кипения для лучшей коагуляции осадка. После отстаивания осадок отфильтровывают через фильтр «белая лента». Осадок и стакан промывают по два раза горячей водой. Промытый осадок смывают с развернутого по стенке стакана фильтра горячей серной кислотой, разбавленной 3 : 97. Той же серной кислотой обмывают платиновую чашку, собирая фильтрат в тот же стакан. К раствору в стакане прибавляют 10 мл серной кислоты, разбавленной 1:1. нагревают содержимое стакана до кипения и прибавляют к нему на кончике шпателя несколько кристаллов аскорбиновой кислоты или же 2 мл свежеприготовленного 5%-ного раствора. К горячему раствору прибавляют 20 мл раствора трило-на Б, кипятят 2 мин, охлаждают, прибавляют 3—4 капли бромфенолового синего и нейтрализуют аммиаком до перехода окраски индикатора в синюю. После этого к раствору прибавляют 10 мл 4 н раствора серной кислоты, охлаждают и доводят водой до 100 мл. Стенки стакана тщательно обмывают дистиллированной водой. В стакан прибавляют пять капель ксиленолового оранжевого и титруют 0,05 М раствором азотнокислого висмута до перехода окраски раствора в красную. Одновременно проводят контрольный опыт через все стадии анализа.
3.3.4. Подсчет результатов анализа
Содержание двуокиси циркония (Хи) в процентах вычисляют по формуле:
V,) . 100
Л16= ъ ’
где
Г4 — титр раствора трилона Б, выраженный в г/мл двуокиси циркония;
V — объем раствора трилона Б в мл;
V} — объем раствора азотнокислого висмута, израсходованный на титрование, в мл;
G — навеска в г;
Ki — соотношение между растворами трилона Б и азотнокислого висмута.
Допускаемые расхождения между результатами параллельных определений не должны превышать 1,0 абс.%.
4. МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА ДВУОКИСИ ЦИРКОНИЯ (ТЕХНИЧЕСКОЙ) И ОГНЕУПОРНЫХ ИЗДЕЛИЙ ИЗ НЕЕ
4.1. Определение потери при прокаливании
Определение потери при прокаливании производят, как указано в разд. 2.1.
4.2. Определение содержания двуокиси кремния
4.2.1. Сущность метода
В основу определения содержания двуокиси кремния положен весовой метод выделения кремниевой кислоты после разложения пробы сплавлением с пиросульфатом калия и обезвоживания в солянокислой среде.
4.2.2. Реактивы и растворы
Калий пиросернокислый по ГОСТ 7172—65.
Натрий углекислый безводный по ГОСТ 83—63.
Кислота соляная по ГОСТ 3118—67 и разбавленая 1:1.
Кислота фтористоводородная (плавиковая кислота) по ГОСТ 10484—73, 40%-ный раствор.
4.2.3. Проведение анализа
Навеску пробы 0,5 г помешают в платиновый тигель, смешивают с 6—7 г пиросульфата калия и сплавляют в электрической печи при 800—850° С в течение 20—25 мин до получения прозрачного сплава. Во избежание разбрызгивания и вспенивания расплава, тигель необходимо нагревать постепенно.
Для ускорения процесса сплавления сплав перемешивают 2—3 раза вращением тигля при помощи щипцов.
Остывший сплав переносят из тигля в стакан, обмывают стенки тигля горячей водой, приливают 30 мл соляной кислоты и нагревают содержимое стакана до растворения сплава.
Если на дне стакана остаются несплавленные частицы пробы, раствор фильтруют через фильтр «белая лента». Остаток на фильтре промывают 3—4 раза горячей водой, переносят в платиновый тигель, сжигают и прокаливают несколько минут. Остаток в тигле сплавляют с 2 г углекислого натрия.
После охлаждения сплав переносят в стакан, растворяют в 20 мл соляной кислоты, разбавленной 1 : 1, и присоединяют раствор к первому фильтрату.
Полученный раствор переводят в фарфоровую чашку и выпаривают на кипящей водяной бане досуха. Чашку с сухим остатком выдерживают на водяной бане в течение 1 ч. После охлаждения в чашку приливают 20—25 мл соляной кислоты, выдерживают 10 мин, добавляют 50—70 мл кипящей воды для растворения солей и отфильтровывают осадок на фильтр диаметром 9 см «белая лента». После этого фильтр промывают 4—5 раз горячей соляной кислотой, разбавленной 1 : 4, затем 7—8 раз горячей водой до полного удаления ионов хлора (реакции с 1%-ным раствором азотнокислого серебра).
Фильтр с осадком помещают в платиновый тигель, озоляют и прокаливают при 1100° С в течение 45 мин. После охлаждения в эксикаторе тигель с осадком взвешивают. Прокаливание повторяют по 10 мин до достижения постоянного веса.
Осадок двуокиси кремния загрязнен примесями двуокисей циркония, гафния, титана, окисей алюминия и железа.
Для получения веса чистой двуокиси кремния прокаленный осадок смачивают несколькими каплями воды, приливают 0,5 мл серной и 4—5 мл фтористоводородной кислот.
Содержимое тигля выпаривают на закрытой электроплите, не доводя до кипения. Для полного удаления серной кислоты тигель прокаливают в течение 10 мин при 1100° С, охлаждают в эксикаторе и взвешивают.
Остаток в тигле после определения содержания двуокиси кремния сплавляют с 2—3 г пиросульфата калия. Остывший сплав растворяют при нагревании в небольшом количестве разбавленной соляной кислоты и присоединяют к фильтрату, полученному после выделения кремниевой кислоты.
Весь раствор переводят в мерную колбу вместимостью 250 мл, разбавляют водой до метки и перемешивают (раствор Д). Раствор используют для определения содержания суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора.
4.2.4. Подсчет результатов анализа
Содержание двуокиси кремния (A\6) в процентах вычисляют по формуле:
v (61— (71) • 100
где
Gi — масса тигля с осадком до обработки фтористоводородной кислотой в г;
G2 — масса тигля с остатком после обработки фтористоводородной кислотой и прокаливания в г;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать:
при содержании двуокиси кремния до 1% —0,15 абс. %;
при содержании двуокиси кремния свыше 1% —0,25 абс. %.
4.3. Определение содержания суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора
4.3.1. Сущность метода
Содержание суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора определяют весовым методом после осаждения указанных компонентов аммиаком и последующего прокаливания осадка до окислов.
4.3.2. Реактивы, и растворы
Реактивы и растворы указаны в п. 2.3.2.
4.3.3. Проведение анализа
Отбирают 100 мл раствора Д (см. п. 4.2.3), что соответствует навеске 0,2 г, помещают в стакан вместимостью 300 мл, приливают 5 мл 20%-него раствора хлористого аммония и воды до 150 мл.
Раствор нагревают до кипения и в присутствии нескольких капель раствора метилового красного осаждают аммиаком цирконий, гафний, титан, железо, алюминий и фосфор. Аммиак прибавляют до перехода окраски индикаторов в желтый цвет. Затем раствор нагревают до кипения, дают осадку гидроокисей осесть, после чего фильтруют через фильтр диаметром 11 см «красная» или «белая лента». Осадок промывают несколько раз горячим 2%-ным раствором азотнокислого аммония. Фильтр с осадком переносят в стакан, в котором производилось осаждение, приливают для растворения осадка 30 мл разбавленной соляной кислоты, растирают фильтр в кашицеобразную массу, разбавляют водой до 100—150 мл и вторично осаждают алюминий, железо, титан, цирконий, гафний и фосфор, как описано выше.
Раствор фильтруют через фильтр диаметром 11 см «красная» или «белая лента» и промывают осадок горячим 2%-ным раствором азотнокислого аммония до удаления ионов хлора (реакция с 1%-ным раствором азотнокислого серебра, подкисленным азотной кислотой). Объединенный фильтрат сохраняют для определения содержания окисей кальция и магния.
Фильтр с осадком помещают во взвешеный платиновый тигель, подсушивают, озоляют и прокаливают 2 ч при 1200° С. После охлаждения в эксикаторе тигель с осадком взвешивают. Прокаливание повторяют по 20 мин до постоянной массы. Осадок используют для определения содержания окиси железа или двуокиси гафния.
4.3.4. Подсчет результатов анализа
Содержание суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора (Хп) в процентах вычисляют по формуле:
Gi ■ 250 * 100
100 - G
где
Gi — масса осадка суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора в г;
250 — исходный объем испытуемого раствора в мл;
100 — аликвотная часть испытуемого раствора в мл;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,50 абс. %.
4.4. Фотоколориметрический метод определения содержания окиси железа
4.4.1. Сущность метода
Метод основан на измерении интенсивности окраски желтого комплексного соединения ионов трехвалентного железа с сульфосалициловой кислотой.
4.4.2. Реактивы и растворы
Реактивы и растворы указаны в п. 2.4.2.
4.4.3. Проведение анализа
Полученный осадок суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора (см. п. 4.3.3) смешивают в том же тигле с 5—6 г пиросульфата калия и сплавляют в электрической печи при 800—850° С в течение 20 мин до получения прозрачного расплава.
Остывший сплав переносят из тигля в стакан вместимостью 200 мл, обмывают стенки тигля водой, приливают 5 мл серной кислоты, разбавленной 1 : 1, и нагревают до растворения сплава. Затем раствор охлаждают, переводят в мерную колбу вместимостью 100 мл, приливают 10 мл 20%-ного раствора сульфосалициловой кислоты, раствор аммиака до устойчивой желтой окраски и прибавляют избыток аммиака 5 мл.
Содержание окиси железа определяют фотоколориметрическим методом, как указано в п. 2.4.3. Определение содержания железа может быть произведено также колориметрическим методом.
Построение калибровочного графика
Калибровочный график строят, как указано в п. 2.4.4.
4.4.4. Подсчет результатов анализа
Содержание окиси железа (Xis) в процентах вычисляют по формуле:
X (? - 100
18 — 2- 0,2 • 1000’
где
G — количество окиси железа, найденное по калибровочному графику, в мг;
2 — коэффициент, приводящий к объему 50 мл;
0,2 — навеска, взятая для определения, в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать:
при содержании окиси железа до 0,10% —0,01 абс. %;
при содержании окиси железа от 0,10 до 0,50% —0,03 абс. %.
4.5. Фотоколориметрический метод определения содержания двуокиси титана
4.5.1. Сущность метода
Метод основан на измерении интенсивности окраски окрашенного в желтый цвет комплекса титана с перекисью водорода.
4.5.2. Реактивы и растворы
Реактивы и растворы указаны в п. 2.6.2.
4.5.3. Проведение анализа
Навеску материала 0,2 г сплавляют в платиновом тигле с 5 г пиросернокислого калия при 800—850° С в течение 20 мии в электрической муфельной печи. Сплав выщелачивают при нагревании 50 мл 5%-ного раствора серной кислоты в стакане вместимостью 100 мл. Раствор охлаждают, фильтруют через фильтр «белая лента» в мерную колбу вместимостью 100 мл, прибавляют 3%-ный раствор перекиси водорода, доводят до метки 5%-ным раствором серной кислоты, перемешивают и определяют содержание двуокиси титана, как указано в п. 2.6.3.
Определение содержания двуокиси титана может быть произведено также колориметрическим методом.
4.5.4. Подсчет результатов анализа
Содержание двуокиси титана вычисляют, как указано в п. 2.6.5.
4.6. Определение содержания окиси алюминия
4.6.1. Сущность метода
Сущность метода изложена в п. 2.7.1.
4.6.2. Реактивы и растворы
Реактивы, растворы, определение титра раствора трилона Б по окиси алюминия, соотношения между растворами трилона Б и сернокислой меди указаны в п. 2.7.2.
4.6.3. Проведение анализа
100 мл раствора Д помещают в стакан вместимостью 300 мл, нейтрализуют 50%-ным раствором едкого натра до покраснения бумаги «конго» и приливают избыток щелочи 5—7 мл. Полученный раствор с осадком кипятят 5 мин, добавляют немного кашицы из фильтровальной бумаги и фильтруют на фильтр «красная» или «белая лента» диаметром 11 см. Фильтрат собирают в коническую колбу вместимостью 500 мл и промывают осадок горячей водой 8—10 раз. В полученном фильтрате определяют содержание окиси алюминия трилонометрическим методом, как указано в п. 2.7.3.
4.6.4. Подсчет результатов анализа
Содержание окиси алюминия вычисляют, как указано в п. 2.7.4.
4.7. Спектральный метод определения содержания двуокиси гафния
Содержание двуокиси гафния определяют, как указано в разд. 2.9, используя для определения прокаленный осадок суммы двуокисей циркония, гафния, титана, окисей алюминия, железа и пятиокиси фосфора, полученный, как указано в п. 4.3.3, или непосредственно пробу, если сумма примесей не превышает 10%.
4.8. Определение содержания пятиокиси фосфора
Содержание пятиокиси фосфора определяют, как указано в разд. 2.12.
4.9. Фотоколориметрический метод определения содержания пятиокиси фосфора (при содержании пятиокиси фосфора до 0,50%)
4.9.1. Сущность метода
Определение содержания пятиокиси фосфора производят фотоколориметрически по синей окраске фосфорно-молибденового комплекса, восстановленного аскорбиновой кислотой. Пробу разлагают сплавлением с едким натром.
4.9.2. Аппаратура, реактивы и растворы Фотоэлектроколориметр ФЭК-М или ФЭК-56. Натрий гидрат окиси (натр едкий) по ГОСТ 4328—66. Кислота серная по ГОСТ 4204—66, разбавленная 1 :2.
Аскорбиновая кислота (витамин С) по ГОСТ 4815—54, 1%-ный раствор; хранится в склянке из темного стекла.
Спирт этиловый ректификованный по ГОСТ 5962—67.
Аммоний молибденовокислый по ГОСТ 3765—72, 5%-ный раствор; готовят следующим образом: 5 г перекристаллизованного молибдата аммония растворяют в 100 мл воды при нагревании до 40—50° С и фильтруют раствор через фильтр «синяя лента». Перекристаллизацию молибдата аммония производят следующим образом: 200 г молибдата аммония растворяют в 400 мл дистиллированной воды при 80—90° С. Раствор фильтруют, охлаждают, прибавляют 500 мл этилового спирта, затем перемешивают и через 1 ч фильтруют через воронку с пористым фильтром, пользуясь вакуумом. Осадок высушивают на воздухе при комнатной температуре.
Фенолфталеин по ГОСТ 5850—72, 1%-ный раствор; готовят следующим образом: 1 г фенолфталеина растворяют в 70 мл этилового спирта и разбавляют дистиллированной водой до 100 мл.
Калий фосфорнокислый однозамещенный по ГОСТ 4198—65.
Стандартный раствор фосфора А; готовят следующим образом: 0,4394 г перекристаллизованного однозамещенного фосфата калия переводят в мерную колбу вместимостью I л, растворяют в воде, доводят до метки водой и перемешивают. 1 мл раствора А содержит 0,0001 г фосфора.
Стандартный раствор фосфора Б готовят следующим образом: 10 мл раствора А пипеткой переводят в мерную колбу вместимостью 100 мл, доводят до метки дистиллированной водой и перемешивают. 1 мл раствора Б содержит 0,00001 г фосфора.
4.9.3. Проведение анализа
В никелевом тигле при температуре 400—450° С расплавляют 3 г едкого натра до прекращения выделения пузырьков (спокойная поверхность расплавленной щелочи означает ее полное обезвоживание). На остывшую поверхность щелочи помещают 0,2 г пробы материала и сплавляют при температуре 600—700° С в течение 20—30 мин. Сплав охлаждают, выщелачивают теплой водой в стакан вместимостью 200—300 мл, кипятят 1—2 мин, охлаждают, переносят в мерную колбу вместимостью 100 мл, доводят до метки водой и перемешивают. Раствор фильтруют через сухой фильтр «белая лента». Первые две порции фильтрата отбрасывают, а из остального фильтрата пипеткой отбирают 10 мл в мерную колбу вместимостью 50 мл, прибавляют две капли раствора индикатора фенолфталеина и нейтрализуют раствором серной кислоты, разбавленным 1 : 2, до изменения цвета раствора. Затем к раствору прибавляют 20 мл воды, 3 мл раствора серной кислоты, разбавленной 1 : 2, приливают 2,5 мл раствора молибдата аммония, обмывают стенки колбы водой и по каплям прибавляют 2 мл раствора аскорбиновой кислоты. Раствор в колбе доводят дистиллированной водой до метки, перемешивают и через 20 мин измеряют оптическую плотность на ФЭК-М или ФЭК-56 с красным светофильтром в кювете с толщиной слоя 50 мм. В качестве раствора сравнения используют раствор контрольного опыта, проведенный через весь ход анализа. Количество фосфора находят по калибровочному графику.
Построение калибровочного графика
В мерные колбы вместимостью 50 мл пипеткой отмеривают 0,4; 0,6; 0,8; 1,0; 1,2; 1,4; 1,6; 1,8 и 2,0 мл стандартного раствора Б, прибавляют при перемешивании 30 мл дистиллированной воды, 3 мл серной кислоты, 2,5 мл раствора молибдата аммония, обмывают стенки колбы водой и по каплям при перемешивании прибавляют 2 мл раствора аскорбиновой кислоты. После этого раствор доводят до метки дистиллированной водой, перемешивают и через 20 мин определяют оптическую плотность на ФЭК-М или ФЭК-56 с красным светофильтром в кюветах с толщиной слоя 50 мм.
В качестве раствора сравнения используют раствор, содержащий все реактивы, кроме стандартного раствора фосфора.
По полученным значениям оптических плотностей строят калибровочный график в координатах: количество фосфора в граммах в 50 мл раствора — оптическая плотность раствора или составляют расчетную таблицу.
4.9.4. Подсчет результатов анализа
Содержание пятиокиси фосфора (Х19) в процентах вычисляют по формуле:
Gi • 100- 100- 2,29
19— Z
где
Gi — количество фосфора, найденное по калибровочному графику при таблице, в г;
G — навеска материала в г;
2,29 — коэффициент пересчета фосфора на пятиокись фосфора; v — аликвотная часть анализируемого раствора в мл;
100 — исходный объем раствора в мл.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,08 абс. %.
Фотоколориметрический метод определения содержания пяти* окиси фосфора является арбитражным.
4.10. Определение содержания двуокиси циркония
Определив содержание суммы двуокисей титана, гафния, окисей алюминия и железа и пятиокиси фосфора, содержание двуокиси циркония (Х20) в процентах вычисляют по разности:
*20 = *17 — (*is Н- К* 4~ -р Х7 + Х10) или
*20 = *17 - (*18 + *4 + *5 + *8 + *19).
Медь сернокислая по ГОСТ 4165—68, 0,05 М раствор; готовят следующим образом: 12,5 г медного купороса (CtiSO* • 5Н2О) растворяют в воде и приливают 2 мл серной кислоты плотностью 1,84 г/см3 на 1000 мл раствора.
Уротропин фармакопейный, 30%-ный водный раствор.
Индикатор 1,2- (пиридил-азо) -2-нафтол («П АН»), 0,2 % -ный спиртовой раствор.
Спирт этиловый ректификованный по ГОСТ 5962—67.
Индикатор кислотный хром темно-синий, 0,5%-ный свежеприготовленный раствор; готовят следующим образом: 0,5 г индикатора растворяют в 20 мл аммиачного буферного раствора и доводят до 100 мл этиловым спиртом.
Аммиачный буферный раствор для приготовления индикатора; готовят следующим образом: 8,25 г хлористого аммония смешивают со 113 мл аммиака плотностью 0,9 г/см3 и разбавляют до 1 л водой.
Индикаторная смесь сухая; готовят следующим образом: 1 г кислотного хром темно-синего растирают в фарфоровой ступке со 100 г хлористого натрия.
Буферный раствор для титрования суммы кальция и магния с pH 10; для приготовления 1000 мл буферного раствора берут 67,5 г хлористого аммония и 570 мл 25%-ного водного раствора аммиака.
Стандартный раствор сернокислого магния, 0,02 н раствор; готовят из фиксанала. Титр его по окиси магния равен 0,0004035 г/мл.
Кальций углекислый по ГОСТ 4530—66, ч. д. а.
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73, 0,025 М раствор
(9,3 г трилона Б на 1 л раствора).
Титр раствора трилона Б по окиси магния и соотношение между растворами трилона Б и сернокислой меди устанавливают, как указано в п. 2.15.2.
Для определения титра раствора трилона Б по окиси кальция в коническую колбу вместимостью 300 мл отбирают 10 мл 0,05 М раствора углекислого кальция, прибавляют 100 мл воды, 30 мл 0,025 Л1 раствора трилона Б, 10 мл аммиачного буферного раствора с pH 10 и пять капель индикатора «ПАН». Избыток трилона Б оттитровывают 0,05 М раствором сернокислой меди до перехода цвета раствора из желто-зеленого в фиолетовый.
Титр раствора трилона Б (Ге), выраженный в г/мл окиси кальция, вычисляют по формуле:
Т = Т' V 8~ Vi - KV-2
Если определение содержания двуокиси гафния не проводилось, вычисляют содержание суммы двуокиси тиркония и гафния в процентах:
(Х2о + *7) = Х17 - (Х18 + Х4 + Хь + Х10) или
(^20 + = ^17 (^18 + 4- Х5 + . )
Допускается определение двуокиси циркония по разности ог 100% при условии определения всех остальных компонентов.
Допускаемые расхождения между результатами параллельных определений содержания двуокиси циркония не должны превышать 0,50 абс. %.
4.11. Определение содержания окиси кальция
Содержание окиси кальция определяют в соответствии с разд. 9, ГОСТ 2642.1—71, используя объединенный фильтрат, полученный в п. 4.3.3.
4.12. Определение содержания окиси магния
Содержание окиси магния определяют, как указано в п. 2.15, используя фильтрат, полученный после выделения щавелевокислого кальция, как указано в разд. 9, ГОСТ 2642.1—71.
4.13. Определение содержания окисей натрия и калия
Содержание окисей натрия и калия определяют, как указано в п. 2.16.
4.14. Определение содержания окисей кальция и магния
4.14.1. Сущность метода
Цирконий, гафний, титан, железо, алюминий и фосфор отделяют от кальция и магния уротропином. Кальций определяют три-лонометрическим титрованием в присутствии индикатора кислотного хром темно-синего, сумму кальция и магния — методом обратного титрования трилона Б раствором сернокислой меди в присутствии индикатора «ПАН». Навеску материала разлагают сплавлением с пиросульфатом калия.
4.14.2. Реактивы и растворы
Калий пиросернокислый по ГОСТ 7172—65.
Кислота соляная по ГОСТ 3118—67.
Кислота серная по ГОСТ 4204—66.
Натрия гидрат окиси (натр едкий) по ГОСТ 4328—66, 20%-ныв раствор.
Натрий хлористый по ГОСТ 4233—66.
Аммоний хлористый по ГОСТ 3773—72, х. ч., 20%-ный раствор.
Аммиак водный по ГОСТ 3760—64, 25%-ный раствор.
Медь сернокислая по ГОСТ 4165—68, 0,05 Л1 раствор; готовят следующим образом: 12,5 г медного купороса (CuSO4 • 5НгО) растворяют в воде и приливают 2 мл серной кислоты плотностью 1,84 г/см3 на 1000 мл раствора.
Уротропин фармакопейный, 30%-ный водный раствор.
Индикатор 1,2- (пиридил-азо) -2-нафтол («ПАН»), 0,2 % -ный
спиртовой раствор.
Спирт этиловый ректификованный по ГОСТ 5962—67.
Индикатор кислотный хром темно-синий, 0,5%-ный свежеприготовленный раствор; готовят следующим образом: 0,5 г индикатора растворяют в 20 мл аммиачного буферного раствора и доводят до 100 мл этиловым спиртом.
Аммиачный буферный раствор для приготовления индикатора; готовят следующим образом: 8,25 г хлористого аммония смешивают со 113 мл аммиака плотностью 0,9 г/см3 и разбавляют до 1 л водой.
Индикаторная смесь сухая; готовят следующим образом: 1 г кислотного хром темно-синего растирают в фарфоровой ступке со 100 г хлористого натрия.
Буферный раствор для титрования суммы кальция и магния с pH 10; для приготовления 1000 мл буферного раствора берут 67,5 г хлористого аммония и 570 мл 25%-ного водного раствора аммиака.
Стандартный раствор сернокислого магния, 0,02 н раствор; готовят из фиксанала. Титр его по окиси магния равен 0,0004035 г/мл.
Кальций углекислый по ГОСТ 4530—66, «ч. д. а».
Трилон Б (комплексон III, двунатриевая соль этилендиамин-тетрауксусной кислоты) по ГОСТ 10652—73, 0,025' М раствор (9,3 гтрилона Б на 1 л раствора).
Титр раствора трилона Б по окиси магния и соотношение между растворами трилона Б и сернокислой меди устанавливают, как указано в п. 2.15.2.
Для определения титра раствора трилона Б по окиси кальция в коническую колбу вместимостью 300 мл отбирают 10 мл 0,05 М раствора углекислого кальция, прибавляют 100 мл воды, 30 мл 0,025 М раствора трилона Б, 10 мл аммиачного буферного раствора с pH 10 и пять капель индикатора «ПАН». Избыток трило-на Б оттитровывают 0,05 М раствором сернокислой меди до пере' хода цвета раствора из желто-зеленого в фиолетовый.
Титр раствора трилона Б (Те), выраженный в г/мл окиси кальция, вычисляют по формуле:
где
Т — титр 0,05 М стандартного раствора хлористого кальция, выраженный в г/мл окиси кальция;
V — объем стандартного раствора хлористого кальция, взятый для определения титра раствора трилона Б, в мл;
Vi — объем раствора трилона Б в мл;
у2 — объем раствора сернокислой меди, израсходованный на титрование избытка трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди.
4.14.3. Проведение анализа
Навеску двуокиси циркония 0,5 г помещают в платиновый тигель, смешивают с 6—7 г пиросульфата калия и сплавляют в электрической муфельной печи при 800—850° С в течение 20—25 мин до получения прозрачного расплава. Нагревание тигля необходимо производить постепенно во избежание разбрызгивания и вспенивания расплава. Для ускорения процесса сплавления перемешивают сплав 2—3 раза вращением тигля при помощи щипцов.
Остывший сплав переводят из тигля в стакан вместимостью 300—400 мл, обмывают стенки тигля горячей водой, приливают 15 мл соляной кислоты плотностью 1,19 г/см3 и нагревают до растворения сплава. Если двуокись циркония стабилизирована окисью магния, то приливают 20 мл 20%-ного раствора хлористого аммония.
После охлаждения раствор нейтрализуют аммиаком до появления устойчивой мути, приливают 30 мл 30%-ного раствора уротропина и нагревают 10—15 мин на закрытой электроплитке (не выше 80°С). Раствор с осадком переводят в мерную колбу вместимостью 200 мл, доводят дистиллированной водой до метки и тщательно перемешивают. Затем раствор фильтруют через сухой фильтр диаметром 11 см «белая лента». Отбросив первые две порции фильтрата в конические колбы вместимостью 250 мл, отбирают две аликвотные части по 50 мл: одну — для определения кальция, а другую — для определения суммы кальция и магния:
а) для определения содержания окиси кальция к отобранному раствору прибавляют 50 мл воды, 15 мл 20%-ного раствора едкого натра, 10 капель 0,5%-ного раствора кислотного хром темно-синего или 0,10—0,15 г сухой индикаторной смеси и титруют 0,025 М растворОхМ трилона Б до перехода цвета раствора из розового в сине-сиреневый;
б) для определения содержания окиси магния к отобранным 100 мл фильтрата приливают 20 мл раствора трилона Б, 10 мл аммиачного буферного раствора с pH 10 и пять капель 0,2%-ного спиртового раствора индикатора «ПАН». Избыток раствора трилона Б титруют раствором сернокислой меди до перехода окраски из желто-зеленой в фиолетовую.
Одновременно проводят контрольный опыт через все стадии анализа.
4.14.4. Подсчет результатов анализа
Содержание окиси кальция (X2i) в процентах вычисляют по формуле:
v V . 200 . Т8 . 100
21 ~ 0,5-50
где
V — объем раствора трилона Б, израсходованный на титро
вание, в мл (с учетом контрольного опыта);
200 — исходный объем раствора в мл;
Т8 — титр раствора трилона Б, выраженный в г/мл окиси кальция;
0,5 — навеска анализируемой пробы в г;
50 — аликвотная часть раствора, взятая для определения в мл.
Содержание окиси магния (Х22) в процентах вычисляют по формуле:
V-*-V1 -Va]r3.100,
где
V — объем раствора трилона Б, взятый с избытком, в мл;
Vt — объем раствора сернокислой меди, израсходованный на титрование избытка раствора трилона Б, в мл;
К — соотношение между растворами трилона Б и сернокислой меди;
Тз — титр раствора трилона Б, выраженный в г/мл окиси магния;
G — навеска пробы, взятая для титрования, в г;
V2 — объем раствора трилона Б, израсходованный на реакцию с кальцием, в миллилитрах, вычисляемый по формуле:
I/ = — Xgl__
Т8. 100 ’
где
A';i — содержание окиси кальция в %;
Д — титр раствора трилона Б, выраженный в г/мл окиси кальция.
Учитывать результаты контрольного опыта обязательно.
Допускаемые расхождения между результатами параллельных определений содержания окиси кальция не должны превышать 0,25 абс. %, окиси магния — 0,15 абс. %.
4.15. Определение содержания окиси железа
4.15.1. Сущность метода
Металлическое железо, присутствующее в пробе, переводят в раствор соляной кислотой и после окисления перекисью водорода образовавшееся трехвалентное железо титруют раствором трехвалентной сернокислой соли титана, используя в качестве индикатора раствор роданида калия.
4.15.2. Реактивы и растворы
Реактивы и растворы указаны в разд. 6, ГОСТ 2642.1—71.
4.15.3. Проведение анализа
Навеску материала 0,5—2,0 г (в зависимости от содержания металлического железа) помещают в коническую колбу вместимостью 300 мл, приливают 60 мл соляной кислоты, разбавленной 1 :1, 2 мл 3%-ного раствора перекиси водорода и кипятят с шариковым воздушным холодильником в течение 7 мин. Раствор охлаждают и титруют образовавшееся трехвалентное железо раствором сернокислого титана, как указано в разд. 6, ГОСТ 2642.1—71.
4.15.4. Подсчет результатов анализа
Содержание окиси железа (Хгз) в процентах вычисляют по формуле:
v V * Т • 0,7 . 100 Л 23- ~ ,
где
V — объем раствора сернокислого титана, израсходованный на титрование, в мл;
Т — титр раствора сернокислого титана, выраженный в г/мл окиси железа;
0,7 — коэффициент пересчета окиси железа на металлическое железо;
G — навеска пробы в г.
Допускаемые расхождения между результатами параллельных определений не должны превышать 0,05 абс. %.
Замена
ГОСТ 5.1177—71 введен взамен ГОСТ 24—60
ГОСТ 61—69 введен взамен ГОСТ 61—5!
ГОСТ 177—71 введен взамен ГОСТ 177—55.
ГОСТ 2642.1—71 введен взамен ГОСТ 2642—60 в части разд. 2.
ГОСТ 3761—72 введен взамен ГОСТ 3761—65.
ГОСТ 3772—74 введен взамен ГОСТ 3772—64.
ГОСТ 3777—69 введен взамен ГОСТ 3777—65.
ГОСТ 3773—72 введен взамен ГОСТ 3772—60.
ГОСТ 3765—72 введен взамен ГОСТ 3765—64.
ГОСТ 4214—70 введен взамен ГОСТ 4214—48.
ГОСТ 4217—73 введен взамен ГОСТ 4217—65.
ГОСТ 4951—71 введен взамен ГОСТ 4951—71.
ГОСТ 5817—69 введен взамен ГОСТ 5817—55.
ГОСТ 6613—73 введен взамен ГОСТ 6613—53.
ГОСТ 6709—72 введен взамен ГОСТ 6709—53.
ГОСТ 10484—73 введен взамен ГОСТ 10484—63.
ГОСТ 10652—73 введен взамен ГОСТ 10652—63.
ГОСТ 10691—63 введен взамен ГОСТ 2817—50 в части кинопленок, фотопленок и фотопластинок.
ГОСТ 11069—74 введен взамен ГОСТ 11064—64.
ГОСТ 19627—74 введен взамен ГОСТ 2549—60.
Огнеупоры и огнеупорные изделия
Редактор С. Г. Вилькина
Переплет художника А. М. Поташева Технический редактор В. Н. Малькова Корректоры Г. М. Фролова и Т. А. Камнева
Сдано в набор 29.03.74 Формат издания 60х90</16 Бумага тип. № 3 42 п. л. Тир. 40 000 (2-й завод 20 001—40 000) Изд. № 3638/02 | Подл, в лея. 27.01.75 36.5 уч.-изд. л. Цена 1 р. 94 к. |
Издательство стандартов. Москва, Д-22. Новопресненский пер.. 3
Великолукская городская типография управления издательств, полиграфии к княжной
торговли Псковского облисполкома, г. Великие Луки, Половская, 13. Зак. 505

{kind=link}