ГОСТ Р ИСО 14155-2014
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
Надлежащая клиническая практика
Clinical investigations. Good clinical practice
ОКС 11.100.20
ОКП 94 000
Дата введения 2015-06-01
Предисловие
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "МЕДИТЕСТ" (ЗАО "МЕДИТЕСТ") на основе собственного аутентичного перевода на русский язык международного стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 4 июня 2014 г. N 497-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14155:2011* "Клинические исследования. Надлежащая клиническая практика" (ISO 14155:2011 "Clinical investigation of medical devices for human subjects - Good clinical practice"), включая техническую поправку ISO 14155:2011 /Соr.1:2011.
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Наименование настоящего стандарта изменено относительно наименования указанного международного стандарта для приведения в соответствие с ГОСТ Р 1.5 (пункт 3.5).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации и действующие в этом качестве межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВЗАМЕН ГОСТ Р ИСО 14155-1-2008, ГОСТ Р ИСО 14155-2-2008
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет ()
Введение
Международная организация по стандартизации (ИСО) является всемирной федерацией национальных организаций по стандартизации (комитетов - членов ИСО). Разработка международных стандартов обычно осуществляется Техническими комитетами ИСО. Каждый комитет-член, заинтересованный в деятельности, для которой был создан Технический комитет, имеет право быть представленным в этом комитете. Международные правительственные и неправительственные организации, имеющие связи с ИСО, также принимают участие в работах. Что касается стандартизации в области электротехники, то ИСО работает в тесном сотрудничестве с Международной электротехнической комиссией (МЭК).
Проекты международных стандартов разрабатываются в соответствии с правилами Директив ИСО/МЭК, Часть 2.
Основная задача Технических комитетов заключается в подготовке международных стандартов. Проекты международных стандартов, принятые Техническими комитетами, рассылаются комитетам-членам на голосование. Их опубликование в качестве международных стандартов требует одобрения не менее 75% комитетов-членов, принимающих участие в голосовании.
Следует иметь в виду, что некоторые элементы настоящего стандарта могут быть объектом патентных прав. ИСО не несет ответственности за идентификацию какого-либо одного или всех патентных прав.
ИСО 14155:2011 был подготовлен Техническим комитетом ИСО/ТК 194 "Биологическая оценка медицинских изделий".
Настоящий стандарт включает техническую поправку ИСО 14155:2011/Сог.1:2011, а также отменяет и заменяет ГОСТ Р ИСО 14155-1-2008 и ГОСТ Р ИСО 14155-2-2008.
1 Область применения
В настоящем стандарте рассмотрена надлежащая клиническая практика планирования, проведения, документального оформления и представления результатов клинических исследований, проводимых с участием человека, для оценки безопасности или функциональных характеристик медицинских изделий в целях регулирования.
Принципы, установленные в настоящем стандарте, также применимы ко всем прочим клиническим исследованиям, и их следует придерживаться, насколько это возможно, с учетом характера клинического исследования и национальных регулирующих требований.
В настоящем стандарте определены общие требования, направленные:
- на защиту прав и обеспечение безопасности и благополучия людей;
- обеспечение использования научных подходов при проведении клинического исследования и обеспечение достоверности результатов клинического исследования;
- определение ответственности спонсора и ответственного исполнителя;
- содействие спонсорам, исследователям, этическим комитетам, регулирующим властям и другим органам, вовлеченным в оценку соответствия медицинских изделий.
Настоящий стандарт не применим к медицинским изделиям для in vitro диагностики.
Примечание - Стандарты, разработанные ИСО/ТК 194, предназначены для применения к медицинским изделиям в целом. Пользователи настоящего стандарта должны оценивать, не применяются ли к рассматриваемому исследуемому изделию(ям) также другие стандарты и (или) требования.
2 Нормативные ссылки
В настоящем стандарте использована нормативная ссылка на следующий международный стандарт*:
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ИСО 14971:2007 Изделия медицинские. Применение менеджмента риска к медицинским изделиям (ISO 14971:2007, Medical devices - Application of risk management to medical devices)
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 неблагоприятное (нежелательное) воздействие изделия; ADE (adverse device effect; ADE): Неблагоприятное событие, связанное с использованием исследуемого медицинского изделия.
Примечание 1 - Данное определение включает неблагоприятные события, вызванные недостатками или неадекватностью инструкции по эксплуатации, размещению, имплантации, установке, а также любой сбой в работе исследуемого медицинского изделия.
Примечание 2 - Данное определение включает любые события, вызванные ошибкой эксплуатации или намеренным неправильным применением исследуемого медицинского изделия.
3.2 неблагоприятное (нежелательное) событие; АЕ (adverse event; АЕ): Любое нежелательное медицинское проявление, непредвиденное заболевание или травма, а также нежелательные клинические признаки, включая отклонение лабораторных показателей от нормы у субъекта, пользователя или любого другого лица, которое может быть и не связанным с исследуемым медицинским изделием.
Примечание 1 - Данное определение включает события, связанные с исследуемым медицинским изделием или изделием для сравнения.
Примечание 2 - Данное определение включает события, связанные с используемыми процедурами.
Примечание 3 - Для пользователей или других лиц данное определение ограничивается событиями, связанными с исследуемым медицинским изделием.
3.3 аудит (audit): Систематическая независимая проверка действий и документов, связанных с клиническими исследованиями, для определения, проводятся ли эти действия и ведутся ли записи данных, анализируется ли точность передаваемых данных в соответствии с CIP, стандартными рабочими процедурами, настоящим стандартом и применимыми нормативными требованиями.
3.4 слепой метод/маскирование (blinding/masking): Метод, при применении которого одной или нескольким участвующим в клиническом исследовании сторонам неизвестно, какое именно лечение назначено субъекту исследования.
Примечание - Простой слепой метод обычно подразумевает, что субъект(ы) не осведомлен(ы) о назначенном лечении. Двойной слепой метод обычно подразумевает, что субъект(ы), исследователь(и), наблюдатель (монитор) и, в некоторых случаях, главный эксперт не осведомлены о назначенном лечении.
3.5 индивидуальная регистрационная карта; CRF (case report forms; CRF): Набор документов в бумажном виде, на электронном или оптическом носителе для каждого субъекта, в которых согласно CIP записана информация, которую необходимо предоставить спонсору.
3.6 клиническое исследование (clinical investigation): Системное исследование с участием одного или более человека в качестве субъекта, проводимое для оценки безопасности или функциональных характеристик медицинского изделия.
Примечание - "Клиническое испытание" или "клиническое изучение" являются синонимами термина "клиническое исследование".
3.7 план клинического исследования; CIP (clinical investigation plan; CIP): Документ, в котором установлены обоснование, цели, проект (схема) исследования и предполагаемый анализ, методология, мониторинг, проведение и ведение записей о клиническом исследовании.
Примечание - Термин "протокол" является синонимом "СIР". Тем не менее, слово "протокол" имеет много различных значений, некоторые из которых не связаны с клиническими исследованиями. Также эти значения в разных странах могут быть различными. Поэтому в настоящем стандарте используется термин CIP.
3.8 отчет о клиническом исследовании (clinical investigation report): Документ, описывающий проект (схему) исследования и его выполнение, статистический анализ и результаты клинического исследования.
3.9 клинические функциональные характеристики (clinical performance): Результат применения медицинского изделия или оценка субъекта(ов) клинического исследования в отношении медицинского изделия, связанная с его предусмотренным назначением при условии правильного применения к соответствующему(им) субъекту(ам) клинического исследования.
3.10 компаратор (comparator): Медицинское изделие, лечебно-диагностическая процедура (например, активный контроль), плацебо или отсутствие лечения, используемые в контрольной группе при клиническом исследовании.
3.11 контрактная исследовательская организация; CRO (contract research organization; CRO): Физическое или юридическое лицо, которое в рамках договора со спонсором выполняет одну или более обязанностей и функций, связанных с проведением клинического исследования.
3.12 координатор-исследователь (coordinating investigator): Исследователь, назначенный спонсором для координации работы в ходе многоцентрового клинического исследования.
3.13 комитет по мониторингу данных; DMC (data monitoring committee; DMC): Независимый комитет, который может быть создан спонсором для оценки через определенные интервалы времени хода клинического исследования, данных по безопасности или важных выходных параметров функциональных характеристик для формирования рекомендаций спонсору о целесообразности продолжения, приостановки, изменения или остановки клинического исследования.
Примечание - Примерами DMC являются "орган по мониторингу данных по безопасности (data safety monitoring board, DSMB)" или "комитет по мониторингу данных по безопасности (data safety monitoring committee, DSMC)".
3.14 отклонение (deviation): Случай (случаи) отступления, умышленного или случайного, от требований CIP.
3.15 недостаток изделия (device deficiency): Несоответствие медицинского изделия требованиям к его идентификации, качеству, надежности, долговечности, безопасности или функциональным характеристикам.
Примечание - Недостатки изделия включают отказы, ошибки эксплуатации и неадекватную маркировку.
3.16 конечная точка (главная) [endpoint(s)]: Важнейший показатель(и), используемый(е) для проверки основной гипотезы клинического исследования.
3.17 конечная точка (цели) (вторичная) [endpoint(s)]: Показатель(и), используемый(е) для проверки дополнительных гипотез клинического исследования.
3.18 этический комитет; ЭК (ethics committee; ЕС): Независимый комитет, ответственный за анализ клинического исследования в целях защиты прав и обеспечения безопасности и благополучия людей, принимающих участие в клиническом исследовании.
Примечание - В рамках настоящего стандарта "этический комитет" является синонимом "комитета по этике исследования", "независимого этического комитета" или "внутренней комиссии по контролю". Нормативные требования, касающиеся этических комитетов или аналогичных органов, зависят от страны и региона.
3.19 гипотеза (hypothesis): Утверждение, подвергающееся проверке, основанное на целях и касающееся безопасности или функциональных характеристик исследуемого медицинского изделия, используемое для разработки клинического исследования, которое может быть подтверждено или опровергнуто на основании результатов клинического исследования и статистических вычислений.
Примечание - Основная гипотеза является определяющим фактором безопасности исследуемого медицинского изделия или параметров его функциональных характеристик и обычно используется для вычисления объема выборки. Также могут быть оценены дополнительные гипотезы, касающиеся других задач исследования.
3.20 независимый (independent): Не вовлеченный в проведение клинического исследования, за исключением специально делегированной ответственности, для предотвращения предвзятого отношения или конфликта интересов.
3.21 процесс получения информированного согласия (informed consent process): Процесс, в ходе которого лицу предоставляется полная информация и берется добровольное согласие на участие в клиническом исследовании.
Примечание - Информированное согласие фиксируется документально с помощью письменной формы информированного согласия с личной подписью и указанием даты.
3.22 исследовательский центр (investigation site): Учреждение или место, в котором проводится клиническое исследование.
Примечание - В рамках настоящего стандарта "исследовательский центр" является синонимом "места исследования".
3.23 исследуемое медицинское изделие (investigational medical device): Медицинское изделие, которое в ходе клинического исследования оценивается на предмет безопасности или результативности функциональных характеристик.
Примечание 1 - Данное определение включает медицинские изделия, уже представленные на рынке, оцениваемые для нового предусмотренного назначения/применения, новой популяции, на предмет использования новых материалов или изменений конструкции.
Примечание 2 - В настоящем стандарте термины "исследуемое медицинское изделие" и "исследуемое изделие" являются взаимозаменяемыми.
3.24 исследователь (investigator): Отдельный член команды исследователей, назначенный и подчиняющийся главному исследователю в исследовательском центре для проведения основных процедур, связанных с клиническим исследованием, или для принятия важных решений, связанных с клиническим исследованием.
Примечание - Отдельный член команды исследователей может также называться "субисследователь" или "соисследователь".
3.25 журнал (брошюра) исследователя; IB (investigator's brochure; IB): Сводное изложение актуальной клинической и неклинической информации об исследуемом медицинском изделии(ях), значимой для клинического исследования.
3.26 законный полномочный представитель (legally authorized representative): Физическое или юридическое лицо, или иной субъект права, обладающий в силу закона правом давать согласие на участие в клиническом исследовании от имени потенциального субъекта исследования.
3.27 сбой (malfunction): Отказ исследуемого медицинского изделия функционировать в соответствии с его предусмотренным назначением при условии, что изделие используется в соответствии с инструкциями по эксплуатации или CIP.
3.28 медицинское изделие (medical device): Любой инструмент, аппарат, прибор, оборудование, приспособление, имплантат, программное обеспечение, материал или другое сходное или связанное изделие, которое:
a) предназначено изготовителем для применения к человеку, отдельно или в сочетании с другими изделиями, для достижения одной или более следующих целей:
1) диагностики, профилактики, наблюдения, лечения или облегчения заболеваний;
2) диагностики, наблюдения, лечения, облегчения или компенсации нарушенных или утраченных физиологических функций;
3) исследования, замещения или изменения анатомического строения или физиологических процессов;
4) поддержания жизнедеятельности;
5) предотвращения или прерывания беременности;
6) дезинфекции медицинских изделий;
b) не оказывает своего основного предназначенного действия внутри или на поверхности тела человека за счет фармакологических, иммунологических или метаболических средств, но которые, тем не менее, могут способствовать предусмотренному функционированию изделия.
Примечание - Термин "медицинское изделие" обычно определяется национальной системой регулирования медицинских изделий. В целях настоящего стандарта в данное определение не входят "медицинские изделия для in vitro диагностики" (см. [1], определение 3.7).
3.29 мониторинг (monitoring): Деятельность, связанная с наблюдением за ходом клинического исследования для проверки того, что данные исследования проводятся и записываются, а отчет формируется в соответствии с CIP, документированными процедурами, настоящим стандартом и применимыми регулирующими требованиями.
3.30 многоцентровые исследования (multicentre investigation): Клинические исследования, которые проводятся в соответствии с единым CIP в двух или более исследовательских центрах.
3.31 цель (objective): Основная цель проведения клинического исследования.
3.32 момент включения в исследование (point of enrolment): Момент времени, в который субъект клинического исследования подписывает и датирует форму информированного согласия.
3.33 главный исследователь (principal investigator): Квалифицированное лицо, ответственное за проведение клинического исследования в исследовательском центре.
Примечание 1 - Если клинические исследования проводятся группой лиц в исследовательском центре, главный исследователь ответственен за руководство данной группой.
Примечание 2 - Является ли ответственным конкретное лицо или институт, может зависеть от особенностей национальной системы регулирования медицинских изделий.
3.34 рандомизация (randomization): Процесс распределения субъектов в группы исследования медицинского изделия или в контрольные группы с использованием установленных признанных статистических методов с целью снижения допустимой погрешности.
3.35 подбор субъектов (recruitment): Активные действия по идентификации субъектов, подходящих для включения в клиническое исследование.
3.36 серьезное неблагоприятное (нежелательное) воздействие изделия; SADE (serious adverse device effect; SADE): Неблагоприятное воздействие изделия, которое приводит к появлению любых последствий, характерных для серьезного неблагоприятного события.
3.37 серьезное неблагоприятное (нежелательное) событие; SAE (serious adverse event; SAE): Неблагоприятное событие, которое:
а) приводит к летальному исходу;
b) приводит к серьезному нарушению здоровья субъекта, которое, в свою очередь, приводит либо:
1) к заболеванию или травме, угрожающей жизни, или
2) увечью, или
3) госпитализации или ее продлению, или
4) медицинскому или хирургическому вмешательству для предотвращения заболевания, угрожающего жизни, или увечья;
c) приводит к травме плода, гибели плода или аномалиям или порокам его развития.
Примечание - Плановая госпитализация по причинам, существовавшим до возникновения серьезного неблагоприятного события, или для проведения процедур, необходимых согласно CIP, без серьезного нарушения здоровья не считаются серьезными неблагоприятными событиями.
3.38 исходные данные (source data): Вся информация в виде оригинальных записей, заверенных копий оригинальных записей о клинических находках, наблюдениях или другой деятельности во время клинического исследования, необходимая для реконструкции и оценивания клинического исследования.
3.39 исходная документация (source documents): Распечатанные документы, документы на оптическом или электронном носителе, содержащие исходные данные.
Примеры - Больничные записи/карты, лабораторные записи, отчетные записи по оборудованию, негативы фотографий, рентгеновские снимки, записи, хранящиеся в исследовательском центре, в лабораториях и в медико-технических отделах, вовлеченных в клинические исследования.
3.40 спонсор (sponsor): Физическое лицо или организация, несущие ответственность и имеющие обязанности по инициации или проведению клинического исследования.
Примечание - Если исследователь инициирует, запускает и несет полную ответственность за клиническое исследование, исследователь также выполняет роль спонсора и определяется как спонсор-исследователь.
3.41 субъект клинического исследования (subject): Лицо, принимающее участие в клиническом исследовании.
Примечание - Субъектом клинического исследования может быть здоровый доброволец или пациент.
3.42 непредвиденное серьезное неблагоприятное (нежелательное) воздействие изделия; USADE (unanticipated serious adverse device effect; USADE): Серьезное неблагоприятное воздействие изделия, характер, степень влияния, тяжесть или последствия применения которого не были идентифицированы в текущей/действующей версии отчета по анализу риска.
Примечание - Ожидаемое серьезное неблагоприятное воздействие изделия (ASADE) является воздействием, характер, степень влияния, тяжесть или последствия применения которого были идентифицированы в отчете по анализу риска.
3.43 ошибка эксплуатации/применения (use error): Выполнение или невыполнение действия, приводящее к функционированию медицинского изделия, отличающемуся от предусмотренного изготовителем или ожидаемого пользователем.
Примечание 1 - К ошибкам эксплуатации/применения относят промахи, упущения и заблуждения.
Примечание 2 - Неожиданная физиологическая реакция субъекта сама по себе не является ошибкой эксплуатации/применения.
[ИСО 14971:2007, определение 2.27]
3.44 уязвимый субъект (vulnerable subject): Лицо, на готовность которого к участию в клиническом исследовании может излишне влиять ожидание, обоснованное или нет, выгоды, связанной с участием или негативной реакцией администрации в случае отказа от участия.
Пример - Лица, ограниченные в дееспособности либо лишившиеся ее из-за нарушений развития или психических заболеваний, лица в домах престарелых, дети, бедные, лица в чрезвычайных ситуациях, этнические меньшинства, лица без определенного места жительства, бродяги, беженцы, лица, неспособные дать информированное согласие. К другим уязвимым субъектам относятся, например, члены групп с иерархической структурой, как, например, студенты университета, служебный больничный и лабораторный персонал, наемные рабочие спонсора, военнослужащие и заключенные.
4 Этические аспекты
4.1 Общие положения
Клиническое исследование должно проводиться в соответствии с этическими принципами, основанными на Хельсинкской Декларации (см. [8]). Эти принципы защищают права и обеспечивают безопасность и благополучие людей, что является наиболее важным аспектом и должно превалировать над интересами науки и общества. Эти принципы следует понимать, соблюдать и применять на каждом этапе клинического исследования.
4.2 Злоупотребление влиянием или мотивация
Спонсор должен избегать злоупотребления влиянием или мотивации субъекта, наблюдателя, исследователя(ей) или других сторон, принимающих участие или обеспечивающих проведение клинического исследования.
Все исследователи должны избегать злоупотребления влиянием или мотивации субъекта, спонсора, наблюдателя, другого исследователя(ей) или других сторон, принимающих участие или обеспечивающих проведение клинического исследования.
4.3 Компенсация и дополнительное медицинское обслуживание
Субъектам клинического исследования может выплачиваться компенсация затрат, связанных с участием в клиническом исследовании (например, транспортных расходов), если это допускается национальными регулирующими требованиями, однако данная компенсация не должна быть настолько большой, чтобы чрезмерно мотивировать субъекта клинического исследования к участию.
Должен быть создан и задокументирован план дополнительного медицинского обслуживания субъектов, пострадавших от неблагоприятных событий, связанных с участием в клиническом исследовании.
Примечание - Данный план может быть объектом национальной системы регулирования медицинских изделий.
4.4 Ответственность
Все стороны, вовлеченные в проведение клинического исследования, должны разделять ответственность за соблюдение этических принципов в соответствии с ролью каждой стороны в клиническом исследовании.
4.5 Обмен информацией с этическим комитетом
4.5.1 Общие положения
Если требования национальных или региональных ЭК являются менее жесткими, чем требования настоящего стандарта, спонсор должен применять требования настоящего стандарта в максимально допустимой степени независимо от любых менее жестких требований и должен регистрировать эти меры.
4.5.2 Предоставление первичных данных в этический комитет
Как минимум, в ЭК должна быть представлена следующая информация и любые дополнения к ней:
a) CIP;
b) IB или эквивалентная документация;
c) форма информированного согласия и любая другая письменная информация, предоставляемая субъектам;
d) процедуры по подбору субъектов и рекламные материалы при их наличии;
e) копия резюме главного исследователя, для которого ЭК осуществляет рассмотрение документов.
Следующие документы могут также при необходимости быть предоставлены в ЭК в зависимости от схемы клинического исследования и национальных или региональных требований:
f) образец или проект CRF, включая все инструменты сбора данных согласно требованиям CIP;
g) документы, касающиеся оплаты и компенсаций, доступных для субъектов;
h) предполагаемые компенсации для учреждения или главного исследователя;
i) документация, касающаяся любого конфликта интересов, включая финансовый, со стороны исследователя;
j) свидетельства страхования клинического исследования.
4.5.3 Информация, получаемая от этического комитета
До начала клинического исследования спонсор должен получить документально подтвержденное одобрение/положительное заключение ЭК с указанием документов и поправок, на основании которых принято данное решение.
Примечание - Спонсор может запросить отчет о голосовании ЭК по клиническому исследованию для документального подтверждения того, что участники группы, проводящей клиническое исследование, не принимали участия в голосовании.
4.5.4 Последующий обмен информацией с этическим комитетом
Следующая информация должна быть предоставлена в ЭК, если это требуется национальными регулирующими требованиями, CIP или ЭК, в зависимости от того, кто из них выдвигает более строгие требования:
a) серьезные неблагоприятные события;
b) запросы на одобрение отклонений, отчеты об отклонениях, если отклонения затрагивают права, безопасность или благополучие субъектов, или научную целостность клинического исследования.
В чрезвычайных ситуациях отклонение от CIP для защиты прав, безопасности и благополучия субъектов-людей может выполняться без предварительного согласования со спонсором и ЭК. Такое отклонение должно быть задокументировано и представлено спонсору и в ЭК так быстро, насколько это возможно:
c) отчеты о ходе работ, включая сводку по безопасности и отклонениям;
d) поправки к любым документам, уже одобренным ЭК.
Примечание - Для несущественных изменений (например, незначительных логистических или административных изменений, замены наблюдателей, телефонных номеров, обновление страховки), не затрагивающих права, безопасность и благополучие субъектов-людей или не затрагивающих цели или критические точки клинического исследования, может быть достаточно простого уведомления ЭК и, если необходимо, регулирующих органов;
e) если применимо, уведомление о приостановке или преждевременном прекращении исследования;
f) если применимо, обоснование и запрос на возобновление клинического исследования после приостановки;
g) отчет о клиническом исследовании или его краткое изложение.
4.5.5 Дополнительная информация, получаемая от этического комитета
Во время клинического исследования до момента практического внедрения от ЭК должна быть получена, как минимум, следующая информация в письменном виде:
a) одобрение/положительное заключение в отношении поправок, указанных в 4.5.4, перечисление d);
b) одобрение запроса на отклонения, которые могут повлиять на права, безопасность и благополучие субъекта клинического исследования или научную целостность клинического исследования, как указано в 4.5.4, перечисление b);
c) одобрение возобновления приостановленного клинического исследования, как указано в 4.5.4, перечисление f), если применимо.
4.6 Уязвимые группы субъектов
Клинические исследования на уязвимых группах населения должны проводиться только в том случае, если они не могут быть проведены на неуязвимых группах населения, при этом необходимо следовать дополнительным процедурам ЭК, если применимо. Данные клинические исследования должны быть разработаны специально с учетом проблем со здоровьем, возникающих в уязвимых группах населения, и предполагать возможность непосредственной пользы их здоровью.
4.7 Информированное согласие
4.7.1 Общие положения
От субъекта клинического исследования должно быть получено информированное согласие в письменной форме, и процесс должен быть документирован до применения к субъекту клинического исследования любых процедур, характерных для клинического исследования, за исключением особых случаев, описанных в 4.7.3.4. Форма информированного согласия состоит из информационной формы (см. 4.7.4) и формы для подписания информированного согласия (см. 4.7.5). Данные формы могут быть объединены в один документ или разделены на два документа.
4.7.2 Процесс получения информированного согласия
Общий процесс получения информированного согласия должен быть документально зафиксирован в CIP и должен:
a) подтверждать, что процесс получения информированного согласия осуществляет главный исследователь или его уполномоченный представитель;
b) включать все аспекты клинического исследования, существенные для принятия субъектом клинического исследования решения об участии в клиническом исследовании;
c) предотвращать любое принуждение или злоупотребление влиянием на желание субъекта клинического исследования принять участие в клиническом исследовании;
d) не нарушать и не производить впечатления нарушения законных прав субъекта;
e) использовать простой не технический язык, понятный субъекту;
f) давать субъекту клинического исследования достаточно времени для прочтения и понимания формы информированного согласия и для принятия решения об участии в клиническом исследовании;
g) включать личную подпись субъекта клинического исследования и главного исследователя или уполномоченного представителя, ответственного за осуществление процесса получения информированного согласия, с указанием даты;
h) обеспечивать субъекта клинического исследования копией формы информированного согласия с подписью и датой и любой другой письменной информацией;
i) демонстрировать, как будет получено и зафиксировано информированное согласие в особых случаях (см. 4.7.3), если субъект клинического исследования не сможет предоставить его самостоятельно;
j) обеспечить предоставление новой важной информации новым и уже участвующим в клиническом исследовании субъектам.
Приведенные выше требования также применяются и к получению информированного согласия от законного полномочного представителя.
4.7.3 Особые случаи получения информированного согласия
4.7.3.1 Общие положения
Положения, приведенные в 4.7.3.2-4.7.3.4, зависят от национальных регулирующих требований.
4.7.3.2 Субъекты клинического исследования, нуждающиеся в законном полномочном представителе
Информированное согласие может быть получено от законного полномочного представителя только в том случае, если субъект клинического исследования не может самостоятельно принять решение об участии в клиническом исследовании (например, новорожденный, ребенок или подросток, субъект, имеющий серьезное заболевание или находящийся без сознания, лица с психическими нарушениями, лица с умственной отсталостью). В данных случаях субъект клинического исследования также должен быть проинформирован о клиническом исследовании в рамках его возможностей понимания.
4.7.3.3 Субъект клинического исследования, не способный читать или писать
Если субъект клинического исследования или законный полномочный представитель не может читать или писать, информированное согласие должно быть получено в ходе контролируемого устного процесса. Во время всего процесса должен присутствовать независимый свидетель. Письменная форма информированного согласия и любая другая информация должны быть громко зачитаны и объяснены потенциальному субъекту клинического исследования или его законному полномочному представителю, и при наличии возможности каждый участник данного процесса подписывает и лично проставляет дату в форме информированного согласия. Свидетель также подписывает и лично проставляет дату в форме информированного согласия, подтверждая, что информация была точно объяснена и что информированное согласие было дано добровольно.
4.7.3.4 Неотложная помощь
Для клинического исследования, включающего неотложную помощь, когда предварительное информированное согласие субъекта клинического исследования невозможно получить из-за медицинского состояния субъекта, требуется информированное согласие законного полномочного представителя, если он есть.
Если от субъекта клинического исследования невозможно получить предварительное информированное согласие, а законный полномочный представитель не доступен, то субъект клинического исследования может быть внесен в список, если только в CIP описана специальная процедура, как указано в А.13, перечисление b).
Должны быть приняты меры по информированию субъекта клинического исследования или его законного полномочного представителя настолько быстро, насколько это возможно:
a) о включении субъекта клинического исследования в клиническое исследование и
b) всех аспектах клинического исследования.
Необходимо получить от субъекта клинического исследования информированное согласие на продолжение участия так быстро, как позволит его медицинское состояние.
Главный исследователь может включить субъект клинического исследования в список без получения информированного согласия от субъекта клинического исследования или его законного полномочного представителя при выполнении следующих условий:
c) потенциальный субъект клинического исследования попал в чрезвычайную ситуацию и явно находится в ситуации, угрожающей жизни;
d) от доступных в настоящее время средств не ожидается существенной клинической пользы;
e) существует реальная вероятность того, что можно избежать угрожающего жизни риска, если будет использовано исследуемое изделие;
f) ожидаемые риски перевешивают возможную пользу от применения исследуемого изделия;
g) невозможно оперативно связаться и проинформировать законного полномочного представителя.
4.7.4 Информация, предоставляемая субъекту
Вся информация, касающаяся клинического исследования, включая, по крайней мере, следующую, должна быть представлена в письменном виде простым не техническим языком, понятным субъекту клинического исследования (или его законному полномочному представителю).
a) Описание и цель:
1) утверждение о том, что клиническое исследование входит в научные исследования;
2) цель клинического исследования;
3) предполагаемая продолжительность клинического исследования и степень вовлеченности и ответственность каждого субъекта клинического исследования в период клинического исследования;
4) описание исследуемого изделия и изделия для сравнения, если оно используется;
5) описание всех процедур, касающихся субъекта;
6) аспекты клинического исследования, относящиеся к экспериментальным;
7) описание клинического исследования, включая описание всех контрольных групп и методов распределения в каждую из групп;
8) предполагаемое число субъектов, принимающих участие в клиническом исследовании.
b) Потенциальная польза:
1) описание реально ожидаемой пользы для субъекта клинического исследования (если ожидаемая прямая терапевтическая польза отсутствует, это необходимо зарегистрировать);
2) описание потенциальной пользы для других.
c) Риски и неудобства для субъекта клинического исследования и, если применимо, эмбриона, плода или грудного ребенка:
1) описание остаточных рисков, определенных в ходе анализа риска;
2) описание рисков, связанных с клиническими процедурами, предусмотренными CIP;
3) сообщение о том, что могут возникнуть неожиданные риски;
4) описание неудобств.
d) Альтернативная процедура (процедуры):
1) информация об альтернативном лечении или процедурах, которые могут быть доступны субъекту клинического исследования и их потенциальной пользе и рисках.
е) Конфиденциальность:
1) заявление, подтверждающее конфиденциальность участия субъекта в клиническом исследовании;
2) заявление, подтверждающее, что записи, идентифицирующие субъект, будут конфиденциальными в той степени, которая допустима по действующему законодательству;
3) заявление, подтверждающее, что субъект клинического исследования понимает, что уполномоченные органы, представители ЭК и представители спонсора, вовлеченные в клиническое исследование, будут иметь прямой доступ к медицинским записям;
4) заявление, показывающее, что результаты клинического исследования могут быть опубликованы без раскрытия данных субъекта.
Примечание - Согласно национальным или региональным регулирующим требованиям могут предъявляться дополнительные требования, касающиеся защиты персональных данных.
f) Компенсация:
1) информация о предоставлении компенсаций, доступных в случае причинения вреда, связанного с участием в клиническом исследовании;
2) информация о дополнительных медицинских мероприятиях для субъектов, пострадавших от неблагоприятных событий в результате участия в клиническом исследовании;
3) информация о финансовой компенсации участия, если применимо.
g) Предполагаемые расходы, если таковые имеются, связанные с участием субъекта клинического исследования в клиническом исследовании.
h) Информация о роли представителя спонсора в клиническом исследовании.
i) Контактные лица:
1) к кому обращаться по вопросам клинического исследования;
2) к кому обращаться в случае причинения вреда;
3) к кому обращаться по вопросам прав субъекта.
j) Заявление о том, что новые данные или основания для любых поправок к CIP, способных повлиять на решение субъекта клинического исследования продолжить участие в исследовании, должны быть доступны субъекту.
k) Заявление о том, что личный врач субъекта клинического исследования проинформирован об участии субъекта клинического исследования в клиническом исследовании с согласия субъекта.
l) Досрочное прекращение исследования:
1) обстоятельства, при которых главный исследователь может прекратить участие субъекта, если применимо;
2) обстоятельства, при которых спонсор может приостановить или досрочно завершить клиническое исследование.
4.7.5 Подписание информированного согласия
Форма для подписания информированного согласия должна содержать следующее:
a) добровольное согласие принять участие в клиническом исследовании и следовать инструкциям исследователя;
b) заявление о том, что при отказе от участия на субъекта клинического исследования не накладывается никаких штрафных санкций;
c) заявление о том, что в случае отказа от продолжения участия в любой момент времени на субъекта клинического исследования не накладывается никаких штрафных санкций;
d) заявление о возможных последствиях прекращения участия;
е) подтверждение получения информации и подтверждение того, что субъект клинического исследования получил ответы на все вопросы;
f) заявление о том, что субъект клинического исследования или его законный полномочный представитель согласен с использованием соответствующих персональных данных субъекта клинического исследования в рамках клинического исследования;
g) заявление о том, что субъект клинического исследования или его законный полномочный представитель согласен, что представители спонсора, уполномоченные органы и представители ЭК будут иметь прямой доступ к медицинским записям субъекта.
4.7.6 Новая информация
Если становится доступной новая информация, которая может существенно повлиять на будущее здоровье и медицинское обслуживание субъектов клинического исследования, данная информация должна быть предоставлена субъекту(ам) клинического исследования в письменном виде. Если необходимо, от всех заинтересованных субъектов необходимо получить информированное согласие на продолжение участия в письменном виде.
5 План клинического исследования
5.1 Общие положения
Все стороны, принимающие участие в проведении клинического исследования, должны иметь квалификацию, подтвержденную обучением, стажировкой или наличием опыта выполнения подобных задач, что должно быть задокументировано соответствующим образом (см. 8.2.1).
5.2 Оценивание риска
До проведения клинического исследования в соответствии с ИСО 14971 должны быть определены уровни рисков, связанных с исследуемым изделием. Анализ риска должен включать или ссылаться на объективный анализ опубликованных и доступных неопубликованных медицинских и научных данных. Отчет об анализе риска, включающий идентификацию остаточных рисков, должен быть включен в IB.
Для принятия решения о начале клинического исследования медицинского изделия необходимо, чтобы остаточный(е) риск(и), идентифицированный(е) в ходе анализа риска, а также риск(и) для субъекта, связанный(е) с клиническими процедурами, необходимыми согласно CIP, были сбалансированы предполагаемой пользой для субъектов.
Данный анализ риска также должен быть использован как основа для определения ожидаемых неблагоприятных воздействий изделия, характеризуемых их природой, сферой распространения, тяжестью и последствиями.
Ожидаемые неблагоприятные воздействия изделия должны быть документированы в CIP (см. А.4), IB (см. В.5) и форме информированного согласия (см. 4.7.4).
Примечание - Эти допущения соответствуют любым зафиксированным требованиям к прогнозируемым и не прогнозируемым SADEs.
5.3 Обоснование проекта клинического исследования
Обоснование проекта клинического исследования должно быть основано на оценке доклинических данных и результатов клинического оценивания.
Клиническое оценивание включает в себя оценку и анализ клинических данных, касающихся безопасности или рабочих характеристик исследуемого изделия, или аналогичного изделия, или методов лечения. Оценивание должно соответствовать предполагаемому назначению исследуемого изделия и предполагаемому методу применения. Данное оценивание относится к научной деятельности, которая должна проводиться со строгостью и объективностью, соответствующими научным стандартам, с использованием принципов клинического оценивания GHTF (см. [6]).
Результаты клинического оценивания должны использоваться для определения и обоснования оптимального проекта клинического исследования. Они также должны помочь идентифицировать соответствующие конечные точки и учитываемые влияющие факторы, а также помочь обосновать выбор изделия(й) для сравнения.
Клиническое исследование должно быть разработано таким образом, чтобы оценить, подходит ли исследуемое изделие для цели(ей) и популяции(й), для которых оно предназначено. Оно должно быть разработано таким образом, чтобы обеспечить уверенность в том, что полученные результаты имеют клиническую значимость и научную достоверность, и отвечают целям клинического исследования.
Примечание - Необходимость проведения клинического исследования для удовлетворения регулирующих требований определена в применимых национальных регулирующих документах.
5.4 План клинического исследования
CIP должен включать информацию, приведенную в приложении А.
CIP и все последующие дополнения к CIP согласуются со спонсором, координатором и главным исследователем и фиксируются с обоснованием каждого дополнения.
5.5 Журнал (брошюра) исследователя
Целью IB является обеспечение главного исследователя достаточным объемом данных по безопасности или рабочим характеристикам, полученных на основании доклинического исследования или клинического исследования, для обоснования описанного в CIP воздействия исследуемого изделия на организм человека.
IB должен корректироваться в ходе клинического исследования, если появляется новая важная информация (например, существенное изменение риска и т.д.).
Главный исследователь должен подтвердить получение IB и всех последующих дополнений и не должен разглашать данную информацию.
IB должен включать информацию, приведенную в приложении В.
5.6 Индивидуальная регистрационная карта
Для фиксирования данных о каждом зарегистрированном субъекте, согласно требованиям CIP, должны быть разработаны CRF. CRF должны включать информацию о состоянии каждого субъекта клинического исследования на момент включения в клиническое исследование и в ходе клинического исследования, а также информацию о любых других воздействиях (см. приложение С).
Процедура должна обеспечивать, чтобы в случае необходимости внесения изменений в CIP спонсор анализировал CRF для определения необходимости внесения поправок в эти формы.
5.7 План мониторинга
Спонсор должен оценивать степень и вид мониторинга, подходящего для клинического исследования, включая стратегию проверки исходных данных, основываясь на таких параметрах, как цель, схема, сложность, объем клинического исследования, критические и предельные значения данных в ходе клинического исследования. Результаты данной оценки должны быть использованы для разработки плана мониторинга.
5.8 Выбор исследовательских центров
До начала клинического исследования квалификация главного исследователя, исследователей и соответствие исследовательского(их) центра(ов) должны быть проверены и документированы в отчете по выбору исследовательского центра. Обоснование выбора исследовательского центра должно быть документировано.
Примечание - Обоснование выбора исследовательского центра может быть основано на предыдущем опыте спонсора и главного исследователя или исследовательского центра.
5.9 Соглашение (соглашения)
Должно быть заключено соглашение между спонсором и руководителем (руководителями)/исследовательским центром(ами) и любыми другими участвующими сторонами (например, исследователями, CRO и внутренними лабораториями), в котором определена ответственность каждого участника клинического исследования. Все соглашения должны быть выполнены в письменной форме, подписаны и датированы всеми участвующими сторонами.
В соглашении должно быть указано, что при участии в клиническом исследовании стороны могут делить ответственность со спонсором.
5.10 Маркировка
На исследуемом изделии, в инструкции по эксплуатации или на упаковке должно быть указано, что исследуемое изделие предназначено исключительно для использования в клиническом исследовании, если это требуется национальными регулирующими документами.
Примечание - См. [3], [4] и национальные или региональные регулирующие документы для дополнительной информации о маркировке.
5.11 Комитет по мониторингу данных
Спонсор должен рассмотреть необходимость создания DMC до начала клинического исследования.
Решение о создании DMC должно быть основано на анализе риска, учитывающем как риски, связанные с применением исследуемого изделия, так и риски, связанные с участием субъекта клинического исследования в клиническом исследовании.
Основные функции DMC должны быть описаны в CIP. Обязанности DMC должны быть подробно описаны отдельными письменными процедурами, устанавливающими частоту заседаний, порядок действий в чрезвычайных ситуациях и документирование таких заседаний.
6 Проведение клинического исследования
6.1 Общие положения
Клиническое исследование должно проводиться в соответствии с CIP.
Клиническое исследование не должно начинаться до получения письменного одобрения/положительного решения ЭК и, если необходимо, соответствующих регулирующих органов стран, в которых проводится клиническое исследование.
6.2 Подготовка исследовательских центров
В начале клинического исследования (см. 8.2.4) спонсором или наблюдателем должен быть проведен и документирован подготовительный визит в каждый участвующий исследовательский центр(ы) или, в качестве альтернативы, проведена встреча с исследователями. Должен быть оформлен регистрационный журнал, содержащий фамилии, инициалы, личные подписи, функции и полномочия каждого руководителя и членов команды в данном исследовательском центре.
6.3 Мониторинг исследовательских центров
Проведение клинического исследования должно подвергаться мониторингу в соответствии с планом мониторинга (см. 8.2.4).
Как правило, существует необходимость мониторинга на месте до, во время и после клинического исследования. Тем не менее, в исключительных случаях спонсор может определить, что обеспечить надлежащее проведение клинического исследования можно с помощью удаленного мониторинга (без посещения исследовательского центра) совместно с такими процедурами, как документирование обучения исследователя, встречи и подробные письменные указания или телефонная связь. В таком случае спонсор должен привести обоснование исключения проверки первичной документации.
6.4 Неблагоприятные события и недостатки изделия
6.4.1 Неблагоприятные события
Все неблагоприятные события во время клинического исследования должны быть своевременно документированы и включены в отчет, как определено в 8.2.5 и 9.8.
Все неблагоприятные события должны быть включены в промежуточный или финальный отчет о клиническом исследовании.
6.4.2 Недостатки изделия
Все недостатки изделия, связанные с идентификацией, качеством, долговечностью, надежностью, безопасностью или рабочими характеристиками исследуемого медицинского изделия, должны быть документированы во время клинического исследования и соответствующим образом обработаны спонсором.
Недостатки изделия, которые не привели к неблагоприятным событиям, но могут привести к медицинским происшествиям:
a) если не будут предприняты соответствующие действия;
b) если не будет выполнено вмешательство, или
c) если случай будет менее благоприятным,
должны быть включены в отчет, как определено в 8.2.5 и 9.8.
6.5 Документы клинического исследования и документирование клинического исследования
6.5.1 Дополнения
IB, CIP, CRF, форма информированного согласия и другая информация о субъекте клинического исследования или другие документы по клиническому исследованию должны по необходимости изменяться в течение клинического исследования, и к каждому изменению документа должно быть приведено обоснование в соответствующем разделе. Предполагаемые дополнения к CIP должны быть согласованы со спонсором и главным исследователем или координатором. Дополнения к СIР и форме информированного согласия должны быть представлены в ЭК или согласованы с ним и уполномоченными органами, если это необходимо (см. 4.5.4). Номер версии и дата изменения должны быть документированы.
6.5.2 Журнал идентификации субъектов
В каждом исследовательском центре должен вестись журнал регистрации всех субъектов, включенных в клиническое исследование, с присвоением идентификационного кода, связанного с их именами, альтернативной идентификацией субъекта клинического исследования или контактной информацией.
Примечание - В зависимости от схемы клинического исследования может регулярно вестись журнал с идентификацией всех лиц, прошедших предварительную проверку на возможность участия в клиническом исследовании.
6.5.3 Исходная документация
Исходная документация должна создаваться и поддерживаться командой данного исследовательского центра в течение всего клинического исследования.
6.6 Дополнительные члены исследовательской команды
Время от времени могут добавляться новые члены исследовательской команды в новые или существующие исследовательские центры. Новый персонал должен приступать к своим обязанностям только после прохождения подготовки по вопросам требований к клиническому исследованию. Данная подготовка должна быть документирована. Должны быть документированы фамилии, инициалы, личные подписи, функции и предусмотренные полномочия нового персонала.
6.7 Неприкосновенность частной жизни субъекта клинического исследования и конфиденциальность данных клинического исследования
Все лица, вовлеченные в любой этап клинического исследования, должны соблюдать конфиденциальность данных. Все данные должны быть защищены от несанкционированного доступа.
Частная жизнь каждого субъекта клинического исследования и конфиденциальность информации должны сохраняться в отчетах и при опубликовании данных.
Руководитель или учреждение должны обеспечить прямой доступ к первичным данным во время и после клинического исследования для мониторинга, аудита, рассмотрения ЭК и инспекции уполномоченными органами. Если необходимо, до начала клинического исследования руководитель или учреждение должны получить разрешение на прямой доступ к исходной информации от субъекта, администрации больницы и национальных регулирующих органов.
6.8 Управление документами и данными
6.8.1 Прослеживаемость документов и данных
Все документы и данные должны создаваться и поддерживаться таким образом, чтобы обеспечивать возможность управления и прослеживаемость. Если необходимо, должна обеспечиваться и документально подтверждаться точность перевода. Вся документация с последующими версиями, касающаяся клинического исследования, должна быть идентифицируемой и прослеживаемой и храниться таким образом, чтобы предоставлять полную историю клинического исследования.
Исследователь должен обеспечивать точность, полноту, разборчивость и своевременность представленных спонсору данных в CRF и всех необходимых отчетах. Если предоставляются копии оригинальной исходной документации, а также распечатки оригинальной электронной исходной документации, они должны быть подписаны с указанием даты членами исследовательской команды с приведением утверждения о том, что они являются корректной копией оригинальной исходной документации.
Если распределение в исследуемые группы лечения проводится с использованием слепого метода/маскирования, это должно быть безопасным на протяжении всего клинического исследования, включая получение и обработку данных. Необходимо следовать установленным процедурам по расшифровке клинического исследования, проводящегося с использованием слепого метода/маскирования.
6.8.2 Запись данных
Данные, приведенные в CRF, должны быть получены из исходной документации и должны быть согласованы с ней, все отклонения должны быть письменно объяснены. В CIP должно быть определено, какие данные могут быть записаны непосредственно в CRF. CRF должны быть подписаны с указанием даты главным исследователем или его полномочным представителем(ями). Любые изменения или исправление данных, приведенных в CRF, должны быть подписаны с указанием даты, расшифровкой подписи и объяснены, если необходимо, и не должны скрывать исходные записи (т.е. следы проверки должны оставаться); это касается как письменных, так и электронных изменений или коррекций.
6.8.3 Электронные клинические системы данных
При использовании электронных клинических баз данных или удаленных электронных клинических систем данных должны применяться письменные процедуры:
a) для определения и документирования требований к электронным клиническим системам данных для получения и обработки данных;
b) верификации и валидации того, что требования к электронным клиническим системам данных согласованы и выполнены;
c) обеспечения полноты, надежности, согласованности и логичности получаемых данных;
d) обеспечения точности отчетов;
e) обеспечения документирования изменения данных и того, что полученные данные не удаляются (т.е. поддерживаются следы проверок, следы данных и следы их редактирования);
f) поддержания системы безопасности для предотвращения как внутреннего, так и внешнего несанкционированного доступа к данным;
g) поддержания списка лиц, имеющих доступ к электронным системам данных, а также периода доступа и прав для каждого пользователя;
h) обеспечения того, что все заполненные CRF подписываются главным исследователем или его полномочным представителем;
i) поддержания надлежащего резервного копирования, хранения и возможности восстановления данных;
j) обучения пользователей правильному использованию системы.
6.9 Учет исследуемых изделий
Доступ к исследуемым изделиям должен быть контролируемым, и исследуемое изделие должно использоваться только в клиническом исследовании и в соответствии с CIP.
Спонсор должен вести записи для документирования физического расположения всех исследуемых изделий, начиная с доставки исследуемых изделий в исследовательский центр до возврата или утилизации.
Главный исследователь или полномочный представитель должен вести записи для документирования получения, использования, возврата и утилизации исследуемых изделий, которые включают:
a) дату получения;
b) идентификацию каждого исследуемого изделия (номер партии/серийный номер или уникальный код);
c) срок годности, если применимо;
d) дату или даты использования;
e) идентификацию субъекта;
f) дату возврата/эксплантации из субъекта клинического исследования исследуемого изделия, если применимо, и
g) дату возврата неиспользованных или неисправных исследуемых изделий или изделий с истекшим сроком годности, если применимо.
Примечание - Письменные процедуры могут требоваться национальными регулирующими документами.
6.10 Учет субъектов
Все субъекты, вовлеченные в клиническое исследование (включая отозванных из клинического исследования или выбывших из наблюдения), должны быть учтены и документированы.
Если субъект клинического исследования отозван из клинического исследования, должна(ы) быть зарегистрирована(ы) причина(ы) этого. Если субъект был отозван из-за проблем, связанных с безопасностью или рабочими характеристиками исследуемого изделия, исследователь должен попросить у субъекта клинического исследования разрешения наблюдать его состояние вне клинического исследования.
6.11 Аудит
Аудит клинического исследования может быть проведен спонсором или третьей стороной, назначенной спонсором для оценки соответствия CIP, письменным процедурам, настоящему стандарту и применимым регулирующим требованиям. Этот аудит может включать все вовлеченные стороны, системы и учреждения и является независимым и отдельным от функций текущего мониторинга или контроля качества.
Аудит полезен:
a) как составная часть программы обеспечения качества, предоставленной спонсором;
b) для оценки результативности мониторинга;
c) для выявления, имеются ли серьезные или неоднократные отклонения от CIP или подозрения в мошенничестве;
d) для приведения исследовательского центра к "готовности к инспекции", т.е. для подготовки исследовательского центра к возможным регулирующим инспекциям, и
е) если он требуется или предполагается регулирующими органами.
Аудиторы должны быть квалифицированными на основе подготовки и опыта для надлежащего проведения аудита.
Аудит клинических исследовательских систем должен проводиться в соответствии с письменными процедурами спонсора или специальным планом относительно того, что проверять, как проверять, с какой частотой проводить аудит и с указанием формы и содержания отчета по аудиту.
План аудита и процедуры аудита клинического исследования спонсора должны учитывать важность клинического исследования, число субъектов клинического исследования, тип и сложность клинического исследования, уровень риска для субъектов и любые выявленные проблемы.
Результаты аудита должны быть документированы и, при необходимости, сообщены соответствующим сторонам.
7 Приостановка, прекращение и завершение клинического исследования
7.1 Приостановка или прекращение клинического исследования
7.1.1 Процедура приостановки или прекращения
Спонсор может приостановить или прекратить (досрочно завершить) любое клиническое исследование в определенном исследовательском центре или все клиническое исследование по существенным и документированным причинам.
Главный исследователь, ЭК или регулирующий орган может приостановить или прекратить участие в клиническом исследовании исследовательского центра, за который они ответственны.
Если возникает подозрение на неприемлемый риск для субъектов во время клинического исследования, или, если это предписано ЭК или регулирующими органами, спонсор должен приостановить клиническое исследование до тех пор, пока оценивается риск клинического исследования. Спонсор должен прекратить клиническое исследование, если подтверждается неприемлемый риск.
Спонсор должен рассматривать возможность прекращения или приостановки участия в клиническом исследовании определенного исследовательского центра или исследователя, если мониторинг или аудит выявляет серьезные или неоднократные отклонения со стороны исследователя.
Если происходит приостановка или досрочное прекращение, сторона, инициировавшая прекращение, должна обосновать свое решение в письменном виде и незамедлительно проинформировать другие стороны, с которыми она напрямую взаимодействует. Главный исследователь и спонсор должны информировать друг друга о любых сообщениях, полученных либо от ЭК, либо от регулирующих органов.
Примечание - Обычными схемами связи являются: "спонсор - главный исследователь" или "спонсор - ЭК" и "спонсор - регулирующие органы".
Если по каким-либо причинам спонсор приостанавливает или прекращает исследование в определенном исследовательском центре, спонсор должен поставить в известность ответственный регулирующий орган и убедиться, что ЭК также поставлен в известность главным исследователем или спонсором. Если приостановка или прекращение были проведены в интересах безопасности, спонсор должен поставить в известность всех других руководителей.
Если происходит приостановка или прекращение:
a) спонсор остается ответственным за обеспечение ресурсов, необходимых для выполнения обязательств по CIP, и существующих соглашений по наблюдению за субъектами, вовлеченными в клиническое исследование, и
b) главный исследователь или полномочный представитель должны своевременно информировать вовлеченных субъектов в своих исследовательских центрах, если это необходимо.
Примечание - Метод и время данного обмена информацией зависит от обстоятельств и возможных рисков.
Все действия, перечисленные в 7.2, также должны быть выполнены.
7.1.2 Процедуры возобновления клинического исследования после временной приостановки
После завершения спонсором анализа причин приостановки, проведения необходимых корректирующих действий и принятия решения о завершении временной приостановки, спонсор должен поставить в известность руководителей, ЭК и, если необходимо, регулирующие органы о причинах данного решения с предоставлением соответствующих данных, поддерживающих данное решение.
Примечание - Обычными схемами связи являются: "спонсор - главный исследователь" или "спонсор - ЭК" и "спонсор - регулирующие органы".
До возобновления клинического исследования должно быть получено одобрение от ЭК и, если необходимо, регулирующих органов.
Если субъекты были поставлены в известность о приостановке, главный исследователь или его полномочный представитель должны поставить их в известность о причинах возобновления.
7.2 Завершение
Процедуры завершения (закрытия) должны проводиться для того, чтобы удостовериться в том, что записи главного исследователя полные, все документы, необходимые для файлов спонсора, получены, оставшиеся клинические материалы утилизированы, идентифицированные ранее проблемы решены и все стороны уведомлены.
а) Записи о завершении включают свидетельства того, что:
1) все необходимые документы являются полными и актуальными;
2) все CRF являются полными;
3) все поставленные вопросы решены;
4) актуальный статус всех текущих неблагоприятных событий документирован;
5) принимаются меры для архивирования и хранения записи, и
6) документировано расположение любых:
a) исследуемых изделий;
b) оставшихся образцов (например, крови или тканей);
c) других клинических исследуемых материалов.
d) Уведомления включают:
1) уведомление ЭК и
2) уведомление регулирующих органов, если необходимо.
7.3 Отчет о клиническом исследовании
После завершения клинического исследования должен быть заполнен отчет о клиническом исследовании в соответствии с применимыми регулирующими требованиями, даже если клиническое исследование было прекращено:
a) отчет о клиническом исследовании должен быть представлен в письменной форме;
b) отчет о клиническом исследовании должен включать идентификацию изделия(й), описание методики и схемы клинического исследования, любые отклонения от CIP, анализ данных совместно со статистической и критической оценками относительно целей клинического исследования;
c) в отчете о клиническом исследовании должны быть учтены данные от всех исследовательских центров и по всем субъектам. Ни один субъект клинического исследования не должен идентифицироваться на основании отчета о клиническом исследовании или опубликованных результатов;
d) если необходимо, отчет о клиническом исследовании должен быть доступным для анализа и комментирования координатору исследования и всем руководителям. Спонсор должен хранить записи, подтверждающие, что отчет о клиническом исследовании был предоставлен для анализа. Если рецензент не согласен со всем или с частью отчета о клиническом исследовании, его комментарии должны быть записаны и переданы другим руководителям;
e) если это необходимо согласно национальным регулирующим требованиям, спонсор и координатор должны подписывать отчет о клиническом исследовании, подтверждая свое согласие с его содержанием. Если координатор не назначен, необходимо получить подпись главного исследователя(ей);
f) в соответствии с применимыми требованиями отчет о клиническом исследовании должен быть предоставлен в ЭК и регулирующие органы.
Примечание 1 - Дальнейшие руководящие указания по содержанию отчета о клиническом исследовании приведены в приложении D.
Приветствуются публикации о положительных или отрицательных результатах клинического исследования для облегчения проведения более поздних исследований, разработки изделий и медицинской помощи.
Примечание 2 - В соответствии с национальными регулирующими требованиями может потребоваться внесение в общую базу данных намерения провести клиническое исследование, а также его результаты.
7.4 Хранение документов
Спонсор и главный исследователь должны хранить документы по клиническому исследованию, как того требуют применимые регулирующие требования. Они должны предпринимать меры по предотвращению случайного или преднамеренного уничтожения данных документов. Главный исследователь или спонсор может передавать обязанности по хранению документов другим лицам/сторонам в исследовательском центре или учреждении спонсора, при этом необходимо документально подтверждать данную передачу.
Примечание - Перечень основных документов по клиническому исследованию, хранящихся в файлах спонсора или исследовательского центра, приведен в приложении Е.
8 Ответственность спонсора
8.1 Обеспечение качества и управление качеством клинического исследования
К процессам клинического исследования должны применяться принципы обеспечения качества и управления качества. Спонсор должен:
a) вводить и поддерживать письменные процедуры, связанные с качеством, для обеспечения уверенности в том, что клиническое исследование разработано, проводится и контролируется, а данные генерируются, документируются, регистрируются и включаются в отчет в соответствии с настоящим стандартом, CIP, любыми последующими дополнениями и любыми другими применимыми стандартами и регулирующими требованиями;
b) поддерживать записи, документирующие соответствие всех сторон, включенных в клиническое исследование;
c) если применимо, обеспечивать выполнение требований к аудитам (см. 6.11), и
d) обосновывать и документировать существенные отклонения от требований настоящего стандарта.
Обеспечение качества и управление качеством клинического исследования могут быть интегрированы в общую систему качества спонсора.
Примечание - Для дополнительной информации см. [1].
8.2 Планирование и проведение клинического исследования
8.2.1 Выбор клинического персонала
До начала клинического исследования спонсор должен:
a) определить, установить и распределить все роли и ответственности, связанные с клиническим исследованием, в одном или более письменном соглашении, как определено в 5.9;
b) выбрать подходящего квалифицированного главного исследователя, как указано в 5.8 и 9.2;
c) выбрать координатора, если необходимо, как например, в случае исследования в нескольких центрах;
d) выявить конфликт интересов между главным исследователем и исследователями, если это требуется национальными регулирующими документами;
e) убедиться, что члены команды в исследовательском центре и их полномочные представители детально идентифицированы в регистрационном журнале, как определено в 6.2;
f) определить или назначить одного или более наблюдателей, или в противном случае принимать ответственность за мониторинг на себя и
g) обеспечить документальное подтверждение подготовки, опыта и научных или клинических знаний всех вовлеченных сторон для проведения клинического исследования надлежащим образом, включая подготовку:
1) по использованию исследуемого изделия(й);
2) процедурам учета изделий (см. 6.9);
3) IB;
4) CIP;
5) CRF и инструкциям по завершению;
6) письменной форме информированного согласия и процесса, а также другой письменной информации, предоставляемой субъекту, и
7) письменным процедурам спонсора, настоящему стандарту и любым применимым регулирующим требованиям;
h) убедиться, что при исследованиях в нескольких центрах все исследователи и все другие вовлеченные стороны получили инструкции по единообразной оценке и документированию клинических и лабораторных данных;
i) убедиться, что любые действия представителя(ей) спонсора в исследовательском центре, связанные с клиническим исследованием, описаны в CIP и форме информированного согласия и что эти действия выполняются таким образом, что они не влияют на целостность данных.
Примечание - Примерами представителей спонсора являются такие лица, как инженер по эксплуатации или торговый представитель, проводящие техническую экспертизу при реализации клинического исследования;
j) рассмотреть необходимость DMC и, если необходимо, создать комитет.
8.2.2 Подготовка документов и материалов
До начала клинического исследования спонсор должен:
a) подготовить документы, как описано в разделах 4, 5 и 6, и убедиться, что они одобрены соответствующими лицами с указанием даты и подписи; если необходимо, должны быть предоставлены копии всем вовлеченным сторонам и получены подписи с указанием даты;
b) обеспечить точность перевода, если необходимо;
c) убедиться, что в ходе клинического исследования поставка исследуемых изделий, как описано в 6.9, доступна в установленные сроки; исследуемые изделия не должны быть доступны главному исследователю до тех пор, пока все требования до начала клинического исследования не будут выполнены;
d) обеспечить страховку, покрывающую расходы на лечение субъектов в случае повреждений, связанных с клиническими исследованиями, в соответствии с национальными регулирующими требованиями, если необходимо;
e) документировать любые финансовые соглашения между главным исследователем или исследовательским центром и спонсором;
f) предоставить в соответствующие регулирующие органы любые необходимые для начала клинического исследования в конкретной стране документы для рассмотрения, принятия или одобрения (согласно применимым регулирующим требованиям);
g) убедиться, что получено и документировано одобрение/положительное решение ЭК и что приняты соответствующие положения для удовлетворения любых условий, выдвинутых ЭК, и
h) убедиться, что любые изменения, которые требуются ЭК или регулирующими органами, выполнены и документированы главным исследователем и в результате их получено одобрение/положительное решение ЭК или регулирующих органов.
8.2.3 Проведение клинического исследования
Спонсор несет ответственность:
a) за учет исследуемых изделий в ходе клинического исследования;
b) документирование связи со всеми сторонами, вовлеченными в клинические исследования, включая ЭК и регулирующие органы;
c) обеспечение того, что клинические исследования должным образом охвачены мониторингом посредством определения масштабов и характера мониторинга, включая стратегии верификации исходных данных на основании таких параметров, как цель, схема, сложность, объем, точки критических значений данных и ожидаемые результаты клинического исследования;
d) обзор отчета(ов) о мониторинге и выполнении любых действий, необходимых согласно отчету(ам) о мониторинге (см. также 8.2.4.7);
е) выполнение оперативных мер, связанных с полным соответствием всем требованиям к клиническому исследованию, и
f) предоставление отчетов о ходе клинического исследования, включая сводки о безопасности и отклонениях, по требованию всех участвующих в анализе ЭК и регулирующих органов.
8.2.4 Мониторинг
8.2.4.1 Общие положения
Целью мониторинга клинического исследования является проверка того, что проведение клинического исследования соответствует принятому CIP, последующим дополнениям, настоящему стандарту и применимым регулирующим требованиям.
8.2.4.2 Квалификация наблюдателей
Наблюдатели должны:
a) быть квалифицированными в области настоящего стандарта посредством подготовки и наличия опыта, а также научных или клинических знаний;
b) обладать знаниями по использованию исследуемого изделия и соответствующим требованиям, CIP и процессам информированного согласия (см. 4.7);
c) быть подготовленными по системам обеспечения и контроля качества спонсора, а также по любым специальным процедурам мониторинга конкретного клинического исследования.
Подготовка должна быть документирована в файлах спонсора.
8.2.4.3 Оценка исследовательского центра
Наблюдатель должен оценивать каждый исследовательский центр для подтверждения того, что главный исследователь:
а) имеет достаточную квалификацию;
b) обладает достаточными ресурсами, включая производственные помещения, лаборатории, оборудование и квалифицированную команду в исследовательском центре;
c) обладает правом доступа к достаточному числу субъектов.
8.2.4.4 Проверка исследовательского центра
Спонсор должен проверять каждый исследовательский центр для обеспечения того, что главный исследователь и команда исследовательского центра:
a) получили и понимают требования и содержание:
1) CIP;
2) IB;
3) форм информированного согласия;
4) CRF;
5) инструкций по эксплуатации;
6) любых письменных соглашений по клиническому исследованию;
b) имеют доступ к достаточному числу исследуемых изделий;
c) подготовлены по применению исследуемого изделия и
d) ознакомлены с ответственностью главного исследователя, как описано в разделе 9.
Примечание - В некоторых случаях вместо или одновременно с подготовительным визитом в исследовательский центр может быть проведена встреча исследователей.
8.2.4.5 Плановые визиты "на место" с целью мониторинга
Наблюдатель должен проводить плановые визиты "на место" с целью мониторинга того, что:
a) поддерживается соответствие CIP, любым последующим дополнениям, настоящему стандарту и регулирующим требованиям; отклонения должны быть согласованы с главным исследователем(ями) или полномочным представителем, их необходимо документировать и сообщить спонсору;
b) в клиническом исследовании принимают участие только уполномоченные лица, как описано в 8.2.1, перечисление е);
c) исследуемое изделие используется в соответствии с CIP или инструкциями по эксплуатации и что, если требуется изменение изделия, его метода применения или CIP, это сообщается спонсору;
d) ресурсы исследовательского центра, включая лаборатории, оборудование и команду исследовательского центра, остаются достаточными на протяжении всего клинического исследования;
e) главный исследователь имеет постоянный доступ к достаточному числу субъектов клинического исследования и исследуемых изделий;
f) от каждого субъекта клинического исследования в момент регистрации или до любых процедур, связанных с клиническим исследованием, получены подписанные, с указанием даты, формы информированного согласия;
g) исходная документация и другие записи клинического исследования точны, полны, актуальны, хранятся и поддерживаются надлежащим образом;
h) CRF и запросы заполнены, своевременно регистрируются и согласуются с исходными данными;
i) вносятся необходимые исправления, дополнения или удаления из CRF с указанием даты и объяснением, если необходимо, и визируются главным исследователем или его полномочным представителем; наблюдатель не должен вносить исправления, дополнения или удаления из CRF;
j) все нежелательные события и недостатки изделия сообщаются спонсору, а все серьезные неблагоприятные события и недостатки изделия, которые могут привести к серьезному неблагоприятному воздействию изделия, сообщаются спонсору без необоснованных задержек;
k) все серьезные неблагоприятные события и отклонения сообщаются в ЭК, если необходимо;
l) хранение и учет исследуемых изделий являются правильными, а также реализуются процессы, обеспечивающие прослеживаемость;
m) все другие необходимые отчеты, уведомления, приложения, заявки на рассмотрение и соглашения хранятся в файлах исследователя и являются точными, полными, своевременными, понятными, датированными и с идентификацией клинического исследования;
n) техническое обслуживание и калибровка оборудования, необходимые для проведения оценки в рамках клинического исследования, проводятся и документируются соответствующим образом, если применимо;
о) действующие нормы лаборатории, сертификация, аккредитация или другие подтверждения присутствуют в файле исследователя, если необходимо;
p) прекращение участия субъекта клинического исследования документировано; наблюдатель должен обсудить данный вопрос с главным исследователем или его полномочным представителем;
q) несоблюдение субъектом клинического исследования требований, приведенных в форме информированного согласия, документировано; наблюдатель должен обсудить данный вопрос с главным исследователем или его полномочным представителем;
r) главный исследователь и команда в исследовательском центре информированы и обладают достаточными знаниями о всех изменениях документов, касающихся клинического исследования, и
s) при необходимости осуществляются и являются эффективными любые корректирующие и предупреждающие действия.
8.2.4.6 Действия по завершению
Наблюдатель должен выполнять действия по завершению, описанные в разделе 7.
8.2.4.7 Отчеты о мониторинге
Все действия по мониторингу должны быть документированы в письменном отчете спонсору [см. также 8.2.3, перечисление d)], который должен включать:
a) дату, идентификацию исследовательского центра, фамилию и инициалы наблюдателя и главного исследователя или других лиц, с которыми устанавливается контакт, и
b) отчет о том, что проверял наблюдатель, и его наблюдения с учетом решения предыдущих вопросов, требующих принятия действий, значимые результаты, факты, отклонения, заключения и рекомендованные меры, которые необходимо предпринять для обеспечения соответствия.
Копия отчета о мониторинге или краткое изложение основных выводов должны быть доведены до сведения главного исследователя в письменном виде.
Примечание - Приведенные выше требования могут также применяться к сообщениям, связанным с клиническим исследованием, в зависимости от процедур спонсора или национальных регулирующих требований.
8.2.5 Оценка безопасности и отчет
Спонсор ответственен за классификацию неблагоприятных событий и постоянную оценку безопасности клинического исследования и должен:
a) анализировать проведенную исследователем оценку всех неблагоприятных событий, определять и документировать в письменном виде их тяжесть и связь с исследуемым изделием; в случае разногласий между спонсором и главным исследователем(ями), спонсор должен передать заинтересованным сторонам, определенным ниже в перечислениях с), d) и е), оба мнения;
b) анализировать все недостатки изделия, определять и письменно документально фиксировать, могут ли они привести к серьезному неблагоприятному воздействию изделия; в случае разногласий между спонсором и главным исследователем(ями), спонсор должен передать заинтересованным сторонам, определенным ниже в перечислениях с), d) и е), оба мнения;
c) сообщать или обеспечивать сообщение главным исследователем в ЭК обо всех серьезных неблагоприятных событиях и недостатках изделия, которые могут привести к серьезному неблагоприятному воздействию изделия, если это требуется национальными регулирующими требованиями, или CIP, или ЭК;
d) сообщать в регулирующие органы в течение определенного периода времени обо всех серьезных неблагоприятных событиях и недостатках изделия, которые могут привести к серьезному неблагоприятному воздействию изделия, если это требуется национальными регулирующими требованиями или CIP;
e) сообщать в DMC, если необходимо, всю информацию, относящуюся к безопасности, в соответствии с письменными процедурами;
f) в случае клинического исследования в нескольких центрах письменно информировать всех главных исследователей о всех серьезных неблагоприятных событиях во всех исследовательских центрах, о которых было сообщено спонсору, и обеспечивать сообщение о них в соответствующие ЭК, если это требуется национальными регулирующими требованиями, CIP или ЭК, в зависимости от того, чьи требования являются более строгими; эта информация должна быть разослана всем руководителям в течение периода времени, определенного на основании предполагаемого риска, определенного в отчете по анализу риска;
g) убедиться, что ЭК и регулирующие органы в курсе существенной новой информации о клиническом исследовании, и
h) в случае серьезного неблагоприятного воздействия изделия и недостатков изделия, которые могут привести к серьезному неблагоприятному воздействию изделия, определять, необходимо ли провести дополнительный анализ риска, и оценивать необходимость проведения корректирующих или предупреждающих действий.
8.2.6 Завершение клинического исследования
Спонсор должен:
a) обеспечивать, чтобы все действия по завершению клинического исследования были проведены надлежащим образом, как описано в разделе 7;
b) обеспечивать статистический анализ данных;
c) создавать отчет о клиническом исследовании и представлять его для анализа, как описано в 7.3, и
d) обеспечивать, чтобы отчет о клиническом исследовании, независимо от того, было ли клиническое исследование завершено или досрочно прекращено, был представлен в ЭК, принимавшим участие исследователям и в регулирующие органы согласно национальным регулирующим требованиям.
8.3 Аутсорсинг обязанностей и функций
Спонсор может передавать отдельные или все обязанности или функции, связанные с клиническим исследованием, включая мониторинг, внешней организации (например, CRO или индивидуальному подрядчику), однако окончательная ответственность за качество и полноту данных клинического исследования должна оставаться на спонсоре. Все требования настоящего стандарта, применяющиеся к спонсору, должны также применяться к внешней организации, поскольку эта организация берет на себя обязанности и функции спонсора, связанные с клиническим исследованием.
Спонсор должен письменно определять все обязанности и функции, связанные с клиническим исследованием, выполняемым внешней организацией, оставив за собой все обязанности и функции, связанные с клиническим исследованием, не предназначенные для передачи или выполнения внешней организацией.
Спонсор должен нести ответственность за верификацию наличия и точное соблюдение во внешних организациях письменных процедур.
8.4 Связь с регулирующими органами
Если требуется, спонсор должен:
a) уведомлять или получать одобрение от регулирующих органов той страны, в которой проводится клиническое исследование;
b) сообщать о ходе и статусе клинического исследования и
c) формировать отчет по безопасности, как определено в 8.2.5.
9 Ответственность главного исследователя
9.1 Общие положения
Задачами главного исследователя являются осуществление и ежедневный менеджмент проведения клинического исследования, а также обеспечение целостности данных и прав, безопасности и благополучия субъектов, вовлеченных в клиническое исследование.
Если спонсор заключает договор на проведение клинического исследования с учреждением, учреждение должно назначать в качестве главного исследователя лицо, обладающее достаточной квалификацией.
9.2 Квалификация главного исследователя
Главный исследователь должен:
a) быть квалифицированным на основе образования, подготовки и опыта для принятия ответственности за проведение клинического исследования надлежащим образом в соответствии с настоящим стандартом; доказательства в отношении такой квалификации главного исследователя и ключевых членов команды в исследовательском центре должны быть предоставлены спонсору в виде актуальных резюме или другой соответствующей документации;
b) иметь опыт в области применения и быть подготовленным по использованию рассматриваемого исследуемого изделия;
c) выявлять возможные конфликты интересов, включая финансовые, которые влияют на проведение клинического исследования или интерпретацию результатов, и
d) быть хорошо осведомленным о методах получения информированного согласия.
9.3 Квалификация исследовательского центра
Главный исследователь должен быть в состоянии продемонстрировать, что предполагаемый исследовательский центр:
a) имеет необходимое число правомочных субъектов, которые могут быть отобраны в согласованный промежуток времени, и
b) имеет одного или более квалифицированного исследователя, квалифицированную команду в исследовательском центре и достаточно оборудования на всю предусмотренную продолжительность клинического исследования.
9.4 Связь с этическим комитетом
Главный исследователь должен:
a) обеспечить спонсора копиями всех сообщений между главным исследователем и ЭК, связанных с клиническим исследованием;
b) выполнять все требования, описанные в 4.5;
c) получать письменное одобрение/положительное заключение ЭК по клиническому исследованию с указанием даты до набора субъектов и внесения любых последующих поправок при необходимости;
d) предоставлять отчетность по безопасности, как определено в 9.8, и
e) своевременно сообщать о любых отклонениях от CIP, которые влияют на права, безопасность или благополучие субъектов или научную целостность клинического исследования, включая те отклонения, которые возникают в чрезвычайных ситуациях, если это требуется ЭК, CIP или национальными регулирующими требованиями.
В определенных обстоятельствах связь с ЭК частично или полностью может осуществлять спонсор, и в этом случае спонсор должен информировать главного исследователя.
9.5 Процесс получения информированного согласия
Главный исследователь должен:
a) выполнять требования, определенные в 4.7;
b) обеспечивать выполнение применимых регулирующих требований и этических принципов к процессу получения информированного согласия и
c) обеспечивать и документировать соответствующую подготовку, если для проведения процесса получения информированного согласия назначается полномочный представитель.
9.6 Соответствие CIP
Главный исследователь должен:
a) письменно указывать свое согласие с CIP;
b) проводить клиническое исследование в соответствии с CIP;
c) создавать и поддерживать исходную документацию в течение всего клинического исследования и делать ее доступной по запросу во время визитов в целях мониторинга или аудита;
d) обеспечить использование исследуемого изделия только авторизованными пользователями, как определено в 6.2, и в соответствии с CIP и инструкциями по эксплуатации исследуемого изделия;
e) предлагать спонсору любые необходимые изменения CIP, или исследуемого изделия, или применения исследуемого изделия;
f) воздерживаться от внесения поправок в CIP без получения согласия спонсора, ЭК и регулирующих органов, если необходимо;
g) документировать и объяснять любые отклонения от одобренного CIP, которые возникают во время проведения клинического исследования;
h) обеспечивать наличие, поддержание и документирование компетентной команды и необходимого оборудования в исследовательском центре во время клинического исследования;
i) обеспечивать актуальность обслуживания и калибровки оборудования для оценки клинического исследования, достаточность проведения и документирования клинического исследования, если это применимо;
j) обеспечивать точность, полноту, разборчивость и своевременность данных, сообщаемых спонсору в CRF и во всех необходимых отчетах;
k) поддерживать записи по учету изделий;
l) допускать и поддерживать проведение спонсором мониторинга и аудита;
m) быть доступным наблюдателю и отвечать на вопросы во время визитов в целях мониторинга;
n) допускать и поддерживать проведение аудита регулирующими органами и ЭК;
o) обеспечивать сохранение всех записей, касающихся клинического исследования, как это определено в 7.4, и
р) подписывать отчет о клиническом исследовании, как определено в 7.3.
9.7 Медицинское обслуживание субъектов клинических исследований
Главный исследователь должен:
a) обеспечивать достаточное медицинское обслуживание субъектов клинического исследования во время и после участия субъекта клинического исследования в клиническом исследовании в случае неблагоприятных событий, как описано в информированном согласии [см. 4.7.4, перечисление f)];
b) информировать субъекта клинического исследования о характере и возможных причинах любых неблагоприятных событий;
с) обеспечивать субъекта клинического исследования необходимыми инструкциями по правильному использованию, обслуживанию, хранению и возврату исследуемого изделия, если оно используется или управляется субъектом;
d) информировать субъекта клинического исследования о любых существенных результатах исследования, возникающих во время клинического исследования, включая необходимость дополнительного медицинского обслуживания, которое может потребоваться;
e) предоставлять субъекту клинического исследования четко определенный порядок действий в случае возможных аварийных ситуаций, связанных с клиническим исследованием, и давать необходимые указания по проведению неотложной помощи, включая процедуры декодирования в случае проведения клинического исследования с использованием слепого метода/маскирования;
f) обеспечивать понятное маркирование клинических записей, указывающее на то, что субъект клинического исследования включен в конкретное клиническое исследование;
g) если необходимо, субъектов, включенных в клиническое исследование, следует снабжать определенными средствами, указывающими на их участие в клиническом исследовании, совместно с идентификацией и информацией по соблюдению мер сопутствующего лечения (должны быть указаны номер телефона и адрес);
h) информировать с согласия пациента или в соответствии с нормами национальных регулирующих требований личного врача субъекта клинического исследования о том, что субъект клинического исследования принимает участие в клиническом исследовании, и
i) делать все возможное для определения причин(ы) досрочного выхода субъекта клинического исследования из клинического исследования с полным соблюдением прав субъекта.
9.8 Отчетность по безопасности
Главный исследователь должен:
a) записывать все неблагоприятные события и наблюдаемые недостатки изделия, а также их оценку;
b) сообщать спонсору без необоснованных задержек о всех серьезных неблагоприятных событиях и недостатках изделия, которые могут привести к серьезному неблагоприятному воздействию; данная информация должна быть оперативной и снабжаться подробным письменным отчетом, как определено в CIP;
c) сообщать в ЭК о всех серьезных неблагоприятных событиях и недостатках изделия, которые могут привести к серьезному неблагоприятному воздействию, если это необходимо согласно регулирующим требованиям, или CIP, или ЭК;
d) сообщать в регулирующие органы о всех серьезных неблагоприятных событиях и недостатках изделия, которые могут привести к серьезному неблагоприятному воздействию, в соответствии с национальными регулирующими требованиями, и
е) снабжать спонсора по его запросу любой дополнительной информацией, связанной с отчетом по безопасности по конкретному событию.
Приложение А
(обязательное)
План клинического исследования
А.1 Общие положения
А.1.1 Введение
В настоящем приложении установлено содержание CIP. Если необходимая информация записана в другой документации, например, в IВ, то такая документация должна быть доступна по запросу и на нее должна быть приведена ссылка в CIP.
CIP и любые последующие дополнения должны включать все пункты, перечисленные в настоящем приложении и, если это нуждается в разъяснении, с расшифровкой каждого пункта.
А.1.2 Идентификация плана клинических исследований
a) наименование клинического исследования;
b) идентификационный номер, определяющий конкретное клиническое исследование, если он есть;
c) версия и дата подписания CIP;
d) сводные данные об истории изменений, в случае внесения поправок;
e) номер версии/выпуска и идентификационный номер, если он есть, а также номер страницы и общее число страниц на каждой странице CIP.
А.1.3 Спонсор
Имя и адрес спонсора клинических исследований.
Примечание - Если спонсор не проживает постоянно в стране (странах), в которой проводится клиническое исследование, то в соответствии с национальным или региональным регулированием может потребоваться указание имени и адреса его представителя в данной стране (странах).
А.1.4 Главный исследователь, координатор и исследовательский центр(ы)
a) фамилия, инициалы, адрес и занимаемая должность:
1) главного исследователя (исследователей);
2) координатора, если назначен;
b) наименование и адрес исследовательского центра (центров), в котором проводятся клинические исследования;
c) наименование(я) и адрес(а) других институтов, вовлеченных в клинические исследования.
Спонсор должен поддерживать актуальный список руководителей, исследовательских центров и институтов. Этот список может храниться отдельно от CIP. Окончательный список должен быть предоставлен вместе с отчетом о клиническом исследовании (см. приложение D).
А.1.5 Общее краткое описание клинических исследований
Сводка или обзор клинических исследований должны включать всю необходимую информацию, касающуюся деталей проекта клинического исследования, такую как критерии включения/исключения, число субъектов, продолжительность клинического исследования, наблюдение, цель(и) и конечная точка(и).
Примечание - Может быть полезно включить блок-схему, показывающую основные этапы клинических исследований, или любую другую информацию, которая может оказаться полезной для проведения клинического исследования.
А.2 Идентификация и описание исследуемого изделия
a) общее описание исследуемого изделия и его предусмотренного назначения;
b) подробные сведения об изготовителе исследуемого изделия;
c) наименование и номер типа или модели, включая, если применимо, версию программного обеспечения и принадлежности, для выполнения полной идентификации;
d) описание того, как достигается прослеживаемость во время и после клинического исследования, например, с помощью присвоения номера серии, номера партии или заводских серийных номеров;
e) предполагаемое использование исследуемого изделия в планируемом клиническом исследовании;
f) популяции пациентов и медицинские показания, для которых предназначено исследуемое изделие;
g) описание исследуемого изделия, включая все материалы, которые будут контактировать с тканями или жидкостями организма. (Описание должно включать подробную информацию о любых лекарственных средствах, тканях человека или животных или их производных, а также другие биологически активные вещества);
h) сводка об обучении и опыте, необходимом для использования исследуемого изделия;
i) описание специальных хирургических и других медицинских процедур, связанных с использованием исследуемого изделия.
А.3 Обоснование плана (схемы) клинического исследования
Обоснование плана (схемы) клинического исследования, которое должно быть основано на результатах проведенного оценивания, как определено в 5.3, должно включать:
a) оценивание результатов соответствующих доклинических испытаний/оценок, проводимых для обоснования использования исследуемого изделия с участием человека в качестве субъекта;
b) оценивание клинических данных, соответствующих предполагаемому клиническому исследованию.
А.4 Риски и польза от исследуемого изделия и клиническое исследование
a) ожидаемая клиническая польза;
b) ожидаемое неблагоприятное воздействие изделия;
c) остаточные риски, связанные с исследуемым изделием, которые идентифицированы в отчете по анализу риска;
d) риски, связанные с участием в клиническом исследовании;
e) возможное взаимное влияние сопутствующих лечебно-диагностических процедур;
f) действия, которые будут предприняты для управления или уменьшения рисков;
g) обоснование соотношения риска и пользы.
Примечание - Процесс менеджмента риска, включающий в себя анализ риска, оценку соотношения риска и пользы, а также управление риском, описан в ИСО 14971.
А.5 Цели и гипотезы клинического исследования
a) цели, основные и дополнительные;
b) гипотезы, основные и дополнительные, которые принимаются или отклоняются на основании статистического анализа данных, полученных в ходе клинического исследования;
c) заявленные свойства и предусмотренные функциональные характеристики, которые подлежат проверке;
d) риски и предполагаемые неблагоприятные воздействия изделия, которые должны быть оценены.
А.6 Проект (схема) клинических исследований
А.6.1 Общие положения
a) описание типа проводимого клинического исследования (например, двойное слепое сравнение, параллельное исследование с или без группы сравнения) с обоснованием выбора;
b) описание мер, направленных на минимизацию или исключение предвзятости и необъективности, включая рандомизацию и использование слепого метода/маскирования;
c) основные и дополнительные конечные точки с обоснованием их выбора и измерения;
d) методы и временные моменты оценки, записи и анализа переменных;
е) оборудование, используемое для оценки переменных клинического исследования, включая порядок обслуживания и калибровки;
f) любые процедуры по замене субъектов.
А.6.2 Исследуемое изделие(я) и изделие(я) для сравнения
a) описание воздействий на исследуемое изделие(я) или изделие(я) для сравнения, если используется;
b) обоснование выбора изделия(й) для сравнения;
c) список всех других медицинских изделий или лекарств, используемых во время клинического исследования;
d) число используемых исследуемых изделий, с обоснованием.
А.6.3 Субъекты клинических исследований
a) критерии включения при выборе субъектов;
b) критерии исключения при выборе субъектов;
c) критерии и процедуры прекращения или приостановки участия субъектов;
d) момент регистрации;
e) общая ожидаемая продолжительность клинического исследования;
f) ожидаемая продолжительность участия каждого субъекта;
g) число субъектов, которых необходимо включить в клиническое исследование;
h) расчетное время, необходимое для набора данного числа субъектов (период регистрации).
А.6.4 Процедуры
a) описание всех процедур, связанных с клиническим исследованием, которым подвергаются субъекты;
b) описание действий, выполняемых представителями спонсора (исключая мониторинг);
c) любые известные или предполагаемые факторы, которые могут ухудшить результаты клинических исследований или отрицательно повлиять на интерпретацию результатов.
Пример - Факторы включают основные характеристики субъекта, сопутствующее лечение, использование других медицинских изделий и субъективные факторы, такие как возраст, пол или образ жизни. Должны быть описаны методы учета этих факторов в ходе клинического исследования, например, с помощью выбора субъектов, использования определенной схемы клинических исследований (например, сортирующая рандомизация) или статистического анализа.
Период наблюдения в ходе клинического исследования должен обеспечивать демонстрацию функциональных характеристик в течение времени, достаточного для обеспечения достоверности испытаний функциональных характеристик исследуемого изделия, а также обеспечения идентификации и оценки за этот период любых рисков, связанных с неблагоприятным воздействием изделия.
В CIP должно быть четко указано, какой медицинский уход, если он необходим, должен быть предоставлен субъекту клинических исследований после завершения клинических исследований.
А.6.5 План мониторинга
Общая схема плана мониторинга, которому необходимо следовать, включая доступ к первичным данным и глубину планируемой проверки первичных данных.
Примечание - Допускается предоставлять подробный план схемы мониторинга отдельно от CIP.
А.7 Статистические аспекты
С учетом А.5 и А.6, описание и обоснование:
a) статистической схемы, метода и аналитических процедур;
b) размера выборки;
c) уровня значимости и статистической достоверности клинического исследования;
d) ожидаемой доли выбывания;
e) критериев отказа/одобрения, применяемых к результатам клинических исследований;
f) указаний по промежуточному анализу, если применимо;
g) критериев завершения клинического исследования на основании статистических параметров;
h) процедур отчета о любых отклонениях от исходного статистического плана;
i) подробностей определения подгрупп для анализа;
j) процедур, учитывающих все данные;
k) обработки недостающих, неиспользуемых или ложных данных, включая данные от выбывших и исключенных субъектов;
l) исключения определенной информации при проверке гипотезы, если необходимо;
m) минимальное и максимальное число субъектов, включенных в исследование в каждом центре, в случае, если клинические исследования проводятся в нескольких центрах.
Для ранних клинических исследований, например клинических исследований для проверки работоспособности, могут быть применимы особые обоснования и размер(ы) выборки.
А.8 Управление данными
a) процедуры, используемые для анализа данных, исключения из баз данных, а также рассмотрения и принятия решения по сомнительным данным;
b) процедуры проверки, валидации и обеспечения безопасности электронных клинических систем управления данными, если применимо;
c) процедуры хранения данных;
d) установленный срок хранения;
е) другие вопросы обеспечения качества в отношении клинических аспектов, если применимо.
А.9 Дополнения к CIP
Описание процедур внесения дополнений в CIP.
А.10 Отклонения от плана клинического исследования
a) заявление, что исследователь не может отклоняться от CIP, за исключением случаев, указанных в 4.5.4, перечисление b);
b) процедуры для записей, создания отчетов и анализа отклонений от CIP;
c) требования к уведомлениям и временные рамки;
d) корректирующие и предупреждающие действия, а также критерии дисквалификации главного исследователя.
А.11 Учет изделий
Описание процедур учета исследуемых изделий, как установлено в 6.9.
А.12 Заявление о соответствии
a) заявление, что клиническое исследование должно быть проведено в соответствии с этическими принципами Хельсинкской Декларации (см. [8]);
b) заявление, подтверждающее соответствие настоящему стандарту, а также любым региональным или национальным регулирующим документам, если необходимо;
c) заявление, что клинические исследования не должны начинаться до получения необходимого одобрения/положительного решения ЭК или регулирующих органов власти, если необходимо;
d) заявление, что должны выполняться любые дополнительные требования, наложенные ЭК или регулирующими органами власти, если необходимо;
e) заявление, определяющее тип страховых выплат, получаемых субъектом, если необходимо.
А.13 Процесс получения информированного согласия
a) описание основного процесса получения информированного согласия, включая процессы предоставления субъектам новой информации, если необходимо;
b) описание процесса получения информированного согласия, если субъект клинических исследований не может его дать; в случае чрезвычайных ситуаций, должны быть включены пункты, перечисленные в 4.7.3.4.
А.14 Неблагоприятные события, неблагоприятное воздействие изделия и недостатки изделия
a) четкие определения неблагоприятных событий и неблагоприятного воздействия изделия;
b) четкое определение недостатков изделия;
c) четкие определения серьезных неблагоприятных событий и серьезного неблагоприятного воздействия изделия и, если применимо, не предполагаемого серьезного неблагоприятного воздействия изделия;
d) период времени, в течение которого главный исследователь должен сообщить о всех неблагоприятных событиях и недостатках изделия спонсору и, если применимо, в ЭК и регулирующие органы власти;
e) детали процесса для создания отчета о неблагоприятных событиях (дата неблагоприятного события, проводимые действия, решение проблемы, оценка тяжести происшествия и взаимосвязи с исследуемым изделием);
f) детали процесса для создания отчета о недостатках изделия;
g) список предполагаемых неблагоприятных событий и предполагаемого неблагоприятного воздействия изделия, а также их вероятный масштаб, способы уменьшения последствий или необходимые действия;
h) сведения о лице, с которым следует контактировать в случае чрезвычайных ситуаций, для сообщения о серьезных неблагоприятных событиях и серьезном неблагоприятном воздействии изделия;
i) информация, касающаяся DMC, если он установлен.
А.15 Уязвимые популяции
a) описание уязвимой популяции;
b) описание особого процесса получения информированного согласия;
c) описание особой ответственности ЭК;
d) описание того, какое медицинское обслуживание будет предоставлено субъектам после завершения клинических исследований, если предусмотрено.
А.16 Приостановка или досрочное завершение клинических исследований
a) критерии и схема приостановки или досрочного завершения всего клинического исследования или клинического исследования в одном или более исследовательском центре;
b) критерии доступа или нарушения кодирования использования слепого метода/маскирования в случае приостановки или досрочного завершения клинического исследования, если клиническое исследование включает использование слепого метода/маскирования;
c) требования к наблюдению за субъектом.
А.17 Принципы опубликования
a) заявление, указывающее, будут ли результаты клинического исследования представлены для опубликования;
b) заявление, в котором приведены условия, при которых результаты клинического исследования будут представлены для опубликования.
А.18 Библиография
Список библиографических ссылок на источники, касающиеся клинического исследования.
Приложение В
Журнал (брошюра) исследователя
В.1 Общие положения
В.1.1 Введение
Если необходимая для IB информация записана в других документах (например, в CIP или в инструкции по эксплуатации), то такие документы должны быть доступны по запросу и на них должна быть приведена ссылка в IB.
IВ, как минимум, должен содержать все пункты, перечисленные в настоящем приложении.
В.1.2 Идентификация IB
a) наименование исследуемого изделия;
b) идентификационный номер документа, если он есть;
c) версия и дата подписания IB;
d) заявление о конфиденциальности, если необходимо;
e) сводка о пересмотрах в случае внесения изменений, если применимо;
f) номер версии/выпуска и идентификационный номер, если он есть, а также номер страницы и общее число страниц на каждой странице IB.
В.1.3 Спонсор/изготовитель
Наименование и адрес спонсора исследования или изготовителя исследуемого изделия.
В.2 Информация об исследуемом изделии
a) сводка литературных источников и оценивание, обосновывающее выбор конструкции (проекта) и предусмотренного назначения исследуемого изделия;
b) заявление в отношении классификации исследуемого изделия, которая принята регулирующими органами, если необходимо;
c) общее описание исследуемого изделия и его компонентов, включая используемые материалы;
d) перечень производственных процессов и связанных валидированных процессов;
e) описание механизма действия исследуемого изделия со ссылками на уместную научную литературу;
f) инструкции производителя по установке и эксплуатации исследуемого изделия, включая любые необходимые требования по уходу и хранению, подготовке к использованию и любому предполагаемому повторному применению (например, стерилизация), любые проверки безопасности или функциональных характеристик перед использованием и любые меры предосторожности, которые необходимо принимать после использования (например, утилизация), если применимо;
g) описание предусмотренных клинических функциональных характеристик.
В.3 Доклинические испытания
Сводная информация о доклинических испытаниях, которые должны быть проведены на исследуемом изделии, включая оценивание результатов этих испытаний, обосновывающая применения его к человеку как к субъекту.
Сводная информация должна содержать или ссылаться на результаты:
a) расчетов конструкции проекта изделия;
b) испытаний in vitro;
c) механических и электрических испытаний;
d) испытаний надежности;
е) валидации программного обеспечения, связанного с функционированием изделия;
f) всех испытаний в отношении функциональных характеристик;
g) испытаний ex vivo;
h) оценивания биологической безопасности.
Примечание - Руководство по биологическому оцениванию медицинских изделий приведено в [2].
В.4 Существующие клинические данные
a) сводная информация предшествующего уместного опыта клинического использования исследуемого изделия, а также медицинских изделий, имеющих аналогичные характеристики, включая те характеристики, которые относятся к другим показаниям к использованию исследуемого изделия;
b) анализ неблагоприятного воздействия изделия, а также вся история изменений или отзывов.
В.5 Менеджмент риска
а) сводная информация об анализе риска, включая идентификацию остаточных рисков;
b) результаты оценки риска;
c) ожидаемые риски, противопоказания, предупреждения и т.д., касающиеся исследуемого изделия.
В.6 Ссылки на регулирующие и иные требования
a) перечень международных стандартов, которые были полностью или частично использованы, если они применялись;
b) заявление о соответствии национальному регулированию, если применимо;
c) перечень ссылок, если уместно.
Приложение С
(справочное)
Индивидуальная регистрационная карта
С.1 Общие положения
CRF вводятся для выполнения CIP, облегчения наблюдения за пациентом, а также для записи данных о субъекте клинического исследования и исследуемом изделии в течение клинического исследования в соответствии с CIP. Они могут существовать в виде печатных документов, документов на оптическом или электронном носителе и могут быть отдельными для каждого субъекта. CRF должны отражать CIP, а также учитывать характер исследуемого изделия.
С.2 Содержание и формат
С.2.1 Общие положения
CRF могут быть составлены так, чтобы отражать все данные отдельной процедуры или отдельного визита, или иметь другую структуру, удобную с клинической точки зрения или с точки зрения хронологии.
Формат CRF должен быть разработан таким образом, чтобы минимизировать ошибки, которые может допустить лицо, вносящее данные и переносящее данные в другие системы.
Категории данных и их формат, перечисленные в настоящем приложении, должны быть учтены при разработке CRF.
С.2.2 Титульный лист/экран при входе в систему
a) фамилия, инициалы спонсора или логотип спонсора;
b) версия и дата подписания CIP (если требуется);
c) номер версии CRF;
d) наименование клинического исследования или идентификационный номер (если применимо).
С.2.3 Верхний или нижний колонтитул CRF
a) наименование клинического исследования или идентификационный номер;
b) номер версии CRF;
c) идентификационный номер исследовательского центра/руководителя;
d) идентификационный номер субъекта клинического исследования и дополнительная идентификация, такая как дата рождения или инициалы, если это допускается национальным регулированием;
e) номер CRF, или дата визита, или номер визита;
f) номер страницы/экрана и общее число страниц/экранов (например, "страница N из nn").
Примечание - С целью избегания многократного введения, возможно заранее напечатать или заранее запрограммировать некоторые из элементов, перечисленных выше.
С.2.4 Типы CRF
Ниже предлагается перечень случаев, для которых могут быть разработаны CRF с целью обеспечения клинического исследования. Данный перечень не является исчерпывающим и предназначен для использования в качестве руководства.
a) скрининг;
b) документальное подтверждение информированного согласия субъекта;
c) включение/исключение;
d) первичный визит:
1) демографические данные,
2) медицинский диагноз,
3) связанные предыдущие медикаментозное лечение или процедуры,
4) дата включения,
5) другие характеристики;
e) вмешательство(а) или лечебно-диагностические процедуры;
f) последующий визит(ы);
g) процедуры в рамках клинического исследования;
h) неблагоприятное событие(я);
i) недостатки изделия;
j) сопутствующее(ие) заболевание(я)/медикаментозное лечение;
k) незапланированный(е) визит(ы);
l) дневник субъекта клинического исследования;
m) исключение субъекта клинического исследования или потеря возможности наблюдения;
n) форма, означающая завершение клинического исследования, подписанная главным исследователем или полномочным представителем главного исследователя;
о) отклонения от CIP.
С.3 Процедурные особенности
Должна быть установлена система, позволяющая давать перекрестные ссылки на версии CRF и CIP.
При проведении исследований в нескольких центрах могут быть разработаны дополнительные CRF для сбора данных в конкретном исследовательском центре.
Приложение D
(справочное)
Отчет о клиническом исследовании
D.1 Общие положения
В настоящем приложении установлено содержание отчета о клиническом исследовании, в котором описывается его проект, исполнение, статистический анализ и результаты клинического исследования.
Приведенный формат может быть использован для промежуточных, ежегодных или окончательных отчетов, если требуется формировать эти отчеты.
D.2 Титульный лист
Титульный лист должен содержать следующую информацию:
a) наименование клинического исследования;
b) идентификацию исследуемого изделия, включая его наименование, модель и т.д., как это необходимо для полной идентификации;
c) если не понятно из наименования исследования, одно предложение, описывающее проект исследования, сравнение, период, метод проведения и популяцию субъектов;
d) фамилию, инициалы и контактную информацию спонсора или представителя спонсора;
e) идентификацию CIP;
f) фамилию, инициалы и отдел координатора, а также других заинтересованных лиц, например, экспертов, биостатистиков, персонала лабораторий;
g) заявление с указанием, проводится ли клиническое исследование в соответствии с настоящим стандартом или любыми другими применимыми руководствами и применимыми регулирующими документами;
h) дату составления отчета;
i) автора(ов) отчета.
D.3 Содержание
Содержание должно включать следующую информацию:
a) номер страницы или информацию о расположении каждого раздела, включая сводку таблиц, рисунков и графиков;
b) список приложений с указанием их расположения.
D.4 Аннотация
Аннотация должна содержать следующие пункты:
a) наименование клинического исследования;
b) введение;
c) цель клинического исследования;
d) описание популяции субъектов клинического исследования;
е) используемый в клиническом исследовании метод;
f) результаты клинического исследования;
g) выводы;
h) дату начала клинического исследования;
i) дату завершения клинического исследования или, если клиническое исследование прервано, дату досрочного завершения.
D.5 Введение
Введение должно содержать краткое указание на места проведения клинических исследований в контексте разработки исследования изделия с указанием критических для данной разработки параметров клинических исследований (например, цели и гипотезы, целевая популяция, процедуры и продолжительность наблюдения).
Должны быть определены и описаны любые руководства, которые выполнялись при разработке CIP, или любые другие соглашения, или результаты совещаний между спонсором и регулирующими органами власти, ответственными за конкретные клинические исследования.
D.6 Исследуемое изделие и методы
D.6.1 Описание исследуемого изделия
Описание исследуемого изделия должно включать следующие пункты:
a) описание исследуемого изделия;
b) предусмотренное назначение исследуемого изделия(й);
c) предыдущее предусмотренное назначение или показания к применению, если есть;
d) любые изменения исследуемого изделия во время клинического исследования или любые изменения относительно IВ, включая:
1) сырье и материалы,
2) программное обеспечение,
3) компоненты,
4) срок годности,
5) условия хранения,
6) руководство по эксплуатации/применению,
7) другие изменения.
D.6.2 План клинического исследования
Должен быть приведен обзор CIP, включая все последующие дополнения с обоснованием каждого дополнения. Обзор должен включать краткое описание следующих пунктов:
a) цели клинического исследования;
b) проект клинического исследования, включая:
1) тип клинического исследования,
2) конечные точки клинического исследования;
c) этические аспекты;
d) данные по обеспечению качества;
e) популяция субъектов клинического исследования, включая:
1) критерии включения/исключения,
2) объем выборки;
f) процедуры и перечень центров, в которых проводятся процедуры;
g) любые сопутствующие процедуры/медикаментозное лечение;
h) продолжительность наблюдения;
i) статистический анализ, включая:
1) гипотезу клинического исследования или критерии принятия/отклонения,
2) расчет объема выборки,
3) методы статистического анализа.
D.7 Результаты
Результаты отчета должны включать следующие пункты:
a) дата начала клинического исследования;
b) дата завершения или приостановки клинического исследования;
c) местонахождение субъектов и исследуемых изделий;
d) демографические данные субъектов;
е) соответствие CIP;
f) анализ, который включает:
1) анализ функциональных характеристик, предусмотренный в СIР;
2) обзор всех неблагоприятных событий и неблагоприятных воздействий изделия, включая обсуждение тяжести, требуемого обращения, решений и соответствующих заключений главного исследователя, касающееся причинно-следственной связи с исследуемым изделием или процедурой;
3) таблицу, в которой указаны все наблюдаемые недостатки изделия, которые могут привести к серьезному неблагоприятному воздействию изделия, а также любые корректирующие действия, предпринятые во время клинического исследования, если они есть;
4) любой необходимый анализ подгрупп в отношении особых популяций (т.е. подгрупп по половой принадлежности, расовых/культурных/этнических подгрупп), если применимо;
5) подотчетность всех субъектов с описанием того, как учитывается в анализе отсутствие данных или отклонения, включая субъектов:
- не прошедших скрининговое обследование,
- вышедших из-под наблюдения,
- исключенных из клинического исследования или в отношении которых участие в клиническом исследовании приостановлено с указанием причин.
D.8 Обсуждение и общие выводы
Выводы должны включать следующие пункты:
a) результаты оценки безопасности или функциональных характеристик, или любые другие конечные точки;
b) оценка рисков и пользы;
c) обсуждение клинической уместности и важности результатов в свете других существующих данных;
d) любая особая польза или особые меры предосторожности, необходимые для отдельных субъектов или групп, считающихся находящимися в зоне риска;
e) любые предпосылки к проведению дальнейших клинических исследований;
f) любые ограничения клинического исследования.
D.9 Аббревиатуры и определения
Должен быть приведен перечень аббревиатур и определений специальных или необычных терминов.
D.10 Этические аспекты
Отчет по этике должен включать следующие пункты:
a) подтверждение того, что CIP и любые дополнения к нему были рассмотрены ЭК (если требуется);
b) перечень всех ЭК, с которыми проводились консультации (может быть приведен в приложении; см. D.13).
D.11 Исследователи и исполнительная структура клинического исследования
Обзор исполнительной структуры должен включать следующие пункты:
a) краткое описание организации клинического исследования;
b) список исследователей, включая их принадлежность к определенной структуре (может быть приведен в приложении; см. D.13);
c) наименование и адрес любых третьих сторон (таких как, центральные лаборатории, CRO, консультанты или другие подрядчики), вносящие свой вклад в клиническое исследование (могут быть приведены в приложении; см. D.13);
d) фамилия, инициалы и адрес спонсора или представителя(ей) спонсора.
D.12 Лист подписей
Должны присутствовать личные подписи спонсора и координатора, подтверждающие их согласие с содержанием отчета. Если координатор не назначен, должна быть получена подпись главного исследователя. Лист подписей может быть отдельным от отчета о клиническом исследовании документом.
D.13 Приложения к отчету
В отчет могут быть включены приложения, содержащие следующую информацию:
a) CIP, включая дополнения;
b) руководство по эксплуатации;
c) список главных исследователей и исследовательских центров, в которых они работают, включая обзор их квалификации или копии их CV;
d) список наименований и адресов третьих сторон (таких как, центральные лаборатории, CRO, консультанты или другие подрядчики), вносящих свой вклад в клиническое исследование;
e) список наблюдателей;
f) список ЭК;
g) таблицы с любыми необходимыми наборами данных, включая:
1) отклонения от CIP, которые могут повлиять на права, безопасность или благополучие субъекта клинического исследования или научную целостность клинических исследований,
2) все неблагоприятные события и неблагоприятные воздействия изделия,
3) исключения и приостановки участия;
h) сертификат прохождения аудита, если применимо.
Приложение Е
(справочное)
Основные документы клинического исследования
Национальные регулирующие органы власти могут требовать от исследовательских центров и спонсора поддерживать документы, список которых приведен в таблицах Е.1, Е.2 и Е.3. Информация, приведенная ниже, может отличаться при различных клинических исследованиях.
Таблица Е.1 - Основные документы клинического исследования до проведения клинического исследования
N | Заголовок документа | Назначение или комментарии | Файлы центра | Файлы спонсора | Ссылка в настоящем стандарте |
Е.1.1 | Брошюра исследователя | Описывается исследуемое изделие, включая инструкции по использованию изделия | X | X | 5.5, приложение В |
Е.1.2 | План клинического исследования | Описываются проект и процедуры клинического исследования | X | X | 5.4, приложение А |
Е.1.3 | Пример маркирования, нанесенного на исследуемое изделие | Утверждает соответствующую маркировку | - | X | 5.10 |
Е.1.4 | CV руководителя: актуальная (последняя), подписанная, с указанием даты | Указывается руководитель. В центре хранятся CV руководителей данного центра; спонсор имеет CV руководителей из всех исследовательских центров | X | X | 4.5.2, перечисление е), 9.2, перечисление а), D.13, перечисление с) |
Е.1.5 | CV ключевых членов команды в исследовательских центрах, актуальные (последние), подписанные, с указанием даты | Указываются ключевые члены команды в исследовательских центрах. В центрах хранятся CV ключевых членов команды в данном исследовательском центре | X | X | 9.2, перечисление а) |
Е.1.6 | CV или другая документация, подтверждающая квалификацию лиц, отличных от приведенных в Е.1.4 и Е.1.5, вносящих существенный вклад в клинические исследования | Документально подтверждается квалификация всех других сторон, вовлеченных в клинические исследования | - | X | 5.1, 8.2.1, 8.2.4.3 |
Е.1.7 | Журнал (брошюра) главного исследователя и ключевых членов команды в исследовательском центре | Документально подтверждается наделение ответственностью с подписью, указанием полномочий и ответственности в рамках клинических исследований | X | X | 6.2, 8.2.1, перечисление е), 8.2.4.5, перечисление b) |
Е.1.8 | Перечень исследовательских центров | Данные о том, кто проводит клинические исследования с указанием имени и адреса | - | X | А.1.4 |
Е.1.9 | Уведомление ЭК, связь и | Предоставляются свидетельства того, что квалифицированный независимый ЭК рассмотрел данные клинического исследования | X | X | 4.5.3, 6.1, 8.2.2, перечисление g), 9.4 |
Е.1.10 | Лист голосования ЭК по клиническим исследованиям | Предоставляются свидетельства того, что исследователь не участвует в голосовании (зависит от регулирующих требований) | X | X | 4.5.3 |
Е.1.11 | Уведомление регулирующих органов, связь и одобрение (если требуется) | Проверяется информация, предоставляемая регулирующим органам. Подтверждается уведомление или одобрение | Х | X | 6.1, 8.2.2, перечисление f) |
Е.1.12 | Подписанное соглашение между руководителем(ями)/ исследовательским цент- ром(ами) и организатором | Демонстрируется понимание ответственности, которая возложена, соответственно, на каждую из сторон | X | X | 5.9, 8.2.1, перечисление а), 8.2.2, перечисление е) |
Е.1.13 | Подписанное соглашение между спонсором и третьими сторонами, например, CRO, центральными лабораториями | Демонстрируется понимание ответственности, которая возложена на каждую из сторон | - | X | 5.9, 8.2.1, перечисление а) |
Е.1.14 | Финансовые соглашения, если они являются отдельным от соглашений по распределению ответственности документом | Предоставляются свидетельства достижения финансовых соглашений между исследователем/ исследовательскими центрами и спонсором (могут заключаться отдельно от подписания других документов в данном центре) | X | X | 8.2.2, перечисление е) |
Е.1.15 | Страховые сертификаты, если применимо | Предоставляются свидетельства доступности для субъектов компенсаций вследствие причинения вреда, связанного с клиническими исследованиями | X | X | 4.3, 4.5.2, перечисление j), 8.2.2, перечисление d) |
Е.1.16 | Записи по транспортированию исследуемого изделия | Проверяется, кому материально принадлежит изделие | X | X | 6.9, 8.2.2, перечисление с), 8.2.3, перечисление а), 8.2.4.5, перечисление I), 9.6, перечисление k) |
Е.1.17 | Записи по транспортированию документации и материалов, связанных с клиническими исследованиями | Проверяется физическая доставка документов и материалов | - | X | 8.2.2, перечисление с), 8.2.4.4, перечисление b) |
Е.1.18 | Образцы утвержденной формы получения информированного согласия, информации для субъектов и рекламных объявлений, включая переводы | Отображается содержание форм получения информированного согласия и информации, предоставляемой субъекту клинических исследований во время клинических исследований | X | X | 4.5, 4.7, 8.2.2, перечисление а) |
Е.1.19 | Лист рандомизации для клинических исследований, проводимых с рандомизацией | Подтверждается, что была проведена рандомизация. В зависимости от проекта клинических исследований, список может быть недоступен в исследовательском центре для обеспечения слепого метода/маскирования | X | X | 6.8.1 |
Е.1.20 | Процедуры декодирования для клинических исследований, использующих слепой метод/маскирование, если применимо | Могут не применяться в исследовательском центре в зависимости от проекта исследований | X | X | 6.8.1, А.6.1, перечисление а), А.16, перечисление b) |
Е.1.21 | Отчет о выборе исследовательского центра | Проверяется, что были рассмотрены квалификация исследователя и исследовательского центра | - | X | 5.8, 8.2.1, перечисление b), 8.2.4.3, 8.2.4.7 |
Е.1.22 | Отчет о контроле перед запуском клинических исследований | Проверяется, что исследователь и команда исследовательского центра были обучены использованию изделия и выполнению CIP | - | X | 6.2, 8.2.4.7 |
Е.1.23 | Повторные сообщения перед запуском клинических исследований; связь с исследовательским центром | Определяются любые выводы или решения, а также выполняемые действия исследовательского центра | X | X | 8.2.4.7 |
Е.1.24 | Индивидуальная регистрационная карта CRF | Бланки, установленные для фиксации собираемых данных | X | X | 5.6, приложение С |
Е.1.25 | Формы отчета о неблагоприятных событиях | Документально фиксируются все нежелательные события, как требуется настоящим стандартом. Формы могут быть или не быть частью CRF | X | X | 5.6, 6.4.1, приложение С |
Е.1.26 | Формы отчета о недостатках изделия | Документально фиксируются все недостатки изделия. Формы могут быть или не быть частью CRF | X | X | 5.6, 6.4.2, приложение С |
Е.1.27 | Фамилия, инициалы/контактная информация наблюдателя (наблюдателей) | Документально фиксируются лица, обеспечивающие постоянное соответствие клинических исследований | X | X | 5.1, 8.2.1, перечисление а), 8.2.1, перечисление f), D.13, перечисление е) |
Е.1.28 | Записи об обучении | Предоставляются свидетельства, что исследователь (исследователи) обучены использованию исследуемого изделия и всем необходимым аспектам клинических исследований | X | X | 8.2.1, перечисление g) |
Е.1.29 | Нормальное(ые) значение(я)/диапазон(ы) показателей при клинических лабораторных исследованиях, необходимых в рамках клинических исследований | Документально фиксируются нормальные значения | X | X | 8.2.4.5, перечисление о) |
Е.1.30 | Подтверждение пригодности оборудования, если необходимо в рамках клинических исследований | Документально фиксируются обслуживание и калибровка оборудования | X | - | 8.2.4.5, перечисление n) |
Е.1.31 | - Сертификация, акредитация* или иное доказательство управления качеством или внешняя оценка качества, | Документально фиксируются компетентность и ответственность подразделений при проведении необходимых испытаний, а также поддержание надежности результатов | X | X | 5.1, 6.11, 8.1, 8.2.1, 8.2.4.5, перечисление о) |
_______________ | |||||
Е.1.32 | Выявление конфликтов интересов | Документально фиксируются конфликты интересов, например, финансовых | X | X | 8.2.1, перечисление d), 9.2, перечисление с) |
| |||||
Таблица Е.2 - Основные документы клинических исследований во время клинических исследований
N | Заголовок документа | Назначение или комментарии | Файлы центра | Файлы спонсора | Ссылка в настоящем стандарте |
Е.2.1 | Дополнения к IB, если есть | Документально фиксируются изменения IB | X | X | 6.5.1 |
Е.2.2 | Дополнения к CIP, если есть | Описываются изменения в плане клинических исследований | X | X | 6.5.1 |
Е.2.3 | Образцы дополнения к форме информированного согласия | - | X | X | 6.5.1 |
Е.2.4 | Положительное решение/одобрение ЭК любых дополнений | - | X | X | 4.5.4, перечисление d), 4.5.5, перечисление а) 8.2.3, перечисление b) 8.2.4.5, перечисление m) 9.4, перечисление с) |
Е.2.5 | Уведомление или одобрение регулирующими органами любых дополнений, если требуется | Проверяется информация, предоставляемая в регулирующие органы. Подтверждается уведомление или одобрение. Регулирование может не требовать наличия данного документа в файлах исследовательского центра и, если необходимо, он может быть оригиналом или копией в зависимости от регулирующих требований | X | X | 6.1, 8.2.2, перечисления f) и h) |
Е.2.6 | CV новых руководителей | Определяется руководитель. В центре хранятся CV руководителей данного центра; спонсор имеет CV руководителей всех исследовательских центров | X | X | 4.5.2, перечисление е), 9.2, перечисление а), D.13, перечисление с) |
Е.2.7 | CV новых ключевых членов команды в исследовательском центре, актуальные (последние), подписанные, с указанием даты | Определяются ключевые члены команды в исследовательском центре. В центре хранятся CV ключевых членов команды в данном исследовательском центре | X | X | 9.2, перечисление а) |
Е.2.8 | Записи по транспортированию исследуемого изделия и записи учета исследуемых изделий | - | X | X | 6.9, 8.2.2, перечисление с), 8.2.3, перечисление а), 8.2.4.5, перечисление I), 9.6, перечисление k) |
Е.2.9 | Записи по транспортированию документов, связанных с клиническими исследованиями | - | - | X | 8.2.4.4, перечисление а) |
Е.2.10 | Отчеты о визитах наблюдателей | Главному исследователю предоставляется обзор ключевых выводов | (X) | X | 8.2.3, перечисление d), 8.2.4.7 |
Е.2.12 | Обновленный список главного исследователя и ключевых членов команды в каждом исследовательском центре, включая подписи, полномочия и ответственность за клинические исследования | Документально подтверждается передача ответственности | X | X | 6.2, 8.2.1, перечисление е), 8.2.4.5, перечисление b) |
Е.2.13 | Подписанные, с указанием даты и полностью заполненные формы информированного согласия | Проверяется получение информированного согласия | X | - | 4.7.1, 7.4, 8.2.4.5, перечисление f), 9.5 |
Е.2.14 | Первичная документация | - | X | - | 6.5.3, 6.8.2, 9.6, перечисление о), 9.7, перечисление f) |
Е.2.15 | CRF, полностью заполненная | Приводятся свидетельства, что данные были собраны, и их достоверность была проверена руководителем | X | X | 6.3, 6.8.1, 6.8.2, 8.2.4.5, перечисление i), 9.6, перечисление j) |
Е.2.16 | Отчеты о неблагоприятных событиях, неблагоприятном воздействии изделия и недостатках изделия | Документально фиксируются возникновение и разрешение неблагоприятного события и неблагоприятного воздействия изделия | X | X | 6.4, 8.2.4.5, перечисления j) и k), 8.2.5, 9.8, D.13, перечисление g) |
Е.2.17 | Исправления CRF | Выявляются любые изменения, дополнения или исправления, внесенные в CRF после даты первоначального подписания | X | X | 6.8, 8.2.4.5, перечисление i), 9.6, перечисление j) |
Е.2.19 | Сообщения руководителя или спонсора, если требуется, о неблагоприятных событиях в ЭК | - | X | X | 4.5.4 |
Е.2.21 | Промежуточные или ежегодные отчеты главного исследователя для ЭК, если применимо | - | X | X | 4.5.4, 8.2.3, перечисление b), 8.2.4.5, перечисление m), 9.4, 9.8 |
Е.2.22 | Журнал наблюдения за субъектами | Находится в файле спонсора только при анонимности субъектов | X | X | 6.5.2 |
Е.2.23 | Журнал идентификации субъектов | - | X | - | 6.5.2 |
Е.2.24 | Журналы учета исследуемых изделий в исследовательском центре, если применимо | Согласуются с записями спонсора по транспортированию и получению | X | X | 6.9, 8.2.3, перечисление а), |
Е.2.25 | Обновленная информация об имени/контактах наблюдателя(ей) | Документально фиксируются лица, обеспечивающие постоянное соответствие клинических исследований. Файлы исследовательского центра содержат только специальную идентификацию наблюдателей | X | X | 8.2.1, перечисление а), 8.2.1, перечисление f), D.13, перечисление е) |
Е.2.26 | Обновления нормальных значений/диапазонов показателей в клинических лабораторных исследованиях, необходимых в рамках клинических исследований | Документально фиксируются изменения нормальных значений в ходе клинических исследований | X | X | 8.2.4.5, перечисление о) |
Е.2.28* | Обновления: | Документально фиксируются корректность испытаний в ходе клинических исследований | X | X | 5.1, 6.11, 8.1, 8.2.1, 8.2.4.5, перечисление о) |
Е.2.29 | Обновления, связанные с выявлением конфликтов интересов | Документально фиксируются конфликты интересов, например, финансовых | X | X | 8.2.1 перечисление d), 9.2, перечисление с) |
________________
* Нумерация соответствует оригиналу. - .
Таблица Е.3 - Основные документы клинических исследований после клинических исследований
N | Заголовок документа | Назначение или комментарии | Файлы центра | Файлы спонсора | Ссылка в настоящем стандарте |
Е.3.1 | Учет исследуемых изделий в каждом исследовательском центре, если применимо | - | X | X | 6.9, 7.2, перечисление а), 9.6, перечисление k), 9.6, перечисление о) |
Е.3.2 | Документальное фиксирование возврата или утилизации исследуемого изделия, если применимо | Документально фиксируется корректная утилизация биологически опасных материалов или других материалов, требующих специальной утилизации | X | X | 6.9, 7.2, перечисление а), 9.6, перечисление k) |
Е.3.3 | Заполненный журнал идентификации субъектов | - | X | - | 6.5.2 |
Е.3.4 | Сертификат, подтверждающий проведение аудита (если требуется или проводится) | - | - | X | 6.11, 8.1 D.13, перечисление h) |
Е.3.5 | Отчет по контролю после завершения | - | - | X | 8.2.4.7 |
Е.3.6 | Уведомление, если необходимо, ЭК руководителем или спонсором о завершении клинических исследований | - | X | X | 4.5.4, 7.1, 7.2, 8.2.6, перечисление d) 9.4 |
Е.3.7 | Уведомление, если необходимо, контролирующих органов главным исследователем или спонсором о завершении клинических исследований | Включается в файлы исследовательского центра только, если главный исследователь обязан уведомить контролирующие органы | X | X | 7.1, 7.2, 8.2.6, перечисление d) |
Е.3.8 | Статистический анализ, выполняемый организатором и отчет о клинических исследованиях | Включается в файлы исследовательского центра только, если это требуется процедурами спонсора | X | X | 7.3, 8.2.6, приложение D |
Приложение F
(справочное)
Категории неблагоприятных событий
В таблице F.1 приведены категории неблагоприятных событий.
Таблица F.1
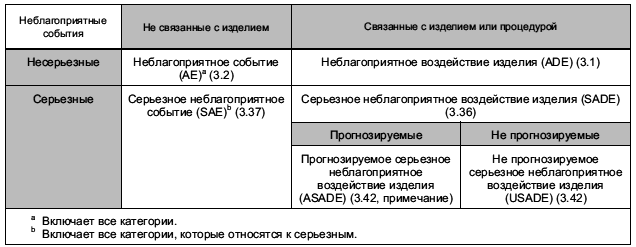
На рисунках F.1 и F.2 приведено руководство с перечнем вопросов, ответив на которые можно определить категорию неблагоприятных событий и недостатков устройства. Однако это руководство не предназначено для иллюстрации взаимосвязей между категориями.
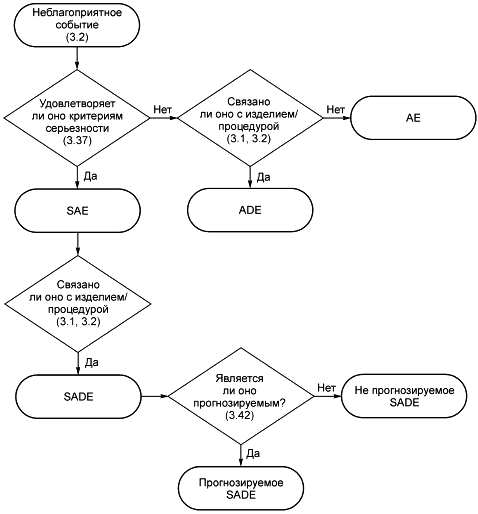
Рисунок F.1 - Деление неблагоприятных событий по категориям
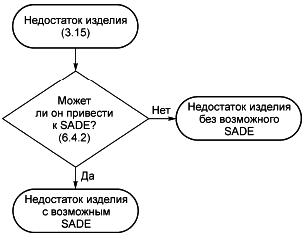
Рисунок F.2 - Деление недостатков изделия по категориям
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации и действующим в этом качестве межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального, межгосударственного стандарта |
ИСО 14971:2007 | IDT | ГОСТ ISO 14971-2011 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: | ||
Библиография
[1] | ISO 13485:2003 | Medical devices - Quality management systems - Requirements for regulatory purposes | |
ИСО 13485:2003 | Медицинские изделия. Системы менеджмента качества. Системные требования для целей регулирования | ||
[2] | ISO 10993 (all parts) | Biological evaluation of medical devices | |
ИСО 10993 (все части) | Изделия медицинские. Оценка биологического действия медицинских изделий | ||
[3] | ISO 15223-1 | Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements | |
ИСО 15223-1 | Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации. Часть 1. Общие требования | ||
________________ | |||
[4] | EN 1041 | Information supplied by the manufacturer of medical devices | |
EH 1041 | Информация, предоставляемая изготовителем медицинских изделий | ||
[5] | Global Harmonization Task Force, [SG1/N29R16:2005] | Information Document Concerning the Definition of the Term "Medical Device", available at: http://www.ghtf.org/documents/sg1/sg1n29r162005.pdf | |
Global Harmonization Task Force, [SG1/N29R16:2005] | Информационный документ, касающийся определения термина "медицинское изделие". Доступно по адресу: http://www.ghtf.org/documents/sg1/sg1n29r162005.pdf | ||
[6] | Global Harmonization Task Force, [SG5/N2R8:2007] | Global Harmonization Task Force, Clinical Evaluation [SG5/N2R8:2007], available at: http://www.ghtf.org/documents/sg5/sg5_n2r8_2007final.pdf | |
Global Harmonization Task Force, [SG5/N2R8:2007] | Клиническая оценка. Доступно по адресу: http://www.ghtf.org/documents/sg5/sg5_n2r8_2007final.pdf | ||
[7] | Global Harmonization Task Force, | Global Harmonization Task Force, Essential Principles of Safety and Performance of Medical Devices [SG1-N41R9:2005], available at: http://www.ghtf.org/documents/sg1/sg1n41r92005. pdf | |
Global Harmonization Task Force, | Изделия медицинские. Руководство по выбору стандартов, поддерживающих важнейшие принципы обеспечения безопасности и эксплуатационных характеристик медицинских изделий. Доступно по адресу: http://www.ghtf.org/documents/sg1/sg1n41r92005. pdf | ||
[8] | Declaration of Helsinki | available at: http://www.wma.net/en/30publications/10policies/b3/index.html | |
Декларация Хельсинки | Доступно по адресу: http://www.wma.net/en/30publications/10policies/b3/index.html | ||
[9] | MEDDEV 2.7.1, Rev. 3, Dec. 2009 | Clinical Evaluation: A Guide for Manufacturers and Notified Bodies, available at: http://ec.europa.eu/consumers/sectors/medicaldevices/files/meddev/2_7_1rev_3_en.pdf | |
MEDDEV 2.7.1, | Клиническая оценка: Руководство для изготовителей и нотифицированных органов. Доступно по адресу: http://ec.europa.eu/consumers/sectors/medicaldevices/files/meddev/2_7_1rev_3_en.pdf | ||
[10] | MEDDEV 2.12-1, Rev. 6, Dec. 2009 | Guidelines on a Medical Devices Vigilance System, available at: http://ec.europa.eu/consumers/sectors/medical-devices/files/meddev/2_12_1-rev_6-12-2009_en.pdf | |
MEDDEV 2.12-1, Rev. 6, Dec. 2009 | Руководящие принципы системы предупреждения и оповещения для медицинских изделий. Доступно по адресу: | ||
[11] | MEDDEV 2.12-2, May 2004 | Guidelines on Post Market Clinical Follow-up, available at: http://ec.europa.eu/consumers/medical_devices/ meddev/2_12-2_05-2004.pdf | |
MEDDEV 2.12-2, May 2004 | Руководящие принципы по сообщению постпродажных клинических наблюдений. Доступно по адресу: | ||
УДК 006.83:006.354 | ОКС 11.100.20 | Р20 | ОКП 94 000 |
Ключевые слова: изделия медицинские, клинические исследования, надлежащая клиническая практика | |||
Электронный текст документа
и сверен по:
, 2015

{kind=link}