ГОСТ Р ИСО 13022-2016
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРОДУКТЫ МЕДИЦИНСКИЕ, СОДЕРЖАЩИЕ ЖИЗНЕСПОСОБНЫЕ ЧЕЛОВЕЧЕСКИЕ КЛЕТКИ
Применение менеджмента риска и требований к методикам обработки
Medical products containing viable human cells. Application of risk management and requirements for processing practices
ОКС 11.020*
ОКП 93 9800
_____________________
* В ИУС N 8 2016 г. ГОСТ Р ИСО 13022-2016 приводится с ОКС 11.100.20,
здесь и далее. - .
Дата введения 2017-01-01
Предисловие
1 ПОДГОТОВЛЕН Автономной некоммерческой организацией "Институт медико-биологических исследований и технологий" (АНО "ИМБИИТ") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 422 "Оценка биологического действия медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 1 марта 2016 г. N 102-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 13022:2012* "Продукты медицинские, содержащие жизнеспособные человеческие клетки. Применение менеджмента риска и требований к методикам обработки" (ISO 13022:2012 "Medical products containing viable human cells - Application of risk management and requirements for processing practices", IDT)
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные и межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Международная организация по стандартизации (ИСО) является всемирной федерацией национальных организаций по стандартизации (комитетов - членов ИСО). Разработку международных стандартов обычно осуществляют технические комитеты ИСО. Каждый комитет - член, заинтересованный в деятельности, для которой был создан технический комитет, имеет право быть представленным в этом комитете. Международные правительственные и неправительственные организации, имеющие связи с ИСО, также принимают участие в работах. ИСО тесно сотрудничает с Международной электротехнической комиссией (МЭК) по всем вопросам электротехнической стандартизации.
Проекты международных стандартов разрабатывают в соответствии с правилами Директив ИСО/МЭК, часть 2.
Основная задача технических комитетов заключается в подготовке международных стандартов. Проекты международных стандартов, принятые техническими комитетами, рассылают комитетам-членам на голосование. Их опубликование в качестве международных стандартов требует одобрения не менее 75% комитетов - членов, принимающих участие в голосовании.
Необходимо обратить внимание на то, что некоторые элементы настоящего стандарта могут быть объектом патентных прав. ИСО не может нести ответственность за идентификацию какого-либо одного или всех патентных прав.
ИСО 13022 был подготовлен Техническим комитетом ИСО/TК 194 "Биологическая оценка медицинских изделий", подкомитетом SC1 "Безопасность тканевых продуктов".
В некоторых медицинских продуктах используют материалы человеческого происхождения. В зависимости от требований национальных нормативных документов эти продукты рассматривают как лекарственные вещества, медицинские изделия или биопрепараты. Материалы из тканей человека применяют в разработке и производстве медицинских продуктов для обеспечения функциональных характеристик, выбор которых мог быть осуществлен ввиду их преимуществ в сравнении с материалами нечеловеческого происхождения, в частности для улучшения регенерации собственных тканей и органов пациента или для замены либо поддержки функции органа.
Медицинские продукты, использующие человеческие материалы, составляют гетерогенную группу. Примеры включают клеточные суспензии, клеточные/матричные конструкции или клетки совместно со сложными медицинскими изделиями, например с оборудованием для диализа.
Хотя медицинские продукты, использующие человеческие материалы, очень разнообразны, опасности, конкретно связанные со всеми человеческими материалами, практически одинаковы:
a) материал может быть контаминирован инфекционными агентами [например, бактерии, плесень, дрожжи, вирусы, инфекционные агенты трансмиссивной губчатой энцефалопатии (Transmissible Spongiform Encephalopathy, TSE), паразиты];
b) материал может быть загрязнен химическими веществами;
c) материал может не подходить для предполагаемого назначения из-за непреднамеренного разложения или деградации, вызванной неправильным обращением на любом этапе производственного процесса;
d) материал может быть опасным для пациента из-за канцерогенного потенциала;
e) после применения могут возникать побочные отрицательные физиологические или анатомические последствия по причине миграции клеток и выделения биологически активных веществ;
f) нарушение и невозможность прослеживаемости;
g) материал может наносить вред пациенту, вызывая иммунную реакцию.
Для учета опасностей, связанных с загрязнением, деградацией, непреднамеренным изменением и/или перепутыванием жизнеспособных человеческих клеток и продуктов, был разработан настоящий стандарт по применению менеджмента риска при производстве медицинских продуктов, использующих жизнеспособный человеческий материал.
Опасности, упомянутые выше, связаны с соответствующими производственными этапами. Основные аспекты, включенные в настоящий стандарт, следующие:
- терминология и определения;
- отбор и исследование доноров (включая живых и трупных доноров);
- получение человеческих материалов;
- обращение с человеческими материалами (включая производство);
- упаковка, хранение и транспортирование человеческих материалов;
- маркировка человеческих материалов;
- риск, связанный с обращением с продуктом при применении;
- рассмотрение рисков и преимуществ в контексте предполагаемого использования.
Настоящий стандарт оказывает поддержку производителям продуктов на основе жизнеспособных человеческих материалов, производящим лекарственные средства, медицинские изделия или биопрепараты.
1 Область применения
В настоящем стандарте определены требования и руководство по методикам обработки и управлению рисками, связанными с жизнеспособными клеточными компонентами продуктов, относящихся к медицинским продуктам, биопрепаратам, медицинским изделиям и активным имплантируемым медицинским изделиям, а также их комбинациям. Настоящий стандарт включает жизнеспособные как аутологичные, так и аллогенные ткани человека, полученные от живых и трупных доноров.
Для производителей медицинских продуктов, содержащих жизнеспособные клетки человека, в настоящем стандарте определены процедуры, используемые при обработке и применении, а также для идентификации опасностей и опасных ситуаций, связанных с такими клетками, для анализа и оценки конечных рисков, для контроля этих рисков и для проверки эффективности данного контроля. В настоящем стандарте определен процесс принятия решения о приемлемости остаточных рисков, принимая во внимание баланс остаточных рисков и ожидаемой медицинской пользы в сравнении с доступными альтернативами.
В настоящем стандарте определены требования и руководства по менеджменту риском, связанным с опасностями, типичными для медицинских продуктов, полученных с использованием жизнеспособных человеческих материалов, такими как:
a) контаминация бактериями, плесенью, дрожжами или паразитами;
b) контаминация вирусами;
c) контаминация агентами, вызывающими трансмиссивную губчатую энцефалопатию (Transmissible Spongiform Encephalopathy, TSE);
d) контаминация материала, вызывающая нежелательные пирогенные, иммуногенные или токсические реакции;
e) деструкция продукта и продукты деструкции, вызванные неправильным обращением;
f) опасности, связанные с канцерогенным потенциалом используемого типа клеток;
g) осложнения, вызванные побочными физиологическими или анатомическим последствиями (это включает непредвиденную миграцию клеток, нежелательное выделение биологически активных веществ, таких как гормоны и цитокины, и непредвиденное взаимодействие между клеточными и неклеточными компонентами продукта);
h) невозможность контроля;
i) осложнения, вызванные тем, что материал вызывает непредвиденную иммунную реакцию.
Для оценки контаминации другими неклассифицированными патогенами могут применять подобные принципы.
Опасности, связанные с генетическими изменениями, находятся за пределами области применения настоящего стандарта и рассматриваются в других документах.
Примечание 1 - Определение "генетически модифицированный" можно найти в АСТМ Ф 2312.
Примечание 2 - В настоящем стандарте не определена система управления качеством для контроля всех этапов производства медицинских продуктов, описанных выше.
Если в странах, где используют медицинский продукт, существуют дополнительные национальные или региональные требования, они также должны быть применимы в дополнение к требованиям настоящего стандарта.
Примечание 3 - Региональные требования могут быть более строгими в отличие от требований, указанных в настоящем стандарте, особенно относительно критериев отбора доноров.
Настоящий стандарт не применим:
- к нежизнеспособным материалам человеческого происхождения;
- жизнеспособным клеткам нечеловеческого происхождения;
- крови и ее компонентам, используемым для переливания, эмбриональным клеткам, цельным органам и костному мозгу, используемым для трансплантации, и другим тканям, которые не подходят под определение "медицинский продукт";
- приборам для диагностики in vitro.
Примечание 4 - Требования по применению настоящего стандарта см. приложение A.
2 Нормативные ссылки
Следующие документы*, частично или в целом, являются необходимыми для применения настоящего стандарта. При датированной ссылке применимо только указанное издание. При ссылке без даты применимо последнее издание указанного документа, включая все поправки.
_______________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ISO 13485:2003 Medical devices - Quality management systems - Requirements for regulatory purposes (Медицинские изделия. Система менеджмента качества. Требования для регулирующих целей)
ISO 14971:2007 Medical devices - Application of risk management to medical devices (Медицинские изделия. Применение менеджмента риска к медицинским изделиям)
ISO 22442-1:2007 Medical devices utilizing animal tissues and their derivatives - Part 1: Application of risk management (Медицинские изделия, использующие ткани животных и их производные. Часть 1. Применение менеджмента риска)
ASTM F 2312 Standard Terminology Relating to Tissue Engineered Medical Products (Стандартная терминология, касающаяся медицинских продуктов, выполненных из тканей BSI PAS 84. Восстановительная медицина. Словарь, июнь 2008)
3 Термины и определения
В настоящем стандарте применены термины и определения, приведенные в ИСО 13485, ИСО 14971, ИСО 22442-1, АСТМ Ф 2312, BSI PAS 84, а также следующие термины с соответствующими определениями:
3.1 медицинское изделие (medical device): Инструмент, устройство, прибор, механизм, приспособление, имплантат, реагент in vitro или калибратор, программное обеспечение, материал или другие подобные либо сопутствующие средства, предназначенные производителем для применения человеком как отдельно, так и в сочетании друг с другом, с одной либо более определенными целями:
a) профилактики, диагностики, мониторинга, лечения или облегчения заболеваний;
b) диагностики, мониторинга, лечения, облегчения или компенсации последствий травмы;
c) исследования, замещения, изменения или поддержания анатомического строения либо физиологических процессов;
d) поддержания жизненно важных функций организма;
e) концептуального контроля;
f) дезинфекции медицинских изделий;
g) получения информации медицинского назначения посредством in vitro исследования проб, взятых из человеческого организма;
при условии, что их основное предусмотренное воздействие на организм человека не реализуется за счет фармакологических, иммунологических или метаболических средств, но может поддерживаться в своей основной функции таковыми.
Примечание 1 - Данное определение разработано Целевой группой по глобальной гармонизации (Global Harmonization Task Force, GHTF).
Примечание 2 - Следующие изделия могут рассматривать в некоторых нормативных документах как медицинские изделия, но в их отношении не выработан единый подход:
a) вспомогательные средства для лиц с ограниченными возможностями или с физическими и умственными недостатками;
b) изделия для лечения/диагностики заболеваний и травм у животных;
c) принадлежности для медицинских изделий (см. примечание 3);
d) дезинфицирующие вещества;
e) изделия, включающие в себя ткани животных или человека, которые могут отвечать требованиям определения, приведенного выше, но при этом являются предметом других нормативных требований.
Примечание 3 - Объектом настоящего стандарта также являются принадлежности, которые специально предназначены производителем для использования в комплекте с "исходным" медицинским изделием с целью обеспечения его предусмотренного применения.
Примечание 4 - Термин "медицинские изделия" включает в себя неактивные и активные медицинские изделия, а также активные имплантируемые медицинские изделия.
Примечание 5 - Адаптировано сообразно ИСО 14971, определение 2.9.
3.2 активное имплантируемое медицинское изделие (active implantable medical device): Активное медицинское изделие, предназначенное для его полного или частичного введения в тело человека хирургическим способом, или с помощью медикаментов, или путем медицинского проникновения в естественные отверстия и для нахождения в данном месте после этой процедуры.
Примечание 1 - Работа активного медицинского изделия основана на источнике электроэнергии или любом другом источнике энергии, отличной от непосредственно производимой телом человека или гравитацией.
Примечание 2 - Адаптировано сообразно ИСО 13485, определение 3.1.
3.3 лекарственный препарат (medicinal product): Вещество или комбинация веществ, представленных как имеющие свойства, обеспечивающие лечение или предотвращение заболевания человека, либо любое вещество или комбинация веществ, которые могут использовать в организме человека или вводить в организм человека либо для сохранения, корректировки или изменения физиологических функций с помощью фармакологического, иммунологического или метаболического воздействия, либо для постановки медицинского диагноза.
Примечание - См. ссылку [34].
3.4 медицинский продукт (medical product): Лекарственный препарат, биопрепарат, медицинское изделие или их комбинация.
3.5 медицинский продукт, содержащий клетки (cell-based medical product): Медицинский продукт, который включает жизнеспособные аутологичные или аллогенные клетки человека, полученные от прижизненного или посмертного донора, прошедший процесс обработки.
Примечание - Медицинский продукт не может комбинироваться с неклеточными компонентами.
3.6 биопрепараты (biologics or biological): Продукт клеточной терапии аутологичного или аллогенного происхождения, в котором клетки размножаются, выращиваются, отбираются, фармакологически обрабатываются или любым другим способом изменяются их биологические характеристики ex vivo для введения в тело человека и который применяется для профилактики, обработки, лечения, диагностики или устранения заболеваний либо повреждений.
Примечание 1 - См. ссылку [57].
Примечание 2 - Разъяснения FDA, содержащие определение биопрепарата можно найти по ссылке http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CBER/ucm133077.htm
3.7 донор (donor): Человеческий источник (прижизненный или посмертный) человеческих клеток или тканей.
Примечание - В случае аутологичного донорства живым донором является пациент.
3.8 забор (procurement): Процесс получения тканей или клеток от донора.
3.9 отходы (residues): Вещества или материалы, остающиеся после процесса производства, такие как клеточный детрит, продукты распада клеточного каркаса, химические вещества, факторы роста или растворители.
3.10 инактивация (inactivation): Процесс, в ходе которого снижается способность трансмиссивных агентов вызывать инфекцию или патогенную реакцию.
Примечание 1 - Эффективность процесса инактивации вирусов и агентов TSE выражена математически в виде коэффициента сокращения (см. ИСО 22442-3, приложение F).
Примечание 2 - Инактивация предназначена для предотвращения инфицирования трансмиссивными агентами и их репликации.
[ИСО 22442-1:2007, определение 3.5]
3.11 производство (manufacture): Любой или все этапы получения, скрининга, испытания, обработки, хранения, этикетирования, упаковки или распространения любых медицинских продуктов на основе клеток или тканей, включая скрининг и испытания для доноров клеток или тканей.
3.12 производитель (manufacturer): Физическое или юридическое лицо, несущее ответственность за проектирование, производство, упаковку и этикетирование медицинского продукта на клеточной основе перед его выпуском в продажу от своего имени независимо от того, выполняет ли эти операции вышеупомянутое лицо или третья сторона от его имени.
3.13 хранение (storage): Обеспечение соответствующих контролируемых условий для медицинского продукта, содержащего клетки, до момента последующей обработки или распространения.
3.14 транспортирование (transport): Перемещение медицинского продукта, содержащего клетки, при соответствующих контролируемых условиях.
3.15 риск (risk): Комбинация вероятности возникновения вреда и тяжести данного вреда.
[Руководство ИСО/МЭК 51:1999, определение 3.2]
3.16 вред (harm): Физическое повреждение или нарушение здоровья человека, или повреждение имущества либо окружающей среды.
[Руководство ИСО/МЭК 51:1999, определение 3.3].
3.17 опасность (hazard): Потенциальный источник вреда.
Примечание 1 - См. ICH Q9.
Примечание 2 - Адаптировано из Руководства ИСО/МЭК 51:1999, определение 3.5.
3.18 остаточный риск (residual risk): Риск, остающийся после принятия мер по контролю рисков.
Примечание 1 - Адаптировано из Руководства ИСО/МЭК 51:1999, определение 3.9.
Примечание 2 - В определении 3.9 Руководства ИСО/МЭК 51:1999 использован термин "меры предосторожности", а не "меры по контролю рисков". В контексте настоящего стандарта "меры предосторожности" - это только один из возможных способов контроля рисков.
3.19 анализ риска (risk analysis): Систематическое использование доступной информации для идентификации опасностей и для определения риска.
[Руководство ИСО/МЭК 51:1999, определение 3.10]
3.20 оценка риска (risk assessment): Общий процесс, включающий анализ риска и оценивание риска.
[Руководство ИСО/МЭК 51:1999, определение 3.12]
3.21 контроль риска (risk control): Процесс принятия решений и выполнения мер по уменьшению рисков до установленных уровней или поддержания рисков на установленных уровнях.
[ИСО 14971:2007, определение 2.19]
3.22 определение риска (risk estimation): Процесс, применяемый для присвоения числовых значений вероятности возникновения вреда и тяжести этого вреда.
[ИСО 14971:2007, определение 2.20]
3.23 оценивание риска (risk evaluation): Процесс сравнения определенного риска с данными критериями риска для определения допустимости риска.
[ИСО 14971:2007, определение 2.21]
3.24 менеджмент риска (risk management): Систематическое применение правил, процедур и практических методов управления для анализа, оценивания, контроля и мониторинга риска.
[ИСО 14971:2007, определение 2.22]
3.25 документация менеджмента риска (risk management file): Совокупность записей и других документов, создаваемых в процессе менеджмента риска.
[ИСО 14971:2007, определение 2.23]
3.26 безопасность (safety): Отсутствие недопустимого риска.
[Руководство ИСО/МЭК 51:1999, определение 3.1]
3.27 степень тяжести (severity): Мера возможных последствий опасности.
[ИСО 14971:2007, определение 2.25]
4 Процесс менеджмента риска
4.1 Общие положения
При рассмотрении менеджмента риском необходимо учитывать два компонента:
a) вероятность возникновения вреда для пациента или пользователя продукта;
b) последствия данного вреда, т.е. насколько тяжелыми они могут быть.
Менеджмент риском крайне важен для клеточных компонентов медицинских продуктов, включающих жизнеспособные клетки или ткани человека, в связи с опасностями, присущими данной группе продуктов, и большим количеством заинтересованных сторон, включая медицинских работников, организации, обеспечивающие медицинский уход, органы управления, промышленность, пациентов и рядовых членов общества. Кроме того, на заинтересованные стороны может влиять доступность данного типа медицинских продуктов (особенно важны клеточные компоненты медицинских продуктов, так как этот исходный материал может быть более редким, чем другие компоненты медицинского продукта). Следовательно, доступность для пациента должны учитывать.
Производитель должен принимать решение о безопасности медицинского продукта на клеточной основе, включая приемлемость рисков, учитывая современный уровень развития науки, для определения возможности размещения медицинского продукта на рынке для его предполагаемого использования. Настоящий стандарт устанавливает процесс, с помощью которого производитель медицинского продукта, содержащего клетки, может идентифицировать опасности, связанные с клеточным компонентом продукта, определить и оценить риски, связанные с этими опасностями, контролировать эти риски и проверять эффективность данного контроля.
Производитель должен обосновывать использование человеческого материала (включая выбор типа и источника клеток и/или тканей), основываясь на приемлемости остаточного риска, учитывая баланс остаточного риска и ожидаемой медицинской пользы по сравнению с доступными альтернативами. При рассмотрении рисков и преимуществ продукта необходимо учитывать влияние хирургических процедур, необходимых для его введения.
К медицинским веществам, составляющим часть продукта, применяют требования Фармацевтической системы качества (ICH Q9 (см. ссылку [20]) и надлежащей производственной практики.
К медицинским изделиям, составляющим часть продукта, применяют требования ИСО 14971 и ИСО 13485.
4.2 Опасности, связанные с клеточными компонентами
4.2.1 Общие положения
Производитель должен установить, документально оформить и поддерживать в течение всего жизненного цикла продукта систематизированный процесс по идентификации опасностей, связанных с клеточным компонентом медицинского продукта, содержащего клетки, определить и оценить сопряженные риски по контролю этих рисков и проверке эффективности контроля.
Этот процесс должен включать следующие элементы:
a) анализ риска;
b) оценивание риска;
c) контроль риска;
d) рассмотрение информации, полученной на стадии производства и после производства.
Соответствующие части процесса менеджмента риска должны быть внедрены в документированный процесс реализации продукта на протяжении всего жизненного цикла продукта.
Примечание - Документированный процесс системы управления качеством может систематически использовать производитель для обеспечения безопасности, в частности с целью ранней идентификации опасностей и опасных ситуаций в сложных медицинских продуктах на клеточной основе.
Соответствие следует определять посредством инспекции определенных документов, таких как документация менеджмента риском. Инспекцию проводят и документально фиксируют уполномоченные сотрудники.
4.2.2 Обязанности
Производитель должен иметь организационную структуру и рабочие процедуры, соответствующие его деятельности. Организационная схема должна четко определять ответственность и схему подотчетности.
Производитель должен обеспечивать доступность адекватных источников и наличие квалифицированного персонала для производства продукта и деятельности по менеджменту риском.
Производитель должен:
a) определить и отразить документально стратегию определения критериев приемлемости риска. Стратегия должна гарантировать, что критерии основаны на применимых национальных или региональных нормативных документах и соответствующих международных стандартах, и учитывать доступную информацию, такую как современный уровень развития науки и известные опасения заинтересованных сторон;
b) проверять соответствие процесса менеджмента риском через плановые интервалы для гарантии эффективности процесса менеджмента риском, а также документально отражать все решения и принятые меры. Данная проверка может быть частью ревизии системы управления качеством.
4.2.3 Документация
4.2.3.1 Деятельность по менеджменту риском должны документально фиксировать. Соответствующим способом может быть документация менеджмента риском, которая включает план менеджмента риском.
Соответствующая информация, входящая в план менеджмента риском или его эквивалент, должна включать следующее:
a) объем запланированных мероприятий по менеджменту риском с определением и описанием медицинских продуктов на клеточной основе и фаз жизненного цикла, к которым применим каждый элемент плана;
b) распределение ответственности и полномочий;
c) требования к ревизии деятельности по менеджменту риском;
d) критерии приемлемости риска, основанные на стратегии производителя по определению приемлемости риска, включая критерии определения приемлемости риска при невозможности оценки вероятности возникновения вреда;
e) действия по верификации;
f) деятельность, связанная со сбором и проверкой соответствующей информации в процессе производства и после производства.
4.2.3.2 Для каждой определенной опасности в документации менеджмента риском или ее аналога должна быть ссылка:
a) на анализ риска;
b) оценивание риска;
c) внедрение, верификации и мониторинга мер по контролю риска;
d) оценку допустимости любого(ых) остаточного(ых) риска(ов) с учетом рассмотрения риска и медицинской пользы при предполагаемом использовании.
4.2.4 Персонал
Персонал, непосредственно вовлеченный в деятельность по производству клеточных компонентов медицинских продуктов на клеточной основе, должен быть достаточно квалифицированным для выполнения этих задач и компетентным для выполнения этих задач с учетом его квалификации, образования и подготовки.
4.3 Анализ риска
4.3.1 Общие положения
Для процесса анализа риска систематически используют доступную информацию для идентификации опасностей и определения сопряженных рисков. Чтобы решить данные задачи, необходимо рассмотреть все фазы жизненного цикла продукта. Особое внимание необходимо уделять забору клеток и/или тканей, а также всем этапам производства, связанным с данными материалами, и, если применимо, сочетаемому элементу в случае комбинированных продуктов.
Внедрение плановой деятельности по анализу риска и результаты анализа риска необходимо отражать в документации менеджмента риском или ее эквиваленте.
Информация по методологическим средствам идентификации и оценивания рисков представлена в ИСО 14971 и Руководстве Фармацевтической системы качества (ICH Q9).
4.3.2 Предполагаемое использование и определение характеристик, связанных с безопасностью клеточного компонента продукта
Необходимо учитывать предполагаемое использование и контакт клеточных компонентов медицинского продукта на клеточной основе с организмом пациента или организмом пользователя. При анализе риска необходимо учитывать количество материала, площадь поверхности контакта и тип(ы) материала, вступающего в контакт с тканями или жидкостями организма, а также тип ткани или жидкости организма, с которыми он контактирует.
Наиболее важен критический анализ:
a) источников клеток (см. приложения C, I, J, K и L);
b) типа клеток и статуса дифференциации клеток (см. приложения D, E, G, H, K, N, O и P);
c) всех аспектов процесса производства (см. приложения E, F, G, H, J, L, M, N и P);
d) конкретных действий, которые могут привести к канцерогенности или непредвиденной иммуногенности клеток (см. приложения E, K, M, N, O и P);
e) возможной перекрестной контаминации (см. приложения C, D, F, H, I, J, L и M);
f) системы контроля материала (см. приложения C, D, F, G, H и I);
g) возможной деструкции (см. приложения D, E, G, H, N, O и P);
h) непредвиденного взаимодействия между клеточными и неклеточными компонентами продукта (см. приложения E и O);
i) клинической оценки и лабораторных исследований (см. приложение P).
4.3.3 Определение опасностей
Возможные опасности, связанные с использованием человеческих клеток и тканей, должны быть идентифицированы и задокументированы. Особое внимание следует уделить следующим опасностям, связанным с клетками и тканями:
a) контаминация бактериями, плесенью, дрожжами или паразитами;
b) контаминация вирусами;
c) контаминация агентами, вызывающими TSE;
d) контаминация материалом, вызывающим нежелательные пирогенные, иммуногенные или токсические реакции;
e) деструкции продукта и продуктов деструкции;
f) отсутствие обратимости воздействия;
g) опасности, связанные с канцерогенным потенциалом используемого типа клеток;
h) нарушение и/или невозможность прослеживаемости;
i) осложнения, вызванные непреднамеренными физиологическими или анатомическими последствиями. Это включает непредвиденную миграцию клеток, нежелательное выделение биологически активных веществ, таких как гормоны и цитокины, и непредвиденное взаимодействие между клеточными и неклеточными компонентами продукта.
При использовании стволовых клеток могут возникать дополнительные опасности. Особое внимание следует уделить испытаниям на канцерогенность и биораспределение. При использовании таких клеток необходимо соблюдать региональные нормативные требования.
Для оценки загрязнения другими неклассифицированными патогенными веществами могут применяться сходные принципы.
4.4 Оценивание риска
Необходимо оценить все идентифицированные риски. Для каждой идентифицированной опасности производитель, используя критерии, определенные в плане менеджмента риска, должен принять решение о необходимости снижения риска. Результаты данного оценивания риска должны быть отражены в документации менеджмента риском или ее эквиваленте.
В приложении B определены основные категории риска, которые необходимо учитывать.
4.5 Контроль риска
4.5.1 Общие положения
Для контроля риска необходимо принять решение и соответствующие меры для снижения риска до определенного уровня или поддержания в определенных пределах. Производитель должен определить меры по контролю риска, которые применимы для снижения риска(ов) до приемлемых уровней. Мероприятия по контролю риска должны быть отражены документально и обоснованы.
Схема в приложении В дает общее представление о процессе менеджмента риском. Если во время процесса менеджмента риском определены дополнительные риски, производитель должен выбрать соответствующие стандарты или нормативные документы и следовать им. Решение должно быть отражено документально и обосновано.
4.5.2 Оценки остаточного риска
После применения мер по контролю рисков необходимо оценить все остаточные риски, используя критерии, определенные в плане менеджмента риском. Таким образом проверяют, не появились ли новые риски в результате любых мер по снижению рисков. Результаты данной оценки должны быть отражены в документации менеджмента риском или ее эквиваленте.
4.6 Оценивание допустимости общего остаточного риска
Оценка допустимости общего остаточного риска должна учитывать баланс между остаточным риском после применения всех мер по контролю и ожидаемой медицинской пользой по сравнению с доступными альтернативами. Если существует остаточный риск, связанный:
- с загрязнением вирусами, паразитами, бактериями, плесенью, грибами или инфекционными агентами, вызывающими TSE; и/или
- опасностями, связанными с канцерогенным или иммуногенным потенциалом используемого типа клеток. В оценке необходимо конкретно рассмотреть риски и преимущество при использовании альтернативных материалов, не связанных с данными рисками, например синтетические материалы, материалы от другого вида животных или аутогенного человеческого материала, и применение альтернативы для той же предполагаемой цели.
Если остаточные риски не удовлетворяют критериям приемлемости риска, общий риск может считаться приемлемым только при его компенсации исключительной пользой и целесообразностью, обоснование которых должно быть задокументировано.
Если применимо, необходимо учитывать оценку конкретного контингента пациентов (например, пациентов с иммунодефицитом).
4.7 Система получения информации на этапе производства и после производства
Производитель должен внедрить, документально оформить и поддерживать систему текущего и систематического сбора и обработки информации о рисках, связанных с применением медицинских продуктов на клеточной основе и сходных продуктов на этапе производства и после производства. Полученная информация должна быть проанализирована, оценена и использована в процесса менеджмента риском.
Примечание - Для этого могут быть использованы принципы контроля за рынком продаж и/или фармаконадзора.
Приложение A
(справочное)
Руководство по применению ИСО 13022
A.1 Общие положения
Менеджмент риском крайне важен для клеточных компонентов медицинских продуктов, включая жизнеспособные клетки или ткани человека, из-за особых характеристик данных продуктов. Они сильно подвержены биологическому загрязнению и крайне чувствительны ко всем видам воздействия, вызывающего повреждение и деградацию клеток. Следовательно, важно, что при установлении риска, связанного с данным материалом, производитель принимает меры по контролю риска или обосновывает в отчете по менеджменту риска, почему этого не было сделано.
A.2 Применение к источникам клеток и тканей человека
В настоящем стандарте рассмотрены такие материалы, как:
a) жизнеспособные клетки и ткани аутологичного и аллогенного происхождения, и
b) жизнеспособные клетки и ткани, полученные от живых и трупных доноров.
A.3 Применение к материалам, полученным третьей стороной
Настоящий стандарт можно применять в том случае, если материалы, используемые производителем, были взяты от человека и подготовлены третьей стороной или субподрядчиком. При рассмотрении рисков, связанных с использованием данных продуктов, производители должны запрашивать от их поставщиков подтверждение того, что оценка применимости человеческого материала основана на соответствующих требованиях настоящего стандарта или использовались альтернативные подходы. Полученная информация должна быть включена в отчет по менеджменту риском, касающийся медицинского продукта, содержащего клетки, и если применимо, то может быть дополнена информацией, поставляемой третьей стороной или субподрядчиком.
Приложение B
(справочное)
Графическое представление части процесса менеджмента риском для медицинских продуктов, содержащих клетки
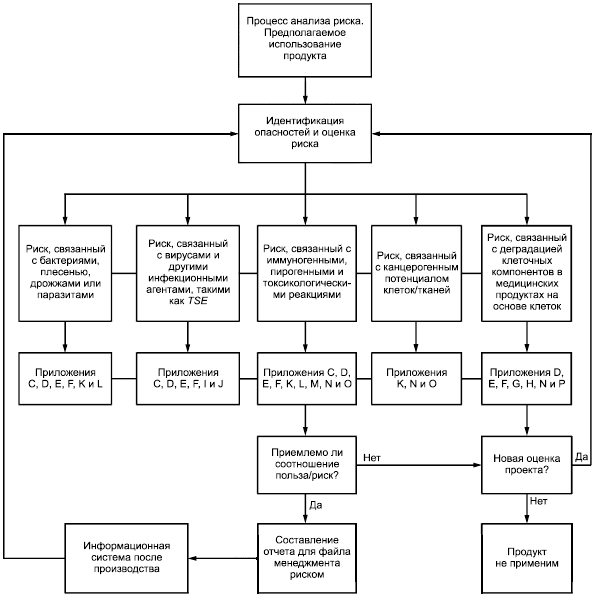
Рисунок B.1 - Графическое представление части процесса менеджмента риском
Приложение C
(обязательное)
Требования к выбору доноров и испытания
C.1 Общие положения
Критерии выбора доноров основаны на анализе рисков, связанных с применением определенных клеток или тканей, и предполагаемом назначении медицинского продукта на клеточной основе. Индикаторы этих рисков должны быть определены путем медицинского осмотра, рассмотрения истории медицинского и поведенческого риска, а также посредством биологических испытаний, патологоанатомических исследований (для трупных доноров) и других подходящих исследований. Доноров исключают из списка потенциальных доноров, если они подпадают под любой критерий из перечисленных в С.2.1, кроме тех случаев, когда их использование документально обосновано в ходе оценки риска, утвержденной ответственным лицом.
Если в стране, на территории которой будут использовать медицинский продукт, существуют дополнительные национальные или региональные критерии, кроме установленных, то они также применимы.
C.2 Трупные доноры
C.2.1 Общие критерии для исключения
a) Неизвестная причина смерти, за исключением тех случаев, когда вскрытие дает информацию о причинах смерти, и ни один из общих критериев исключения, определенных в настоящем пункте, не применим.
b) История заболевания неясной этиологии.
c) Наличие в настоящее время или в анамнезе онкологических злокачественных заболеваний. Возможными исключениями для данного критерия являются первичная базальноклеточная карцинома, карцинома in situ шейки матки и некоторые первичные опухоли центральной нервной системы, которые могут быть приемлемыми при наличии известной клинической истории донора.
d) Системные инфекции, которые не контролируются на момент забора материала, включая бактериальные заболевания, системные вирусные, грибковые или паразитарные инфекции либо существенные локальные инфекции в забираемых тканях и клетках.
e) ВИЧ (вирус иммунодефицита человека), острый или хронический гепатит В (за исключением лиц с подтвержденным иммунным статусом), гепатит C и HTLVI/II (вирус Т-клеточного лейкоза человека I/II) в анамнезе, клинически или лабораторно подтвержденный, а также риск заражения или свидетельства факторов риска данных инфекций.
f) Хроническое системное аутоиммунное заболевание, которое может повлиять на качество забираемых клеток или тканей, в анамнезе.
g) Признаки того, что результаты лабораторных исследований образцов донорской крови будут недействительны по причине:
1 случаев гемодилюции, если получить образцы крови до проведения этой процедуры невозможно; или
2 лечения иммуносупрессорами.
h) Свидетельства любых других факторов риска инфекционного заболевания на основе оценки риска с учетом истории перемещения донора и его контактов, а также локальной распространенности инфекционных заболеваний.
i) Наличие на теле донора физических признаков, которые могут указывать на риск инфекционного(ых) заболевания(ий).
j) Прием внутрь или воздействие веществ (таких как цианид, свинец, ртуть, золото), которые могут быть переданы реципиенту в дозе, которая может быть опасна для его здоровья.
k) Недавняя вакцинация живым ослабленным вирусом, если считается, что существует риск заражения.
I) Трансплантаты из ксенотканей.
m) Риск передачи заболеваний, вызываемых инфекционным агентом TSE.
Данный риск характерен, например:
1) для людей с диагностированной болезнью Крейтцфельдта-Якоба, или вариантной болезнью Крейтцфельдта-Якоба, или имеющих в семейном анамнезе заболевание Крейтцфельдта-Якоба неятрогенного характера;
2) людей с быстро прогрессирующей деменцией или дегенеративными неврологическими заболеваниями, включая заболевания неизвестного происхождения, в анамнезе;
3) реципиентов гормонов, полученных из гипофиза человека (например, гормонов роста) и реципиентов с пересаженной роговицей, склерой и dura mater, а также лиц, которые перенесли недокументированное нейрохирургическое вмешательство (при котором могла использоваться dura mater). Для вариантной болезни Крейтцфельдта-Якоба рекомендуется использовать дальнейшие меры предосторожности.
C.2.2 Дополнительные критерии для детского посмертного донорства
Дети, рожденные от матери, инфицированной ВИЧ или подпадающей под любой критерий исключения из описанных выше, должны быть исключены из списка доноров, до тех пока нельзя будет точно исключить риск передачи инфекции.
Дети в возрасте младше 18 мес, рожденные от матерей, инфицированных ВИЧ, а также при наличии гепатита B, C или вируса HTLVI/II, или с риском наличия такой инфекции, которые получали грудное молоко матери в течение предыдущих 12 мес, не могут быть донорами вне зависимости от результатов аналитических исследований.
Дети, рожденные от матерей, инфицированных ВИЧ, при наличии гепатита В, С или вируса HTLVI/II, или с риском наличия такой инфекции, которые не получали грудное молоко матери в течение предыдущих 12 мес и для которых проведение аналитических исследований, медицинского осмотра и обзор медицинской документации не подтвердили наличие вышеперечисленных инфекций, могут быть донорами.
C.3 Прижизненное донорство. Общие критерии отбора
C.3.1 Аутологичный живой донор
При заборе тканей и клеток следует применять тот же минимальный набор требований биологического тестирования, что и для аллогенного живого донора. Положительные результаты лабораторных исследований не обязательно препятствуют хранению, обработке и реимплантации тканей или клеток либо других продуктов, полученных из них, если имеется соответствующее изолированное хранение для обеспечения того, что нет риска перекрестной контаминации с другими трансплантатами, и/или нет риска загрязнения случайными веществами, и/или перепутывания.
C.3.2 Аллогенный живой донор
Аллогенных живых доноров должны отбирать на основе состояния их здоровья и медицинского анамнеза, который собирают с помощью анкетирования и интервьюирования доноров, проводимого квалифицированным и обученным медицинским персоналом. Такая оценка должна включать значимые факторы, которые могут повлиять на выявление и исключение лиц, донорство которых может представлять риск здоровью других, такой как вероятность передачи заболевания или риск их собственному здоровью. При любом донорстве процесс забора не должен влиять на здоровье донора или его ухудшать. В случае забора пуповинной крови или амниотической мембраны это правило применимо как к матери, так и к ребенку.
Критерии отбора аллогенных живых доноров должны быть установлены и отражены документально банком тканей (а также врачом, проводящим трансплантацию, в случае непосредственной передачи реципиенту), основываясь на конкретных донорских тканях или клетках, а также на физическом состоянии донора, медицинском и поведенческом анамнезе и результатах клинических исследований и лабораторных испытаний, определяющих состояние здоровья донора.
До- и послеродовой забор клеток требует оценки соответствующего риска для ребенка во время послеродового осмотра. Для забора клеток пуповинной крови до или после родов, а также амниотической мембраны мать рассматривают в качестве донора.
Следует применять те же критерии исключения, что и для трупных доноров, за исключением C.2.1, a).
В зависимости от забираемых типов клеток или тканей могут применять другие специфические критерии исключения, такие как:
a) беременность (за исключением доноров клеток пуповинной крови и амниотической мембраны и доноров-сибсов гемопоэтических прогениторных клеток);
b) грудное вскармливание;
c) гемопоэтические прогениторные клетки при возможности передачи наследственных заболеваний.
C.4 Биологические испытания, необходимые для доноров
Следующие биологические испытания должны проводить для всех доноров в качестве минимальных требований:
a) ВИЧ 1 и 2 - анти-ВИЧ-1, 2;
b) гепатиты B - HBsAg (поверхностный антиген гепатита B), HBc (антитела к сердцевине вируса гепатита B);
c) гепатиты C - anti-HCV-Ab (антитела к вирусу гепатита C);
d) сифилис - (см. далее).
Исследование на антитела к вирусу Т-клеточного лейкоза человека (HTLV) I/II следует проводить в соответствии с национальными нормативными документами.
Если anti-HBc положителен, а HBsAg отрицателен, то необходимы дополнительные исследования с оценкой риска для определения возможности клинического использования донорского материала.
Следует использовать валидированный алгоритм лабораторных исследований для исключения наличия активной инфекции Treponema pallidum. Нереактивный тест, специфичный или неспецифичный, допускает использование тканей и клеток. Если проводят неспецифичный тест, реактивный результат не препятствует забору или получению, если специфичный тест на Treponema pallidum, проводимый для подтверждения, не реактивен. Для донора, чей образец реактивен на Treponema pallidum при специфичном испытании, необходимо проведение оценки риска для определения допустимости клинического использования.
Могут потребоваться дополнительные исследования в зависимости от анамнеза донора и характеристик забираемых тканей или клеток, например RhD (D-антиген резус-группы), HLA (лейкоцитарный антиген человека), малярия, ЦМВ (цитомегаловирус), токсоплазмоз, EBV (вирус Эпштейна-Барра), Trypanosoma cruzi.
В отношении аутологичных доноров см. C.3.1.
C.5 Требования для определения биологических маркеров
C.5.1 Общие положения
Испытания должны проводить в квалифицированной лаборатории.
Примечание - Квалифицированной лабораторией является аккредитованная, лицензированная, зарегистрированная или авторизованная в соответствии с местными требованиями лаборатория.
Используемый тип испытаний должен быть утвержден для данных целей в соответствии с текущим уровнем развития науки. Биологические испытания должны проводить на сыворотке или плазме донора и ни в коем случае на других жидкостях или выделениях, таких как вода или стекловидное тело, если конкретно клинически не обосновано использование утвержденного испытания для таких жидкостей.
C.5.2 Гемодилюция
Если потенциальные доноры потеряли кровь и недавно получали от доноров кровь, компоненты крови, коллоиды или кристаллоиды, испытания на крови потенциальных доноров могут быть недействительными из-за гемодилюции образца крови. В следующих случаях необходимо использовать алгоритм оценки степени гемодилюции:
- забор образцов крови до наступления смерти: если кровь, компоненты крови и/или коллоиды были введены в течение 48 ч до забора крови или кристаллоиды были введены за 1 ч до забора образцов;
- забор образцов крови после наступления смерти: если кровь, компоненты крови и/или коллоиды были введены в течение 48 ч до смерти или кристаллоиды были введены за 1 ч до смерти.
Банк тканей может принимать ткани и клетки от доноров, которые были исследованы на определенные инфекционные заболевания, используя образец крови, который предположительно разведен более чем на 50%, только если испытания утверждены для использования такого образца или если доступен образец, который был взят до переливания/введения.
C.5.3 Забор образцов от трупных доноров
В случае трупных доноров образцы крови должны быть получены непосредственно перед смертью, или, если это невозможно, забор образцов должен быть в течение 24 ч после смерти.
C.5.4 Забор образцов от живых доноров
a) В случае живых доноров (за исключением, по практическим причинам, аллогенных доноров стволовых клеток костного мозга и стволовых клеток периферической крови) образцы крови должны быть получены в момент забора материала или, если это невозможно, в течение семи дней после забора материала (это "образец донорского материала").
b) Если ткани и клетки аллогенных живых доноров могут храниться в течение длительного периода времени, требуется повторный забор образцов и проведение испытаний через 180 дней. При повторных испытаниях образец донорского материала может быть получен за 30 дней до и через 7 дней после забора материала.
c) Если ткани и клетки аллогенного живого донора не могут храниться в течение длительного периода времени и повторное получение образцов, следовательно, невозможно, то применяется пункт а).
Если для живых доноров (за исключением доноров стволовых клеток костного мозга и стволовых клеток периферической крови) "образец донорского материала", как определено в a), дополнительно исследуют методом амплификации нуклеиновых кислот (nucleic acid ampliication technique, NAT) на ВИЧ 1 и 2, HBV (вирус гепатита B) и HCV, испытания повторных образцов крови в соответствии с b) не требуется. Повторные испытания также не требуются, если обработка включает этап инактивации, который был утвержден для упомянутых вирусов.
В случае забора стволовых клеток костного мозга и стволовых клеток периферической крови образцы крови должны быть взяты для исследования в течение 30 дней до забора материала.
В случае неонатальных доноров биологические исследования могут быть проведены на матери донора для предотвращения необязательных медицинских процедур для грудного ребенка.
Приложение D
(справочное)
Руководство по забору тканей
D.1 Общие положения
Материал клеток и тканей должен быть получен от идентифицируемых должным образом доноров при получении их согласия. Специальные принципы применяют для аутологичных, живых и трупных доноров.
D.2 Согласие и идентификация доноров
До процедуры получения тканей и клеток должно быть получено согласие на процедуру и документально отражено, как и кем был достоверно идентифицирован донор.
D.3 Оценка донора для аллогенных доноров
Должна быть собрана и записана соответствующая медицинская и поведенческая информация донора в соответствии с требованиями, описанными в D.5.
Для получения соответствующей информации следует использовать различные соответствующие источники, включая, по меньшей мере, собеседование с донором для живых доноров и следующее, если применимо:
a) медицинская документация донора;
b) собеседование с лицом, хорошо знавшим донора (для посмертных доноров);
c) собеседование с лечащим врачом;
d) собеседование с врачом-терапевтом;
e) протокол результатов вскрытия.
Кроме того, в случае посмертного донорства и в случае прижизненного донорства при наличии обоснования должен быть проведен медицинский осмотр тела для выявления любых признаков, которые сами по себе могут быть достаточным основанием для исключения из списка доноров или которые должны быть рассмотрены в свете медицинского и личного анамнеза донора. Полная документация донора должна быть пересмотрена и проанализирована на предмет соответствия требованиям, предъявляемым к донорам, и подписана квалифицированным медицинским работником.
D.4 Процедура получения тканей и клеток
Процедура получения должна соответствовать типу донора и типу забираемых тканей или клеток. Должны быть установлены процедуры для защиты безопасности прижизненного донора. Процедуры получения должны сохранять те свойства тканей или клеток, которые необходимы для их окончательного клинического использования, и в то же время минимизировать риск микробиологического загрязнения в течение процесса.
Для забора органов и тканей от трупных доноров площадь доступа должна быть ограничена. Необходимо использовать локальное стерильное поле, ограниченное стерильной простыней. Персонал, осуществляющий забор, должен быть одет соответственно типу получения. Обычно это подразумевает, что персонал вымыт, одет в стерильную одежду и стерильные перчатки, а также имеет защиту для лица и хирургические маски.
В случае трупных доноров место вскрытия должно быть отражено документально и определен временной интервал между смертью и вскрытием, чтобы гарантировать сохранение необходимых биологических и/или физических свойств тканей и/или клеток.
Любые негативные события во время забора, которые нанесли или могут нанести вред живому донору, а также результат какого-либо расследования по определению причин необходимо отразить документально и рассмотреть.
Необходимо установить правила и процедуры для сведения к минимуму риска загрязнения тканей или клеток персоналом, который может быть инфицирован трансмиссивными заболеваниями.
Для забора тканей и клеток должны использовать стерильные инструменты и устройства. Инструменты или устройства должны быть соответствующего качества, валидированы и нормативно сертифицированы и содержаться в рабочем состоянии для забора тканей и клеток. Если применяют инструменты многократного использования, соответствие утвержденному процессу чистки и стерилизации для удаления инфекционных агентов должно быть отражено документально. Необходимо внедрить систему, которая позволит обеспечить контроль всех инструментов или устройств для забора до прихода донора или до момента получения тканей или клеток.
Весь соответствующий персонал должен пройти надлежащее обучение по использованию таких устройств. Необходимо сохранять документацию, подтверждающую, что весь соответствующий персонал прошел надлежащее обучение по использованию таких устройств.
D.5 Документация донора
D.5.1 Для каждого донора должна быть заведена история, содержащая:
a) личность донора, т.е. имя, фамилию и дату рождения (если в донорстве участвуют мать и ребенок, то имя, фамилию и дату рождения матери и имя, если известно, и дату рождения ребенка);
b) возраст, пол, медицинский и поведенческий анамнез (собранной информации должно быть достаточно для применения критериев исключения, если необходимо);
c) время с момента смерти до момента забора;
d) выводы на основании осмотра тела, если применимо;
e) формулу гемодилюции, если применимо;
f) форму информированного согласия/разрешения, если применимо;
g) клинические данные, результаты лабораторных исследований и результаты других проведенных испытаний;
h) результаты вскрытия, если проводилось (для тканей и клеток, которые не могут храниться в течение длительного периода времени, должен быть записан предварительный устный отчет о вскрытии);
i) для доноров гемопоэтических прогениторных клеток пригодность донора выбранному реципиенту;
j) при независимом донорстве, когда организация, ответственная за забор имеет ограниченный доступ к данным реципиента, необходимо предоставить организации, проводящей трансплантацию, данные о доноре, достаточные для подтверждения пригодности донора.
D.5.2 Организация, проводящая забор, должна создавать отчет о заборе, который передается вместе с тканями.
Организация, проводящая забор, должна создать отчет о заборе, который передает в банк тканей. Данный отчет должен содержать, по меньшей мере, следующее:
a) идентификацию, наименование и адрес банка тканей, который получает клетки или ткани;
b) данные личности донора (включая информацию, как и кем была установлена личность донора);
c) описание и идентификацию забранных тканей и клеток (включая образцы для исследований);
d) личность лица, ответственного за забор, включая подпись;
e) дату, время (если применимо, время начала и окончания) и место забора, а также примененные стандартные рабочие процедуры, включая любые происшествия; если применимо, то условия окружающей среды в учреждении, в котором проводили забор (описание физической области, в которой происходил забор материала);
f) для посмертного донорства условия, в которых хранится труп, т.е. заморожен (или нет), время начала и окончания заморозки;
g) идентификационный номер/номер партии использованных реагентов и растворов для транспортирования.
Отчет должен также содержать дату и время смерти. Все записи должны быть четкими и читаемыми, защищенными от посторонних поправок; они должны храниться и быть доступными в данном состоянии в течение всего установленного периода хранения в соответствии с законами о защите данных.
Приложение E
(обязательное)
Требования к обращению с клетками и тканями во время производства
E.1 Общие положения
Процесс производства медицинских продуктов, содержащих клетки, должен быть тщательно разработан и утвержден для обеспечения стабильности продукции. Все требования должны быть определены и обоснованы.
Если в стране, в которой будут использовать медицинский продукт, существуют дополнительные национальные или региональные требования свыше определенных в данном приложении, то применимы такие требования.
Примечание - Руководства по специальным требованиям к обращению с клетками и тканями во время производства можно найти по ссылкам [38] и [46].
E.2 Спецификации
Требуется наличие спецификаций для используемых клеток и тканей, всех производственных добавок, промежуточного и медицинского продукта. Спецификации должны быть документально зафиксированы.
E.3 Документация
Требуется наличие процедуры, отраженной документально, с подробным описанием производства клеточных компонентов и конечного продукта. Должен быть описан тип манипуляций, необходимых для обработки клеток и поддержания физиологической функции клеток. Должна быть подготовлена блок-схема всего процесса, начиная с биологической жидкости, ткани или органа либо с клеточного фонда, с указанием критических этапов и промежуточных продуктов (например, промежуточные партии клеток), а также рабочих параметров, параметров технологического контроля и критериев приемки.
E.4 Комбинированные медицинские продукты на клеточной основе
Производство медицинского продукта, состоящего из клеток и матриц/устройств/каркасов, требует дополнительного внимания относительно взаимодействия между клетками/матрицей/каркасом и вопросов качества, связанных с этими компонентами.
Матрицы/устройства/каркасы должны соответствовать своим нормативным документам и стандартам.
Необходимо обратить внимание на биодеградируемые материалы, которые могут, например, потенциально изменить окружающую среду (например, повысить значение pH) клеток во время производства или после введения.
E.5 Производственное помещение
Производственное помещение должно быть пространственно отделено от места забора. Если в одном производственном помещении обрабатывают разные тканевые и клеточные продукты, существует повышенный риск перекрестного загрязнения во время каждого этапа производства, например через рабочее оборудование. Следовательно, необходимо внедрить соответствующие меры по чистке и контролю для предотвращения перекрестного загрязнения.
Помещение и оборудование для производства медицинских продуктов на клеточной основе должны быть соответствующими и аттестованными для асептического производства. Рекомендуется по возможности использовать отдельное, специфическое для продукта или одноразовое оборудование.
Для требований, относящихся к забору, см. приложение D.
E.6 Процедуры обращения с клетками
E.6.1 Общие положения
Все процедуры обращения с клетками должны быть обоснованы с учетом их предполагаемого использования. Влияние манипуляций клеток или тканей на всех этапах процесса производства рассматривают в процессе менеджмента риском. Значимость изменений (например, источник клеток, исходные материалы, производственные платформы, производственные зоны) требует особого внимания.
Все клеточные процедуры должны проводить в соответствии с документально подтвержденными стандартными рабочими процедурами. Различные этапы процесса должны быть утверждены. Должен быть определен соответствующий контроль. Микробиологический контроль является ключевым аспектом контроля процесса и менеджмента риском всех клеточных процедур. По возможности следует проводить контроль клеток, выращенных in vitro, на избранных этапах производства. Культура должна быть исследована на любое микробное загрязнение в соответствии с процедурами выращивания культивирования и характеристиками роста клеток.
Руководство по специальным требованиям к обращению с клетками и тканями во время производства можно найти по ссылке [38].
E.6.2 Отделение органа/ткани
Процедура отделения органа и/или ткани должна быть описана с упоминанием используемого метода и примененных ферментов и среды. Следует учитывать степень повреждения тканей для сохранения предназначенной функциональной целостности клеточных препаратов и минимизации клеточных примесей в продукте (клеточный детрит, перекрестное загрязнение с другими типами клеток).
Необходимо тщательно рассмотреть контроль среды, в которой проводят отделение органа/ткани. Может быть полезным использовать ламинарный шкаф класса A.
E.6.3 Изоляция нужных клеточных популяций
Процедура, используемая для изоляции, и/или очищения, и/или обогащения нужной клеточной популяции, должна быть документально отражена надлежащим образом. Процесс, включая оборудование и реагенты, должен быть утвержден с учетом эффективности процесса для получения нужной клеточной популяции.
E.6.4 Культивирование клеток
Процесс культивирования клеток должен быть утвержден, а его эффективность рассмотрена с учетом предполагаемого использования. Этапы обработки должны быть разработаны соответствующим образом для защиты целостности и функции клеток. Любые манипуляции во время культивирования должны быть подробно отражены документально и тщательно контролироваться в соответствии с установленными процедурами контроля процесса. Продолжительность культивирования клеток и максимальное число пассажей клеток должны быть четко определены и утверждены. Должны быть определены соответствующие генотипические и фенотипические характеристики исходных клеточных культур, установленных линий клеток и полученных клеточных клонов, а также установлена их стабильность с учетом долговечности культуры. Должны быть продемонстрированы стабильность и воспроизводимость процесса культивирования клеток, а условия культивирования клеток, включая среду и продолжительность, - оптимизированы, определены и утверждены с учетом предполагаемой клинической функции продукта.
Особое внимание необходимо обратить на потенциал роста клеток в ответ на добавленные факторы роста, так как субпопуляции клеток могут иметь преимущество в росте при определенных условиях культивирования in vitro.
Обзор научной литературы можно применять при оценке возможности размножения вирусных загрязнителей во время культивирования клеток.
E.6.5 Изменение клеток
К клеткам можно применять различное воздействие (например, физическое, химическое и/или генетическое). Метод, используемый для изменения клеток, должен быть полностью описан. Процесс должен быть утвержден, а его эффективность оценена с точки зрения предполагаемого использования.
В случае генетических изменений клеток применяют соответствующие стандарты и руководства.
E.6.6 Клетки, культивируемые в или на матрице/устройстве/каркасе
Если клетки выращены непосредственно внутри или на матрице/устройстве/каркасе, качество комбинированного медицинского продукта на клеточной основе зависит от должным образом контролируемого процесса производства (см. E.1-E.5) и качества комбинируемого материала. Для таких продуктов необходимо учитывать влияние матрицы/устройства/каркаса на рост клеток, их функцию и целостность. Следует также учитывать влияние, которое клетки могут оказывать на изделие (например, на скорость распада) (см. приложение M).
E.7 Контроль в ходе процесса
Процесс производства должны контролировать посредством процедур внутри самого процесса на уровне критических этапов или промежуточных продуктов. Промежуточные клеточные или тканевые продукты являются продуктами, которые могут быть изолированы во время процесса; спецификации этих продуктов должны быть установлены для гарантии воспроизводимости процесса и стабильности конечного продукта. Должны быть описаны испытания и критерии приемки.
E.8 Определение партии
Целью определения партии является обеспечение стабильности и контроля. Должно быть предоставлено четкое определение производственной партии от источников клеток или тканей до маркировки конечного контейнера (т.е. размер, число пассажей/дупликаций клеток, стратегии сбора, система нумерации партий). В случае аутологичного материала произведенный продукт следует рассматривать как партию.
Приложение F
(обязательное)
Требования к упаковке и маркировке
F.1 Общие положения
Все полученные клетки и ткани, промежуточные и конечные продукты должны хранить и упаковывать стерильными для минимизации риска загрязнения и распада. Используемые контейнеры должны подходить для хранения и транспортирования соответствующих биологических материалов. Контейнеры должны защищать и сохранять безопасность и качество тканей, содержащихся в них. Должны быть установлены спецификации для всех частей упаковки.
F.2 Спецификации
Требуется наличие спецификаций для всех компонентов упаковки (например, тара и упаковка, а также первичная упаковка, вторичная и внешняя упаковка). Спецификации должны быть оформлены документально.
F.3 Документация
Требуется детальное описание процесса(ов) упаковки и упаковочных материалов.
F.4 Химические свойства упаковочных контейнеров
Первичная упаковка и материалы, которые вступают в непосредственный контакт с тканями должны подходить для этих целей. Медицинские материалы могут быть предпочтительной формой упаковки благодаря их биосовместимости и требований к контролю качества; тем не менее такой контроль может быть недостаточен и должен быть обоснован. Загрязнение продукта вымываемыми или выделяемыми из упаковочного материала веществами, которые являются токсичными, иммуногенными или пирогенными, необходимо избегать любыми способами.
Контейнер также должен быть инертным по отношению к продукту (например, диметилсульфоксид в продукте склонен вызывать вымывание веществ из некоторых форм упаковки).
F.5 Физические свойства упаковочных контейнеров
Контейнеры должны препятствовать разрушению клеток, тканей или конечного продукта из-за условий окружающей среды (например, температуры, света).
F.6 Загрязнение
Упаковка должна препятствовать загрязнению, вызываемому лицами, ответственными за упаковку и транспортированию тканей.
F.7 Маркировка
F.7.1 Для обеспечения контроля и однозначной идентификации тканей, или клеток, или конечного продукта, все транспортные контейнеры должны иметь этикетку.
На каждую упаковку, содержащую ткани, должна быть нанесена этикетка в момент забора ткани. Процедуры упаковки и маркировки должны быть определены и документально отражены, и на реальной практике необходимо следовать этим процедурам.
На первичном контейнере, содержащем ткани, должна быть, по меньшей мере, этикетка, идентифицирующая донора и/или код донора, а также тип тканей внутри контейнера. Если размер первичного контейнера позволяет, то на нем должна быть этикетка со следующей информацией:
a) личность донора и/или код донора и тип тканей;
b) дата и, если возможно, время забора материала;
c) предупреждения об опасности;
d) тип любых добавок (если используют);
e) в случае аутологичного донорства на этикетке должно быть указано "только для аутологичного использования"; в случае целевого донорства этикетка должна определять предназначенного реципиента.
Если на первичный контейнер нельзя нанести этикетку для включения всей информации, определенной в списке выше, эта информация должна быть включена в сопроводительную документацию, прикрепленную к первичному контейнеру. Аналогично образцы тканей или крови, находящиеся в контейнерах для испытаний, должны быть надлежащим образом маркированы для обеспечения их соотнесения с определенным донором и для предотвращения ошибки. Также на этих образцах должна присутствовать конкретная информация о времени и месте забора тканей.
F.7.2 Если ткани транспортируют третьи лица (например, транспортно-экспедиторская компания), каждый транспортный контейнер должен быть снабжен, по меньшей мере, следующей информацией:
a) маркировка "Осторожно", "Ткани и клетки" и "Обращаться с осторожностью";
b) идентификатор учреждения, из которого получена упаковка (включая адрес и номер телефона), и инициалы, фамилия лица в данном учреждении или уполномоченного представителя, с которым необходимо связаться в случае возникновения проблем;
c) идентификация компании, выполняющей обработку, банка тканей или другого учреждения назначения (включая адрес и номер телефона) и инициалы, фамилия лица или уполномоченного представителя, с которым необходимо связаться для доставки контейнера;
d) дата и время начала транспортирования с учетом его условий, необходимых для поддержания безопасности и качества тканей или клеток;
e) дополнительный текст - "Не облучать";
f) если известно, что донор положителен на определенные инфекционные заболевания, необходимо добавить следующий текст: "Биологическая опасность";
g) в случае аутологичного донорства тканей или клеток необходимо добавить следующий текст - "Только для аутологичного использования";
h) любые дополнительные спецификации, касающиеся условий хранения (например, "Не замораживать").
F.8 Требования к первичной упаковке
F.8.1 Первичная упаковка, используемая после забора или во время обработки (промежуточных или конечных продуктов)
После забора или других этапов процесса производства, все ткани и клетки, промежуточные продукты, включающие клетки или конечные продукты должны быть упакованы таким образом, чтобы свести к минимуму риск загрязнения. Они должны храниться при температурах, сохраняющих необходимые характеристики и биологические функции клеток или тканей. Упаковка должна также препятствовать загрязнению лиц, ответственных за упаковку и транспортирование тканей и клеток. Первичная упаковка включает в себя стерильный и плотный контейнер, который соответствует продукту и утвержден для предназначенного использования.
Предпочтительно, чтобы первичная упаковка была медицинским материалом (см. F.4). Пользователь должен утвердить выбранную форму упаковки, что позволит удостовериться в ее соответствии заданным спецификациям, в частности с учетом требований физической безопасности (если необходимо, устойчивость к низким температурам), химических свойств (отсутствие взаимодействия с контактируемыми продуктами) и микробиологических требований (стерильность и отсутствие пирогенного материала).
Необходимо привести описание типа тары и способа укупорки. Должна быть продемонстрирована совместимость тары и укупорки с продуктом, а также приведена информация о процедурах стерилизации контейнера и крышки. Может потребоваться дополнительная информация, если компоненты упаковки использованы при транспортировании и/или процедурах применения.
F.8.2 Первичная упаковка для образцов крови, полученных от донора
У донора клеток и тканей берут образцы крови для проведения соответствующих лабораторных исследований. Упаковка, используемая для забора и хранения образцов крови в банке крови, должна быть утверждена как подходящая для этой цели. Любые дополнительные образцы тканей или крови для исследований должны быть четко маркированы для обеспечения идентификации донора и включать отметку о времени и месте взятия образца.
F.8.3 Первичная упаковка для образцов, используемых для контроля качества продукта
Упаковка, используемая для сбора и хранения образцов полученных материалов, промежуточных и конечных продуктов, предназначенных для контроля качества, должна быть плотной и подходящей для типа образца и анализа. В частности, тип контейнера, способ его укупорки, тип и количество или концентрация вспомогательных материалов (среды), если применимо, должны быть обозначены по необходимости для предназначенного образца. Упаковка должна быть утверждена как соответствующая для предполагаемого использования. Отличия упаковки, используемой для продукта и для образцов в целях контроля качества, должны быть обоснованы.
F.9 Транспортный контейнер
Упакованные клетки или ткани следует перевозить в контейнере, соответствующем целям транспортирования биологических материалов и обеспечивающем безопасность и качество содержащихся в нем тканей или клеток. Данная упаковка обязательна для всех продуктов человеческого происхождения на всех этапах транспортирования и необходима для некоторых из этих продуктов во время забора, обработки и хранения для гарантии обеспечения асептических условий первичной упаковки (например, двойная упаковка для головки бедренной кости).
Для транспортирования продуктов в жидком состоянии внешняя упаковка должна быть герметичной, и в контейнер должно быть помещено достаточное количество абсорбирующего материала для предотвращения любого возможного выхода жидкости. В транспортном контейнере должна находиться первичная и внешняя упаковка. Обычная сопроводительная документация может быть помещена между внешним и транспортным контейнером. Транспортный контейнер должен быть устойчив к ударам, поддерживать стерильность первичной упаковки и исключать несанкционированный доступ.
Вне зависимости от статуса транспортируемого продукта (полученный материал, промежуточный или конечный продукт), этикетка контейнера должна включать информацию, указанную в F.7.
F.10 Упаковка отходов с биологическим риском от забора материалов или их обработки
Потенциально зараженные или зараженные отходы должны быть помещены в одноразовые контейнеры, плотно запечатаны, соответственно маркированы и подготовлены к сожжению. При потенциально зараженных жидких отходах контейнер должен содержать дезинфицирующее вещество.
Приложение G
(справочное)
Руководство по транспортированию
G.1 Общие положения
В данном приложении приведено руководство по транспортированию полученных клеток или тканей, промежуточных продуктов и транспортированию конечного продукта.
Все действия по транспортированию должны проводить в безопасных и контролируемых условиях, которые гарантируют сохранение свойств клеток, тканей и продукта, необходимых для предполагаемого использования, и предотвращать разрушение и загрязнение продукта. Выбранные виды транспорта должны подходить для данной цели с учетом биологических и логистических требований. Вид транспорта должен быть утвержден. Это необходимо закрепить письменно в соответствии с внутренним законодательством.
Методы транспортирования, описанные в настоящем стандарте, применяют относительно международного или национального транспортирования производителем или его представителем, без ущерба действующим нормативным актам, касающимся инфекционных агентов [например, IATA (International Air Transport Association), ADR (Accord Europeen Relatif au Transport International des Marchandises Dangereuses par Route)], продуктов, перечисленных ниже:
a) ткани, клетки, образцы крови, полученные при заборе от донора;
b) образцы для внутреннего и внешнего контроля качества тканей;
c) клетки для банка биологических продуктов и банка крови.
Руководство, описанное в данном приложении, не применяют при транспортировании отходов с биологическим риском и образцов для контроля окружающей среды в учреждении, в котором проходит процесс.
Продукт следует транспортировать в условиях, которые:
1) обеспечивают его безопасное хранение и целостность;
2) обеспечивают его доставку в течение определенного времени;
3) соответствуют требованиям гигиены и безопасности для окружающей среды и лиц, ответственных за транспортирование.
Данное приложение применяют при проведении следующих операций:
- подготовка контейнера;
- перевозка (или транспортирование);
- получение продукта производителем и пользователем.
G.2 Спецификации
Спецификации транспортирования должны определить тип транспортного контейнера и способ его идентификации, а также включение образцов, при наличии, и отчет о заборе для компании, выполняющей обработку.
G.3 Утверждение
Рабочее утверждение транспортирования ткани должно учитывать требования, касающиеся внутренней температуры контейнера, температуры окружающей среды, а также другие биологические, химические и физические параметры, важные для защиты клеток, или тканей, или продукта. Если применимо, необходимо учитывать сезонные колебания температуры. В том случае, если, например, ткань или продукт следует поддерживать в определенном диапазоне температур во время транспортирования, рекомендуется, чтобы внутрь упаковки был помещен соответствующий поверенный датчик температуры для контроля и документальной фиксации внутренних температур.
В заранее определенных правилах и условиях транспортирования должны быть отмечены его сроки, которые обеспечивают сохранение необходимых свойств клеток и тканей.
Если в контейнер добавлен твердый диоксид углерода или жидкий азот либо использованы эвтектические аккумуляторы холода или тепла, они должны быть в достаточном количестве, чтобы поддерживать в контейнере предусмотренные температуры в течение времени, вдвое большего, чем было указано вначале. Их положение в контейнере должно равномерно поддерживать температуру по всему объему контейнера.
G.4 Документация
Дата и, если применимо, время комплектации упаковки в организации, выполняющей забор, и доставки производителю или компании, выполняющей обработку, должны быть отражены документально.
G.5 Перекрестное загрязнение
Необходимо предотвратить доступ к упаковке и ее содержимому посторонних третьих лиц во время транспортирования. Если необходимо, следует использовать герметизирующий внешний транспортный контейнер. Отклонения или происшествия, случившиеся за время транспортирования, должны быть сообщены организации, ответственной за забор, и производителю или компании, ответственной за обработку.
G.6 Контракты
Контракты на транспортирование должны заключать с квалифицированными транспортно-экспедиторскими компаниями. Транспортно-экспедиторская компания должна гарантировать точное соблюдение определенных, утвержденных условий транспортирования. Организации, ответственные за забор, могут передавать ответственность за транспортирование полученных тканей или клеток производителю, компании, ответственной за обработку, или третьим лицам. В таких случаях должны быть подписаны письменные соглашения, определяющие ответственность третьих лиц и подробно описывающие процедуры, которым необходимо следовать.
G.7 Подготовка транспортного контейнера
Должны быть установлены процедуры, определяющие следующее:
a) условия подготовки контейнера;
b) маркировка контейнера;
c) инструкции по транспортированию;
d) документы, передаваемые лицу, ответственному за транспортирование.
В тех случаях, когда транспортные контейнеры содержат жидкости, сухой лед или жидкий азот, этикетки и документация должны соответствовать конкретным письменным требованиям, установленным для данных случаев.
Учреждение, ответственное за транспортирование, или производитель должны проверять соответствие транспортного контейнера соответствующим спецификациям.
G.8 Транспортирование продукта
Между организацией, ответственной за транспортирование, и организацией получателя должно быть заключено подробное соглашение, описывающее обязанности по всем вопросам процесса транспортирования.
Относительно графика и маршрута должны быть установлены инструкции, содержащие следующее:
a) вид транспорта (включая, например, конкретное оборудование, обслуживание, гигиену);
b) маршрут и график движения;
c) контроль соответствующих параметров окружающей среды, например температуры.
G.9 Обязанности производителя
Производитель должен осуществлять все операции, касающиеся транспортирования исходных материалов к конечному продукту. Эти задачи могут быть также переданы одному или более субподрядчикам. Производитель ответственен за проверку того, что субподрядчик достаточно квалифицирован для выполнения задачи.
Если транспортирование осуществляет производитель, оно должно происходить в соответствии с процедурами, описывающими упаковку транспортируемого продукта, условия и способ транспортирования, маршрут, максимально допустимую задержку между отправлением и доставкой, обязанности всех лиц, участвующих в транспортировании и контроль доставки. Условия транспортирования должны быть выбраны с учетом критериев безопасности и условий хранения, соответствующих транспортируемому продукту.
Персонал, ответственный за транспортирование, должен получить соответствующие инструкции, касающиеся сопряженных рисков (например, инфекционных заболеваний) и типа транспортируемого продукта. Должны быть установлены стандартные рабочие процедуры, четко описывающие меры, принимаемые для защиты продукта, и проведена аттестация ответственных за транспортирование лиц.
Если в экстренном случае обоснована исключительная необходимость задействовать субподрядчика, который не обладает надлежащей квалификацией, производитель должен дать субподрядчику точные письменные инструкции по необходимым для продукта условиям транспортирования.
G.10 Материал и гигиена
Материалы и оборудование, используемые для транспортирования продукта (например, средства передвижения, емкости с температурным контролем, контейнеры) должны соответствовать своему назначению и должны быть утверждены при необходимости с учетом определенных требований к транспортированию каждого типа продукта. В частности, следует учитывать продолжительность транспортирования, поддержание необходимых температур хранения во время транспортирования, специальный или укомплектованный транспорт, гигиену и защиту. Материалы и оборудование должны подвергать профилактическому осмотру на постоянной основе, проводимому в соответствии с определенными процедурами.
Любые неисправные материалы или оборудование должны быть исключены из логистической цепочки либо, по меньшей мере, четко маркированы как неисправные, пока они ожидают ремонта или утилизации.
Устройство регистрации данных для контроля температуры должно иметь соответствующий диапазон и соответствующую точность для отображения температур хранения и транспортирования продукта. Его следует калибровать и поверять через регулярные интервалы.
Транспортные контейнеры и вторичная упаковка многократного использования должны оставаться чистыми и подвергаться чистке и дезинфекции в соответствии с описанием, приведенным в процедурах.
Средства передвижения, используемые для наземного транспортирования, должны регулярно чистить и обеззараживать в случае загрязнения продуктом.
G.11 Упаковка и маркировка транспортного контейнера
См. информацию, приведенную в приложении F.
G.12 Документация, касающаяся транспортирования
Одновременно с передачей контейнера перевозчику или его представителю должна быть передана специальная транспортная форма.
В порядке подготовки к любым авариям или происшествиям, которые могут случиться во время транспортирования, перевозчику должны быть даны четкие инструкции, точно определяющие:
a) характер опасности, представляемой транспортируемыми продуктами, а также меры предосторожности, которые необходимо принять при столкновении с ней;
b) меры, которые необходимо принять для персонала и продукта в случае поломки или износа упаковки, особенно если продукт вышел за пределы упаковки.
Эти инструкции должны включать список лиц, с которыми следует связаться при необходимости [имя, адрес, и номер(а) телефона].
G.13 Условия и продолжительность транспортирования
Транспортирование продуктов на клеточной основе должны осуществлять в сжатые сроки, по мере возможности. Продолжительность и условия транспортирования следует контролировать либо с помощью системы, которая гарантирует поддержание температур, либо посредством жесткого соблюдения этапов транспортирования (например, задержки и условий хранения). Этот контроль, в частности, включает информацию относительно:
a) установленной продолжительности транспортирования в соответствии с конечным пунктом назначения;
b) маршрута между учреждением, ответственным за доставку, и пунктом назначения продукта и, в конечном счете, местом промежуточного хранения до прибытия продукта в место его окончательного хранения.
G.14 Доставка и контроль в месте получения
Должно быть проверено соответствие определенным условиям транспортирования, в особенности, относительно:
a) целостности контейнера и этикетки;
b) условий контроля соответствующих параметров окружающей среды (например, температуры) во время транспортирования;
c) продолжительности транспортирования.
Любые отклонения или несоответствия, выявленные во время транспортирования, должны быть надлежащим образом отражены. Если отклонения зафиксированы, рекомендуется, чтобы данная информация передавалась отправителю и в организацию или компанию, ответственную за транспортирование. Должны быть проведены расследования для определения причин и предприняты корректирующие или профилактические действия.
G.15 Получение клеток или тканей
Когда ткани или клетки доставлены в организацию, ответственную за ткани, должна быть документально отражена проверка на предмет того, что вся партия, включая условия транспортирования, упаковку, маркировку, сопутствующую документацию и образцы, соответствует требованиям настоящего стандарта и спецификациям получающей организации.
Каждая организация должна гарантировать, что полученные ткани и клетки будут находиться в карантине до тех пор, пока они, вместе с сопутствующей документацией, не будут проинспектированы или определенным образом утверждены на соответствие требованиям. Обзор соответствующей информации о доноре и заборе и, следовательно, приемлемости донорства должны проводить определенные лица.
Каждая организация, ответственная за ткани, должна иметь документально отраженные правила и спецификации, на соответствие которым проверяют каждую партию тканей и клеток, включая образцы. Они должны включать технические требования и другие критерии, которые организация, ответственная за ткани, считает важными для поддержания приемлемого качества. Организация, ответственная за ткани, должна иметь документированные процедуры управления и отделения несоответствующих партий или партий с неполными результатами испытаний для гарантии отсутствия риска загрязнения других обрабатываемых, сохраняемых или хранящихся тканей и клеток.
Данные, регистрируемые организацией, ответственной за ткани, должны включать:
a) согласие/разрешение, включающее цель(и), для которой(ых) могут использовать ткани и клетки (т.е. терапевтические, или исследовательские либо терапевтическое использование и исследования) и любые конкретные инструкции по утилизации, если ткани или клетки не используют для целей, на которые получено согласие;
b) всю необходимую документацию по забору и анамнезу донора в соответствии с D.5;
c) результаты физических исследований, лабораторных исследований и других испытаний (такие как протокол вскрытия);
d) для аллогенных доноров соответствующим образом документально подкрепленный обзор полной оценки донора по критериям отбора, сделанный уполномоченным и обученным лицом;
e) в случае клеточных культур, предназначенных для аутогенного использования, документацию по возможности проявления у реципиента лекарственных аллергий (таких как аллергия на антибиотики).
Приложение H
(справочное)
Руководство по хранению
H.1 Общие положения
Требования данного приложения применимы как к хранению полученных клеток или тканей, так и клеточных компонентов конечного продукта.
Полученный клеточный материал, подвергшийся обработке, может представлять собой:
a) суспензию первичных культивируемых клеток, используемую непосредственно для медицинского продукта на клеточной основе;
b) первичные клетки, культивированные в течение нескольких пассажей перед использованием для медицинского продукта, содержащего клетки;
c) клетки четко определенной системы банков клеток, состоящей из посевной серии, рабочей посевной серии, главного банка клеток и рабочего банка клеток.
Должны быть установлены и применены процедуры контроля зоны хранения для предотвращения любых ситуаций, которые могут негативно повлиять на функциональность и целостность тканей и клеток. Все процессы хранения должны проводить в контролируемых условиях.
Условия хранения должны обеспечивать жизнеспособность, плотность, чистоту, стерильность, функциональность и активность клеток.
Должна быть установлена соответственно контролируемая система хранения клеток для обеспечения надлежащего поддержания и получения клеток, тканей и медицинских продуктов на клеточной основе без какого-либо нарушения их предполагаемых конечных характеристик. Идентификацию следует проверять с помощью соответствующих генетических и/или фенотипических маркеров, а пропорцию клеток, имеющих эти маркеры идентификации, оценивают как индикатор предполагаемой клеточной популяции.
H.2 Помещение
Помещение для хранения должно быть достаточного размера с защищенным доступом. Оно должно быть оборудовано и поддерживаться таким образом, чтобы гарантировать качество и контроль хранимых продуктов. Помещение должно быть чистым и сухим. Если необходимо, должны контролировать определенные пределы температуры и влажности.
H.3 Персонал
Доступ в помещение должен иметь только надлежащим образом обученный и утвержденный персонал.
H.4 Спецификации
Должны быть определены условия хранения для гарантии качества клеток или тканей с учетом идентификации, чистоты, жизнеспособности и функциональности клеток. Необходимо предотвращать загрязнение микробиологическими веществами, а также перекрестное загрязнение клетками или нуклеиновыми кислотами иного происхождения.
H.5 Проверка и утверждение
Оборудование или устройства для хранения должны быть пригодными, а условия хранения утверждены.
Рекомендуется вести документацию, которая подтверждает пригодность оборудования или устройств, используемых для хранения тканей или клеток. Спецификации, выбранные для оборудования для хранения, должны гарантировать, что оборудование способно поддерживать необходимую контролируемую среду. Должна существовать процедура, описывающая установленные условия, обязанности и спецификации для утверждения хранения, переноса или распределения каждого типа тканей и клеток.
Должно быть определено максимальное время хранения для каждого типа условий хранения, основываясь на данных стабильности в режиме реального времени (и, если применимо, ускоренных испытаний), используя соответствующие методы испытания. Выбранный период должен быть ограничен, кроме всего прочего, возможным ухудшением функциональных свойств тканей и клеток.
H.6 Контроль и документация
Необходимо обеспечить, чтобы все процедуры, связанные с хранением тканей и клеток, были документально отражены в ходе стандартных рабочих процедур, а условия хранения соответствовали определенным требованиям.
Должны быть использованы системы непрерывного контроля для гарантии того, что необходимые условия окружающей среды поддерживают в течение срока хранения. Соответствующие данные следует записывать и периодически анализировать. Необходимо использовать системы сигнализации, которые способны предупреждать персонал до наступления критических условий. Рекомендуется разработка правил и процедур для аварийного переноса тканей или клеток в предусмотренное альтернативное устройство или помещение для хранения и для альтернативных методов контроля в случае отказа оборудования или источника питания.
Должны быть документально отражены все этапы обращения с тканями или клетками, которые требуют поддержания контролируемых температур или других параметров хранения. Система документации должна гарантировать контроль условий хранения тканей или клеток.
H.7 Перекрестное загрязнение
Для предотвращения перекрестного загрязнения помещение для хранения должно быть специально отведенным. Оно должно быть физически отделено от области забора и/или области производства.
Независимо от используемого метода хранения должны быть предусмотрены отдельные области для хранения неиспытанных клеток и тканей (область карантина) и испытанных материалов, готовых к передаче. Эти различные области должны быть четко обозначены.
Кроме того, должны быть определены и отведены физически отделенные области для хранения клеток или тканей, которые не соответствуют обычным спецификациям.
Загрязненные ткани и клетки, например те, которые имеют положительные маркеры на инфекционные заболевания, должны быть четко и заметно маркированы таким способом, который указывает на их конкретные характеристики. Например, загрязненный продукт может быть маркирован символом "биологическая опасность" (см. ИСО 7010). При необходимости эти клетки хранят в подходящей изолированной камере для хранения с целью предотвращения риска перекрестного загрязнения или путаницы. В частных случаях хранения в емкостях с жидким азотом эти продукты должны хранить только в паровой фазе.
Переносчики для подготовки генетически модифицированных клеток должны хранить в обозначенных и отведенных областях отдельно от клеток и тканей.
H.8 Хранение неклеточных материалов
В помещениях для хранения клеток и тканей области, предназначенные для хранения рабочих и расходных материалов, оборудования многократного использования (например, стекла) и вспомогательных продуктов, применяемых при обработке тканей и клеток, должны быть отделены от областей хранения тканей и клеток.
H.9 Хранение отходов с биологическим риском/биологической опасностью
Зараженные или потенциально зараженные отходы, полученные во время процессов хранения тканей и клеток, должны быть надлежащим способом отделены с учетом конкретных письменных требований для рисков инфицирования.
H.10 Стабильность
Для следующих материалов должен быть определен срок хранения клеток или тканей при определенных условиях хранения:
a) все хранимые промежуточные материалы, если применимо;
b) компоненты комбинированных медицинских продуктов на клеточной основе;
c) клетки или ткани;
d) конечный продукт.
Кроме того, для конечного продукта должен быть определен соответствующий срок годности при использовании (после открытия транспортного контейнера). Должны быть определены все условия хранения, включая диапазон температур. Условия транспортирования и хранения должны быть подтверждены экспериментальными данными с точки зрения поддержания целостности клеток и стабильности продукта в течение определенного периода срока действия. Если применимо, должны быть документально отражены соответствующие методы замораживания и размораживания.
Из-за сложного характера медицинских продуктов на клеточной основе требования к стабильности должны определять на индивидуальной основе. По возможности, стабильность должны оценивать как для клеточных, так и для неклеточных компонентов до объединения и совместно в качестве конечного продукта в конечной упаковке.
H.11 Условия хранения конечного продукта
Возможности хранения продуктов на основе клеток или тканей, как правило, ограничены. Тем не менее некоторые продукты могут допускать хранение, например, в замороженном виде. Для медицинских продуктов такого типа применяют все соответствующие разделы данного приложения.
Приложение I
(обязательное)
Требования к системе контроля
I.1 Общие положения
Следует использовать систему, которая обеспечивает полный контроль процесса от исходного материала до пациента и наоборот, сохраняя конфиденциальность пациента и донора. Это необходимо для контроля безопасности и эффективности медицинских продуктов на клеточной основе. Установление и поддержание данной системы следует реализовать таким образом, чтобы обеспечить согласованность, соответствие с требованиями контролируемости процесса и тщательность выполнения процедур.
I.2 Специальные требования
Для обеспечения анонимности донора должна быть установлена двухуровневая система, связывающая требуемый контроль процессов от донорства и забора клеток до производителя и пользователя (больницы или врача).
В организации, ответственной за ткани, должна быть связь между донором и полученными материалами. На стороне производителя должна быть связь между полученными материалами и продуктом. В больнице или у врача должна быть связь между продуктом и реципиентом.
Системы должны обеспечивать полный контроль процессов от донора до реципиента посредством анонимных кодирующих систем. Производители должны рационально устанавливать свои кодирующие системы, основанные на кодирующей системе организации, ответственной за ткани, и разработанные для обеспечения контроля процесса донорства продукта пациенту. Для целей обслуживания пациентов подходящими средствами могут быть системы штрих-кодов и снимаемых этикеток.
Руководство можно найти по ссылке [29].
Требования к маркировке определены в F.7.
Приложение J
(обязательное)
Меры по снижению риска, связанного с заражением вирусами и другими инфекционными агентами, такими как TSE*
________________
* В бумажном оригинале слово "TSE" в наименование Приложения J выделено курсивом. - .
J.1 Общие положения
Для поддержания характеристик медицинских продуктов на клеточной основе обычно живые клетки не могут быть подвергнуты воздействию физических или химических методов, которые эффективны для инактивации или удаления инфекционных агентов, таких как вирусы или агентов TSE. Пока методы инактивации или удаления инфекционных агентов не утверждены, безопасность медицинских продуктов на клеточной основе зависит от надлежащего выбора и тестирования доноров и пригодности исходных материалов, используемых во время обработки. Должны быть установлены строгие требования к источникам и критерии приемки для всех материалов человеческого происхождения для обеспечения того, что клетки или ткани, используемые в качестве исходного материала для медицинских продуктов на клеточной основе, не заражены, а материалы, используемые при обработке, не привносят инфекционных агентов.
Для медицинских продуктов на клеточной основе можно использовать непосредственно первичную культуру основных клеток, или выращенные клетки, или ткани, в течение нескольких циклов культивированные на протяжении нескольких пассажей, поэтому обширные испытания невозможны. Следовательно, необходимо использовать соответствующий режим исследования доноров, такой как в Европе (см. ссылку [35]) и США (см. ссылку [53]). Подробности приведены в приложении C. Объединение клеток может увеличить риск передачи заболевания. Если используют клеточные линии, по возможности, должны быть установлены соответствующие охарактеризованные главный банк клеток (Master Cell Bank, MCB) и рабочий банк клеток (Working Cell Banks, WCB).
При разработке и производстве медицинских продуктов на клеточной основе необходимо принимать соответствующие меры для минимизации риска передачи вируса; кроме того, необходимо учитывать риск заражения TSE, а также другие аспекты.
J.2 Источник заражения вирусом/TSE
J.2.1 Человеческие ткани и клетки
Процесс производства медицинских продуктов на клеточной основе обычно не включает конечную стерилизацию, этапы очистки, этапы удаления и/или инактивации вирусов. Следовательно, должны быть четко определены строгие требования к источникам, а также критерии приемки для всех материалов человеческого и животного происхождения в соответствии с предполагаемым использованием.
Обычно ткани и клетки человека могут содержать вирусы. Это патогенные вирусы человека, которые могут расти в условиях обработки и в отдельных случаях при росте клеток. Как правило, все вирусы, передающиеся через кровь, рассматривают как потенциальные контаминанты. Дополнительно следует рассматривать тканеспецифичные вирусы, такие как вирусы герпеса (CMV, EBV), связанные с клетками крови.
Необходимо учитывать риск заражения человеческих тканей агентом вариантной болезни Крейтцфельдта-Якоба (variant Creutzfeldt-Jakob disease, vCJD) и, если применимо, использовать соответствующий отбор доноров (см. cсылки [35] и [53]).
Производство медицинских продуктов на клеточной основе должно соответствовать принципам надлежащей производственной практики. Область производства должна быть физически отделена от области забора. Если в одной и той же области производства обрабатывают и хранят различные тканевые и клеточные продукты, существует повышенный риск перекрестного загрязнения на каждом этапе процедуры, например через оборудование для обработки или контейнеры для хранения, такие как емкости с жидким азотом. Следовательно, должны быть внедрены надлежащие меры контроля для предотвращения перекрестного загрязнения.
Лечебные продукты из аутогенных клеток могут также разрабатывать от вирус-положительных (например, ВИЧ, гепатит B, гепатит C) пациентов. Важно проверить, увеличиваются ли вирусы во время культивирования клеток ex vivo. Кроме того, должны быть внедрены строгие принципы предотвращения перекрестного загрязнения между вирус-положительными и вирус-отрицательными клеточными культурами.
J.2.2 Технологические добавки
Для получения, отбора, культивирования или даже генетической или фенотипической модификации клеток требуются различные материалы, такие как другие клетки, ферменты, антитела, цитокины, сыворотка и антибиотики. Каждое вещество, используемое во время процедуры, должно быть четко определено и оценено на предмет его соответствия предполагаемому применению. Качество биологически активных добавок в культуральной среде, таких как факторы роста, цитокины и антитела, должно быть документально отражено; кроме того, следует дать оценку их качества и безопасности. Необходимо контролировать даже безопасность аминокислот. Если они получены от материалов животного происхождения, должно быть приведено документальное подтверждение, что нет риска заражения животным TSE.
Для распространения клеток часто используют фетальную бычью сыворотку (fetal bovine serum, FBS). Хорошо известно, что FBS может быть заражена вирусами (см. ссылки [41], [61], [62]). Свиной трипсин, часто используемый для культивирования клеток, связан с риском заражения. Испытания сыворотки или трипсина, например заявленные поставщиками в сертификате об анализе (Certificate of Analysis, CoA), являются недостаточными для гарантии отсутствия заражения вирусами; причинами ограниченной значимости испытаний является чувствительность используемых методов выявления и малый объем, который может быть использован для испытаний. Необходимо избегать использования этих компонентов, если иное не обосновано и документально не отражено. Если обосновано, следует использовать инактивированную сыворотку и/или инактивированный трипсин. Если в качестве добавки к среде применяют человеческую сыворотку, отсутствие загрязнения вирусами должно быть обосновано соответствующими способами.
При использовании материалов FBS производитель должен продемонстрировать:
a) соответствие ИСО 22442 (все части стандарта); или
b) сертификат соответствия, принятый EDQM (European Directorate for the Quality of Medicines and Health Care, Европейским директоратом по качеству лекарственных средств и здравоохранению), или эквивалент.
Поставщик должен предоставить надлежащую документацию, демонстрирующую качество и безопасность по отношению к вирусам и TSE всех исходных материалов человеческого или животного происхождения. Очень важно учитывать этот момент с самого начала разработки продукта и вести документацию на все исходные материалы во время различных циклов разработки продукта.
J.3 Методы снижения риска
J.3.1 Тестирование донора человеческих тканей и клеток
Разработаны конкретные требования к выбору и тестированию доноров, например в Европе (см. ссылку [35]) и в США (см. ссылку [53]). Подробности приведены в приложении C. Статус вирусного инфицирования любого клеточного образца, включая аутогенные клетки, должен быть известен для обеспечения возможности соответствующего отделения инфицированных клеток и установления соответствующих мер безопасности при обслуживании и хранении клеток и материалов.
J.3.2 Испытания банков клеток
Если использованы клеточные линии, по возможности, должны быть установлены соответствующие охарактеризованные главный банк клеток (Master Cell Bank, MCB) и рабочий банк клеток (Working Cell Banks, WCB). Банки клеток, включая характеристику и исследования клеток, должны соответствовать Руководству ICH Q5D (см. ссылку [17]).
J.3.3 Отбор соответствующих исходных материалов и/или реагентов
Для получения, отбора, культивирования или модификации клеток требуются различные исходные материалы или реагенты. Качество биологических материалов должно быть документально отражено, и каждое вещество, используемое в процедуре, должно быть точно определено и оценено в соответствии с предполагаемым использованием.
Следует учитывать риск загрязнения случайными веществами, и особенно в оценке риска необходимо рассматривать все исходные материалы и/или реагенты человеческого или животного происхождения. Основной стратегией является избегание использования биологических материалов, если это возможно.
При невозможности, необходимо учитывать следующее.
При росте in vitro основных клеток сыворотка является соответствующей добавкой к среде, и многие клетки невозможно успешно культивировать без сыворотки. Если для снижения риска заражения рассматривают применение бычьей сыворотки, следует использовать только фетальную бычью сыворотку и, если возможно, инактивированные продукты FBS. Поставщики должны документально подтвердить, что проводимые процедуры инактивации эффективны в отношении инактивации вирусов и что перед инактивацией были проведены соответствующие испытания, которые продемонстрировали, за исключением BVDV, что сыворотка свободна от определяемых бычьих вирусов. Было показано, что гамма-излучение 30 кГр или выше эффективно для инактивации вирусов (см. ссылки [31], [36], [40]).
Если используют FBS, должны быть приняты меры для снижения риска TSE. Необходимо учитывать соответствующие стандарты, такие как ИСО 22442 (все части стандарта) и руководства (см. ссылку [41]).
Для технологических добавок животного происхождения необходимо учитывать возможную чувствительность пациента, такую как аллергические реакции и/или образование антител.
Риск заражения бычьими вирусами во время культивирования клеток возможно снизить, используя в качестве добавок к среде сыворотку человека, однако при этом необходимо принимать соответствующие меры для предотвращения заражения вирусами человека. В случае лечебных продуктов из аутогенных клеток сыворотка пациента может подходить для использования в качестве добавки к клеточной культуре.
Для культивирования клеток часто используют трипсин. Он может быть получен из свиных тканей и, следовательно, заражен свиными вирусами, особенно свиным парвовирусом и/или свиным цирковирусом типа 1/2. Оба вируса обладают высокой устойчивостью к тепловой и химической инактивации. Следовательно, если возможно, необходимо применять инактивированный свиной трипсин растительного происхождения или рекомбинантный трипсин.
Материалы, полученные от человека или животных, включая клетки, которые функционируют как поддержка для роста и адгезии, например питающие клетки, должны быть оценены и/или утверждены на их соответствие предполагаемому использованию. Если применяют клетки животных, они должны исследоваться на отсутствие выявляемых видоспецифичных вирусов с использованием соответствующих утвержденных проб. Можно ознакомиться с методическими документами на предмет выбора соответствующих вирусов для установления соответствующих режимов испытания (см. ссылки [16], [32], [54], [55]).
Если исходные материалы, реагенты и/или наполнители имеют лицензию на продажу или упомянуты в Фармакопее, необходимо сделать соответствующие ссылки. Если применимы другие методические документы, такие как для моноклональных антител (см. ссылки [30], [56]), или для медицинских продуктов, выделенных из плазмы (см. ссылку [42]), или для оценки вирусной безопасности лекарственных биопрепаратов (см. ссылку [16]), они также должны быть учтены. Такие материалы следует использовать в производстве медицинских продуктов на клеточной основе, только если их вирусная/TSE безопасность надлежаще продемонстрирована (см. ссылку [41]).
Приложение K
(справочное)
Руководство, касающееся опасностей, вызванных канцерогенным потенциалом человеческих клеток или тканей, используемых при производстве медицинских продуктов
В каждом отдельном случае необходимо учитывать, при возможности, риск вызвать образование опухолей из-за неопластической трансформации клеток реципиента и клеток медицинских продуктов на клеточной основе. Общепринятые исследования канцерогенности могут быть невыполнимы. Исследования канцерогенности могут проводиться in vitro и/или in vivo. Решение о проведении испытания следует принимать в каждом отдельном случае на соответствующих моделях и с учетом научной литературы. Исследования канцерогенности предпочтительно должны проводиться на клетках, которые находятся на пределе текущего культивирования клеток или даже за этим пределом. Ткани, выявленные как содержащие примененные клетки или выраженные продукты в исследованиях на биораспределение, должны также анализироваться с особым вниманием в ходе исследования канцерогенности.
Исследования на генотоксичность не считаются необходимыми для человеческих медицинских продуктов на клеточной основе, пока характер любого выраженного продукта не продемонстрирует непосредственное взаимодействие с дезоксирибонуклеиновой кислотой (ДНК) или другим хромосомным материалом.
Приложение L
(справочное)
Руководство, касающееся микробиологической контаминации
L.1 Общие положения
Для успешного применения продуктов на клеточной основе критически важно удостовериться в том, что медицинские продукты на клеточной основе свободны от занесенных микробных агентов (вирусов, микоплазмы, бактерий, грибов). Следует проводить анализ риска для оценки вероятности реактивации латентных (интегрированных, бессимптомных) форм занесенных агентов.
В отношении микробиологической контаминации необходимо рассматривать два основных источника. Может быть инфицирован сам клеточный материал и может произойти заражение на любом этапе процесса производства.
Для предотвращения микробиологической контаминации могут также применяться другие приложения настоящего стандарта.
L.2 Выявление микробиологического загрязнения исходного материала клеток или тканей
Для определения микробиологического статуса клеточного материала, по применимости, должна быть внедрена конкретная микробиологическая скрининговая программа для клеток, поступающих для использования в продукте. Эта скрининговая программа должна быть адаптирована к типу клеток и использовать утвержденные пробы, способные выявлять инфекционные агенты человека с соответствующей чувствительностью. Необходимо учитывать компоненты среды, которые могут повлиять на пробу (например, антибиотики).
Если клетки получены от нездоровых тканей, должны быть определены критерии приемки, соответствующие продукту и его предполагаемому использованию.
L.3 Предотвращение микробиологического загрязнения продукта во время производства
Для менеджмента риском крайне важно учитывать, что все этапы процесса производства могут вносить вклад в микробиологическое загрязнение конечного продукта. Следовательно, во время процесса разработки продукта необходимо принять соответствующие меры.
Необходимо провести скрупулезные испытания на предмет отсутствия бактерий, грибов и микоплазмы на уровне конечного продукта. В тех случаях, когда короткий срок годности продукта препятствует испытанию на отсутствие бактерий, возможны альтернативные утвержденные методы испытания, если они обоснованы.
Работа в контролируемой соответствующим образом окружающей среде и асептические методы производства обязательны для предотвращения микробиологического загрязнения. Руководство можно найти в ИСО 13408.
Приложение M
(справочное)
Руководство, касающееся потенциального негативного влияния неклеточных осадков продукта
M.1 Общие положения
Неклеточные осадки продукта могут появиться на любом этапе процесса производства (например, за счет использования добавок во время забора материала, растворов для хранения, растворов для криоконсервации, вымываемых продуктов, технологических добавок и/или промежуточных продуктов).
M.2 Неклинические испытания
Для выявления и оценки данных типов примесей могут быть применимы приемы и методы, определенные в ИСО 10993.
Любой материал, который может внести в продукт продукты распада на любом этапе процесса производства, например биоразлагаемые материалы, должен быть тщательно охарактеризован с учетом этих свойств, а влияние продуктов распада на клеточные компоненты должно быть оценено.
M.3 Комбинированные продукты
Неклеточные компоненты должны быть охарактеризованы в контексте их требуемой функции в конечном продукте. Это структурные компоненты, предназначенные для поддержания клеточных компонентов, например каркасы или мембраны, для которых должны быть определены и описаны химические и физические свойства, такие как пористость, плотность, микроскопическая структура и размер частиц, в соответствии с типом веществ и предполагаемым использованием согласно ИСО 10993-18 и ИСО 10993-19.
Приложение N
(обязательное)
Требования, касающиеся потенциального негативного влияния клеточных компонентов на медицинский продукт
N.1 Общие положения
Современный уровень развития науки показывает, что поведение биологически активных объектов, которыми являются клетки и ткани, непредсказуемо. Для расширения кругозора о потенциальном негативном влиянии продуктов на клеточной основе могут применять различные пробы in vitro и in vivo. Решение о наиболее подходящем испытании или комбинации испытаний принимают отдельно в каждом случае.
N.2 Доклинические испытания на негативное влияние
Доскональное изучение, применяемое во время неклинических испытаний, должно учитывать характер медицинского продукта на клеточной основе и быть пропорционально ожидаемому риску, связанному с клиническим использованием.
Вариативность медицинских продуктов на клеточной основе должна быть отражена в доклинических исследованиях.
Целью доклинических исследований являются демонстрация подтверждения принципа действия и определение фармакологического и токсикологического эффектов, фиксирующих человеческий ответ не только до начала клинических исследований, но также в течение клинической разработки. Задачи данных исследований включают следующее:
a) получение информации для выбора безопасных доз для клинических исследований;
b) получение информации для обеспечения безопасности, осуществимости и эффективности способа введения и схемы применения;
c) получение информации для подтверждения продолжительности воздействия и продолжительности времени наблюдения для выявления негативных реакций;
d) идентификация органов-мишеней с точки зрения токсичности;
e) идентификация параметров контроля пациентов, получающих данную терапию.
Доклинические исследования должны проводить на соответствующих животных моделях. Если не могут быть разработаны соответствующие животные модели, исследования in vitro могут заменить исследования на животных. Должны быть обоснованы аргументация доклинических исследований и критерии, используемые для выбора конкретной модели животных. Уровень экспрессии биологически активных молекул, путь введения и исследуемая доза должны отражать предполагаемое клиническое использование на людях.
Следует учитывать рекомендации Руководства ICH S6 (см. ссылку [21]). Количество животных, их пол, частота и продолжительность контроля должны быть соответствующими для выявления возможных негативных эффектов.
Необходимость репродуктивных исследований зависит от медицинских продуктов на клеточной основе и должна рассматриваться отдельно в каждом случае.
Приложение O
(справочное)
Руководство по описанию клеточных компонентов медицинских продуктов
O.1 Общие положения
Должна быть установлена полная характеристика клеточного компонента с точки зрения идентификации, чистоты, эффективности, жизнеспособности и соответствия предназначенному использованию. Характеристика должна быть разработана так, чтобы позволить проводить текущий контроль, который применяется для выпуска активных веществ и конечного продукта, а также контроль, проводимый на нескольких этапах процесса для гарантии стабильности партии.
Если биологически активные молекулы (например, факторы роста, цитокины) присутствуют в качестве компонента или осадка от производственного процесса медицинских продуктов на клеточной основе, они должны быть соответствующим образом описаны. Их сопряженные риски должны быть оценены.
Руководство по неклинической характеристике можно найти по ссылке [38].
O.2 Идентификация
Идентификация клеточных компонентов, в зависимости от популяции и происхождения клеток, должна быть охарактеризована с точки зрения фенотипических и/или генотипических профилей (генетический статус/дифференциальный статус). При рассмотрении фенотипа клеток, если обосновано, могут использовать соответствующие маркеры. Эти маркеры могут основываться на экспрессии гена, презентации антигена, биохимической активности, ответе на экзогенные стимулы, способности производить биологически активные или другие измеримые молекулы и т.д. Для адгезивных клеток совместно с другими испытаниями полезным инструментом может являться морфологический анализ.
Если применимо, необходимо предоставить описание процедур, которые могут привести к изменениям характеристик продукта, включая адгезию, абсорбцию, деградацию, представление компонентов в питательной среде.
Для клеточных компонентов аллогенного происхождения, если применимо, идентификация должна включать маркеры гистосовместимости и идентификацию генетических полиморфизмов с конкретными ссылками на предназначенное использование.
O.3 Чистота клеток
По сравнению с химическими веществами для продуктов на клеточной основе чистота является относительным термином. Сложно определить абсолютную чистоту такого продукта. По отношению к клеточным компонентам продуктов на клеточной основе термин "чистота клеток" используют для описания характеристик продукта, связанных с популяцией(ями) клеток, необходимой(ых) для выполнения требований, определенных для предназначенного использования продукта.
Общая популяция клеток может содержать другие клетки, которые имеют различное происхождение и/или степень дифференциации или могут не относиться к предназначенной клеточной популяции. Предусмотренные и непредусмотренные клеточные популяции, представленные в медицинском продукте, должны быть определены, а их количество в конечном продукте следует контролировать соответствующими мерами. Критерии приемки для количества клеток должны быть установлены в каждом случае отдельно.
Что касается смесей клеток и тканей, в тех случаях, когда желаемая биологическая активность и эффективность продукта требует использования сложной смеси клеток или тканей, смесь клеток и/или ткани должна быть охарактеризована, а ее состав - контролироваться соответствующими методами текущего контроля и испытаниями при выпуске продукта.
O.4 Примеси, полученные в ходе процесса
Во время производства медицинского продукта на клеточной основе в конечный продукт может попадать различное количество примесей, связанных с продуктом и процессом. Любые реагенты, известные как вредные для человека, должны быть исследованы в конечном продукте (или в отдельных компонентах, если иное невозможно) и установлены критерии приемки. Должны быть обоснованы допустимые пределы.
Для примесей, связанных с клетками, см. также приложение N.
Для примесей, связанных с матрицей/каркасом/устройством, применяют ИСО 10993.
Для примесей, связанных с обработкой и производством, см. приложение M.
O.5 Жизнеспособность
Независимо от типа клеток популяция клеток может загрязняться нежизнеспособными клетками. Так как жизнеспособность клеток является важным параметром для целостности продукта и непосредственно связана с биологической активностью, необходимо определять соотношение между жизнеспособными и нежизнеспособными клетками и устанавливать соответствующие спецификации.
O.6 Эффективность
O.6.1 Общие положения
Если предназначенное использование продукта связано с определяемой и измеряемой биологической активностью, испытания эффективности должны быть частью процесса менеджмента риском. Испытания эффективности должны проводить, если это действительно требуется и когда применимые системы исследований подходят для получения достоверных результатов, отражающих ситуацию in vivo.
В соответствии с Руководством ICH Q6B (см. ссылку [18]) эффективность определена как количественное измерение биологической активности, основанной на свойствах продукта, которые связаны с соответствующими биологическими свойствами. Настоятельно рекомендуется, чтобы разработка подходящей пробы эффективности начиналась в процессе разработки продукта. Предпочтительно, чтобы подходящая проба эффективности уже была внедрена на момент производства материала для первых клинических исследований, и она должна быть утверждена до проведения основных клинических исследований, если не обосновано иное. Выпуск партии и спецификации срока годности с точки зрения эффективности должны быть определены и уточнены во время разработки продукта, если применимо.
Проба, демонстрирующая биологическую активность, должна быть основана на предполагаемом биологическом эффекте, который в идеале должен быть связан с клиническим ответом.
В основном можно выделить два типа проб эффективности:
1) пробы in vitro, использующие системы клеток, и
2) пробы in vivo, использующие животные модели.
Системы in vitro являются более предпочтительными, хотя пробы эффективности in vivo могут также быть полезными, особенно при доступности экспериментальных животных моделей.
Для определения эффективности продукта необходимо контролировать основные клеточные функции, такие как жизнеспособность, самообновление, смерть и дифференциация, во время производства, оценок стабильности и в момент выпуска, используя суррогатные маркеры и соответствующие технологии, например микроматричный анализ профиля экспрессии генов, потоковый цитометрический иммунофлуоресцентный анализ, клонирование клеток, полимеразная цепная реакция (ПЦР) и многие другие.
Обычно маркеры чистоты и маркеры эффективности не должны быть смешаны в одной пробе, однако этот подход может быть применим при утверждении.
В ссылке [39] обращено внимание на медицинские продукты на клеточной основе для иммунотерапии, однако принципы, включая подготовку эталонного препарата, также полезны для менеджмента риском других продуктов на клеточной основе. Как описано в указанных руководствах, для надлежащего определения эффективности продуктов во время разработки может потребоваться комбинация нескольких методов. Некоторые пробы могут быть востребованы для контроля изменений процесса, в то время как другие больше подходят для испытаний на момент выпуска продукции.
Эффективность стволовых клеток при этом не рассматривают.
O.6.2 Клетки для восстановления/регенерации тканей
Испытания in vivo могут либо проводиться на соответствующей животной модели, имитирующей предназначенное клиническое восстановление/регенерацию ткани, либо основываться на способе действия (например, эктопическая модель). Проба in vitro может быть основана на экспрессии маркеров, для которых продемонстрировано, что они напрямую или косвенно (суррогатные маркеры) связаны с предназначенной биологической активностью, таких как маркеры клеточной поверхности, маркеры активации, профиль экспрессии конкретных генов. Также в качестве основного принципа испытания эффективности можно использовать физиологический ответ при определенных условиях, таких как дифференциация определенных типов клеток и/или выделение ткани специфичных белков (например, внеклеточных матричных компонентов). Производители должны тем не менее гарантировать, что метод описания характеристических свойств соответствует предполагаемому биологическому эффекту in vivo. Пробу эффективности следует проводить, используя определенное количество клеток, и, если возможно, количественно оценивать по сравнению с подходящим эталонным препаратом. Эффективность может быть вычислена на основании измеренного эффекта за определенный период времени.
O.6.3 Клетки с преимущественно метаболической/фармакологической функцией
Клетки, содержащиеся в медицинском продукте на клеточной основе, могут быть химически обработаны или модифицированы in vitro для экспрессии некоторых требуемых белков, таких как факторы роста, антигены клеточной поверхности или другие молекулы, для поддержания биологического ответа настолько долго, насколько необходимо в новой микросреде. Следовательно, разрабатываемая проба эффективности должна быть способна оценивать связанные с активностью аспекты активного вещества, которые могут не полностью состоять из неповрежденных жизнеспособных клеток, но также из других компонентов.
Если предназначенная биологическая функция медицинского продукта на клеточной основе, в первую очередь, основана на способности клеток выделять конкретные молекулы, например для восстановления нарушения обмена веществ, стимуляции роста или выведения метаболитов, то проба его эффективности будет основана на определении выделяемых активных молекул и ожидаемой биологической активности. Она может проводиться с использованием общепринятых надежных количественных и качественных аналитических методов (анализ белков, идентификация нуклеиновых кислот, жидкостная хроматография высокого разрешения и т.д.). Те же молекулы можно также оценивать на предмет функционирования в системах животных моделей, предполагая, что активное вещество выделяется из медицинского продукта на клеточной основе в биологические жидкости (плазма, цереброспинальная жидкость, моча или тканевая жидкость).
O.6.4 Клетки для иммунотерапии
Пробы на эффективность медицинских продуктов на клеточной основе, предназначенных для иммунотерапевтического использования, основаны на сложных иммунных механизмах, которые могут быть осложнены образованиями мультиантигена и присущей вариативностью исходного материала. Специальное руководство по медицинским продуктам на клеточной основе для иммунотерапии приведено в ссылке [39].
Приложение P
(справочное)
Клиническая оценка и испытания
Для установления качества и безопасности продукта на клеточной основе используют доклинические испытания, как описано в приложениях C, J, K, L, M, N и O. Окончательное подтверждение рабочие характеристики получают в ходе тщательно контролируемых клинических исследований.
Ожидается, что для медицинских продуктов на клеточной основе частью обычной практики станет специальное клиническое наблюдение, включая лабораторные исследования. Должны быть разработаны не интервенционные исследования безопасности после авторизации таким образом, чтобы максимально использовать данные этих стандартных лабораторных исследований.
Выявление ранних осложнений (инфекционные заболевания, осложнения, связанные с сопряженными хирургическими процедурами) и поздних осложнений (злокачественные заболевания, развивающиеся заболевания) обычно требует разных подходов. Кроме того, они должны рассматриваться совместно с возможным постепенным повышением или снижением эффективности введенного продукта с течением времени. Схема соответствующих исследований должна также учитывать такую динамику и надлежащую медицинскую практику, которая может требовать определенного расписания процедур, регулирования лечения и лабораторных исследований для индивидуального применения к отдельным пациентам.
Любое непрерывное гуманное применение и наблюдение за пациентами, на которых воздействовал продукт в клинических испытаниях, должно быть описано и служить основой для разработки исследований долгосрочного наблюдения/безопасности после авторизации. Продолжительность и вид наблюдения безопасности должны быть установлены в соответствии с существующими руководствами и в каждом случае отдельно.
Для продуктов на клеточной основе, являющихся медицинскими изделиями, следует применять требования ИСО 14155.
Для продуктов на клеточной основе, являющихся медицинскими продуктами, должны применять требования соответствующих руководств ICH и надлежащей клинической практики (Good Clinical Practice, GCP).
Руководство по специальным требованиям, касающимся клинической оценки и испытаний, можно найти по ссылке [38].
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов ссылочным национальным и государственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ISO 13485 | IDT | ГОСТ ISO 13485-2011 "Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования" |
ISO 14971 | IDT | ГОСТ ISO 14971-2011 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" |
ISO 22442-1 | IDT | ГОСТ Р ИСО 22442-1-2011 "Изделия медицинские, использующие ткани и их производные животного происхождения. Часть 1. Менеджмент риска" |
АSТМ F 2312 | - | * |
* Соответствующий национальный стандарт отсутствует. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - IDT - идентичные стандарты. | ||
Библиография
Международные
[1] ИСО 7010 Графические символы. Цвета безопасности и знаки безопасности. Зарегистрированные знаки безопасности
[2] ИСО 10993 (все части), Биологическая оценка медицинских изделий
[3] ИСО 13408-1 Асептическая обработка изделий медицинского назначения. Часть 1. Общие требования
[4] ИСО 13408-2 Асептическая обработка изделий медицинского назначения. Часть 2. Фильтрация
[5] ИСО 13408-3 Асептическая обработка изделий медицинского назначения. Часть 3. Лиофилизация
[6] ИСО 13408-4 Асептическая обработка изделий медицинского назначения. Часть 4. Методики очистки на месте
[7] ИСО 13408-5 Асептическая обработка изделий медицинского назначения. Часть 5. Стерилизация на месте
[8] ИСО 13408-6 Асептическая обработка изделий медицинского назначения. Часть 6. Системы изоляторов
[9] ИСО 13408-7 Асептическая обработка изделий медицинского назначения. Часть 7. Альтернативные технологии для нестандартных медицинских изделий и комбинаций продуктов
[10] ИСО 14155 Клинические исследования медицинских изделий для людей. Надлежащая клиническая практика
[11] ИСО 22442-2 Медицинские изделия, использующие ткани животных и их производные. Часть 2. Контроль источников, отбора и обработки
[12] ИСО 22442-3:2007 Медицинские изделия, использующие ткани животных и их производные. Часть 3. Валидация уничтожения и/или инактивации вирусов и агентов заразной спонгиозной энцефалопатии (transmissible spongiform encephalopathy, TSE)
[13] ИСО/TO 22442-4 Медицинские изделия, использующие ткани животных и их производные. Часть 4. Принципы уничтожения и/или инактивации агентов заразной спонгиозной энцефалопатии (transmissible spongiform encephalopathy, TSE) и оценка с целью валидации данных процессов
[14] ИСО/МЭК Руководство 51:1999 Аспекты безопасности. Руководство по их включению в стандарты
[15] ICH E6 Примечания к руководству по надлежащей клинической практике http://www.ich.org
[16] ICH Q5A Примечания к руководству по качеству биотехнологической продукции: оценка вирусной опасности биотехнологической продукции, полученной из клеточных линий человеческого или животного происхождения
[17] ICH Q5D Получение и описание клеточных субстратов, используемых для производства биотехнологических/биологических продуктов
[18] ICH тема Q6B Этап 4 Примечания к руководству по спецификациям. Процедуры испытаний и критерии приемки для биотехнологических/биологических продуктов ICH Q7, Good manufacturing practice guide for active pharmaceutical ingredients
[19] ICH Q7 Руководство по надлежащей производственной практике для активных фармацевтических ингредиентов
[20] ICH Q9 Руководство по промышленному производству. Менеджмент риска и качества, Июнь 2006
[21] ICH S6 Руководство Доклиническая оценка безопасности лекарственных препаратов, полученных с помощью биотехнологий
[22] USC Title 21 Пища и лекарства
[23] WHO 2008 Руководящие принципы для трансплантации клеток, тканей и органов человека
[24] Global Harmonization Task Force (GHTF) - Study Group 1 (SG1), Document No. N029R11, 2002-02-02
Европа
[25] EН 166 Персональное средство для защиты глаз. Спецификации
[26] EН 511 Защитные перчатки от холода
[27] EН 1251-3 Криогенные сосуды. Переносные сосуды с вакуумной изоляцией, объемом не более 1000 литров. Часть 3. Эксплуатационные требования
[28] EН 13458-3 Криогенные сосуды. Статичные сосуды с вакуумной изоляцией. Часть 3. Эксплуатационные требования
[29] CWA 15849 Кодирование информации и прослеживаемость тканей и клеток человека
[30] CPMP/BWP/269/95, rev. 3 Примечания к руководству по медицинским продуктам, полученным из плазмы. http://www.ema.europa.eu
[31] CPMP/BWP/1793/02 Примечания к руководству по использованию бычьей сыворотки при производстве биологических медицинских продуктов для человека
[32] CPMP/BWP/3354/99 Примечания к руководству по производству и контролю качества животных иммуноглобулинов и иммунных сывороток для использования человеком
[33] CPMP/BWP/41450/98 Указания по рассмотрению терапии соматическими клетками человека
[34] Directive 2001/83/EC of the European Parliament and of the Council on the community code relating to medicinal products for human use, OJ L 311, 2004-11-28
[35] Directive 2006/17/EC implementing Directive 2004/23/EC of the European Parliament and of the Council as regards certain technical requirements for donation, procurement and testing of human tissues and cells, OJ L 38, 2006-02-09
[36] EMEA/410/01, rev. 3 Примечания к руководству по минимизации риска переноса агентов спонгиозной энцефалопатии медицинскими продуктами для людей и животных, адаптированные Комитетом по патентованным лекарственным препаратам (Committee for Proprietary Medicinal Products, CPMP) и Комитетом по медицинским продуктам для животных (Committee for Veterinary Medicinal Products, CVMP). (2004/C24/03).
[37] EMEA/149995 Руководство по безопасности и эффективности наблюдения. Менеджмент риска современных терапевтических медицинских продуктов
[38] EMEA/CHMP/410869, Руководство по медицинским продуктам, основанным на клетках человека
[39] EMEA/CHMP/BWP/271475 Руководство по испытанию эффективности медицинских продуктов для иммунотерапии, основанных на клетках и предназначенных для лечения рака
[40] EMEA/CVMP/743/00 Руководство по требованиям и контролю, применяемым к бычьей сыворотке, используемой для производства иммунологических медицинских продуктов для животных
[41] Ph. Eur. Monograph Bovine Serum (02262) European Pharmacopeia Chapter 5.2.8: Minimizing the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products http://online.edqm.eu/entry.htm (requires access licence)
[42] Rules Governing Medicinal Products, volume 3, 3AB4a Production and quality control of monoclonal antibodies. http://www.ema.europa.eu
Япония
[43] Pharmaceutical and Food Safety Bureau (PFSB)/Ministry of Health, Labour and Welfare (MHLW) Notifications No.0208003 (8/02/2008) Guidelines on ensuring quality and safety of products derived from engineered autologous human cells/tissues; and PFSB/MHLW Notifications No.0912006 (12/09/2008), Guidelines on ensuring quality and safety of products derived from engineered allogenic human cells/tissues
Примечание - Английский перевод данного документа в настоящее время осуществляют.
США
[44] АСТМ Ф 2027-08 Стандартное руководство по описанию и испытанию исходных или начальных биоматериалов для медицинских продуктов, выполненных из тканей
[45] АСТМ Ф 2149-01 (2007) Стандартный метод испытания для автоматического анализа клеток - метод подсчета с использованием электрочувствительной зоны и определение размера клеток в суспензии клеток одного вида
[46] АСТМ Ф 2210-02 Стандартное руководство по обработке клеток, тканей и органов для использования в медицинских продуктах, выполненных из тканей
[47] АСТМ Ф 2211-04 Стандартная спецификация медицинских продуктов, выполненных из тканей (Tissue Engineered Medical Products, TEMP)
[48] АСТМ Ф 2383-05 Стандартное руководство по оценке побочных веществ в медицинских продуктах, выполненных из тканей (Tissue Engineered Medical Products, TEMP)
[49] АСТМ Ф 2385-05 Стандартное руководство по оценке побочных веществ в медицинских продуктах, выполненных из тканей (Tissue Engineered Medical Products, TEMP)
[50] АСТМ F2386-04 Стандартное руководство по хранению медицинских продуктов, выполненных из тканей (Tissue Engineered Medical Products, TEMP)
[51] АСТМ Ф 2451-05 Стандартное руководство по оценке имплантируемых изделий, предназначенных для восстановления или регенерации суставных хрящей
[52] АСТМ Ф 2739-08 Стандартное руководство по количественному анализу жизнеспособности клеток в клеточных каркасах из биоматериала
[53] U.S. FDA: Guidance for Industry, Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps)
http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/ucm091345.pdf
[54] U.S. FDA: Guidance for Industry, Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications
http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/UCM202439.pdf
[55] U.S. FDA: Human Cells, Tissues, and Cellular and Tissue-Based Products; Donor Screening and Testing; and Related Labeling 6/19/2007 Final Rule Questions and Answers
http://www.fda.aov/BioloaicsBloodVaccines/TissueTissueProducts/QuestionsaboutTissues/ucm136397.htm
[56] U.S. FDA: Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use. Доступно по адресу: dationsforManufacturers/UCM153182.pdf
[57] U.S. FDA: Application of Current Statutory Authorities to Human Somatic Cell Therapy Products and Gene Therapy Products, 1993-10-14, 58 FR 53248 (Federal Register notice)
[58] 21 CFR 1271: Human Cells, Tissues and Cellular and Tissue-Based Products:
http://www.accessdata.fda.aov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=1271
[59] Cellular & Gene Therapy Guidances Documents:
http://www.fda.aov/BioloaicsBloodVaccines/GuidanceComplianceReaulatoryInformation/Guidances/CellularandGeneTherapy/default.htm
[60] Vaccines, Blood & Biologics - Tissue & Tissue Products:
http://www.fda.aov/BioloaicsBloodVaccines/TissueTissueProducts/default.htm
[61] Wessman, S.J. Vaccine cell substrates: bovine and porcine virus considerations. Dev Biol (Basel), 2006; 123: pp.273-280
[62] Robertson J.S. Bovine serum - regulatory issues. Dev Biol (Basel), 2006; 123: pp.269-279
УДК 615.006.354 | ОКС 11.020 | ОКП 93 9800 |
Ключевые слова: медицинское изделие, продукт медицинский, инактивация, контроль риска, донор | ||
Электронный текст документа
и сверен по:
, 2016

{kind=link}