ГОСТ Р ИСО 11138-1-2000
Группа Р26
ГОСУДАРСТВЕННЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Стерилизация медицинской продукции
БИОЛОГИЧЕСКИЕ ИНДИКАТОРЫ
Часть 1
Общие требования
Sterilization of health care products. Biological indicators. Part 1. General requirements
ОКС 11.080
ОКП 94 5120
Дата введения 2001-07-01
1 ПОДГОТОВЛЕН Ассоциацией инженеров по контролю микрозагрязнений (АСИНКОМ), Московской медицинской академией им. И.М.Сеченова и Испытательным лабораторным центром Московского городского центра дезинфекции
ВНЕСЕН Техническим комитетом по стандартизации ТК 383 "Стерилизация медицинской продукции" Госстандарта России
2 ПРИНЯТ И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Госстандарта России от 10 августа 2000 г. N 205-ст
3 Настоящий стандарт содержит аутентичный текст международного стандарта ИСО 11138-1-94 "Стерилизация медицинской продукции. Биологические индикаторы. Часть 1: Общие требования"
4 ВВЕДЕН ВПЕРВЫЕ
ВНЕСЕНА поправка, опубликованная в ИУС N 4, 2001 год
Поправка внесена изготовителем базы данных
Введение
Биологические индикаторы используются для оценки эффективности процесса стерилизации медицинской продукции и других целей. К медицинской продукции относят лекарственные средства и медицинские изделия.
Применение биологических индикаторов определено изготовителем и указано в маркировке. Использование несоответствующих индикаторов может привести к ошибочным результатам.
Биологические индикаторы должны всегда использоваться в комбинации с физическим и/или химическим контролем эффективности процесса стерилизации. Если колебания физико-химических показателей процесса стерилизации выходят за допустимые пределы, процесс стерилизации всегда должен рассматриваться как неудовлетворительный независимо от результатов контроля с помощью биологических индикаторов.
Эффективность биологических индикаторов может зависеть от условий хранения до момента их применения, методов применения или действий после завершения процесса. Поэтому необходимо выполнять рекомендации изготовителя в отношении хранения и применения. Биологические индикаторы должны быть переданы для лабораторного контроля как можно скорее после их использования в процессе стерилизации. Не допускается использовать биологические индикаторы после истечения срока годности, указанного изготовителем.
Оценка эффективности процессов стерилизации с помощью биологических индикаторов должна выполняться персоналом, подготовленным соответствующим образом.
1 Область применения
1.1 Настоящий стандарт устанавливает общие требования к производству, маркировке и работе оборудования при изготовлении биологических индикаторов и суспензий, применяемых для валидации и контроля процессов стерилизации.
Примечание - Конкретные требования к биологическим индикаторам для определенных видов стерилизации - по ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
Стандарт не содержит требований к продукции, непосредственно инокулированной микроорганизмами, методикам культивирования жизнеспособных микроорганизмов, а также к биологическим индикаторам, в которых используют более одного вида или штамма микроорганизмов, инокулированных в носителе.
2 Нормативные ссылки
В настоящем стандарте использованы ссылки на следующие стандарты:
ГОСТ Р ИСО 9002-96* Системы качества. Модель обеспечения качества при производстве, монтаже и обслуживании
______________
* На территории Российской Федерации действует ГОСТ Р ИСО 9001-2008, здесь и далее по тексту. - .
ГОСТ Р ИСО 11138-3-2000 Стерилизация медицинской продукции. Биологические индикаторы. Биологические индикаторы для стерилизации влажным теплом
ГОСТ Р ИСО 11138-2-2000 Стерилизация медицинской продукции. Биологические индикаторы. Биологические индикаторы для стерилизации окисью этилена
3 Определения
В настоящем стандарте применяют термины с соответствующими определениями:
3.1 биологический индикатор - БИ (biological indicator - BI): Готовый к применению инокулированный носитель в первичной упаковке, обеспечивающий определенную резистентность (устойчивость) к конкретному режиму стерилизации.
3.2 носитель (carrier): Удерживающий материал, на который нанесены тест-микроорганизмы.
3.3 первичная упаковка (primary pack): Система, предохраняющая инокулированный носитель от повреждения и контаминации, но не препятствующая проникновению стерилизующего агента (агентов).
3.4 вторичная упаковка (secondary pack): Упаковочная тара, в которую помещены биологические индикаторы для транспортирования и хранения.
3.5 инокулированный носитель (inoculated carrier): Носитель, на который нанесено определенное количество тест-микроорганизмов.
3.6 тест-микроорганизм (test organism): Микроорганизм, используемый для изготовления инокулированных носителей.
3.7 число живых тест-микроорганизмов (viable test organism count): Число живых тест-микроорганизмов в единице объема суспензии или на инокулированном носителе, определяемое по росту дискретных колоний при определенных условиях культивирования.
3.8 инактивация (inactivation): Потеря тест-микроорганизмами способности к росту, прорастанию и/или размножению при определенных условиях культивирования.
3.9 условия культивирования (culture conditions): Указанная изготовителем комбинация условий для обеспечения роста, прорастания и/или размножения тест-микроорганизмов, включающая питательную среду, продолжительность и температуру инкубации.
3.10 признанная коллекция культур (recognized culture collection): Международная коллекция, находящаяся под юрисдикцией Будапештского договора о международном признании коллекций микроорганизмов для целей патентования и регулирования.
3.11 величина (параметр) ; Величина десятикратного сокращения (
value): Время выдержки или поглощенная доза излучения, необходимые для уменьшения популяции тест-микроорганизмов в 10 раз при определенных условиях обработки.
3.12 кривая выживания (survivor curve): Графическое представление зависимости процесса инактивации от времени выдержки (поглощенной дозы излучения) при определенных условиях.
3.13 устройство для испытания процесса (process challenge device): Устройство, моделирующее наихудшие условия воздействия стерилизующего агента (агентов) на объект среди стерилизуемых объектов.
Примечания
1 Устройство для испытания процесса должно быть построено таким образом, чтобы биологический индикатор можно было расположить в месте, наиболее труднодоступном для стерилизующего агента (агентов).
2 Конструкция устройства для испытания процесса стерилизации зависит от вида стерилизуемых объектов и процедуры стерилизации. Биологический индикатор не должен влиять на функционирование устройства.
3 В некоторых устройствах вместо биологического индикатора может быть использован инокулированный носитель.
3.14 колониеобразующая единица - КОЕ (colony-forming unit - CFU): Видимая колония микроорганизмов, выросшая из одной клетки или из группы клеток.
3.15 автономный биологический индикатор (self-contained biological indicator): Биологический индикатор, первичная упаковка которого содержит питательную среду, необходимую для выращивания тест-микроорганизмов.
3.16 окно выживания-гибели (survival-kill window): Интервал дозы излучения или времени выдержки в процессе стерилизации при определенных условиях, в течение которых осуществляется переход от прорастания всех биологических индикаторов (экспозиция выживания) к отсутствию прорастания всех биологических индикаторов (экспозиция гибели).
3.17 номинальная популяция (nominal population): Заданное число микроорганизмов.
Примечание - Обнаруженное число микроорганизмов будет отличаться от номинальной популяции микроорганизмов в зависимости от погрешности использованных методик инокуляции и методов обнаружения жизнеспособных клеток.
3.18 резистомер (resistometer): Оборудование, предназначенное для создания комбинации определенных физико-химических параметров процесса стерилизации с определенными допусками.
4 Общие требования к продукции, производству и маркировке
4.1 Производственный контроль и контроль качества
4.1.1 Все операции, предусмотренные настоящим стандартом, должны контролироваться в соответствии с требованиями ГОСТ Р ИСО 9002.
4.1.2 Необходимо предусматривать регистрацию компонентов производства.
Производственные компоненты должны включать все материалы и элементы, входящие в состав или находящиеся в прямом контакте с суспензией тест-микроорганизмов, инокулированным носителем или биологическим индикатором.
4.1.3 Конечный продукт, поставляемый изготовителем (суспензия, инокулированный носитель или биологический индикатор), не должен содержать тест-микроорганизмов других видов в количестве, способном ухудшить потребительские свойства продукции. Соблюдение этого требования при производстве обеспечивается валидацией производства, управлением, текущим контролем и записью производственных параметров.
4.2 Тест-микроорганизмы
4.2.1 Штаммы тест-микроорганизмов не должны требовать мер изоляции при работе с ними.
4.2.2 Тест-микроорганизмы должны принадлежать к определенному штамму, взятому из обязательной коллекции культур, и однозначно идентифицироваться с эталонным номером этой коллекции культур.
4.2.3 Если штамм предназначенного для использования тест-микроорганизма не принадлежит к обязательной коллекции культур, изготовитель несет ответственность за отнесение конкретного штамма к обязательной коллекции.
4.2.4 Для каждой серии суспензии тест-микроорганизмов необходимо:
а) зарегистрировать происхождение инокулята отнесением его к соответствующей коллекции культур;
б) удостоверить его идентичность и чистоту.
Методы, используемые для поддержания культур тест-микроорганизмов, должны обеспечить предохранение от контаминации и вредных изменений их неотъемлемых качеств. Контрольные тесты являются специфическими для каждого штамма тест-микроорганизма и должны быть документированы и валидированы изготовителем.
4.3 Суспензии тест-микроорганизмов
4.3.1 Питательная среда и условия инкубирования, используемые для суспензии тест-микроорганизмов, должны быть определены изготовителем. Эти условия должны обеспечивать последовательное воспроизводство суспензий тест-микроорганизмов в соответствии с требованиями настоящего стандарта, ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
4.3.2 Методы подсчета микроорганизмов и последующей обработки должны обеспечить отсутствие остатков питательной среды в суспензии, используемой для инокуляции носителей. Указанные остатки могут быть помехой при работе с инокулированным носителем или биологическим индикатором.
Если изготовитель показал, что остатки питательной среды не оказывают вредного влияния на работу с инокулированным носителем или биологическим индикатором, указанное требование не учитывается.
4.3.3 Изготовители суспензий тест-микроорганизмов или биологических индикаторов должны письменно подтвердить, что биологические индикаторы и суспензия тест-микроорганизмов получены из культуры, взятой из коллекции культур.
4.3.4 Если суспензия тест-микроорганизмов предназначена для приготовления инокулированных носителей или инокулированного продукта, каждый контейнер, содержащий суспензию тест-микроорганизмов, должен сопровождаться информацией, содержащей:
a) название тест-микроорганизма;
b) название или аббревиатуру коллекции культуры, откуда был получен тест-микроорганизм, и ссылку на номер штамма;
c) номинальный объем суспензии в миллилитрах (или в граммах, если тест-микроорганизмы представлены не в форме суспензии);
d) единый код, по которому может быть восстановлена история изготовления;
e) число жизнеспособных клеток в миллилитре суспензии;
f) рекомендуемые условия хранения;
g) срок годности или срок хранения;
h) название изготовителя, торговую марку, адрес и другие средства идентификации;
i) инструкции по утилизации.
4.3.5 По требованию потребителя изготовитель обязан представить информацию о характеристиках резистентности и других характеристиках суспензии. Эти данные должны быть согласованы между покупателем и изготовителем.
4.3.6 Условия хранения суспензий тест-микроорганизмов и срок их годности должны быть указаны изготовителем. Эти условия должны контролироваться в период хранения. Они должны поддерживать суспензии тест-микроорганизмов в таком состоянии, чтобы они соответствовали требованиям настоящего стандарта, ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
4.3.7 Должно быть установлено число жизнеспособных микроорганизмов в суспензии.
По требованию потребителя показатель прорастаемости тест-микроорганизмов определяют через процентное отношение числа жизнеспособных тест-микроорганизмов, обнаруженных с помощью микроскопии.
4.3.8 Изготовитель должен гарантировать транспортирование третьей стороне в контролируемых условиях, сравнимых с условиями хранения суспензий тест-микроорганизмов.
4.4 Носитель, первичная упаковка и конструкция
4.4.1 Носитель и первичная упаковка не должны содержать загрязнений (физических, химических или микробиологических), которые могли бы ухудшить характеристики биологического индикатора.
4.4.2 Носитель и первичная упаковка не должны разрушаться в предусмотренном для них процессе стерилизации, вследствие чего могли бы измениться характеристики инокулированного носителя.
Носитель должен выдерживать транспортирование в первичной и вторичной упаковках, а также обеспечивать работу с ним в месте использования без повреждений.
Конструкция носителя и/или первичной упаковки должна предусматривать:
a) минимизацию потерь начальной концентрации тест-микроорганизмов во время транспортирования, обращения с ним и хранения в течение срока годности;
b) соответствие устройству для испытания процесса стерилизации.
4.4.3 Соответствие требованиям 4.4.2 должно проверяться путем визуального контроля носителя и первичной упаковки, подвергшихся воздействию стерилизации при максимальных значениях и скоростях изменения физико-химических параметров процесса стерилизации.
Примечание - Пределы изменения этих свойств приведены в соответствующих частях ГОСТ Р ИСО 11138.
4.4.4 Во время и после процесса стерилизации носитель и первичная упаковка не должны поглощать или выделять никаких веществ в количествах, способных замедлить рост малого числа выживших тест-микроорганизмов в условиях выращивания при попадании этих веществ в среду выращивания.
Порядок выполнения тестов на проверку соответствия этим требованиям приведен в приложении F.
4.4.5 По требованию потребителя изготовитель обязан предоставить сведения о предельных размерах носителя.
4.5 Инокулированные носители
4.5.1 При изготовлении одной партии носителей должен использоваться только один штамм тест-микроорганизмов.
4.5.2 Инокулированные носители готовят путем нанесения на них суспензии тест-микроорганизмов с последующим высушиванием в контролируемых условиях.
4.5.3 Условия, при которых осуществляется инокуляция, должны быть разработаны, валидированы и проконтролированы, чтобы гарантировать отсутствие в инокулированном носителе посторонних микроорганизмов, которые могут ухудшить характеристики продукции в соответствии с ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
4.5.4 Каждый носитель должен содержать одно и то же количество клеток тест-микроорганизма.
4.5.5 Условия хранения инокулированных носителей и срок годности определяет изготовитель. Эти условия должны контролироваться в период хранения. Они должны соответствовать требованиям к инокулированным носителям, изложенным в ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
4.5.6 При упаковывании инокулированных носителей с целью превращения их в биологические индикаторы упаковка не должна изменять номинальную популяцию и препятствовать проведению работ с конкретными носителями.
4.5.7 Каждую серию инокулированных носителей должна сопровождать информация, содержащая:
a) название "инокулированные носители";
b) название тест-микроорганизма;
c) руководство по применению, специальные данные, касающиеся питательной среды и условий, необходимых для роста тест-микроорганизмов после воздействия процесса стерилизации;
d) название коллекции культур, откуда был взят тест-микроорганизм, и ссылка на номер штамма;
e) число тест-микроорганизмов на одном инокулированном носителе;
f) номер серии или специальный код, по которым может быть восстановлена история производства;
g) характеристики резистентности инокулированных носителей к воздействию процесса стерилизации, которому они должны подвергаться, включая условия тестирования и методы определения указанных характеристик;
h) число инокулированных носителей во вторичной упаковке;
i) рекомендуемые условия хранения;
j) срок годности инокулированных носителей;
k) название изготовителя, торговая марка, адрес или другие средства идентификации;
l) процесс стерилизации, для контроля которого предназначен инокулированный носитель;
m) инструкции по утилизации и уничтожению.
4.6 Биологические индикаторы
4.6.1 Биологические индикаторы должны быть изготовлены путем упаковки отдельных инокулированных носителей в первичную упаковку.
4.6.2 Первичная упаковка должна быть сконструирована, изготовлена и валидирована так, чтобы находящийся в ней биологический индикатор отвечал требованиям ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
4.6.3 Первичная упаковка должна быть сконструирована, изготовлена и валидирована так, чтобы в период хранения и транспортирования в соответствии с инструкциями изготовителя биологический индикатор и инокулированный носитель были защищены от контаминации и потери инокулята из носителя.
4.6.4 Условия помещения в первичную упаковку должны быть четко определены, валидированы и проконтролированы, чтобы инокулированный носитель не содержал микроорганизмы, иные, чем тест-микроорганизмы, и которые могли бы неблагоприятно повлиять на характеристики продукта так, как это определено стандартами ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3 соответственно.
4.6.5 Первичная упаковка должна быть валидирована в соответствии с ее назначением и соответствующим международным или национальным стандартом.
4.6.6 Каждая первичная упаковка для биологического индикатора должна иметь этикетку, содержащую следующую информацию:
a) название тест-микроорганизма;
b) номер серии биологического индикатора;
c) срок годности биологического индикатора;
d) указание на процесс стерилизации, которому соответствует биологический индикатор;
e) название изготовителя, торговую марку, адрес и другие данные.
4.6.7 Биологические индикаторы должны быть упакованы во вторичную упаковку для транспортирования и хранения.
4.6.8 Вторичная упаковка должна быть снабжена этикеткой, содержащей следующую информацию:
a) название "биологические индикаторы";
b) информацию, указанную в 4.6.6;
c) название коллекции культур, откуда был взят тест-микроорганизм, и ссылку на номер штамма;
d) число тест-микроорганизмов, находящихся в каждом биологическом индикаторе, как это определено для серии инокулированных носителей;
e) количество биологических индикаторов во вторичной упаковке;
f) рекомендуемые условия хранения;
g) резистентность тест-микроорганизмов в инокулированном носителе, находящемся в первичной упаковке, включая условия тестирования и методы, используемые для определения указанных характеристик;
h) руководство по применению, особенно данные, касающиеся питательной среды и условий, при которых выращиваются тест-микроорганизмы после воздействия факторов стерилизации;
i) инструкции по утилизации и уничтожению.
Текст этикетки должен быть на русском языке.
4.6.9 Каждая вторичная упаковка должна быть снабжена копией сертификата для каждой серии биологического индикатора, который должен включать следующую информацию:
a) информацию, указанную в 4.6.8;
b) характеристики резистентности индикаторов к воздействию процесса стерилизации, для контроля которого предназначены инокулированные носители;
c) ссылку на настоящий стандарт и другие соответствующие международные стандарты.
4.6.10 Каждая вторичная упаковка должна быть снабжена письменными инструкциями по работе с биологическими индикаторами и по определению числа выживших микроорганизмов, а также следующими требованиями:
a) биологические индикаторы должны храниться при условиях, указанных изготовителем;
b) биологические индикаторы данной серии не должны использоваться после истечения срока хранения;
c) после выдержки в проверяемом цикле стерилизации биологические индикаторы должны быть изучены на наличие выживших микроорганизмов в течение времени, указанного изготовителем;
d) при проверке индикаторов для оценки выживших тест-микроорганизмов должны использоваться методы и условия, предписанные изготовителем. Если применяются альтернативные методы, то они должны быть валидированы.
4.7 Автономные биологические индикаторы
4.7.1 Автономные биологические индикаторы должны соответствовать всем требованиям настоящего стандарта.
4.7.2 Автономные биологические индикаторы должны быть достаточно прочными, чтобы переносить транспортирование во вторичной упаковке, а также обращение без повреждений в месте использования.
Конструкция автономного биологического индикатора должна обеспечивать:
a) минимизацию потерь исходного инокулята тест-микроорганизма во время транспортирования и обращения с ним;
b) возможность использования его совместно с устройством для испытания процесса стерилизации.
4.7.3 Во время и после завершения процесса стерилизации материалы, из которых изготовлен автономный биологический индикатор, не должны удерживать или выделять какие-либо вещества в таком количестве, которое может ингибировать рост малого количества выживших тест-микроорганизмов при заданных условиях культивирования (см. приложение F).
5 Определение резистентности
5.1 Требования к определению резистентности
5.1.1 Резистентность (устойчивость) каждой серии биологических индикаторов должна быть показана экспериментально, чтобы подтвердить соответствие требованиям ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
5.1.2 Контроль резистентности (5.4 и 5.5) должен включать определение числа выживших тест-микроорганизмов и определение характеристик резистентности комбинацией двух или более следующих методов:
a) определение величины путем построения кривой выживаемости;
b) определение величины методом анализа частичного прорастания;
c) расчет окна выживания-гибели с использованием рассчитанной величины и проверка характеристик выживания-гибели.
5.1.3 Величины, определенные такими методами, должны находиться в пределах, указанных в ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3 соответственно. По крайней мере две из этих величин должны быть приведены на этикетке вторичной упаковки и в сертификате, сопровождающем каждую серию инокулированных носителей (4.5.7).
Примечание - Соответствующие разделы ГОСТ Р ИСО 11138 могут потребовать дополнительных определений (например, величины для стерилизации биологических индикаторов влажным теплом, как предусмотрено ГОСТ Р ИСО 11138-3).
5.2 Расчет окна выживания-гибели
Окно выживания-гибели может быть рассчитано с использованием одной из величин , определенной согласно приложениям В и D по следующим формулам:
Время (доза) выживания ![]() ,
,
где - число тест-микроорганизмов на одном носителе, указанное в маркировке.
Время (доза) гибели ![]() .
.
5.3 Определение числа выживших тест-микроорганизмов
Порядок определения числа выживших тест-микроорганизмов приведен в приложении А.
5.4 Определение величины
5.4.1 Данные для расчета величины для биологических индикаторов должны быть получены в соответствии с приложением В (т.е. построением кривой выживания с использованием прямого подсчета тест-микроорганизмов) и/или приложением С (анализ частичного прорастания или метод наиболее вероятного числа).
5.4.2 Величина рассчитывается в соответствии с приложением В и/или D.
5.4.3 Могут быть использованы другие методы, но при этом должна быть продемонстрирована их эквивалентность эталонному методу.
5.5 Определение характеристик выживания-гибели
Характеристики выживания-гибели определяются и проверяются в соответствии с приложением Е.
ПРИЛОЖЕНИЕ А
(обязательное)
Определение числа живых тест-микроорганизмов
А.1 Инокулированные носители должны быть исследованы на наличие выживших тест-микроорганизмов в соответствии с А.2-А.4 или альтернативным методом. При использовании альтернативного метода должно быть известно соотношение рекомендуемого и альтернативного методов.
А.2 Следует использовать не менее четырех проб из каждой партии, серии или экспозиции. Каждая проба должна быть помещена в вымывающую питательную среду соответствующего объема. Тест-микроорганизмы вымываются от инокулированных носителей с помощью принятой методики (например, встряхиванием со стеклянными бусами, перемешиванием в гомогенизаторе, ультразвуковом или другим соответствующим методом).
A.3 Суспензия должна разбавляться в соответствующей стерильной разбавляющей жидкости так, чтобы в дозах, предназначенных для посева в чашках, находилось от 30 до 300 колониеобразующих единиц (КОЕ). Указанные дозы стерильной жидкости должны быть либо перемешаны с расплавленным питательным агаром, либо посеяны в чашки с твердой питательной средой. Изготовители биологических индикаторов должны указать или обеспечить поставку соответствующей среды и/или привести полные данные для приготовления такой среды.
А.4 Помещенные в чашки пробы должны инкубироваться при температуре и в течение времени, установленных изготовителем.
Примечания
1 Обычно температура и время инкубации составляют от 55 до 60 °С не менее 48 ч для термофильных организмов и от 30 до 37 °С не менее 48 ч для мезофильных организмов.
2 Высыхание питательных сред при повышенных температурах инкубации может неблагоприятно влиять на рост.
А.5 После соответствующего периода инкубации должно быть подсчитано количество колониеобразующих единиц в чашках и по ним вычислено среднее количество выживших тест-микроорганизмов.
ПРИЛОЖЕНИЕ В
(обязательное)
Метод кривой выживания
Примечание - Идеальная кривая выживания является линейной во всем диапазоне инактивации. На практике имеют место отклонения от этого идеала, однако линейность должна сохраняться в приемлемых пределах. Построение кривой выживания методом прямого подсчета позволяет представить резистентность популяции численностью более 5х10, в то время как метод MPN (приложение С) позволяет представить резистентность популяции численностью менее 5х10
. Хорошая корреляция между величинами
, полученными этими двумя методами, позволяет утверждать, что серьезных отклонений от линейности кривой выживания не наблюдается.
B.1 Исследуемые пробы подлежат обработке при ступенчатом возрастании экспозиции (выдержки) в определенных условиях. Должен быть установлен диапазон изменения экспозиции. Каждая экспозиция или доза должны отличаться от предыдущей на постоянную величину.
Примечание - Требования к характеристикам аппаратуры приведены в ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
B.2 Должны быть использованы не менее пяти экспозиций, которые должны включать:
a) одну экспозицию, в которой проба не подвергается воздействию стерилизующего агента (стерилизующий агент может отсутствовать или может быть заменен на газ, не вызывающий летального действия);
b) снижение популяции до 0,01% исходного инокулята, по крайней мере, при одной экспозиции.
B.3 В каждой экспозиции должно быть использовано не менее четырех инокулированных носителей в каждом определении. При каждой экспозиции должно быть то же самое количество повторностей.
B.4 Отобранные пробы должны быть обработаны в течение 2 ч после завершения каждой экспозиции, чтобы удалить тест-микроорганизмы из носителя. Определение числа выживших тест-микроорганизмов должно быть проведено в специальных условиях культивирования по методам, установленным изготовителем.
В.5 Пользуясь полученными данными, строится кривая зависимости логарифма выжившей популяции от времени в минутах или уровня дозы в линейном виде кривая регрессии, пользуясь методом наименьших квадратов. Точки с данными выживаемости менее 0,5 логарифма начальной популяции не должны включаться в регрессионный анализ. По отрицательному наклону линии регрессии рассчитывается величина в минутах или в единицах поглощенной дозы.
В.6 Коэффициент регрессии линейности кривой выживания должен быть не менее 0,8.
ПРИЛОЖЕНИЕ С
(обязательное)
Фракционный негативный анализ (метод MPN для последовательного определения величины с использованием ограниченного метода Спирмана-Карбера)
Примечание - Фракционные негативные данные применяются для определения влияния процесса стерилизации на характеристики резистентности тест-микроорганизмов с помощью метода наиболее вероятных величин - MPN (Most Probable Number).
C.1 Контролируемые пробы должны быть подвергнуты кратным экспозициям в определенных условиях воздействия всех переменных факторов процесса стерилизации, исключая продолжительность или уровень дозы воздействия, которые остаются постоянными. В каждой экспозиции должно быть не менее 20 повторностей. Каждый период экспозиции или уровень дозы должен отличаться от предыдущего на постоянный интервал. В каждой экспозиции должно использоваться одно и то же количество повторностей.
Примечание - Требования к работе испытательного оборудования приведены в ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
С.2 После 2 ч экспозиции каждый инокулированный носитель в асептических условиях помещается в пробирку, содержащую адекватное количество соответствующей стерильной питательной среды. Объем среды должен быть одинаков для каждой повторности. Если питательная среда включена изготовителем в состав биологического индикатора, то изготовитель должен приложить инструкции по культивированию, а также выдать требования к питательной среде или указать на возможность ее приобретения и/или привести исчерпывающие данные для ее приготовления.
С.3 Инокулированные носители инкубируются при температуре, рекомендованной изготовителем. Культуры исследуются на рост тест-микроорганизмов по истечении времени инкубации, рекомендуемого изготовителем. Рост тест-микроорганизма может быть установлен по мутности питательного бульона, поверхностному росту на бульоне или осаждению на дне пробирки в зависимости от характеристик тест-микроорганизма.
С.4 Результаты следует записывать в виде отношения инокулированных носителей с выжившими тест-микроорганизмами к общему числу инокулированных носителей, подвергнутых воздействию каждой сублетальной экспозиции.
ПРИЛОЖЕНИЕ D
(обязательное)
Расчет величины с использованием ограниченного метода Спирмана-Карбера
D.1 Расчет величины
Примечание - Ограниченный метод Спирмана-Карбера (Spearman-Karber) требует, чтобы последовательные экспозиции отличались друг от друга на постоянный интервал и в каждой экспозиции использовалось одно и то же число реплик (биологических индикаторов)
.
D.1.1 Чтобы охватить фракционно-негативную область, нужно выбрать интервал экспозиции или уровня доз (,
,...,
). Начальная экспозиция
выбирается так, чтобы стерилизующий эффект был равен нулю или
=0. Последняя экспозиция
выбирается такой, чтобы был обеспечен полный стерилизующий эффект или
. Тест считается действительным, если в экспозиции, предшествующей
, отсутствуют отрицательные (стерильные) единицы (
=0) и в экспозиции, следующей за
, присутствуют только отрицательные единицы (
). При этом в двух промежуточных интервалах между
и
(где 0<
<
) должны быть две значимые пробы. Затем рассчитывается средняя экспозиция до достижения стерильности или ограниченного показателя Спирмана-Карбера по формуле
![]() ,
,
где - средняя экспозиция до достижения стерильности (оценка ограниченного показателя Спирмана-Карбера);
- первая экспозиция, в которой наблюдаются все стерильные реплики,
![]() ;
;
- интервал времени или дозы между экспозициями;
- число реплик (биологических индикаторов) в каждой экспозиции;
- число стерильных реплик в каждой экспозиции;
- самая длительная экспозиция, в которой не наблюдаются отрицательные биологические индикаторы,
=0;
- экспозиция, предшествующая
;
- сумма стерильных реплик (
) во всех экспозициях между
и
.
D.1.2 Величина рассчитывается по формуле
![]() ,
,
где - среднее значение экспозиции до достижения стерильности, полученное из предыдущей формулы;
- среднее число живых спор в расчете на один биологический индикатор, определенное по всему количеству живых спор.
D.2 Расчет дисперсии , стандартного отклонения и доверительного интервала с помощью метода Спирмана-Карбера
Примечание - Расчет по методу Спирмана-Карбера позволяет установить вариацию и, в свою очередь, рассчитать стандартное отклонение и доверительный интервал.
D.2.1 Дисперсия , т.е.
![]() рассчитывается по формуле
рассчитывается по формуле
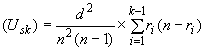 ,
,
где - интервал времени или дозы между экспозициями;
- число проб (реплик) в каждой экспозиции;
- число стерильных реплик в каждой экспозиции;
![]() - сумма величин, полученная умножением
- сумма величин, полученная умножением на
![]() для каждой экспозиции и между
для каждой экспозиции и между и
.
D.2.2 Стандартное отклонение ![]() рассчитывается по формуле
рассчитывается по формуле
![]() .
.
D.2.3 Пользуясь верхним и нижним доверительными пределами (доверительные пределы для
равны
![]() ;
; =0,95), рассчитываются нижний и верхний доверительные пределы для
:
нижний доверительный предел
![]() ;
;
верхний доверительный предел
![]() .
.
ПРИЛОЖЕНИЕ Е
(обязательное)
Характеристики выживания-гибели
Примечание - Контроль характеристик выживания-гибели с помощью большого числа биологических проб является дополнительным средством, гарантирующим постоянство характеристик всех единиц в каждой данной серии.
E.1 Значения величины , вычисленные либо из кривой выживания, либо путем использования метода фракционного негативного анализа (приложения В и D), должны использоваться для расчета времени (дозы) выживания и времени (дозы) гибели.
Е.2 Для получения надежных величин времени (дозы) выживания и времени (дозы) гибели следует использовать не менее 50 проб.
Е.3 Для характеристики времени (дозы) выживания и времени (дозы) гибели необходимо не менее 50 проб. Характеристика выживания - это фиксированная экспозиция времени (дозы), которой соответствует определенное количество выживших организмов в каждой единице пробы. Характеристика времени (дозы) гибели - это фиксированная экспозиция времени (дозы), которой соответствует полная гибель всех организмов в каждой единице пробы.
Е.4 Характеристики выживания - гибели должны определяться в резистомере биологических индикаторов. Периоды времени/дозы выживания - гибели определяются следующим образом:
Время (доза) выживания ![]() ,
,
где - исходное количество организмов.
Время (доза) гибели ![]() .
.
Примечание - Число проб, взятых для каждой экспозиции, будет зависеть как от производительности, так и от рабочих характеристик используемого резистомера биологических индикаторов. Иногда следует выполнить несколько предварительных экспозиций для времени/доз как для выживания, так и гибели, чтобы определить необходимое общее количество проб.
ПРИЛОЖЕНИЕ F
(справочное)
Определение ингибирования роста материалами носителей и первичных упаковок во время стерилизации
F.1 Материалы
F.1.1 Суспензия тест-микроорганизмов одного штамма, приготовленная тем же способом, что и тест-микроорганизмы, предназначенные для инокуляции носителей. Суспензия должна состоять из микроорганизмов известной популяции, определенная по числу живых микроорганизмов, позволяющая выделить их в количестве от 10 до 100 единиц.
F.1.2 Резистомер должен соответствовать требованиям, определенным в последующих частях настоящего стандарта.
F.1.3 Питательная среда - как определено условиями культивирования.
F.1.4 Установка для инкубирования с контролем температуры - как определено условиями инкубирования.
F.2 Метод определения
F.2.1 Представительное количество из 12 неинокулированных носителей разделить на шесть групп по два носителя. Приготовить девять контейнеров с питательной средой (средой роста).
F.2.2 Упаковать каждый из двух носителей каждой из этих трех групп в материал, используемый для изготовления биологических индикаторов, и подвергнуть их стерилизации.
F.2.3 Установить рабочие условия в резистомере по ГОСТ Р ИСО 11138-2 и ГОСТ Р ИСО 11138-3.
F.2.4 После завершения процесса как можно быстрее, но не позднее чем через 120 мин, развернуть носители и в асептических условиях перенести в питательную среду, не подвергая их при этом немедленной обработке. Записать время, необходимое для полной передачи.
F.2.5 Поместить одну группу по два носителя в каждый из трех контейнеров с питательной средой, предварительно инкубированной при соответствующей температуре. Инкубировать питательную среду при установленной температуре в течение 2 ч, чтобы позволить ингибирующим веществам десорбироваться из носителей. Извлечь питательную среду из инкубатора и инокулировать ее таким количеством суспензии тест-микроорганизмов, которое содержит от 10 до 100 тест-микроорганизмов. Вернуть инокулированную питательную среду в инкубатор и инкубировать ее в течение времени, установленного изготовителем для регенерации биологических индикаторов при нормальных условиях их использования.
F.2.6 Отрицательный контроль: поместить одну группу по два носителя, не подвергнутых стерилизации, в каждый из трех контейнеров с питательной средой, инкубированной в течение 2 ч, инокулировать их тест-микроорганизмами в количестве от 10 до 100 и инкубировать в течение периода времени, установленного изготовителем, и в тех же условиях, упомянутых выше.
F.2.7 Положительный контроль: инкубировать три контейнера с питательной средой в течение 2 ч, инокулировать их тест-микроорганизмами в количестве от 10 до 100 единиц и инкубировать так же, как указано выше.
F.2.8 После окончания установленного периода регенерации извлечь все девять контейнеров из инкубатора и исследовать их на наличие живых организмов в соответствии с инструкциями изготовителя.
F.2.9 Записать результаты наличия и отсутствия роста тест-микроорганизмов.
F.3 Интерпретация результатов
F.3.1 Если отсутствие роста отмечается в одном или более позитивных контрольных образцах, испытание считается недействительным.
Примечание - Отсутствие роста в позитивном контроле может объясняться ошибкой контроля популяции тест-микроорганизмов или несоответствующими условиями регенерации (питательная среда, температура и т.д.).
F.3.2 Если отсутствие роста наблюдается в одном или более отрицательных контрольных образцов, носитель не может считаться пригодным для изготовления инокулированных носителей или биологических индикаторов.
Примечание - Отсутствие роста в отрицательных контрольных образцах при наличии роста в положительных контрольных образцах может означать, что материал, из которого изготовлен носитель, сам является ингибитором роста тест-микроорганизмов.
F.3.3 Если отсутствие роста наблюдается в одном или более испытаний носителей, подвергнутых стерилизации, носитель не может считаться пригодным для изготовления инокулированных носителей или биологических индикаторов.
Примечание - Отсутствие роста может объясняться высоким уровнем адсорбции/абсорбции стерилизующего агента или деградацией материала носителя во время процесса стерилизации
F.4 Определение ингибирования роста, вызванного материалами упаковки
Образцы материала первичной упаковки могут быть испытаны тем же способом, что и носитель (F.1-F.3).
Образец части материала первичной упаковки должен быть достаточным для погружения в питательную среду объемом, равным удвоенному объему питательной среды, необходимой для нормального контакта с инокулированным носителем, или для автономных биологических индикаторов в объеме, эквивалентном для нормального контакта с питательной средой.
Электронный текст документа
и сверен по:
М.: ИПК Издательство стандартов, 2000
Редакция документа с учетом
изменений и дополнений подготовлена

{kind=link}