ГОСТ ISO 11135-2017
Группа Р26
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
Стерилизация медицинской продукции
ЭТИЛЕНОКСИД
Требования к разработке, валидации и текущему управлению процессом стерилизации медицинских изделий
Sterilization of health-care products. Ethylene oxide. Requirements for the development, validation and routine control of a sterilization process for medical devices
МКС 11.080.01
Дата введения 2018-09-01
Предисловие
Цели, основные принципы и основной порядок проведения работ по межгосударственной стандартизации установлены в ГОСТ 1.0-2015 "Межгосударственная система стандартизации. Основные положения" и ГОСТ 1.2-2015 "Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены"
Сведения о стандарте
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "Фармстер" (ООО "Фармстер") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 5
2 ВНЕСЕН Федеральным агентством по техническому регулированию и метрологии
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 1 июня 2017 г. N 51)
За принятие проголосовали:
Краткое наименование страны по МК (ИСО 3166) 004-97 | Код страны по | Сокращенное наименование национального органа по стандартизации |
Армения | AM | Минэкономики Республики Армения |
Беларусь | BY | Госстандарт Республики Беларусь |
Казахстан | KZ | Госстандарт Республики Казахстан |
Киргизия | KG | Кыргызстандарт |
Россия | RU | Росстандарт |
Таджикистан | TJ | Таджикстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 22 августа 2017 г. N 938-ст межгосударственный стандарт ГОСТ ISO 11135-2017 введен в действие в качестве национального стандарта Российской Федерации с 1 сентября 2018 г.
5 Настоящий стандарт идентичен международному стандарту ISO 11135:2014* "Стерилизация медицинской продукции. Этиленоксид. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" ("Sterilization of health-care products. Ethylene oxide. Requirements for the development, validation and routine control of a sterilization process for medical devices", IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
Международный стандарт разработан Техническим комитетом ISO/ТС 198 "Стерилизация медицинских изделий" Международной организации по стандартизации (ISO).
Официальные экземпляры международного стандарта, на основе которого подготовлен настоящий стандарт, и международные стандарты, на которые даны ссылки, имеются в Федеральном информационном фонде стандартов.
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
6 ВЗАМЕН ГОСТ ISO 11135-2012
Информация об изменениях к настоящему стандарту публикуется в ежегодном информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячном информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Стерильное медицинское изделие - это медицинское изделие, которое не содержит жизнеспособных микроорганизмов. Медицинские изделия, производимые в стандартных производственных условиях в соответствии с требованиями системы менеджмента качества ISO (см., например, ISO 13485), могут до стерилизации содержать на себе микроорганизмы, хотя и в малых количествах. Такие изделия нестерильны. Цель стерилизации состоит в инактивации микробиологических контаминантов, чтобы таким образом превратить нестерильные изделия в стерильные.
Кинетика инактивации чистой культуры микроорганизмов физическими и/или химическими агентами, используемыми для стерилизации медицинских изделий, в общем случае наилучшим образом может быть описана экспоненциальной зависимостью между числом выживших микроорганизмов и степенью их обработки этиленоксидом (далее - ЭО); это означает, что всегда имеется определенная вероятность выживания микроорганизмов вне зависимости от степени проведенной обработки. Для каждой обработки вероятность выживания определяется количеством и устойчивостью микроорганизмов, а также окружающей средой, в которой организм находится во время обработки. Из этого следует, что стерильность любого медицинского изделия из совокупности изделий, подвергнутых стерилизационной обработке, не может быть гарантирована, а стерильность обработанной совокупности изделий определяется в терминах вероятности присутствия на изделии жизнеспособного микроорганизма.
ISO 11135 устанавливает требования, которые при условии их выполнения обеспечат процесс этиленоксидной стерилизации, предназначенный для стерилизации медицинских изделий, с достаточной микробоцидной активностью. Более того, соответствие требованиям гарантирует, что валидации, выполняемые в соответствии с настоящим стандартом, обеспечат продукты, отвечающие определенным требованиям к стерильным продуктам с высокой степенью достоверности. Определение данной вероятности является задачей уполномоченных органов и может отличаться на территориях разных стран (смотрите, например, EN 556-1 и ANSI/AAMI ST67).
Общие требования к системам менеджмента качества для проектирования, разработки, производства, монтажа и обслуживания даны в ISO 9001, а специальные требования к системам менеджмента качества для производств медицинских изделий даны в ISO 13485. Стандарты систем менеджмента качества признают, что результативность некоторых процессов, используемых в производстве и переработке, не может быть полностью проверена последующим осмотром и испытанием продукта. Стерилизация является примером именно такого процесса. По этой причине процессы стерилизации валидируются для использования, характеристики процессов стерилизации подлежат рутинному мониторингу, а стерилизационное оборудование подлежит регулярному обслуживанию.
Экспозиция правильно валидированному и точно контролируемому процессу стерилизации не является единственным фактором, связанным с обеспечением надежной гарантии того, что продукт стерилен и в этом отношении пригоден к использованию по назначению. Поэтому уделено внимание еще нескольким факторам, включающим в себя:
a) микробиологическое состояние входящих сырьевых материалов и/или компонентов;
b) валидацию и текущее управление любыми процедурами очистки и дезинфекции, используемыми с продуктом;
c) управление окружающей средой, в которой продукт производится или перерабатывается, собирается и упаковывается;
d) управление оборудованием и процессами;
e) управление персоналом и его гигиеной;
f) способы упаковки и материалы, в которые продукт упаковывается;
g) условия хранения продукта.
Типы контаминации продуктов, подвергающихся стерилизации, могут быть разными, и это влияет на эффективность процесса стерилизации. Продукты, использованные в учреждениях здравоохранения и направляемые на повторную стерилизацию в соответствии с инструкциями производителя (см. ISO 17664), являются особым случаем. Для таких изделий имеется вероятность наличия широкого ряда контаминирующих микроорганизмов и остаточных неорганических и/или органических загрязнений, несмотря на использование процессов очистки. Следовательно, нужно уделять особое внимание валидации и контролю процессов очистки и дезинфекции, используемых при повторной обработке. Загрузка из смешанных продуктов является общим случаем в учреждениях здравоохранения, а проходящие через стерилизацию объемы диктуются историческим и прогнозируемым запросом на стерильный продукт.
Требования являются обязательной частью ISO 11135, соответствие которому декларируется. Руководство, представленное в справочных приложениях, не является обязательным и не должно служить в качестве контрольного списка для аудиторов. Руководство, представленное в приложении D, содержит разъяснения и описания методов, которые считаются приемлемыми для достижения соответствия требованиям к промышленности и учреждений здравоохранения.
Руководство, приведенное в приложении D, предназначено для лиц, имеющих базовые познания о принципах ЭО-стерилизации. Могут применяться и иные методы, отличные от представленных в руководстве, если они эффективны для достижения соответствия требованиям настоящего стандарта.
Разработка, валидация и текущее управление процессом стерилизации состоят из ряда отдельных, но взаимосвязанных действий, таких как, например, калибровка, техническое обслуживание, определение продукта, определение процесса, аттестация установленного оборудования, аттестация функционирующего оборудования и аттестация эксплуатируемого оборудования. В то время как действия, требуемые ISO 11135, были сгруппированы и представлены в определенном порядке, ISO 11135 не требует их выполнения именно в том порядке, в котором они представлены. Требуемые действия не обязательно должны быть последовательными, так как программа разработки и валидации может быть итерационной. Есть вероятность, что выполнение этих различных действий потребует участия ряда отдельных лиц или организаций, каждая из которых берет на себя исполнение одного или нескольких действий. Настоящий стандарт не оговаривает, какие именно лица или организации могут выполнять эти действия.
Важно, чтобы безопасность пациента обеспечивалась минимизацией воздействия ЭО и его побочных продуктов в ходе нормального использования медицинского изделия. ISO 10993-7 устанавливает пределы для ЭО и этиленхлоргидрина (далее - ЕСН); тем не менее, не установлены пределы экспозиции для этиленгликоля (далее - EG), поскольку оценка риска показывает, что при контролируемых остатках ЭО присутствие биологически значимых остатков этиленгликоля маловероятно.
1 Область применения
1.1 Охват
Настоящий стандарт устанавливает требования к разработке, валидации и текущему контролю процесса этиленоксидной стерилизации медицинских изделий как в промышленности, так и в учреждениях здравоохранения и учитывает сходные и различающиеся черты этих двух областей применения.
Примечание 1 - Общими чертами являются общая потребность в системах менеджмента качества, обучение персонала и правильные средства обеспечения безопасности. Основная разница заключается в уникальных физических и организационных условиях в учреждениях здравоохранения, а также в начальном состоянии медицинских изделий, предоставляемых для стерилизации.
Примечание 2 - Учреждения здравоохранения отличаются от производств медицинских изделий физическим устройством зон обработки, используемым оборудованием и доступностью персонала с адекватным уровнем подготовки и практического опыта. Основной функцией учреждения здравоохранения является забота о пациенте; переработка медицинских изделий - это только одно из множества действий, выполняемых для поддержки этой функции.
Примечание 3 - С точки зрения начального [исходного] состояния медицинских изделий, их производители, как правило, стерилизуют большие количества сходных медицинских изделий, произведенных из первичного сырья. Учреждения здравоохранения, с другой стороны, должны обрабатывать как новые, так и многоразовые медицинские изделия различного назначения с различными уровнями биологической нагрузки, по этим причинам они сталкиваются с дополнительными проблемами очистки, оценки, подготовки и упаковки медицинского изделия перед стерилизацией. В настоящем стандарте приведены альтернативные подходы и рекомендации, специфичные для медицинских учреждений.
Примечание 4 - Газообразный ЭО и его смеси являются эффективными стерилизующими агентами, которые используются в основном для тепло- и/или влагочувствительных медицинских изделий, которые не могут быть стерилизованы влажным теплом.
Примечание 5 - Несмотря на то, что область применения настоящего стандарта ограничена медицинскими изделиями, он оговаривает специфические требования и дает руководства, которые могут быть применимы и к другим продуктам здравоохранения.
1.2 Исключения
1.2.1 Настоящий стандарт не устанавливает требования к разработке, валидации и рутинному контролю процесса инактивации возбудителей спонгиоформных энцефалопатий, таких как скрепи, коровья губчатая энцефалопатия и болезнь Крейтцфельда-Якоба. Конкретные рекомендации были разработаны в отдельных странах для обработки материалов, потенциально загрязненных такими возбудителями.
Примечание - См. ISO 22442-1, ISO 22442-2 и ISO 22442-3.
1.2.2 Настоящий стандарт не детализирует специфическое требование к обозначению медицинского изделия как "стерильного".
Примечание - Следует обращать внимание на национальные или региональные требования к обозначению медицинского изделия как "стерильного". Например, см. EN 556-1 и ANSI/AAMI ST67.
1.2.3 Настоящий стандарт не устанавливает систему менеджмента качества для управления всех стадий производства медицинских изделий.
Примечание - Эффективная реализация определенных и документированных процедур обязательна для разработки, валидации и текущего управления процессом стерилизации медицинских изделий. Такие процедуры, как правило, считаются элементами системы менеджмента качества. Настоящий стандарт не требует наличия полной системы управления качеством в процессе производства или переработки. Обязательные элементы имеются в соответствующих местах текста (см., в частности, раздел 4). Внимание обращается на стандарты для систем управления качеством (например, ISO 13485), которые контролируют все этапы производства или переработки медицинских изделий. Национальные и/или региональные положения об обеспечении медицинскими изделиями могут требовать внедрение полной системы менеджмента качества и оценку этой системы третьей стороной.
1.2.4 Настоящий стандарт не устанавливает требования к охране труда, связанные с проектированием и эксплуатацией предприятий, занимающихся ЭО-стерилизацией.
Примечание 1 - Для получения более полной информации об охране труда смотрите примеры, приведенные в разделе "Библиография". Могут также существовать национальные или региональные требования.
Примечание 2 - ЭО является токсичным, воспламеняемым и взрывоопасным. Следует обратить внимание на возможное существование в некоторых странах нормативных документов, устанавливающих требования безопасности к обращению с ЭО и к помещениям, в которых он используется.
1.2.5 Настоящий стандарт не распространяется на стерилизацию инжекцией ЭО или его смесей непосредственно в упаковки или в гибкие камеры.
Примечание - См. ISO 14937, где рассматриваются эти процессы.
1.2.6 Настоящий стандарт не рассматривает аналитические методы для определения уровней остаточного ЭО и/или продуктов его реакции.
Примечание 1 - Больше информации можно получить из ISO 10993-7.
Примечание 2 - Следует обращать внимание на возможное существование национальных или региональных правил, определяющих ограничения для уровня остаточного ЭО, присутствующего на поверхности или внутри медицинских изделий.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие международные стандарты*.
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ISO 10012, Measurement management systems - Requirements for measurement processes and measuring equipment (Системы управления измерениями. Требования к измерительным процессам и измерительному оборудованию)
ISO 10993-7, Biological evaluation of medical devices - Part 7: Ethylene oxide sterilization residuals (Оценка биологическая медицинских устройств. Часть 7. Остатки при стерилизации оксидом этилена)
ISO 11138-1:2006, Sterilization of health care products - Biological indicators - Part 1: General requirements (Стерилизация медицинской продукции. Биологические индикаторы. Часть 1. Общие требования)
ISO 11138-2:2009, Sterilization of health care products - Biological indicators - Part 2: Biological indicators for ethylene oxide sterilization processes (Стерилизация медицинской продукции. Биологические индикаторы. Часть 2. Биологические индикаторы для стерилизации оксидом этилена)
ISO 11140-1, Sterilization of health care products. Chemical indicators - Part 1: General requirements (Стерилизация медицинской продукции. Химические индикаторы. Часть 1. Общие требования)
ISO 11737-1, Sterilization of medical devices - Microbiological methods - Part 1: Determination of apopulation of microorganisms on products (Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукте)
ISO 11737-2, Sterilization of medical devices - Microbiological methods - Part 2: Tests of sterility performed in the definition, validation and maintenance of a sterilization process (Стерилизация медицинских изделий. Микробиологические методы. Часть 2. Испытания на стерильность, проводимые при определении, валидации и обслуживании процесса стерилизации)
ISO 13485:2003/Cor 1:2009, Medical devices - Quality management systems - Requirements for regulatory purposes - Technical Corrigendum 1 (Изделия медицинские. Система менеджмента качества. Требования для регулирующих целей. Техническая поправка 1)*
_______________
* Заменен на ISO 13485:2016.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 аэрация (aeration): Часть процесса стерилизации, во время выполнения которой оксид этилена и/или продукты его реакции десорбируются из медицинских изделий до достижения заранее определенных уровней.
Примечание 1 - Это может быть выполнено внутри стерилизатора и/или в отдельной камере или помещении.
3.2 зона аэрации (aeration area): Камера или помещение, в котором выполняется аэрация.
3.3
бионагрузка (bioburden): Популяция жизнеспособных микроорганизмов на или в изделии и/или защитной системе стерилизации. [ISO/TS 11139:2006, 2.2] |
3.4
биологический индикатор (biological indicator): Испытательная система, содержащая жизнеспособные микроорганизмы, обеспечивающие определенную устойчивость процессу стерилизации. [ISO/TS 11139:2006, 2.3] |
3.5
калибровка (calibration): Совокупность операций, при определенных условиях устанавливающих соотношение между количественными значениями, показываемыми измерительным прибором или измерительной системой, или значениями, представленными материальными измерениями или контрольным материалом, и соответствующими значениями, определяемыми стандартами. [ISO/TS 11139:2006, 2.4] |
3.6
химический индикатор (chemical indicator): Испытательная система, обнаруживающая изменения в одном или нескольких предопределенных параметрах процесса, основанная на химических или физических изменениях, вызываемых воздействием процесса. [ISO/TS 11139:2006, 2.6] |
3.7 кондиционирование (conditioning): Обработка продукта в рамках цикла стерилизации, но до впуска оксида этилена, для достижения заранее определенных температуры и влажности.
Примечание 1 - Эта часть цикла стерилизации может выполняться как при атмосферном давлении, так и под вакуумом.
Примечание 2 - [см. предварительное кондиционирование (3.27)].
3.8
Примечание 1 - Для целей настоящего стандарта значение [ISO/TS 11139:2006, 2.11] |
3.9
разработка (development): Действие, в результате которого разрабатывается спецификация. [ISO/TS 11139:2006, 2.13] |
3.10 точка росы (dew point): Температура, при которой давление насыщенного водяного пара равно парциальному давлению водяного пара в атмосфере.
Примечание 1 - Любое охлаждение атмосферы ниже точки росы будет приводить к конденсации воды.
3.11
установить (establish): Определить по теоретической оценке и подтвердить экспериментом. [ISO/TS 11139:2006, 2.17] |
3.12 время инжекции этиленоксида (ЭО) (ethylene oxide (ЕО) injection time): Продолжительность фазы, начиная с первого впуска ЭО (смеси) в камеру до завершения инжекции.
3.13
время экспозиции (exposure time): Период, в течение которого параметры процесса поддерживаются в рамках специфицированных допусков. [ISO/TS 11139:2006, 2.18] Примечание 1 - Для расчета летальности цикла - это период стерилизации между концом инжекции ЭО и началом удаления ЭО. |
3.14
сбой (fault): Один или несколько параметров процесса вышли за пределы их специфицированных допусков. [ISO/TS 11139:2006, 2.19] |
3.15 очистка (flushing): Процедура, с помощью которой оксид этилена удаляется из загрузки и из камеры путем либо множественных последовательных впусков профильтрованного воздуха, инертного газа или пара и откачек камеры стерилизатора, либо непрерывным пропусканием потока профильтрованного воздуха, инертного газа или пара через загрузку и камеру стерилизатора.
3.16 фракционный цикл (fractional cycle): Цикл, в котором время экспозиции ЭО газа уменьшено по сравнению со временем, специфицированным в процессе стерилизации.
3.17 половинный цикл (half cycle): Цикл, в котором время экспозиции ЭО газа уменьшено на 50% по сравнению со временем, специфицированным в процессе стерилизации.
3.18 учреждение здравоохранения (health care facility): Государственные или частные организации и учреждения, предназначенные для ухода, поддержания здоровья, предотвращения и лечения заболеваний и травм.
Пример - Учреждение здравоохранения может быть, например, больницей, поликлиникой, домом престарелых, службой продленного ухода, автономным хирургическим центром, амбулаторией или стоматологическим кабинетом.
3.19
продукт для здравоохранения (health care product): Медицинские изделия, включая медицинские изделия для диагностики in vitro, или лекарственные средства, включая биофармацевтические. [ISO/TS 11139:2006, 2.20] |
3.20
аттестация установленного оборудования; IQ (installation qualification, IQ): Процесс получения и документирования доказательств того, что оборудование было поставлено и смонтировано в соответствии с его спецификацией. [ISO/TS 11139:2006, 2.22] |
3.21
медицинское изделие (medical device): Любой инструмент, аппарат, прибор, машина, приспособление, имплант, реагент in vitro или калибратор, программные средства, материал или иное подобное изделие, предназначенное производителем для использования (отдельно или в сочетании), на людях с одной или несколькими определенными целями, такими как: - диагностика, профилактика, мониторинг, лечение, облегчение заболевания; - диагностика, мониторинг, лечение, облегчение или компенсация травмы; - исследования, пересадка, модификация или поддержка органов или физиологического процесса; - поддержание или обеспечение жизнедеятельности; - контроль зачатия; - дезинфекция медицинских изделий; - получение информации для медицинских целей посредством рассмотрения in vitro образцов, полученных из человеческого организма, и не выполняющее своего основного действия внутри или на человеческом теле с помощью фармакологических, иммунологических или метаболических средств, но функции которого могут быть поддержаны таковыми средствами. [EN ISO 13485:2012, 3.7] |
3.22
микроорганизм (microorganism): Организм микроскопических размеров, включая бактерии, грибы, простейшие и вирусы. Примечание 1 - Специфический стандарт может не требовать демонстрации эффективности процесса стерилизации в инактивации всех видов микроорганизмов, указанных в приведенном выше определении, для валидации и/или рутинного контроля процесса стерилизации. [ISO/TS 11139:2006, 2.26] |
3.23
аттестация функционирующего оборудования; OQ (operational qualification, OQ): Процесс получения и документирования доказательств того, что смонтированное оборудование работает в заданных пределах параметров при его использовании в соответствии с инструкциями по эксплуатации. [ISO/TS 11139:2006, 2.27] |
3.24 подход "массовой гибели" (overkill approach): Подход с использованием процесса стерилизации, обеспечивающий логарифмическую редукцию спор (SLR) равной минимум 12 в биологическом индикаторе, имеющем устойчивость равную или превышающую бионагрузку продукта.
3.25
параметрический выпуск (parametric release): Декларация о том, что продукт стерилен, базирующаяся на записях, демонстрирующих нахождение параметров процесса в пределах специфицированных допусков. Примечание - Этот метод выпуска не включает в себя использование биологических индикаторов. [ISO/TS 11139:2006, 2.29] |
3.26
аттестация эксплуатируемого оборудования; PQ (performance qualification, PQ): Процесс получения и документирования доказательств того, что оборудование, установленное и работающее в соответствии с инструкциями по эксплуатации, постоянно работает в соответствии с заданными критериями и поэтому выпускает продукцию, отвечающую спецификациям. [ISO/TS 11139:2006, 2.30] |
3.27 предварительное кондиционирование (preconditioning): Обработка продукта перед циклом стерилизации в помещении или в камере для достижения специфицированных пределов температуры и относительной влажности.
3.28
устройство контроля процесса; УКП (process challenge device, PCD): Устройство, предназначенное для обеспечения свидетельств устойчивости процесса стерилизации и используемое для оценки результатов процесса. [ISO/TS 11139:2006, 2.33] Примечание 1 - Для целей настоящего стандарта УКП может быть продуктом, имитатором продукта или иным устройством, инокулированным непосредственно или косвенно. См. 7.1.6 и D.7.1.6 (приложение D). Примечание 2 - В этом стандарте устанавливается различие между внутренним УКП и внешним УКП. Внутренний УКП используется для демонстрации того, что требуемый уровень обеспечения стерильности (SAL) достигнут. УКП, расположенный внутри границ продукта или внутри товарной упаковки, является внутренним, тогда как УКП, расположенный между товарными упаковками или на внешних поверхностях загрузки, является внешним. Внешний УКП является элементом, предназначенным для использования в микробиологическом мониторинге рутинных производственных циклов. |
3.29
параметр процесса (process parameter): Оговоренное [специфицированное] значение переменной процесса. Примечание - Спецификации процесса стерилизации включают в себя параметры процесса и их допуски. [ISO/TS 11139:2006, 2.34] |
3.30
переменная процесса (process variable): Условие процесса стерилизации, изменение которого изменяет микробицидную эффективность. Пример - Время, температура, давление, концентрация, влажность. [ISO/TS 11139:2006, 2.35] |
3.31 категория обработки (processing category): Набор различных продуктов или семейств продуктов, которые можно стерилизовать вместе.
Примечание - Все продукты, входящие в категорию, были определены как представляющие равную или меньшую нагрузку на процесс стерилизации, чем устройство нагрузки процесса для этой группы.
3.32
продукт (product): Результат процесса. [ISO 9000:2005, 3.4.2] Примечание - Согласно стандартам стерилизации, продуктом является материальный объект, который может быть исходным (сырьевым) материалом, промежуточным звеном (соединением), сборочным узлом и продуктом для здравоохранения. |
3.33 семейство продукта (product family): Группа процессных характеристик продуктов, позволяющая им быть стерилизованными с использованием определенных условий процесса.
3.34 объем загрузки продукта (product load volume): Определенное пространство внутри полезного объема камеры, занимаемое продуктом.
3.35
признанная коллекция культур (recognized culture collection): Депозитарий в соответствии с Будапештским договором о Международном признании депонирования микроорганизмов для целей патентной процедуры и регламентирования. [ISO/TS 11139:2006, 2.38] |
3.36
контрольный микроорганизм (reference microorganism): Штамм микроорганизма, полученный из признанной коллекции культур. [ISO/TS 11139:2006, 2.39] |
3.37
повторная аттестация (requalification): Повторение части валидации в целях подтверждения продолжающейся приемлемости специфицированного процесса. [ISO/TS 11139:2006, 2.40] |
3.38 многоразовое медицинское изделие (reusable medical device): Медицинское изделие, предназначенное или признаваемое изготовителем подходящим для повторных обработок и многократного использования.
Примечание - Этим оно отличается от медицинского изделия, которое предназначено или признано изготовителем пригодным только для однократного применения.
3.39
питающие среды (services): Питание из внешних источников, необходимое для функционирования оборудования. Пример - Электроэнергия, вода, сжатый воздух, канализация. [ISO/TS 11139:2006, 2.41] |
3.40 одноразовое медицинское изделие (single use medical device): Медицинское изделие, которое предназначено или признано изготовителем пригодным только для однократного применения.
3.41
специфицировать (specify): Подробно описывать в утвержденном документе. [ISO/TS 11139:2006, 2.42] |
3.42
логарифмическая редукция спор; SLR (Spore-log-reduction SLR): Логарифм начальной популяции спор, [ISO 14161:2009, 3.19] Примечание - Описание снижения количества спор в биологическом индикаторе или на инокулированном объекте в результате экспозиции в специфицированных условиях. Для прямого подсчета: | |
логарифмическая редукция | (1) |
где
Для отрицательной фракции: | |
логарифмическая редукция | (2) |
где
Если выживших спор нет, истинное значение логарифмической редукции не может быть рассчитано. Это значение может быть выражено как "не более чем" | |
3.43
стерильный (sterile): Не содержащий жизнеспособных микроорганизмов. [ISO/TS 11139:2006, 2.43] |
3.44
стерильная барьерная система (sterile barrier system): Минимальная упаковка, которая предотвращает попадание микроорганизмов и позволяет предоставить асептический продукт в точке использования. [ISO/TS 11139:2006, 2.44] |
3.45
стерильность (sterility): Состояние, характеризующееся отсутствием жизнеспособных микроорганизмов. Примечание 1 - На практике невозможно доказать абсолютное отсутствие микроорганизмов. Примечание 2 - [см. стерилизация (3.47)]. [ISO/TS 11139:2006, 2.45] |
3.46
уровень обеспечения стерильности; SAL (sterility assurance level, SAL): Вероятность наличия единичного жизнеспособного микроорганизма на продукте после стерилизации. Примечание - Уровень обеспечения стерильности (SAL) имеет количественное значение, как правило, порядка 10 [ISO/TS 11139:2006, 2.46] |
3.47
стерилизация (sterilization): Валидированный процесс, используемый для освобождения продукта от всех жизнеспособных микроорганизмов. Примечание 1 - В процессе стерилизации природа отмирания микроорганизмов описывается экспоненциальной функцией. Следовательно, наличие жизнеспособных микроорганизмов на каждом отдельном изделии может быть выражено в терминах вероятности. В то время как эта вероятность может быть снижена до очень малых чисел, она никогда не может быть доведена до нуля [см. ISO/TS 11139:2006, 2.47]. Примечание 2 - [см. уровень обеспечения стерильности (3.46)]. [ISO/TS 11139:2006, 2.47] |
3.48 цикл стерилизации (sterilization cycle): Обработка в герметичной камере, включающая удаление воздуха, кондиционирование (если используется), введение стерилизующего агента (оксида этилена), инертный газ (если используется), выдержку в оксиде этилена, удаление оксида этилена, очистку (если используется) и впуск воздуха/инертного газа.
3.49
стерилизационная загрузка (sterilization load): Продукт(ы), которые должны быть или были стерилизованы вместе с использованием одного процесса стерилизации. [ISO/TS 11139:2006, 2.48] |
3.50
процесс стерилизации (sterilization process): Последовательность действий или операций, необходимая для достижения специфицированных требований к стерильности. [ISO/TS 11139:2006, 2.49] Примечание - Это серия действий или операций включает в себя предварительную подготовку (при необходимости), экспозицию окиси этилена при определенных условиях с последующей обработкой, необходимой для удаления окиси этилена и его побочных продуктов. Они не включают какую-либо очистку, дезинфекцию или упаковочные операции, которые предшествуют процессу стерилизации. |
3.51 специалист по стерилизации (sterilization specialist): Лицо с техническими знаниями об используемой технологии стерилизации и ее влиянии на материалы и микроорганизмы.
3.52
стерилизующий агент (sterilizing agent): Физический или химический объект, или комбинация объектов, имеющих достаточную бактерицидную активность для достижения стерильности при определенных условиях. [ISO/TS 11139:2006, 2.50] |
3.53
кривая выживаемости (survivor curve): Графическое представление инактивации популяции микроорганизмов с увеличением экспозиции микробоцидного агента при указанных условиях. [ISO/TC 11139:2006, 2.51] |
3.54
испытание на стерильность (test for sterility): Техническая операция, определенная в Фармакопее, выполняемая на изделии после воздействия на него процесса стерилизации. [ISO/TC 11139:2006, 2.53] |
3.55
проверка стерильности (test of sterility): Техническая операция, выполняемая как часть разработки, валидации и реаттестации для определения присутствия или отсутствия жизнеспособных микроорганизмов на продукте или его части. [ISO/TC 11139:2006, 2.54] |
3.56 полезный объем камеры (usable chamber volume): Определенное пространство внутри камеры стерилизатора, не ограниченное фиксированными или подвижными частями и способное принять стерилизационную загрузку.
Примечание - Объем, оставляемый для обеспечения циркуляции газа вокруг загрузки внутри камеры, не включается в полезный объем камеры.
3.57
валидация (validation): Документированная процедура получения, регистрации и интерпретации результатов, необходимая для демонстрации того, что процесс неизменно выдает продукт, соответствующий заранее определенным спецификациям. [ISO/TC 11139:2006, 2.55] |
3.58 исходный материал (virgin material): Материал, который не был ранее использован или подвергнут иной обработке, кроме обработки, необходимой для его первоначального производства.
4 Системы менеджмента качества
4.1 Документирование
4.1.1 Должны быть установлены процедуры по разработке, валидации, текущему контролю и выпуску продукции после стерилизации.
4.1.2 Документы и записи, требуемые настоящим стандартом, должны быть рассмотрены и одобрены специально назначенными лицами (см. 4.2.1). Документы и записи должны управляться в соответствии с применимыми положениями ISO 13485.
4.2 Ответственность руководства
4.2.1 Ответственность и полномочия за осуществление и удовлетворение требований, установленных настоящим стандартом, должны быть четко определены и описаны. Ответственность должна быть возложена на квалифицированный персонал в соответствии с применимыми положениями ISO 13485.
4.2.2 Если требования настоящего стандарта выполняются организациями с раздельными системами менеджмента качества, то должны быть установлены ответственность и полномочия каждой из таких организаций.
Если учреждение здравоохранения заключает контракты на стерилизацию изделий медицинского многократного применения со сторонней организацией, то ответственность за валидацию и выпуск стерилизованного продукта несет учреждение здравоохранения.
4.3 Выпуск продукции
4.3.1 Должны быть установлены процедуры закупок. Эти процедуры должны соответствовать применимым положениям ISO 13485.
4.3.2 Должны быть установлены процедуры идентификации и прослеживания продукта. Эти процедуры должны соответствовать применимым положениям ISO 13485.
4.3.3 Должна быть определена и описана система калибровки всего оборудования, соответствующая применимым статьям ISO 13485 или ISO 10012, включающая в себя также средства измерений для проведения испытаний, используемые для обеспечения соответствия требованиям настоящего стандарта.
4.4 Измерение, анализ и улучшение. Управление несоответствующей продукцией
Должны быть установлены процедуры по управлению несоответствующей продукцией, а также процедуры по коррекции, корректирующим и предупреждающим действиям. Такие процедуры должны удовлетворять применимым положениям ISO 13485.
5 Характеризация стерилизующего агента
5.1 Общие требования
Цель этих действий - определение стерилизующего агента, демонстрация его микробоцидной эффективности, идентификация факторов, влияющих на микробоцидную эффективность, оценка эффекта воздействия стерилизующего агента на материалы, определение требований безопасности для персонала и защиты окружающей среды. Эти действия могут быть выполнены при испытаниях модели (прототипа) системы. В таких случаях итоговая спецификация на оборудование (см. 6.3) должна быть соотносимой с результатами экспериментальных исследований, выполненных при испытаниях прототипа системы. Для целей настоящего стандарта стерилизующий агент - ЭО.
5.2 Стерилизующий агент
Спецификация стерилизующего агента должна включать в себя, если необходимо, условия хранения, обеспечивающие сохранение свойств ЭО в рамках его спецификаций в течение всего заявленного срока хранения.
5.3 Микробоцидная эффективность
Данные микробоцидной эффективности должны быть разработаны если предполагается использовать ЭО вне пределов диапазона широко известных смесей или если предполагается использование нового разбавителя.
Примечание - Инактивация микроорганизмов ЭО была всесторонне описана в литературе. Эта литература дает знание того, каким образом переменные процесса влияют на инактивацию микроорганизмов. Ссылка на эти общие исследования по микробиологической инактивации не требуется настоящим стандартом.
5.4 Воздействие на материалы
Эффекты воздействия стерилизующего агента на широкий ряд материалов, используемых для изготовления медицинских изделий, были всесторонне описаны в литературе, и эта документация имеет ценность для тех, кто разрабатывает и производит медицинские изделия, которые должны стерилизоваться ЭО. Настоящий стандарт не требует проводить специфические исследования о воздействии на материалы, но требует проведения исследований о воздействии ЭО на продукт (см. раздел 7).
5.5 Безопасность и окружающая среда
5.5.1 Для ЭО и его растворителей (если таковые используются) должны иметься либо паспорт безопасности материала (MSDS), либо иная аналогичная информация по безопасности. Необходимые меры, обязательные для защиты здоровья и обеспечения безопасности персонала должны быть идентифицированы.
5.5.2 Потенциальное влияние работы стерилизатора на окружающую среду должно быть оценено и должны быть установлены меры по охране окружающей среды. Эта оценка, включая потенциальное воздействие и меры, направленные на управление, должна быть задокументирована.
5.5.3 Пользователи ЭО должны соблюдать соответствие требованиям локальных, национальных и международных нормативов в отношении эмиссии (выбросов) и утилизации ЭО и его разбавителей, а также любых субпродуктов.
6 Характеризация процесса и оборудования
6.1 Общие требования
6.1.1 Цель этих действий - полностью определить весь стерилизационный процесс и оборудование, необходимое для обеспечения безопасности и воспроизводимости стерилизационного процесса.
6.1.2 Если для стерилизации продукта был использован существующий процесс, то эти действия не нужны, однако процесс и оборудование должны быть пересмотрены, чтобы убедиться, что идентифицированные в 6.2 и 6.3 переменные были включены в спецификацию процесса для рутинного производства.
6.2 Характеризация процесса
6.2.1 Характеризация процесса должна включать в себя как минимум следующее:
a) определение фаз, которые необходимы для процесса ЭО стерилизации;
b) определение переменных процесса для каждой фазы;
c) документирование параметров процесса.
Примечание - Данные, разработанные при определении продукта (раздел 7), могут повлиять на характеризацию процесса стерилизации.
6.2.2 Процесс стерилизации включает в себя следующие фазы:
a) предварительное кондиционирование (если используется);
b) цикл стерилизации;
c) аэрацию (если используется).
6.2.3 В число процессных переменных для предварительного кондиционирования (если оно используется) включается, как минимум, следующее:
a) время;
b) температура;
c) влажность;
d) время переноса.
6.2.4 В число процессных переменных для цикла стерилизации включается, как минимум, следующее:
a) время экспозиции;
b) температура;
c) влажность;
d) концентрация ЭО;
e) давление.
6.2.5 В число процессных переменных для аэрации (если оно используется) включается, как минимум, следующее:
a) время;
b) температура.
Примечание - В фазе аэрации эти параметры рассматриваются как процессные переменные только в том случае, когда аэрация принимает участие в обеспечении микробоцидной эффективности процесса стерилизации (см. AAMI TIR 16:2009).
6.3 Характеризация оборудования
6.3.1 Спецификации оборудования должны быть разработаны и задокументированы. Эти спецификации должны включать в себя:
a) зону предварительного кондиционирования (если используется);
b) стерилизатор;
c) зону аэрации (если используется).
Примечание - На некоторые аспекты конструкции оборудования могут влиять требования национальных или региональных нормативных документов или стандартов.
6.3.2 В спецификацию включается как минимум следующее:
a) описание оборудования вместе с необходимыми дополнительными устройствами, в том числе описание материалов конструкции;
b) описание средств, с помощью которых осуществляется подача стерилизующего агента в камеру;
c) описание средств, с помощью которых осуществляется подача иных газов, включая пар, в камеру;
d) описание приборной оснастки для мониторинга, управления и записи данных процесса стерилизации, включая характеристики датчиков и точек их размещения;
e) неисправности и ошибки, распознаваемые стерилизационным оборудованием;
f) устройства безопасности, включая безопасность персонала и защиту окружающей среды;
g) требования к монтажу, включая спецификации необходимых питающих сред и требования к контролю выбросов.
6.3.3 Программное обеспечение, используемое для управления и/или мониторинга стерилизационного процесса, должно быть подготовлено и валидировано в соответствии с требованиями элементов системы управления качеством, обеспечивающими документированное свидетельство того, что программное обеспечение соответствует проектной спецификации.
Примечание - Для дополнительной информации следует обратить внимание на требования ISO 90003.
6.3.4 Средства мониторинга и управления процессными переменными должны быть определены и специфицированы.
6.3.5 Должны быть предусмотрены средства, обеспечивающие гарантию того, что сбой в функции контроля не приведет к сбою записи переменных процесса таким образом, что неэффективный процесс будет отмечен как эффективный.
Примечание - Это может быть достигнуто как использованием независимых систем управления контроля и мониторинга, так и перекрестными проверками между контролем и мониторингом, которые определяют любые несоответствия или индуцируют сбой.
7 Определение продукта
7.1 Общая информация
7.1.1 Цель этого действия - определить продукт, подлежащий стерилизации, включая микробиологическое качество продукта до стерилизации и способ упаковки и подготовки к стерилизации.
7.1.2 Определение продукта должно быть выполнено до внедрения нового или модифицированного продукта, упаковки или конфигурации загрузки. Следует рассмотреть демонстрацию эквивалентности (со ссылкой на нагрузку процесса) ранее валидированного продукта, упаковки или конфигурации загрузки, чтобы соблюсти требование к выполнению определения продукта. Любая демонстрация эквивалентности должна быть задокументирована.
7.1.3 Продукт должен быть рассчитан так, чтобы он позволял выполнить удаление воздуха (если нужно), обеспечивал проникновение пара, влажности, ЭО, и удаление ЭО в конце процесса.
7.1.4 Упаковка должна быть рассчитана так, чтобы она позволяла выполнить удаление воздуха (если нужно), обеспечивала проникновение пара, влажности, ЭО и удаление ЭО в конце процесса.
7.1.5 Конфигурация загрузки должна быть рассчитана так, чтобы она позволяла выполнить удаление воздуха (если нужно), обеспечивала проникновение пара, влажности, ЭО и удаление ЭО в конце процесса.
7.1.6 Должно быть продемонстрировано, что специфицированный процесс стерилизации эффективен при обработке трудно поддающихся стерилизации мест внутри продукта. Это может быть достигнуто путем выполнения определения процесса и валидации нового продукта, или через демонстрацию эквивалентности ранее валидированного продукта, или использованием внутреннего УКП, используемого для определения уровня обеспечения стерильности продукта при воздействии специфицированного процесса стерилизации (см. 8.6 и D.8.6).
7.2 Безопасность продукта, качество и характеристики
7.2.1 Должно быть подтверждено, что продукция и ее упаковка отвечают установленным требованиям безопасности, качества и обеспечения характеристик после применения установленного процесса стерилизации с использованием процессных параметров с допусками, которые были определены как имеющие наибольшее влияние на продукцию/упаковку.
Примечание - Управление проектированием является одним из аспектов, который рассматривается в ISO 14971.
7.2.2 Если разрешены многократные циклы стерилизации, последствия такой обработки продукта и его упаковки должны быть оценены.
7.2.3 Биологическая безопасность продукта после воздействия процесса стерилизации должна устанавливаться в соответствии с действующими частями ISO 10993.
7.2.4 Должны быть установлены средства для снижения остаточных уровней ЭО до такой степени, чтобы обработанные продукты соответствовали требованиям ISO 10993-7.
7.3 Микробиологическое качество
7.3.1 Должна быть определена и поддерживаться система, гарантирующая, что микробиологическое качество и чистота продукта, представленного для стерилизации, контролируются и не ухудшают эффективность процесса стерилизации.
Примечание - Бактериальные эндотоксины не разрушаются в ЭО-процессе. Руководство по испытаниям для бактериальных эндотоксинов предоставляется в ANSI /AAMI ST72/ и в применимой фармакопее.
7.3.2 Для медицинских изделий одноразового использования оценка бионагрузки через определенные интервалы времени должна осуществляться в соответствии с ISO 11737-1. Для медицинских изделий многократного использования должна быть выполнена оценка эффективности специфицированного процесса очистки и (если применимо), процесса дезинфекции.
Примечание - Требования к информации, предоставляемой для повторной обработки многократно стерилизуемых изделий, приведены в ISO 17664. Информация относительно оценки эффективности очистки и дезинфекции дается в соответствующих частях ISO 15883.
7.4 Документирование
Результаты определения продукта должны быть задокументированы изготовителем медицинского изделия.
8 Определение процесса
8.1 Цель данного действия - получение спецификации процесса, который может быть применен для стерилизации определенного продукта (см. раздел 7), во время валидационных исследований.
8.2 Процесс стерилизации, соответствующий определенному изделию, должен быть установлен. Определенный продукт может быть новым или модифицированным продуктом, упаковкой или конфигурацией загрузки.
8.3 Действия по определению процесса должны выполняться в стерилизационной камере (камере для разработки или производственной камере), прошедшей процедуры аттестации установленного оборудования (IQ) и аттестации функционирующего оборудования (OQ) (см. 9.2 и 9.3).
8.4 Документация и записи должны поддерживать валидность параметров процесса и соответствующих процессных переменных, определенных в ходе характеризации процесса (см. 6.2).
8.5 Степень микробиологической инактивации, обеспечиваемой специфицированным циклом стерилизации для специфической микробиологической нагрузки, должна быть определена с помощью одного из методов, описанных в приложениях А или В, или альтернативным способом, демонстрирующим, что продукт достигает требуемого уровня обеспечения стерильности.
8.6 Биологические индикаторы (БИ), используемые как часть установления процесса стерилизации, должны:
a) соответствовать требованиям ISO 11138-2:2006, раздел 5 и пункт 9.5;
b) иметь как минимум такую же устойчивость к ЭО, как и бионагрузка продукта, подлежащего стерилизации,
c) располагаться в соответствующем УКП.
Пригодность УКП, используемого для определения процесса, валидации или рутинного контроля и мониторинга, должна быть определена. УКП должен представлять для стерилизационного процесса нагрузку, равную или превышающую естественную нагрузку в местах продукта, наиболее трудно поддающихся стерилизации.
Примечание - Информацию о выборе биологических индикаторов, их использовании и интерпретации результатов смотрите в ISO 14161.
8.7 Поставляемые на коммерческой основе биологические индикаторы, используемые для определения процесса стерилизации, должны соответствовать требованиям 8.6 и всех применимых статей ISO 11138-1.
8.8 Если химические индикаторы используются как часть определения стерилизационного процесса, то они должны соответствовать требованиям ISO 11140-1.
Химические индикаторы не должны использоваться как единственное средство установления стерилизационного процесса и не должны использоваться как средство индикации достижения требуемого уровня обеспечения стерильности.
8.9 Если проверка стерильности выполняется во время установления процесса стерилизации, такая проверка должна соответствовать требованиям ISO 11737-2.
9 Валидация
9.1 Общая информация
9.1.1 Целью валидации является демонстрация того, что стерилизационный процесс, установленный в ходе определения процесса (см. раздел 8), может эффективно и с хорошей воспроизводимостью выполняться на продукте в стерилизационной загрузке. Валидация состоит из нескольких определенных этапов: аттестации установленного оборудования (IQ), аттестации функционирующего оборудования (OQ) и аттестации эксплуатируемого оборудования (PQ). Валидационные испытания не должны начинаться, пока процедуры и/или протоколы не будут одобрены.
9.1.2 Аттестация установленного оборудования (IQ) выполняется для демонстрации того, что оборудование и любые дополнительные устройства были поставлены и смонтированы в соответствии с их спецификациями.
9.1.3 Аттестация функционирующего оборудования (OQ) выполняется для демонстрации способности оборудования обеспечивать выполнение требований к его специфицированным расчетным характеристикам.
9.1.4 Аттестация эксплуатируемого оборудования (PQ) - это этап валидации, использующий продукт для демонстрации того, что оборудование постоянно работает в соответствии с заранее установленными критериями приемлемости, и что процесс выдает продукт, являющийся стерильным и отвечающим специфицированным требованиям.
Аттестации установленного оборудования (IQ) и функционирующего оборудования (OQ) могут быть однократными мероприятиями для конкретного оборудования, применяемого при выполнении процесса стерилизации. Аттестацию эксплуатируемого оборудования (PQ) следует проводить для каждого нового валидируемого процесса и/или продукта с целью подтверждения того, что процесс удовлетворяет идентифицированным критериям приемлемости и способен обеспечивать необходимый уровень обеспечения стерильности.
9.2 Аттестация установленного оборудования (IQ)
9.2.1 Оборудование
9.2.1.1 Оборудование, используемое в стерилизационном процессе, включая любые вспомогательные устройства, должно соответствовать своим расчетным (проектным) характеристикам.
9.2.1.2 Стерилизационное оборудование должно соответствовать требованиям применимых стандартов по безопасности.
9.2.1.3 Рабочие процедуры для оборудования должны быть специфицированы. Эти рабочие процедуры должны включать (но не ограничиваться):
a) пошаговые рабочие инструкции;
b) условия возникновения сбоев/ошибок/неисправностей, способы их отображения и действия, которые должны быть при этом выполнены;
c) инструкции для обслуживания и калибровки;
d) подробную контактную информацию по технической поддержке.
9.2.2 Аттестация установленного оборудования
9.2.2.1 Монтаж оборудования и всех связанных с ним питающих сред должен выполняться согласно архитектурным и инженерным чертежам. Монтаж должен соответствовать требованиям всех соответствующих национальных и локальных нормативных документов.
9.2.2.2 Должна быть специфицированная инструкция по монтажу, включающая в себя инструкции по охране здоровья и технике безопасности для персонала.
9.2.2.3 Условия безопасного хранения ЭО должны быть специфицированы для сохранения его качества и состава согласно спецификации.
9.2.2.4 Перед выполнением аттестации установленного оборудования необходимо подтвердить статус калибровки всех приборов, используемых во время аттестации.
9.2.2.5 Во время выполнения аттестации установленного оборудования должны быть актуализированы все монтажные чертежи, в том числе чертежи трубной обвязки и вспомогательного оборудования по состоянию "как есть".
9.2.2.6 Изменения, внесенные в системы в ходе выполнения аттестации установленного оборудования, должны быть оценены с точки зрения их воздействия на проект и на спецификации процесса и задокументированы в файл проекта.
9.3 Аттестация функционирующего оборудования (OQ)
9.3.1 Перед выполнением аттестации функционирующего оборудования необходимо подтвердить статус калибровки всех приборов (включая все контрольные приборы), используемых для мониторинга, контроля, индикации и записи процесса стерилизации (см. 4.3.3).
9.3.2 Аттестация функционирующего оборудования должна продемонстрировать, что установленное (смонтированное) оборудование удовлетворяет своим рабочим спецификациям.
9.4 Аттестация эксплуатируемого оборудования (PQ)
9.4.1 Общая информация
9.4.1.1 Аттестация эксплуатируемого оборудования состоит из микробиологической аттестации и аттестации физических характеристик, и выполняется на оборудовании, используемом для стерилизации продукта.
9.4.1.2 Аттестация эксплуатируемого оборудования должна выполняться при внедрении нового или модифицированного продукта, упаковки, конфигурации загрузки, оборудования или параметров процесса, если только нет документированного доказательства эквивалентности ранее валидированному продукту, упаковке, конфигурации загрузки, оборудованию или параметрам процесса (см. 7.1.2, 7.1.6 и 12.5).
9.4.1.3 Аттестация эксплуатируемого оборудования должна использовать продукт или материал, представительный в отношении рутинно стерилизуемого продукта, для демонстрации того, что оборудование постоянно работает в соответствии с заранее установленными критериями приемлемости, и что процесс выдает продукт, отвечающий установленным требованиям к уровню обеспечения стерильности.
9.4.1.4 Способ представления продукта для стерилизации, включая конфигурацию загрузки, должен быть документирован.
Примечание - Если в ходе валидации использовался товарный продукт, пункт 7.2 дает информацию относительно качества продукта, необходимого для его использования пациентами, а пункт 11.4 дает информацию относительно требований к выпуску стерильного продукта.
9.4.1.5 Загрузка, используемая в аттестации эксплуатируемого оборудования, должна быть представительной в отношении рутинно стерилизуемого продукта, и должна быть определена на основе наиболее трудной для стерилизации рутинной загрузки.
9.4.1.6 Для учреждений, работающих с широко различающимися конфигурациями загрузки, степень, до которой изменения влияют на процесс стерилизации, должна быть оценена. Должно быть продемонстрировано, что весь продукт, подвергнутый процессу стерилизации, достиг требуемого уровня обеспечения стерильности.
9.4.1.7 Если используется материал, отличный от продукта, он должен представлять для процесса стерилизации нагрузку, как минимум, равную или большую по сравнению с продуктом.
9.4.1.8 Если (одни и те же) загрузки неоднократно используются в валидационных циклах, они должны аэрироваться в промежутках между циклами, чтобы соответствовать требованиям к охране труда персонала, и чтобы гарантировать, что остатки ЭО в загрузках не повлияют на биологическую оценку в последующих микробиологических исследованиях.
9.4.1.9 Если в ходе аттестации эксплуатируемого оборудования используются химические индикаторы, они должны соответствовать требованиям ISO 11140-1 и должны применяться в связке с микробиологическим и физическим мониторингом.
9.4.1.10 Биологические индикаторы, используемые при микробиологической аттестации эксплуатируемого оборудования, должны соответствовать требованиям ISO 11138-1:2006 (раздел 5) и ISO 11138-2:2009 (пункт 9.5).
9.4.2 Аттестация эксплуатируемого оборудования - микробиологические исследования
9.4.2.1 Микробиологическая аттестация эксплуатируемого оборудования (MPQ) должна продемонстрировать, что после применения процесса стерилизации все специфицированные требования к стерильности удовлетворены. Исследования должны выполняться в производственной камере с использованием определенных параметров процесса, выбираемых для получения меньшей летальности, чем предусматривает специфицированный процесс стерилизации.
9.4.2.2 Микробиологическая аттестация эксплуатируемого оборудования должна подтвердить эффективность определенного процесса в отношении комбинации продукта/конфигурации загрузки в производственной камере.
9.4.2.3 Летальность цикла должна определяться путем использования одного из методов, описанных в приложениях А или В либо с применением альтернативного метода, способного продемонстрировать достижение требуемого уровня обеспечения стерильности.
9.4.2.4 Если определение процесса выполнялось не в производственной, а в разработочной камере, микробиологическая аттестация эксплуатируемого оборудования должна включать в себя по меньшей мере три фракционных или три половинных цикла, выполняемых в производственной камере, которые подтвердят данные, полученные в разработочной камере.
9.4.2.5 Если используются половинные циклы с подходом "массовой гибели" [см. B.1.2, перечисление a) (приложение B)], то после их выполнения не должно быть ни одного позитивного внутреннего УКП.
Положительные внешние УКП допустимы, если они продемонстрировали большую устойчивость, чем внутренние УКП, обеспечивающие ситуацию "худшего случая" для рутинной обработки. Тем не менее, внутренние УКП должны дать отрицательный результат.
9.4.2.6 Если используется расчет по циклу "массовой гибели" [см. B.1, перечисление b) (приложение B)] или расчет с использованием биологического индикатора/биологической нагрузки (см. приложение A), то возможно обнаружение нескольких выживших внутренних УКП, но подсчитанный уровень обеспечения стерильности должен совпадать со специфицированным значением (см. ISO 14161).
9.4.3 Аттестация эксплуатируемого оборудования - физические исследования
9.4.3.1 Физические исследования, выполняемые в ходе аттестации эксплуатируемого оборудования, должны продемонстрировать:
a) что специфицированные критерии приемлемости выполняются во всем объеме загрузки на протяжении периода, предусмотренного спецификацией рутинного процесса;
b) воспроизводимость процесса.
Физические исследования должны включать в себя как минимум три плановых квалификационных цикла, выполняемых последовательно в рамках одного испытания, в которых должны быть удовлетворены все специфицированные критерии приемлемости. Физические исследования могут проводиться во время выполнения микробиологических исследований. Если физические исследования выполняются параллельно с микробиологическими в минимум трех циклах, тогда необходимо выполнение как минимум еще одного цикла физических исследований с полными спецификациями рутинного процесса.
Если сбой (неудачное выполнение) связан с факторами, не имеющими отношения к эффективности валидируемого процесса, он может быть документирован как не относящийся к характеристикам процесса без необходимости выполнения трех дополнительных последовательных циклов. Примерами таких сбоев могут быть (но не ограничиваются приведенными) перебои в подаче электропитания, перебои в подаче других сред или неисправности внешнего оборудования мониторинга.
9.4.3.2 Физические исследования должны подтвердить, что:
a) минимальная температура продукта, подаваемого в процесс стерилизации и/или определенные условия, позволяющие ее достигнуть, установлены;
b) температура и влажность загрузки в конце заданного времени предварительного кондиционирования (если оно применяется) установлены;
c) специфицированное максимальное время между окончанием предварительного кондиционирования (если оно применяется) и началом выполнения стерилизационного цикла является достаточным;
d) температура и влажность загрузки в конце заданного времени кондиционирования (если оно применяется) установлены;
e) влажность в камере записывается, если был предусмотрен параметрический выпуск (продукта);
f) газообразный ЭО был введен в стерилизационную камеру;
g) рост давления и количество использованного ЭО или концентрация ЭО в камере были установлены [см. 9.5.4, перечисление f)]. Если предусмотрен параметрический выпуск, см. также 9.5.4, перечисление b).
h) во время стерилизационного цикла температура и влажность (если она записывалась) в камере и другие параметры процесса, где это применимо, были установлены;
i) температура продукта в загрузке во время экспозиции была установлена;
j) во время аэрации (если она использовалась) температура продукта в загрузке была установлена.
9.5 Рассмотрение (анализ) и утверждение результатов валидации
9.5.1 Цель этой деятельности - проанализировать и документировать результаты обзора валидационных данных для подтверждения их приемлемости относительно утвержденных валидационных процедур/протоколов процесса стерилизации и утверждения спецификации стерилизационного процесса.
9.5.2 Информация, собранная или полученная во время определения продукта, определения процесса и всех аттестаций, в том числе результаты инкубирования биологических индикаторов, должна быть записана и проверена на приемлемость. Результаты этой оценки должны быть записаны.
9.5.3 Должен быть подготовлен отчет о валидации. Отчет должен быть рассмотрен и утвержден назначенным ответственным лицом (лицами).
9.5.4 Валидационный отчет должен описывать продукт, использованный при аттестации, или дать ссылку на его описание, определенные конфигурации загрузки и документированную спецификацию процесса ЭО-стерилизации, а также содержать следующие данные:
Примечание - Для практических целей должны быть определены скорость изменения давления и время (с допусками), необходимые для достижения специфицированного изменения давления.
a) минимальная температура продукта, подаваемого в процесс стерилизации и/или определенные условия, позволяющие ее достигнуть;
b) предварительное кондиционирование (если используется):
1) время пребывания в камере/зоне, температура и влажность в камере/зоне;
2) температура и влажность стерилизационной загрузки;
3) максимальное время между выемкой загрузки из камеры/зоны предварительного кондиционирования и началом выполнения стерилизационного цикла;
c) уровни вакуума и скорость откачки воздуха (если используется):
1) время удержания в вакууме (если используется);
Примечание - Скорость откачки обычно специфицируется либо как минимальное допустимое время откачки, либо как максимально допустимое время откачки, реже как время, специфическое для каждого цикла.
d) продувка инертным газом (если используется):
1) давление ( или окончательное давление) и скорость (
/время), необходимые для достижения давления, ассоциируемого с газом/паром;
2) глубина ( или окончательное давление) и скорость (
/время), необходимые для достижения вакуума;
3) количество повторений и любые разбросы в успешных повторах;
e) фазы выдержки кондиционирования и увлажнения (если используются):
1) уровни давления и/или скорость достижения вакуума, или уровни относительной влажности (что из этого контролируется и отслеживается);
2) количество импульсов пара/вакуума (если используется);
3) время;
4) температура камеры;
5) температура и влажность загрузки в конце кондиционирования;
f) инжекция ЭО и экспозиция:
1) повышение давления (), время инжекции ЭО, окончательное давление в фазе инжекции ЭО;
2) доказательство того, что газообразный ЭО был введен в стерилизационную камеру, полученное за счет повышения давления и наличия одного из следующих признаков:
i) массы введенного ЭО [см. D.10.2, перечисление i), (приложение D)];
ii) прямого измерения концентрации ЭО;
iii) объема использованного ЭО
3) температура камеры стерилизатора;
4) время экспозиции;
5) температура стерилизационной загрузки;
6) индикация удовлетворительной работы системы циркуляции газа в камере (если используется) во время экспозиции;
g) продувка после экспозиции (если используется):
1) глубина ( или окончательное давление) и скорость (
/время) достижения вакуума;
2) давление ( или окончательное давление) и скорость (
/время) достижения давления, ассоциируемого с инертным газом/воздухом/паром;
3) количество повторений и любые разбросы в успешных повторах;
h) аэрация (если используется):
1) время и температура в камере/помещении;
2) изменения давления (если они есть) в камере и/или помещении;
3) скорость воздухообмена или газообмена, если используется иной газ;
4) температура стерилизационной загрузки.
9.5.5 Если должен использоваться параметрический выпуск, валидационный отчет должен также содержать:
a) значение влажности в камере (с допусками), полученное прямым измерением во время кондиционирования;
b) значение концентрации (с допусками) ЭО, определенное непосредственным анализом атмосферы в камере с использованием аналитических методов, для установления процессной спецификации для рутинной обработки. Отбор проб должен выполняться через определенные промежутки времени для верификации требуемых условий во время экспозиции ЭО;
c) температура в камере; записывается в двух отдельных точках мониторинга.
9.5.6 Спецификация процесса для рутинной обработки, включая параметры процесса и их допуски, должна быть установлена на базе документации, полученной в ходе валидации. Эта спецификация процесса также должна включать в себя критерии для определения стерилизуемого ЭО продукта как изделия, подходящего для этой обработки и одобренного для выпуска.
10 Рутинный мониторинг и управление
10.1 Цель рутинного мониторинга и управления - продемонстрировать, что продукт подвергался валидированному и специфицированному стерилизационному процессу.
10.2 Данные каждого цикла стерилизации должны записываться и сохраняться для демонстрации того, что спецификация процесса была соблюдена. Эти данные должны включать в себя как минимум следующее:
Примечание - Для практических целей могут быть определены скорость изменения давления и время (с допусками), необходимые для достижения специфицированного изменения давления.
a) минимальная температура продукта, вносимого в процесс стерилизации и/или определенные условия, используемые для акклиматизации загрузки;
b) температура и влажность в зоне предварительного кондиционирования (если используется), контролируемые и записываемые в специфицированной точке;
c) время начала предварительного кондиционирования загрузки и ее выемки из зоны предварительного кондиционирования (если используется) для каждой загрузки;
d) время, прошедшее с момента извлечения загрузки из зоны предварительного кондиционирования (если используется) до начала цикла стерилизации;
e) влажность в камере во время кондиционирования и выдержка фаз влажности через давление, рост давления () и/или через прямой мониторинг;
f) время кондиционирования;
g) индикация удовлетворительной работы системы циркуляции газа в камере (если используется) во время инжекции и во время экспозиции;
h) температура и влажность в камере на протяжении всего цикла стерилизации;
i) если в качестве первичного средства контроля используется измерение давления, то требованием ко вторичному средству является только подтверждение ввода ЭО в камеру путем:
1) измерения массы использованного ЭО [см. D.10.2, перечисление i), (приложение D];
2) прямого измерения концентрации ЭО в стерилизационной камере;
3) измерения объема использованного ЭО;
j) время инжекции ЭО;
k) время инжекции инертного газа (если используется);
I) время экспозиции;
m) время, потребовавшееся для откачки стерилизационной камеры;
n) время и изменения давления во время продувки (очистки) после экспозиции;
о) время, температура, изменения давления (если были) во время аэрации.
10.3 Если для рутинного мониторинга используются биологические индикаторы, они должны соответствовать требованиям, изложенным в 8.6 и 8.7.
Если УКП, используемое при рутинном выпуске, отличается от УКП, использованного в ходе валидационных микробиологических исследований, оно должно иметь устойчивость процессу по меньшей мере равную сопротивляемости УКП, использованного в микробиологических исследованиях.
10.4 Если для рутинного мониторинга используются химические индикаторы, они должны соответствовать требованиям, изложенным в 8.8.
Химические индикаторы не могут заменять биологические индикаторы для выпуска продукта и не могут поддерживать обоснование параметрического выпуска продукта.
10.5 Если выполняется параметрический выпуск, то должны записываться и сохраняться следующие данные:
a) температура в камере, замеряемая как минимум в двух отдельных точках на протяжении всего цикла стерилизации;
b) влажность в камере во время кондиционирования, определяемая прямым измерением;
c) концентрация ЭО, определяемая прямым анализом атмосферы в камере с использованием аналитических методов через определенные интервалы времени, достаточные для верификации требуемых условий на протяжении времени экспозиции.
11 Выпуск продукта после стерилизации
11.1 Критерии, определяющие соответствие процесса стерилизации, используемого для стерилизации определенной конкретной стерилизационной загрузки, должны быть документированы. Критерии должны включать в себя:
a) подтверждение того, что данные, записанные во время рутинной обработки, соответствуют спецификации процесса;
b) подтверждение того, что отсутствует рост контрольных организмов во всех биологических индикаторах (если используются).
Примечание - Формальный выпуск загрузки после стерилизации может потребовать приложения результатов других испытаний (например, на остаточный ЭО, на эндотоксины, результаты физических испытаний и т.п.) прежде, чем продукт сможет быть выпущен в цепочку распределения.
11.2 Если процесс не отвечает всем критериям соответствия, указанным выше, причина несоответствия должна быть расследована. Если требуется ремонт или замена оборудования, то требуется выполнение необходимых аттестаций перед тем, как процесс можно будет использовать вновь.
11.3 Если один или несколько критериев соответствия, перечисленных в 11.1, не удовлетворены, продукт должен рассматриваться как несоответствующий и дальнейшая работа с ним должна проводиться согласно применимым статьям ISO 13485. В случае, когда обнаруживается позитивный биологический индикатор, выпуск продукта на основании приемлемых результатов проверки продукта на стерильность недопустим.
Несоответствие должно расследоваться согласно документированным процедурам.
11.4 Если в валидационных исследованиях используется продаваемый продукт, требования к выпуску такого продукта в продажу должны быть разработаны до начала валидационных исследований. Важно оценить влияние повторных экспозиций в ходе валидационных процессов стерилизации на функциональность продукта и упаковки, а также оценить уровни остаточного ЭО и/или продуктов реакции до выпуска продукта.
Если в валидационных микробиологических исследованиях используется продаваемый продукт, должны быть установлены процедуры, гарантирующие, что перед выпуском на рынок продукт был подвергнут процессу стерилизации при полной экспозиции с формальным рассмотрением его приемлемости.
Примечание - Относительно выпуска единичного лота смотрите приложение Е.
12 Поддержание эффективности процесса
12.1 Общая информация
12.1.1 Продолжительная эффективность системы для обеспечения нужного состояния продукта, представленного на стерилизацию (см. 7.3.1), должна быть продемонстрирована.
12.1.2 Точность и надежность измерительных приборов, используемых для управления, контроля и мониторинга стерилизационного процесса, следует периодически поверять в соответствии с 4.3.3.
12.2 Обслуживание оборудования
12.2.1 Профилактическое обслуживание должно планироваться и выполняться согласно документированным процедурам. Все процедуры должны соответствовать как рекомендациям изготовителя, так и соответствующим требованиям национальных, региональных или местных нормативных документов.
12.2.2 Оборудование может использоваться для обработки продукта только после того, как все специфицированные операции по обслуживанию будут успешно выполнены и документированы.
12.2.3 Записи об обслуживании следует сохранять (см. 4.1.2).
12.2.4 Схема обслуживания, процедуры обслуживания и записи о проведении обслуживания должны регулярно пересматриваться уполномоченным лицом через специфицированные интервалы времени, а результаты пересмотра должны документироваться.
12.3 Повторная аттестация
12.3.1 Аттестации установленного, функционирующего и эксплуатируемого оборудования и последующие повторные аттестации должны пересматриваться ежегодно для определения объема повторной аттестации, если таковая необходима. В аттестацию должна входить оценка необходимости повторного подтверждения уровня обеспечения стерильности продукта путем выполнения микробиологических исследований. Результат такого пересмотра, включая обоснование принятых решений, должен документироваться.
12.3.2 Повторная аттестация стерилизационного процесса, выполняемая с помощью специфицированного оборудования, должна проводиться через определенные интервалы времени по специфицированным критериям приемлемости и в соответствии с документированными процедурами. Периодичность переаттестаций должна быть обоснована.
12.3.3 Если в результате повторной аттестации выяснится, что стерилизационный процесс более не обеспечивает требуемого уровня стерильности продукта, причина этого должна быть расследована, после чего должны быть предприняты корректирующие и/или предупредительные действия. В рамках такого расследования должен быть рассмотрен эффект достижения специфицированного уровня обеспечения стерильности ранее обработанных загрузок, а также выполнен анализ риска их пригодности к использованию. Если расследование показывает, что требуемый уровень обеспечения стерильности более не может быть достигнут, тогда проводится новая аттестация эксплуатируемого оборудования с выполнением физических и микробиологических исследований с целью восстановления требуемого уровня обеспечения стерильности продукта. Расследование и последующие действия должны быть записаны.
12.3.4 Записи о пересмотре данных повторной аттестации, отчеты и записи о последующих корректирующих действиях должны сохраняться (см. 4.1.2).
12.4 Оценка изменений
12.4.1 Любые изменения производственных процессов, продукта, стерилизационного оборудования и/или стерилизационного процесса, должны быть оценены с точки зрения их способности повлиять на эффективность стерилизационного процесса.
12.4.2 Пригодность внутреннего и/или внешнего УКП для бионагрузки продукта по результатам внесенных изменений должна быть повторно подтверждена (см. 8.6 и 10.3 соответственно).
12.4.3 Загрузка и ее конфигурация в связи с внесенным изменением должны быть переоценены на его пригодность, а результат этой переоценки должен быть задокументирован в соответствии с требованием 4.1.2.
12.4.4 Аттестация процесса стерилизации должна пересматриваться каждый раз после внесения в процесс, стерилизационное оборудование или продукт любого изменения, которое могло бы изменить эффективность процесса (см. 8.2).
12.4.5 Влияние изменения должно быть рассмотрено как определение степени, в которой [из-за него] необходимо [новое] выполнение определения продукта и проведение всех видов аттестаций.
12.4.6 Результат оценки, включая обоснование принятых решений, должен быть документирован.
12.5 Оценка эквивалентности процесса
12.5.1 Эквивалентность процесса
Стерилизационное оборудование, имеющее одинаковые процессные параметры, проходя аттестацию установленного оборудования и аттестацию функционирующего оборудования, должно пройти аттестацию:
a) теми же методами, что использовались при аттестации оригинальной камеры;
b) с уменьшенным микробиологическим исследованием, которое должно продемонстрировать достижение требуемого уровня микробиологической летальности, и с физическим исследованием, которое должно продемонстрировать единообразие распределения температуры и влажности в загрузке и управляемость этих параметров производственной камерой. Обоснование такой урезанной аттестации должно быть записано и документировано.
Влияние различных географических местоположений на продукт и загрузку должно быть определено.
12.5.2 Продукт
12.5.3 Продукт может быть добавлен к валидированному процессу, если он считается равным или меньшим по бионагрузке, чем существующий, прошедший аттестацию продукт или внутреннее УКП. Должен быть выполнен технический обзор, сравнивающий продукт-кандидат с существующим продуктом или УКП, использованным при валидации рассматриваемого ЭО процесса. Результат такого обзора, включая обоснование принятых решений, должен быть документирован. К продукту по-прежнему должны быть применимы требования 7.2.
Приложение A
(обязательное)
Определение степени летальности стерилизационного процесса - подход с использованием биологического индикатора/биологической нагрузки
A.1 Общая информация
A.1.1 Этот подход сочетает знание сопротивляемости биологического индикатора данному процессу стерилизации со знанием популяции бионагрузки и ее сопротивляемости для установления параметров стерилизационного процесса (время экспозиции процесса).
Использование этого метода требует, чтобы уровни бионагрузки продукта демонстрировали относительное постоянство во времени, а устойчивость бионагрузки должна быть равной или меньшей, чем устойчивость биологического индикатора [см. D.8.6 (приложение D)].
Устойчивость внутреннего УКП демонстрируется путем исполнения цикла стерилизации с разными длительностями экспозиции или путем экспозиции биологических индикаторов с количественно отличающимися популяциями при одном и том же времени экспозиции, с последующим определением уровня летальности (степени инактивации через расчет -значения) стерилизационного цикла. Знание уровня летальности биологического индикатора, популяции и относительной сопротивляемости бионагрузки позволяет установить время экспозиции так, чтобы уровень обеспечения стерильности был предсказуемым.
Должно быть уделено внимание влиянию упаковки и удалению ЭО из УКП.
Руководство по применению этого подхода можно найти в ISO 14161.
A.1.2 Условия, используемые для восстановления биологического индикатора в аттестационных исследованиях, включая продолжительность инкубации, должны быть установлены и документированы. Инкубационный период должен учитывать возможность задержанного роста спор, подвергнутых воздействию ЭО. Обратитесь к ISO 14161 для получения дополнительной информации об инкубационном периоде для биологических индикаторов.
A.1.3 После разных по длительности экспозиций ЭО, отличающихся по популяциям биологических индикаторов при сохранении прочих параметров одинаковыми, летальность процесса может быть определена следующими способами:
a) прямым подсчетом;
b) фракционным негативным анализом;
c) комбинацией способов a) и b), описанных выше.
Примечание - Метод фракционного негативного анализа использует данные о росте/отсутствии роста, полученные от процедуры восстановления контрольных микроорганизмов после воздействия на них фрагментарных периодов экспозиции газа, или от градационных популяций контрольных микроорганизмов, подвергнутых единому фрагментарному периоду экспозиции.
A.2 Процедура
Дополнительное руководство по данному разработочному процеосу можно получить в AAMI TIR 16 и в ISO 14161, оба этих источника рассматривают разработку процесса в деталях.
Приложение B
(обязательное)
Консервативное определение уровня летальности стерилизационного процесса - подход "массовой гибели"
B.1 Общая информация
B.1.1 Этот подход к определению процесса основан на инактивации контрольных микроорганизмов, и он широко применялся в учреждениях здравоохранения (см. ISO 11138-2). Стерилизационные процессы, аттестованные по этому методу, зачастую консервативны и используют обработку, превышающую степень, необходимую для достижения специфицированных требований к стерильности.
Руководство по этому методу приведено в ISO 14161.
B.1.2 Консервативное определение процесса требует использования одного из подходов, описанных ниже:
a) подход половинного цикла: с целью подтверждения минимально необходимого времени экспозиции в целом должны быть выполнены три последовательных эксперимента, имеющих результатом полную инактивацию биологических индикаторов (содержащих популяцию не менее чем 10 и, где это возможно, помещенных в УКП). Установленное для процесса стерилизации время экспозиции должно быть по крайней мере в два раза больше этого минимального времени.
Для демонстрации адекватности техники восстановления биологических индикаторов, экспонированных ЭО, также должен быть выполнен фракционный цикл короткой продолжительности, после которого могут быть восстановлены выжившие биологические индикаторы;
Примечание - Этот короткий цикл может также быть использован для демонстрации относительной сопротивляемости биологического индикатора, УКП и бионагрузки продукта.
b) подход расчета цикла: рутинные процессные параметры, обеспечивающие логарифмическую редукцию спор (SLR) в биологическом индикаторе как минимум 12, должны быть установлены с использованием одного из методов, установленных в А.1.3 (приложение А). Количество циклов при этом диктуется используемым методом.
B.1.3 Условия, используемые для регенерации биологических индикаторов в ходе аттестации, должны быть установлены и документированы. При установлении инкубационного периода следует принимать во внимание возможность отсроченного роста спор, экспонированных ЭО. Более подробное руководство по выбору длительности инкубации биологических индикаторов можно найти в ISO 14161.
B.1.4 Должно быть показано, что бионагрузка продукта такова, что общее время ее полной инактивации меньше, чем время полной инактивации расположенного биологического индикатора продукта (внутреннего УКП).
B.2 Процедура
B.2.1 Создайте нагрузку для стерилизационного процесса, УКП, содержащее известное число микроорганизмов с известной устойчивостью к ЭО путем размещения биологических индикаторов в продукте или путем инокуляции тех мест в продукте, где наиболее сложно достичь условий стерилизации. Если место размещения биологической нагрузки отличается от места, где наиболее сложно достичь условий стерилизации, то его связь с наиболее тяжелым местом должна быть установлена.
B.2.2 Используйте УКП, продемонстрировавшее свою эквивалентность или даже большую микробиологическую устойчивость процессу стерилизации, чем продукт, отвечающий требованиям. Следует уделить внимание влиянию упаковки и удалению стерилизующий агента из УКП.*
_______________
* Текст документа соответствует оригиналу. - .
B.2.3 Разместите выбранное УКП в соответствии с B.2.1 и B.2.2 (приложение B) внутри стерилизационной загрузки или на ней, в зависимости от возможностей.
B.2.4 Экспонируйте стерилизационную загрузку ЭО при условиях, рассчитанных на обеспечение меньшей летальности, чем обеспечивает специфицированный процесс стерилизации.
B.2.5 Для подхода с расчетом цикла - если инактивация известного количества микроорганизмов была подтверждена в соответствии с A.1.3 (приложение A), определите степень воздействия обработки путем экстраполяции до известной расчетной вероятности выживаемости микроорганизмов, учитывая требуемый уровень обеспечения стерильности.
Приложение C
(справочное)
Датчики температуры, относительной влажности и количество биологических индикаторов
C.1 Датчики температуры
Во время аттестации функционирующего оборудования рекомендуется использовать один датчик на 2,5 м для установления температурной карты помещения или камеры, которая выявит горячие или холодные зоны. Поэтому мониторинг должен включать в себя больше, чем центр помещения/камеры и участки около дверей.
Для проведения аттестации эксплуатируемого оборудования требуется использовать один датчик на 1 м объема, занимаемого продуктом. Минимальное количество используемых датчиков - три. Для проведения аттестации эксплуатируемого оборудования датчики относительной влажности должны располагаться внутри упаковок (где это возможно), находящихся внутри загрузки. Этого можно достичь, размещая датчики внутри стерильной барьерной системы или между упаковками с отдельными предметами.
Результат расчета должен быть округлен до ближайшего большего числа.
Таблица C.1 обеспечивает руководство по выбору количества датчиков температуры.
Таблица C.1 - Минимальное рекомендуемое количество датчиков температуры
Объем, м | Количество для аттестации функционирования оборудования (полезный объем камеры/помещения) | Количество для аттестации эксплуатируемого оборудования (объем загрузки с продуктом) | ||||
Предвари- | Кондициони- | Аэрация | Предвари- | Кондициони- | Аэрация | |
| 3 | 3 | ||||
10 | 4 | 10 | ||||
15 | 6 | 15 | ||||
20 | 8 | 20 | ||||
25 | 10 | 25 | ||||
30 | 12 | 30 | ||||
35 | 14 | 35 | ||||
40 | 16 | 40 | ||||
50 | 20 | 50 | ||||
100 | 40 | 100 | ||||
Пример - Во время выполнения аттестации функционирующего оборудования помещения предварительного кондиционирования с полезным объемом камеры 70 м: 70/2,5=28.
Пример - Во время выполнения аттестации эксплуатируемого оборудования с объемом загруженного продукта 2 м: 2/1=2. При этом количество датчиков должно быть как минимум три (минимально допустимое для использования).
C.2 Датчики влажности
Рекомендуется использовать один датчик на каждые 2,5 м для установления карты влажности зоны продукта, который реагирует на потенциальные колебания уровней влажности. Минимальное количество датчиков - два.
Для выполнения аттестации в эксплуатируемом состоянии датчики влажности должны быть расположены внутри упаковок (где это возможно), находящихся внутри загрузки. Это можно достичь, поместив датчик внутри стерильной барьерной системы или между упаковками с отдельными предметами.
Таблица C.2 обеспечивает руководство по выбору количества датчиков влажности.
Таблица C.2 - Минимально рекомендуемое количество датчиков влажности
Объем, м | Количество для аттестации функционирования оборудования (полезный объем камеры/помещения) | Количество для аттестации эксплуатируемого оборудования (объем загрузки с продуктом) | ||||
Предвари- | Кондициони- | Аэрация | Предвари- | Кондициони- | Аэрация | |
| 2 | непри- | 2 | непри- | ||
10 | 4 | 4 | ||||
15 | 6 | 6 | ||||
20 | 8 | 8 | ||||
25 | 10 | 10 | ||||
30 | 12 | 12 | ||||
35 | 14 | 14 | ||||
40 | 16 | 16 | ||||
50 | 20 | 20 | ||||
100 | 40 | 40 | ||||
Пример 1 - Во время выполнения аттестации функционирующего оборудования камеры с полезным объемом 6 м: 6/2,5=2,4. Количество используемых датчиков должно быть не меньше 3.
Пример 2 - Во время выполнения аттестации эксплуатируемого оборудования с продуктом объемом 60 м: 60/2,5=24. Количество используемых датчиков должно быть не меньше 24.
C.3 Биологические индикаторы
Минимальное рекомендуемое количество биологических индикаторов должно быть следующим:
a) для микробиологических исследований аттестации эксплуатируемого оборудования с объемом продукта в загрузке до 10 м используются 3 биологических индикатора на 1 м
объема продукта, но не менее 5 биологических индикаторов.
b) для микробиологических исследований аттестации эксплуатируемого оборудования с объемом продукта в загрузке более 10 м используется 1 дополнительный биологический индикатор на каждый дополнительный кубический метр объема, превышающего 10 м
.
Если биологические индикаторы используются для рутинного контроля, используйте половину количества индикаторов, использовавшихся во время микробиологических исследований аттестации эксплуатируемого оборудования, но не более 30.
Результат расчета должен быть округлен до ближайшего целого числа.
Таблица C.3 обеспечивает руководство по выбору количества биологических индикаторов/УКП.
Таблица C.3 - Примеры минимально рекомендуемого количества биологических индикаторов/УКП
Объем загрузки продукта, м | Микробиологические исследования аттестации эксплуатируемого оборудования | Текущее управление (если используется) |
| 5 | 3 |
10 | 30 | 15 |
15 | 35 | 18 |
20 | 40 | 20 |
25 | 45 | 23 |
30 | 50 | 25 |
35 | 55 | 28 |
40 | 60 | 30 |
50 | 70 | 30 |
100 | 120 | 30 |
Фактическое количество используемых биологических индикаторов/УКП будет зависеть от следующих факторов:
a) выбранный метод микробиологической аттестации (см. приложение А или приложение С);
b) объем продукта;
c) тип камеры (разработочная или производственная).
При использовании процедуры Стамбо-Мэрфи-Кохрейна и подхода с расчетом цикла массового уничтожения рекомендуемое количество биологических индикаторов/УКП может базироваться на объеме продукта, подлежащего стерилизации. Если используется этот подход, то указано минимальное количество 10 биологических индикаторов/УКП, смотрите [38].
Пример 1 - Для объема загрузки продукта 3 м: 3x3=9. Количество используемых биологических индикаторов должно быть не менее 9 для микробиологических исследований аттестации эксплуатируемого оборудования. Для рутинного контроля: 9/2=4,5. Количество биологических индикаторов должно быть не менее 5.
Пример 2 - Для объема загрузки продукта 18 м:10x3+(18-10)х1=38. Количество используемых биологических индикаторов должно быть не менее 38 для микробиологических исследований аттестации эксплуатируемого оборудования. Для рутинного контроля: 38/2=19. Количество биологических индикаторов должно быть не менее 19.
Приложение D
(справочное)
Руководство по применению нормативных требований
Руководство, приводимое в данном приложении, не должно служить контрольным списком при оценке соответствия с настоящим стандартом. Это руководство предназначено для оказания поддержки в достижении единообразного понимания и применения настоящего стандарта путем предоставления разъяснений и приемлемых методов для достижения соответствия со специфицированными требованиями. Могут быть использованы и иные методы, отличающиеся от приведенных в руководстве, при условии, что их использование обеспечивает достижение соответствия с требованиями настоящего стандарта.
Примечание - Для облегчения поиска ссылок нумерация статей в этом приложении соответствует нумерации статей в нормативной части настоящего стандарта.
D.1 Область применения
Дополнительное руководство не приводится.
D.2 Нормативные ссылки
В настоящем стандарте использованы ссылки на международные стандарты. В случае ссылки на стандарт в целом она не датируется, в случае ссылки на конкретный пункт стандарта она датируется.
D.3 Термины и определения
Дополнительное руководство не приводится.
D.4 Система менеджмента качества
Примечание - Поскольку область применения ISO 13485 сфокусирована на изготовителях медицинских изделий, учреждения здравоохранения могут использовать стандарты менеджмента качества, применимые к этим организациям.
D.4.1 Документирование
Обратитесь к ISO 13485.
D.4.2 Ответственность руководства
D.4.2.1 Требования к ответственности и полномочиям установлены в ISO 13485:2003, пункт 5.5, а требования к человеческим ресурсам - в ISO 13485:2003, пункт 6.2.
В ISO 13485 требования к ответственности руководства относятся к обязательствам руководства, фокусированию внимания на заказчике, политике в области качества, планированию, ответственности, полномочиям и коммуникациям, а также к пересмотру, осуществляемому руководством.
Каждая организация должна устанавливать процедуры для определения необходимости в обучении, и обеспечивать уверенность в том, что персонал прошел обучение, достаточное для адекватного исполнения своих обязанностей.
D.4.2.2 Разработка, валидация стерилизационного процесса и текущее управление им может включать в себя участие нескольких различных сторон, каждая из которых отвечает за определенный элемент. Важно, чтобы соответствующие процедуры четко определяли ответственность сторон за соблюдение требований настоящего стандарта. Это особенно важно, когда стороны привлекаются к исполнению специфических функций.
Даже когда элементы стерилизационного процесса выполняются привлеченными по контракту сторонами, важно отметить, что в конечном счете изготовитель медицинского изделия несет ответственность за валидацию, выпуск и распространение стерилизованного продукта на рынке. Когда учреждение здравоохранения заказывает по контракту стерилизацию многократно используемых медицинских изделий, то ответственность за валидацию и выпуск стерилизованного продукта несет само учреждение здравоохранения.
Дальнейшее руководство доступно в ISO 14937:2009, приложение Е.4.2.2.
D.4.3 Реализация продукта
Примечание - В ISO 13485 требования к реализации продукта относятся к жизненному циклу продукта исходя из определения требований пользователя, проектирования и разработки, закупки, контроля производства, калибровки устройств мониторинга и средств измерения.
D.4.3.1 Требования к закупке установлены в ISO 13485:2003, пункт 7.4. В частности, следует отметить, что требования ISO 13485, пункт 7.4 к верификации закупаемого продукта относятся к продукту и услугам, влияющим на качество процесса, осуществляемого вне организации.
Процедуры закупки в учреждении здравоохранения должны гарантировать, что медицинские изделия многократного применения поставляются с валидированными инструкциями по их очистке, дезинфекции, стерилизации и аэрации, как оговорено в ISO 17664.
Подлежит верификации и возможность исполнения предписанных процедур очистки, дезинфекции, стерилизации и аэрации в учреждении здравоохранения.
D.4.3.2 Требования к идентификации и прослеживаемости установлены в ISO 13485:2003, пункт 7.5.3.
Для тех учреждений, которые не полностью соответствуют требованиям ISO 13485 (например, учреждений здравоохранения), процедуры идентификации продукта и поддержания прослеживаемости должны включать в себя этикетирование каждого изделия или упаковки перед стерилизацией с идентификатором контроля лота, включающим в себя следующую информацию:
a) идентификатор стерилизатора или его код;
b) дата стерилизации;
c) номер цикла (например, номер текущего дневного цикла или порядковый номер цикла на стерилизаторе);
d) идентификатор лица, собравшего упаковку.
Включение идентификатора лица, собравшего упаковку, позволяет провести расследование в случае, если возникают проблемы. Информация об идентификаторе лота позволяет персоналу в случае отзыва изделий установить изделия, стерилизованные с помощью специфического цикла, и выявить источник проблемы.
D.4.3.3 Требования к калибровке устройств мониторинга и средств измерения установлены в ISO 13485:2003, пункт 7.6.
D.4.4 Измерения, анализ и улучшение - управление несоответствующей продукцией
Процедуры управления несоответствующей продукцией и корректирующие действия установлены в ISO 13485:2003, пункт 8.3 и пункт 8.5.2 соответственно.
D.5 Характеризация стерилизующего агента
D.5.1 Общая информация
Дополнительное руководство не приводится.
D.5.2 Стерилизующий агент
ЭО - это газ с высокой проникающей способностью, проходящий сквозь большинство упаковочных материалов, включая полимерные. Широко известные составы включают в себя чистый ЭО и его смеси с двуокисью углерода или с азотом.
Примечание - Для газовых смесей ЭО с двуокисью углерода, азотом или другими инертными газовыми смесями скорость молекулярной диффузии ЭО в полимерные материалы может зависеть от процентного содержания объема газовых молекул ЭО в стерилизующем агенте, что в результате способно приводить к более длительному времени экспозиции для достижения желаемого снижения логарифма микробиологических спор.
Условия хранения и срок хранения ЭО должны соответствовать рекомендациям изготовителя ЭО и всем применимым нормативным требованиям. Это особенно важно для предварительно приготовленных газовых смесей, где их расслоение может стать проблемой.
D.5.3 Микробоцидная эффективность
Дополнительное руководство не приводится.
D.5.4 Воздействие на материалы
Дополнительное руководство не приводится.
D.5.5 Безопасность и окружающая среда
D.5.5.1 ЭО - токсичный, легко воспламеняющийся и взрывоопасный газ, по этой причине обращение с ним и его использование требуют исключительной осторожности. Пределы по взрывоопасности составляют от 2,6% до 100% объема в воздухе.
Там, где это осуществимо практически, ЭО стерилизационные циклы должны работать внутри негорючих зон на протяжении полного цикла стерилизации с целью минимизации риска взрыва. Это требует удаления воздуха из камеры перед впуском в нее ЭО. Для 100% стерилизационных циклов этого можно достичь путем создания глубокого вакуума или путем нескольких частичных вакуумных откачек, каждая из которых сопровождается впуском инертного газа, например, азота. Это удаляет воздух из камеры, позволяя осуществить безопасный впуск ЭО. По завершении фазы экспозиции ЭО необходимо удалить газообразный ЭО из камеры, пока содержание в ней газа не упадет ниже предела взрывоопасной концентрации, равного 2,6%. Такой уровень достигается путем нескольких пост-вакуумных откачек, каждая из которых сопровождается впуском азота.
Использование негорючих смесей стерилизующего агента может увеличить безопасность за счет снижения риска пожара или взрыва. Эти смеси могут также облегчить достижение соответствия со специфическими для разных стран требованиями к безопасности. Негорючие смеси производятся путем смешивания легко воспламеняемого газообразного ЭО с одним или несколькими инертными газами. Воспламеняемость такой смеси можно оценить, измеряя относительные пропорции ЭО, воздуха, газа-разбавителя (например, и т.п.), инертного газа (например, азота) и водяного пара в стерилизаторе. Следует уделить внимание тому, чтобы не возникало возможности разделения ЭО-смеси, поскольку это может привести к проблемам с безопасностью и качеством.
ЭО-стерилизаторы должны устанавливаться в специально предназначенных для них помещениях. Органы оперативного управления стерилизатором должны монтироваться вне этого помещения так, чтобы операторы могли задавать или изменять параметры программы, не входя в помещение со стерилизатором. Все воздушные потоки из зоны доступа к стерилизатору должны отводиться наружу и соответствовать применимым нормативным требованиям.
До извлечения продукта из стерилизатора должны быть предприняты меры предосторожности, гарантирующие, что операторы не подвергаются воздействию уровней концентрации ЭО, превышающих соответствующие пределы экспозиции сотрудников (допустимый предел экспозиции и предел кратковременной экспозиции) из-за выхода остатков газа из загрузки. Когда продукты, стерилизованные инертными смесями ЭО, по завершении цикла не вынимаются из стерилизатора немедленно, концентрация ЭО в стерилизаторе может привести к проблемам с безопасностью персонала.
D.5.5.2 Принципы системы менеджмента окружающей среды могут быть применимы к ЭО-процессу стерилизации. ISO 14001 дает спецификации для систем менеджмента окружающей среды. ISO 14040 обеспечивает руководство по планированию исследований для оценки жизненного цикла.
D.5.5.3 Остаточный газ должен отводиться через системы обработки газообразного ЭО, например, через каталитический окислитель, мокрый кислотный скруббер (газопромыватель) или тепловой окислитель в соответствии с локальными требованиями и законодательством по контролю эмиссий.
При выборе разбавителя следует учитывать его влияние на разрушение озонового слоя, а также способы утилизации его побочных продуктов.
D.6 Характеризация процесса и оборудования
В учреждениях здравоохранения характеризация процесса и оборудования обычно относится к области ответственности изготовителя оборудования. Администрация учреждения здравоохранения должна осуществлять контроль на месте для убеждения, что оборудование, которое она закупает, соответствует требованиям национальных, региональных и местных нормативных документов и пригодно для использования в стерилизации продуктов, требующих ЭО-стерилизации. Администрация учреждения здравоохранения должна убедиться, что учреждение имеет инфраструктуру, необходимую для правильной работы со стерилизационным оборудованием и для достижения эффективной стерилизации медицинских изделий.
D.6.1 Общая информация
Дополнительное руководство не приводится.
D.6.2 Характеризация процесса
D.6.2.1 Дополнительное руководство не приводится.
D.6.2.2 Сопротивление микроорганизмов деактивации ЭО зависит от содержания в них влаги. При уровнях влажности, ниже 30% микробиологическая устойчивость для некоторых продуктов может возрасти. По этой причине общепринята практика контроля и мониторинга влажности атмосферы, в которой экспонируется продукт, с целью уравновешивания содержания влаги в микроорганизмах местным условиям. Следует уделить особое внимание упакованным продуктам, чтобы убедиться, что повышенная относительная влажность не повлияет на функциональность продукта и целостность его упаковки. Одним из путей, помогающих поддерживать нужную влажность продукта, является предварительное кондиционирование продукта при определенных температуре и влажности. Такое предварительное кондиционирование может уменьшить продолжительность стерилизационного цикла. В учреждениях здравоохранения повышенное содержание влаги в продуктах может быть вызвано также недостаточной сушкой после очистки.
Нагрев и увлажнение продукта используются для установления воспроизводимых температуры и содержания влаги до экспозиции ЭО. Исследования, устанавливающие минимальное время выдержки в камерах/помещениях предварительного кондиционирования гарантируют, что в стерилизационной загрузке достигнуты требуемые условия. Должны предприниматься меры предосторожности, чтобы избежать усиленной конденсации воды в стерилизационной загрузке.
Несмотря на то, что выполнение предварительного кондиционирования в отдельной камере, помещении или ячейке является общепринятой практикой, стерилизационные циклы могут быть рассчитаны и на достижение требуемых диапазонов температуры и влажности в загрузке в ходе фазы кондиционирования непосредственно в стерилизационной камере. Для минимизации риска повышенной конденсации рекомендуется, чтобы температура загрузки поддерживалась на уровне, превышающем температуру точки росы процесса во время предварительного кондиционирования и фазы кондиционирования процесса стерилизации.
Фактические диапазоны температуры и влажности внутри стерилизационной загрузки должны быть продемонстрированы в ходе выполнения аттестации эксплуатируемого оборудования.
Где это применимо, должно быть установлено максимальное время между извлечением загрузки из предварительного кондиционирования и пуском стерилизационного цикла. Общей практикой является время переноса порядка 60 минут или менее:
a) когда продукт помещается в стерилизационную камеру без предварительного кондиционирования, следует обратить внимание на вероятность повышенной конденсации в продукте и упаковке;
b) остатки ЭО и продуктов его реакции могут быть опасными.
Для изготовителя продукта, подлежащего стерилизации, очень важно позаботиться о возможном появлении остатков ЭО в продукте. Температура, время выдержки, усиленная (принудительная) циркуляция нагретого воздуха, характеристики загрузки, материалы продукта и упаковки - все это влияет на эффективность аэрации, и все уставки и допуски должны приниматься в расчет при оценке остаточных уровней, как подчеркивается в ISO 10993-7. Аэрация может выполняться внутри стерилизатора, в отдельных зонах, или в комбинации того и другого. Для учреждений здравоохранения аэрация в камере является в большей степени обычной практикой, чем аэрация в отдельной комнате, из-за опасности экспозиции ЭО. В учреждениях здравоохранения повторно обработанные изделия, стерилизованные ЭО, должны быть тщательно аэрированы перед работой с ними или использованием их по назначению в соответствии с рекомендациями изготовителей медицинского изделия и жесткой камеры стерилизатора. Неадекватно аэрированные изделия и упаковки могут выделять ЭО, что может привести к поражению больных и персонала учреждения здравоохранения.
D.6.2.3 Время переноса включает в себя каждый шаг переноса продукта во время предварительного кондиционирования и заключительного переноса продукта в стерилизатор для пуска цикла.
D.6.2.4 Ниже приводится перечень фаз, которые могут входить в цикл стерилизации, вместе с факторами, которые следует учитывать для каждой фазы:
a) удаление воздуха:
1) глубина ( или терминальное давление) и скорость (
/время) достижения вакуума;
b) проверка камеры на утечку (испытание, выполняемое либо под вакуумом для субатмосферных циклов, либо под вакуумом или при избыточном давлении для суператмосферных циклов), если применимо:
1) период стабилизации и/или время удержания;
2) изменение давления;
c) добавление инертного газа (если используется);
1) глубина ( или терминальное давление) и скорость (
/время) достижения заданного давления при впуске инертного газа;
d) кондиционирование (если используется):
1) во время фазы кондиционирования - подъем давления ( или терминальное давление) или % относительной влажности и скорость (
/время) достижения давления при впуске пара;
2) количество впусков пара/вакуумных откачек, если применимо;
e) инжекция ЭО:
1) давление, рост давления () и скорость (
/время) достижения специфицированного давления при впуске ЭО и корреляция методов, используемых для мониторинга концентрации ЭО;
2) давление, рост давления () и скорость (
/время) достижения специфицированного давления при впуске любого инертного газа (если используется);
f) поддержание специфицированных условий в течение периода экспозиции:
1) дифференциал давления, используемый для подачи стерилизующий агента или инертного газа (если используется);*
_______________
* Текст документа соответствует оригиналу. - .
2) температура в стерилизационной камере;
g) удаление ЭО:
1) глубина ( или терминальное давление) и скорость (
/время) достижения вакуума для удаления ЭО;
h) очистка [продувка] (если используется);
1) рост давления и скорость достижения давления:
2) глубина ( или терминальное давление) и скорость (
/время) достижения вакуума для удаления ЭО;
3) количество повторных впусков и любые отклонения при успешных повторах;
i) впуск воздуха/инертного газа:
1) давление ( или терминальное давление) и скорость (
/время) достижения давления при впуске воздуха или инертного газа;
2) количество повторных впусков и любые отклонения при успешных повторах;
3) уравнивание давления с атмосферным с помощью впуска воздуха.
D.6.2.5 Скорость рециркуляции должна быть специфицирована при оценке остаточных уровней продукта.
D.6.3 Характеризация оборудования
D.6.3.1 При характеризации оборудования должны учитываться следующие факторы:
а) характеризация зоны предварительного кондиционирования.
Предварительное кондиционирование может выполняться в отдельной зоне предварительного кондиционирования (в камере, ячейке или комнате). Необходимо увлажнение паром, поскольку увлажнители, работающие по принципу дисперсии ненагретой воды в виде аэрозоли (например, увлажнители с вращающимся диском и небулайзеры), могут являться потенциальным источником микробной контаминации.
Зона предварительного кондиционирования (если используется) должна иметь следующие характеристики и возможности мониторинга:
- адекватная циркуляция воздуха для обеспечения единообразия распределения температуры и влажности в полезном объеме и для обеспечения поддержания этого единообразия в загруженной комнате или камере;
- оборудование для обнаружения воздушного потока, системы тревог или индикаторы, осуществляющие мониторинг циркуляционной системы для обеспечения соответствия с заранее заданными допусками;
- средства регистрации времени помещения загрузки внутрь и ее извлечения из зоны предварительного кондиционирования;
- средства мониторинга температуры и влажности в ячейке/комнате;
- средства управления температурой и влажностью в ячейке/комнате.
b) характеризация стерилизатора.
Камера стерилизатора должна иметь следующие характеристики и возможности мониторинга:
- средства мониторинга времени, давления в камере, температуры и влажности (если изменения влажности контролируются считыванием показаний с датчика);
- средства управления временем, давлением в камере, температурой и влажностью, если изменения влажности контролируются считыванием показаний с датчика (если датчики прикреплены к оборудованию, убедитесь, что в ходе выполнения аттестаций функционирующего и эксплуатируемого оборудования была сделана корреляция со скоростью роста давления);
- если влажность не контролируется по показаниям датчика - средства мониторинга и управления впусками пара;
- если применяется параметрический выпуск продукта - аналитические приборы для прямого анализа (измерения) влажности во время кондиционирования и концентрации ЭО в фазе экспозиции [см. раздел 9, а также 9.5.5 и D.9.5.5 (приложение D)];
- система контроля впуска газообразного ЭО в камеру;
- средства, демонстрирующие, что газообразный ЭО впущен в камеру. Например измерение температуры газообразного ЭО, перетекающего из испарителя в камеру стерилизатора. Эта система может контролировать концентрацию ЭО во время выполнения фазы экспозиции.
- средства обнаружения и предупреждения об отклонениях параметров цикла таким образом, чтобы корректирующие действия могли быть предприняты своевременно.
c) характеризация зоны аэрации.
Зона аэрации (камера, ячейка или комната) может быть использована для удаления остатков ЭО из продукта/упаковки. Единообразие распределения температур, подача свежего воздуха и рециркуляция воздуха имеют важное значение для обеспечения постоянных и воспроизводимых результатов. Зона аэрации должна иметь следующие характеристики и возможности мониторинга:
- оборудование для обнаружения воздушного потока, системы тревог или индикаторы, следящие за системой воздухообеспечения для обеспечения ее работы в пределах заданных допусков и поддержания адекватного воздушного потока в загруженной комнате или камере;
- оборудование, обеспечивающее рециркуляцию воздуха;
- средства мониторинга температуры в помещении;
- средства управления температурой в помещении.
D.6.3.2 Спецификация оборудования должна пересматриваться для обеспечения выполнения требований нормативных документов, в том числе в части безопасности, чтобы технические характеристики оставались в рамках пригодности к использованию по назначению, а также для обеспечения инфраструктуры и среды, необходимых для работы оборудования.
При подготовке спецификации оборудования должны быть рассмотрены следующие аспекты:
a) если подача ЭО к стерилизатору осуществляется из общего бака-хранилища, который периодически пополняется, то такой бак должен быть оборудован средствами отбора проб для анализа, средствами опорожнения бака от ЭО и средствами очистки в случае контаминации или усиленного накопления полимеров;
b) система впуска ЭО в стерилизатор должна быть снабжена испарителем для предотвращения впуска жидкого ЭО в камеру стерилизатора;
c) температура газообразного ЭО, перетекающего из испарителя в камеру стерилизатора, должна измеряться для демонстрации того, что был получен именно газообразный ЭО;
d) пар должен использоваться только для увлажнения загрузки и не должен рассматриваться как стерилизующий агент. Устойчивость характеристик системы подачи пара может быть определена периодическим анализом воды, питающей бойлер, или анализом конденсата;
e) для измерения температуры в камере должны использоваться минимум два датчика. Камеры большого объема могут быть оснащены более чем двумя датчиками для того, чтобы обеспечить получение системами мониторинга и контроля данных, отражающих температуру во всем объеме камеры во время ее использования.
Примечание - Целью использования двух отдельных датчиков является предотвращение ошибочного принятия вышедшего за рамки спецификаций процесса в качестве нормального в случае отказа одного датчика. Сравнение показаний двух раздельных датчиков позволяет обнаружить выход из строя одного из них. Для удовлетворения этого требования может использоваться двухканальный датчик.
f) важно обеспечить поддержание единообразных условий внутри камеры стерилизатора во время обработки продукта. Этого можно достичь усиленной (принудительной) циркуляцией газа. Если используется система циркуляции газа, она должна быть оборудована устройством мониторинга для индикации случаев, когда циркуляция становится неэффективной, поскольку устройства, которые отслеживают и показывают только состояние "включен" насоса или вентилятора в таких системах функционально недостаточны;
g) зоны, используемые для хранения баллонов, баков или картриджей с ЭО или с газовыми смесями ЭО, должны быть изолированными и иметь вентиляцию;
h) там, где условия окружающей среды подвержены изменениям температуры, выходящим за пределы диапазонов, рекомендованных поставщиками, зона хранения контейнеров с ЭО должна быть обеспечена средствами контроля температуры.
Может оказаться невозможной калибровка средств управления и мониторинга в реальных условиях обработки, например, датчиков влажности. Результаты калибровки этих приборов должны быть коррелированы с исследованиями, сделанными в ходе аттестаций. Условия обработки могут пагубно влиять на некоторые типы датчиков, в том числе на датчики влажности. Датчики могут требовать замены после повторяющихся экспозиций в условиях обработки по причине необратимой порчи материалов, используемых в настоящее время в качестве чувствительных элементов. Может оказаться необходимым внедрение программы более частого обслуживания таких датчиков, чем периодичность, рекомендованная изготовителем/поставщиком датчика.
D.6.3.3 Дополнительное руководство не приводится
D.6.3.4 Дополнительное руководство не приводится
D.6.3.5 Если имеется необнаруженный сбой системы управления или мониторинга, стерилизационная загрузка может быть выпущена, хотя она и была обработана с нарушением необходимых параметров обработки. Для того, чтобы предотвратить подобную возможность, общепринятой практикой является использование "избыточных" датчиков для многих критических параметров. Обычным возможным использованием таких избыточных датчиков является:
a) использование одного датчика для управления, а второго - для мониторинга и записи отчета;
b) использование обоих датчиков (или среднего значения их показаний) и для управления, и для мониторинга; такая система обязательно должна автоматически генерировать сигнал о сбое, если разница между показаниями двух датчиков превысит заранее заданное значение;
c) использование двухканальных датчиков и для управления, и для мониторинга; такая система обязательно должна автоматически генерировать сигнал о сбое, если разница между показаниями двух каналов измерения превысит заранее заданное значение.
D.7 Определение продукта
D.7.1 Общая информация
D.7.1.1 Определение продукта включает в себя документирование важной информации о медицинском изделии, подлежащем стерилизации (например, о новом или модифицированном продукте).
Определение продукта для медицинского изделия включает в себя само медицинское изделие, стерильную барьерную систему, содержащую внутри себя изделие, а также любые принадлежности, инструкции или иные предметы, заключенные в упаковочную систему. В определение также включается описание заданных функций изделия и доступных процессов его изготовления и стерилизации. Процесс определения продукта должен также учитывать, является ли рассматриваемое изделие новым, или это лишь часть существующего семейства продуктов.
Ниже перечислено то, что должно рассматриваться как часть определения продукта:
a) физические атрибуты медицинского изделия (состав и конфигурация);
b) заданная функциональность (назначение) медицинского изделия;
c) предназначено ли медицинское изделие для однократного применения, или это изделие для многократного использования;
d) характеристики конструкции, способные повлиять на выбор процесса стерилизации (например, содержит ли оно батареи, волоконную оптику, микросхемы или микрокомпьютеры);
e) сырьевые материалы/условия изготовления, способные повлиять на микробиологическое качество (например, материалы природного происхождения);
f) требуемый уровень обеспечения стерильности;
g) упаковка;
h) конфигурация загрузки; требования для специфической загрузки или для смешанной конфигурации загрузки, или несколько вариантов приемлемых конфигураций загрузки;
i) совместимость с ЭО или с газовой смесью и условиями обработки (предварительное кондиционирование, стерилизация и процесс аэрации).
D.7.1.2 Для сравнения нового или модифицированного продукта с валидированным продуктом и/или УКП, которое использовалось для валидации уже существующего процесса стерилизации, должен выполняться технический пересмотр (техническая экспертиза). Конструкция и конфигурация нового или модифицированного продукта должна быть тщательно проверена на наличие любых отличающихся свойств, способных повлиять на проникновение ЭО, тепла или влаги. Для изготовителей медицинских изделий такое сравнение должно также включать в себя проверку факторов, способных повлиять на изначальную бионагрузку продукта, с учетом местоположения производственных помещений, типов используемых сырьевых материалов, источников получения этих материалов и способов производства. Для модифицированных продуктов многократного использования это сравнение должно включать в себя оценку эффективности очистки продукта.
Если новый или модифицированный продукт продемонстрировал эквивалентность уже существующему медицинскому изделию или УКП, стерилизационные характеристики которого уже известны, то этот новый или модифицированный продукт может рассматриваться как часть семейства продукта или часть категории обработки.
Примечание - AAMI TIR 28 [26] является полезным руководством по минимизации риска внедрения нового или модифицированного продукта, который представляет собой большую нагрузку на стерилизационный цикл, чем ранее валидированный продукт/УКП.
Если конфигурация, плотность или конфигурация загрузки продукта-кандидата и его упаковки могут представлять собой большую нагрузку на стерилизационный процесс, чем ранее валидированный продукт, то следует выполнить исследования проникновения ЭО, тепла, влаги и/или летальности цикла.
В качестве части технического пересмотра (технической экспертизы) должны быть рассмотренные перечисленные далее вопросы. Если ответом на любой из них будет "да", тогда может оказаться необходимой дополнительная оценка нового или модифицированного продукта с целью определения, является ли он более трудным для стерилизации, чем ранее валидированный продукт:
a) по сравнению с ранее валидированным продуктом - имеет ли новый или модифицированный продукт:
1) более ограниченные внутренние проходы или камеры;
2) меньше отверстий;
3) большую площадь внутренних поверхностей;
4) больше матовых поверхностей или закупоренных полостей;
5) больше закрывающихся частей;
6) более длинные или более узкие каналы;
7) изменения или отличия, способные уменьшить перенос тепла, влажности или ЭО;
8) бионагрузку или устойчивость бионагрузки, заметно превышающую те же характеристики контрольного продукта (из-за производственных условий, обращения, процессов очистки или используемых материалов);
9) содержание материалов или структур, способных ухудшить свои свойства под воздействием предполагаемой обработки или предполагаемого способа стерилизации;
b) по сравнению с ранее валидированным продуктом - имеет ли упаковка нового или модифицированного продукта:
1) какие-либо изменения упаковочных элементов, включая инструкции или защитные барьеры;
2) любые дополнительные непроницаемые защитные барьеры (например, контейнер, коробка, трафарет, которые могут ограничить проникновение ЭО или влаги);
3) изменения пористости упаковочных материалов (например, изменение удельного веса, покрытия);
4) уменьшение площади поверхности вентилируемого материала или отверстий в нижних слоях (например, за счет клейкой ленты или вторичной этикетки, изменения размера внешней этикетки);
5) увеличенный уровень бионагрузки на продукт;
6) изменение количества барьерных слоев;
c) по сравнению с ранее валидированным продуктом - имеет ли конфигурация загрузки нового или модифицированного продукта:
1) значительные отличия от ранее валидированной конфигурации контрольной загрузки;
2) значительные отличия по объему абсорбирующих материалов;
3) значительные отличия в плотности от ранее валидированной конфигурации контрольной загрузки;
4) значительные отличия в общем объеме загрузки.
D.7.1.3 Наличие либо закрытых пространств, либо матированных поверхностей должно оцениваться с точки зрения выбора внутреннего УКП, которое могло бы быть использовано в последующих аттестационных исследованиях летальности.
D.7.1.4 Основной функцией стерильной барьерной системы для стерилизованного медицинского изделия является обеспечение уверенности в том, что продукт останется стерильным вплоть до момента его использования. Стерильная барьерная система должна быть способной выдержать условия процесса и сохранять свою целостность во время выполнения стерилизации, чтобы гарантировать качество продукта.
При выборе упаковочной системы для продукта, подлежащего стерилизации, должны рассматриваться ее определенные основные конструкционные и производственные факторы в соответствии с выбранным стерилизационным процессом. Для обеспечения проникновения ЭО проницаемость упаковки для определенных условий стерилизующей среды имеет наибольшую важность. Поскольку удаление воздуха является частью ЭО-процесса стерилизации, упаковочная система должна также позволять газам как проходить внутрь, так и выходить наружу из упаковки во время изменений давления в ходе впуска газа и откачки без повреждений, разрывов или нарушений целостности швов.
Способность стерильной барьерной системы защищать продукт в ходе обычного обращения с ним и распределения должна быть продемонстрирована. Должны быть получены доказательства того, что стерильная барьерная система способна выдержать стерилизационный процесс не теряя своей способности защищать продукт. Валидация стерильной барьерной системы должна учитывать потенциальные нагрузки, которым она может подвергаться в ходе выполнения ЭО-процесса стерилизации. Рассмотрению подлежат, в том числе, уровни вакуума/давления, скорость изменения давления, температура и т.п. Общепринятой практикой является демонстрация пригодности стерильной барьерной системы путем ее экспозиции нескольким процессам стерилизации [см. D.7.2.1 и D.7.2.2 (приложение D)].
Упаковка более детально рассматривается в ISO 11607-1 и ISO 11607-2.
D.7.1.5 Конфигурация загрузки в камере может влиять как на проникновение в продукт тепла, влаги, газообразного ЭО, так и на удаление ЭО из продукта. Конфигурация загрузки должна быть определена в ходе валидации для обеспечения адекватной температуры, влажности и проникновения/удаления ЭО в процессе обработки.
D.7.1.6 УКП является устройством, внутри которого расположена микробиологическая нагрузка. Примеры способов разработки УКП для демонстрации эквивалентности включают в себя, но не ограничиваются перечисленным ниже:
a) размещение микробиологической нагрузки между кольцами, сальниками, прокладками или ребрами поршня (пробки) шприца;
b) размещение микробиологической нагрузки в середине канала трубки, которая затем вновь присоединяется к изделию с помощью скрепляющего растворимого агента или коннектора для восстановления его целостности;
c) размещение микробиологической нагрузки в серии упаковок или оберток.
Для использования в учреждениях здравоохранения были рекомендованы несколько конструкций УКП.
Примечание - Дополнительную информацию смотрите в ANSI/AAMI ST41. См. также D.8.6 (приложение D) для получения дополнительной информации о внутренних и внешних УКП.
Для подготовки внутреннего УКП микробиологическая нагрузка может быть инокулирована на продукт непосредственно или косвенно. Прямая инокуляция выполняется путем нанесения жидкой суспензии спор на продукт. Косвенная инокуляция выполняется путем размещения инокулированного носителя либо внутри упаковки, либо внутри продукта.
Ниже перечислены различные способы подготовки УКП:
a) инокуляция продукта: продукт, подлежащий стерилизации, используется для приготовления УКП и инокулируется прямо или косвенно;
b) инокуляция имитатора продукта: имитатор продукта используется для приготовления УКП и инокулируется непосредственно или косвенно. Имитатор продукта состоит из частей медицинского изделия или из комбинации компонентов, известных как представляющие наибольшую устойчивость процессу, и в то же время адекватно представляющих все продукты, входящие в семейство;
c) инокулированный объект: упаковка, ее часть или трубка, которые используются для приготовления УКП и инокулируются непосредственно или косвенно.
Примечание - Непосредственная инокуляция споровой суспензией может приводить к переменной сопротивляемости инокулированного продукта благодаря поверхностным явлениям или иным факторам окружающей среды, а также к окклюзии спор на поверхности или внутри продукта. По этой причине важно обеспечить научное обоснование или результаты валидации такой практики, чтобы гарантировать, что устойчивость инокулированного продукта будет разумно коррелировать с рутинным продуктом. Восстановление инокулята также должно быть валидировано, если устойчивость измеряется техникой подсчета на пластинах. Смотрите работу Джиллиса и Шмидта [30], Уэста [40] и ISO 11737-1 для получения дополнительной информации.
Средство демонстрации эквивалентности ранее валидированному продукту или внутреннему УКП - это сравнение относительных степеней инактивации биологических индикаторов, расположенных в точке наибольшей трудности внутри нового или модифицированного продукта с ранее валидированным продуктом/мастер-продуктом [см. D.8.6 и D.12.5.2 (приложение D)], когда тот и другой продукты обрабатываются фракционным циклом. Исследования эквивалентности должны сравнивать новый или модифицированный продукт с внутренним УКП, используемым для валидации процесса. Если для этого сравнения используется УКП, устойчивость УКП должна быть оценена как часть ежегодного пересмотра.
D.7.2 Безопасность продукта, качество и характеристики
D.7.2.1 Важен выбор материала, который выдерживает химические и физические изменения, вызываемые воздействием ЭО и/или любых разбавителей на протяжении всего ожидаемого диапазона изменений условий стерилизации. Свойства материалов, необходимые для удовлетворения требований к характеристикам продукта, например, физическая прочность, проницаемость, физические размеры и эластичность, оцениваются после стерилизации, чтобы убедиться, что материалы по-прежнему пригодны для использования. Эффекты деградации из-за воздействия процесса стерилизации, такие как волосные трещины и охрупчивание, могут потребовать внимательного рассмотрения. Там, где это необходимо, оценке могут подвергаться эффекты воздействия многократных процессов стерилизации.
Демонстрация того, что специфицированный стерилизационный процесс не оказывает пагубного воздействия на правильное функционирование продукта, может быть осуществлена путем выполнения испытаний на функциональность или иных подходящих испытаний медицинского изделия и его упаковки. Эти испытания могут выполняться после экспозиции в стерилизаторе или в других камерах искусственного климата, имитирующего специфицированный процесс, и могут допускать как простой визуальный осмотр, так и выполнение серии специализированных испытаний.
Элементы, способные повлиять на безопасность, качество или рабочие характеристики, включают в себя:
a) изменения давления в цикле, которые могут влиять на стерильную барьерную систему и целостность швов;
b) влияние длительности экспозиции ЭО, температуры, влажности и, если применимо, любых газов-разбавителей, присутствующих в используемой стерилизующей смеси;
c) включение новых материалов, о которых известно, что они могут сохранять больше остаточного ЭО;
d) характеристики упаковки;
e) наличие смазочных материалов, особенно на матированных зонах поверхностей;
f) необходимость разборки или очистки медицинского изделия;
g) факторы, влияющие на безопасность (например, выщелачиваемые материалы, или батарейки, или герметично закрытые жидкости, которые могут вытечь или взорваться);
h) количество стерилизационных циклов.
Медицинские изделия, содержащие потенциальные источники возгорания (например, батареи), должны стерилизоваться с применением процесса, который не использует взрывоопасных смесей ЭО ни в одной части цикла.
D.7.2.2 Оценка воздействия многократных стерилизационных циклов может выполняться с использованием рутинного процесса стерилизации продукта/упаковки. Эффект воздействия повторной стерилизации и любой необходимой предварительной обработки на материалы, функциональность и безопасность продукта должен быть оценен.
Для многократно используемых медицинских изделий должны иметься и выполняться инструкции изготовителя по их повторной обработке. Инструкции должны включать в себя рекомендуемые параметры процесса стерилизации и ограничения на количество циклов стерилизации, которым может быть подвергнуто медицинское изделие многократного применения. Если применимо, должны быть выполнены испытания и проверки для оценки функциональности медицинского изделия многократного использования после прохождения стерилизации. Заявленное изготовителем медицинского изделия допустимое число циклов должно рассматриваться как максимально допустимое. Должна иметься действующая система, извещающая о достижении максимально допустимого количества циклов.
Примечание - См. ISO 17664 для получения дополнительной информации.
D.7.2.3 Дополнительное руководство не приводится.
D.7.2.4 Правильная аэрация жизненно необходима для контроля остатков ЭО в медицинских изделиях после ЭО-стерилизации. Следует уделять внимание размещению контрольных образцов, предназначенных для оценки остатков, внутри загрузки, учитывая при этом места, наиболее трудные для удаления ЭО.
Локальная окружающая среда, нормы безопасности и охраны здоровья могут потребовать дополнительных предосторожностей в отношении экспозиции работников при работе со стерилизованными ЭО продуктами, даже если остатки в продукте соответствуют ISO 10993-7.
Для учреждений здравоохранения: если информация изготовителя об аэрации медицинского изделия недоступна, учреждение здравоохранения должно установить для такого изделия процесс аэрации, используя данные или знания о продукте, его материалах и конструкции. Процесс аэрации должен устанавливаться, основываясь на наиболее затруднительных для аэрации продуктах или семействах продуктов.
D.7.3 Микробиологическое качество
D.7.3.1 Руководство по проверке на бактериальные эндотоксины приводится в ANSI/AAMI/ST72 и применимых фармакопеях.
D.7.3.2 В учреждениях здравоохранения внимание к микробиологическому качеству должно включать в себя наличие строгих процедур по сбору и обращению с бывшими в употреблении медицинскими изделиями многократного применения, а также процедур валидации и контроля процессов очистки многоразовых медицинских изделий в соответствии с инструкциями изготовителя этих изделий.
При использовании подхода с бионагрузкой (см. приложение А) испытания бионагрузки должны выполняться по меньшей мере ежеквартально. Период мониторинга может быть расширен в соответствии с результатом документированного анализа риска, рассматривающего следующие аспекты: использование семейств продукта, исторические данные, статистический анализ, частота изготовления и конструкция продукта.
D.7.4 Документирование
После завершения определения продукта должно быть документировано следующее:
a) описание конфигурации продукта и то, как он должен быть представлен на процесс ЭО-стерилизации (упаковка и конфигурация загрузки). Спецификация должна также включать в себя (или ссылаться на соответствующий документ) требуемый уровень обеспечения стерильности наряду с доказательствами (или с оценкой) совместимости продукта с процессом;
b) результат сравнения между новым или модифицированным продуктом и существующим валидированным продуктом (продуктами). Этот результат должен четко демонстрировать, что сложность продукта, материалы, упаковка и конфигурация загрузки были подвергнуты оценке;
c) доказательства или оценка бионагрузки продукта и ее сопротивляемости по отношению к внутреннему УКП;
d) документированное заключение о том, что новый или модифицированный продукт пригоден для включения в семейство продукта/категорию обработки с особой ссылкой на достижение специфицированного уровня обеспечения стерильности в текущих валидационных исследованиях. Это заключение должно включать в себя (или ссылаться на них) любые результаты дополнительных испытаний, выполненных для поддержки текущих валидационных исследований и на любые дальнейшие испытания, выполненные для подтверждения/аттестации рутинного выпуска продукта из существующего валидационного цикла (например, проверка остатков или функциональные испытания).
Упомянутая документация подлежит утверждению и сохранению с возможностью последующего извлечения.
D.8 Определение процесса
D.8.1 Дополнительное руководство не приводится.
D.8.2 Результат действий по определению процесса - это детальная спецификация процесса стерилизации.
Выбор процесса стерилизации, который будет использоваться для стерилизации медицинского изделия, должен включать в себя рассмотрение всех факторов, способных повлиять на эффективность процесса. Приниматься во внимание должно следующее:
- доступность стерилизационного оборудования;
- диапазон условий, которые могут быть достигнуты внутри доступного стерилизационного оборудования;
- стерилизационные процессы, уже используемые для других продуктов;
- стерилизующий агент, который предполагается использовать (например, 100% ЭО или ЭО, смешанный с газом-разбавителем);
- ограничения продукта (например, температура, влажность, чувствительность к давлению);
- требования к остаточным уровням ЭО и/или его продуктов реакции;
- результаты экспериментов, выполненных при разработке процесса.
В ходе определения процесса изготовитель будет использовать микробиологические исследования и другие аналитические инструменты для поддержки установления подходящего стерилизационного процесса для медицинского изделия.
Подлежащие установлению параметры стерилизационного процесса включают в себя следующее:
a) диапазон температур в помещении для предварительного кондиционирования (если используется);
b) диапазон относительной влажности в помещении для предварительного кондиционирования (если используется);
c) уставка и диапазон времени в помещении для предварительного кондиционирования (если используется);
d) уровни давления, вакуума и скорости изменения давления в камере стерилизатора;
e) если используется - подтверждение того, что рециркуляция в камере работает во время выдержки под воздействием стерилизующего агента;
f) уставка и диапазон времени в камере стерилизатора;
g) уставка и диапазон контроля влажности (давление или % относительной влажности) в среде внутри камеры стерилизатора;
h) уставка и диапазон давления инжекции ЭО и газа-разбавителя (если используется); сюда должна включаться концентрация ЭО, если на камере стерилизатора установлен газоанализатор;
i) время выдержки под воздействием ЭО;
j) уставка очистки (продувки) внутреннего пространства камеры от газа перед выемкой загрузки из стерилизационной камеры (если используется);
k) уставка и диапазон температуры внутри аэрационного помещения (если используется);
I) уставка и диапазон времени внутри аэрационного помещения (если используется);
m) параметры скорости потока воздуха и воздухообмена.
Примечание - В качестве справочной информации при разработке стерилизационных процессов приложения А и В дают требования к определению летальности цикла.
Для учреждений здравоохранения: для медицинских изделий многократного применения, которые будут неоднократно обрабатываться в учреждении здравоохранения, предполагается предоставление изготовителем валидированных инструкций по повторной обработке, которые основаны на части определения процесса. Затем на учреждение здравоохранения переходит ответственность за пересмотр этой документации и за подтверждение, что оно может соблюдать инструкции изготовителя медицинского изделия, используя собственное оборудование и собственные процессы стерилизации. Процедуры закупки учреждения здравоохранения должны требовать, чтобы перед закупкой медицинского изделия, стерилизуемого ЭО, инструкции по повторной обработке этого изделия были подвергнуты оценке для подтверждения совместимости изделия с оборудованием и стерилизационными процессами, используемыми в учреждении. См. также ISO 17664.
Если изготовитель медицинского изделия или упаковки предоставляет инструкции по повторной обработке, которые недостаточно специфичны или непригодны (например, ЭО-процесс с подачей 100% ЭО, в то время как учреждение здравоохранения использует смесь ЭО с газом-разбавителем), учреждение должно либо выполнить валидацию, или оценить пригодность собственного метода повторной обработки, основываясь на данных о воздействии на материалы и инструкции по обработке других изделий. Если учреждение здравоохранения не может валидировать продукт или оценить пригодность собственного способа его повторной обработки, оно не должно выполнять повторную обработку такого изделия.
D.8.3 Разработочная камера обычно представляет собой сосуд (емкость) меньшего размера, чем производственная камера, и может использоваться для проведения исследований для поддержки валидации.
Использование разработочной камеры не отменяет подтверждение результатов исследований путем выполнения аттестации эксплуатируемого оборудования (PQ) в производственной камере.
D.8.4 При установлении определения процесса важно рассмотрение воздействия выбранных параметров обработки и их допусков на безопасность и функциональность продукта и его упаковки. Поскольку в стерилизационном процессе задействовано много параметров (температура, влажность, изменения и скорость изменений давления, концентрация ЭО и время), непрактичной будет оценка допусков всех возможных комбинаций всех переменных. Сначала нужно определить те переменные, которые могут оказать наибольшее влияние, и именно их нужно оценивать.
Данные, поддерживающие эту деятельность, могут быть собраны из альтернативных исследований, например, из валидации продукта и его упаковки, из испытаний стабильности продукта и его упаковки, испытаний ускоренного старения и т.п. Кроме того, данные могут быть получены из результатов специфического нагрузочного цикла (циклов) с контрольным заражением, выполненных в разработочной или производственной камере.
D.8.5 Дополнительное руководство не приводится.
D.8.6 Для демонстрации пригодности биологического индикатора может быть использовано несколько разных подходов.
Подход 1
Этот подход основан на использовании научного обоснования того, что микроорганизмы, найденные на продукте, представляют собой меньшую нагрузку, чем контрольные микроорганизмы. Этот подход пригоден, если:
a) биологические индикаторы, используемые в УКП, соответствуют описанным в ISO 11138-2:2006, раздел 5 и пункт 9.5:
b) бионагрузка продукта постоянна и с малой вероятностью может содержать микроорганизмы с высокой устойчивостью.
При этом подходе должны быть доступны данные тренда бионагрузки, и они должны демонстрировать постоянство бионагрузки в отношении количества и типов микроорганизмов. Производственные процессы и материалы, контактирующие с продуктом, также подлежат оценке с целью убеждения, что потенциальные источники бионагрузки идентифицированы и контролируются.
Подход 2
Этот подход заключается в использовании проверки на стерильность продукта и УКП, выполняемой после фракционного цикла. Результаты этого исследования должны обеспечить средства сравнения летальности с использованием данных о выживаемости из проверок на стерильность продукта и упаковки.
Обычно при этом подходе образцы продуктов и биологические индикаторы/УКП, которые будут проверяться на стерильность, подвергаются воздействию фракционного цикла (циклов) с намерением получить отрицательный рост во всех проверках на стерильность продукта и отсутствие выживших контрольных микроорганизмов в биологических индикаторах/УКП.
Подход 3
Этот подход может применяться в случаях, когда:
a) бионагрузка продукта равна или превышает нагрузку, представленную биологическим индикатором внутри УКП;
b) бионагрузка продукта содержит микроорганизмы с высокой устойчивостью;
c) в УКП используется биологический индикатор с меньшей популяцией, чем требуется в соответствии с ISO 11138-2:2006, пункт 9.3.
В этом третьем подходе исследование летальности бионагрузки и УКП может базироваться на методах прямого подсчета и/или методах фракционного негативного анализа. См. ISO 14161.
Если наблюдаются признаки того, что бионагрузка, предоставляемая продуктом, превышает нагрузку, создаваемую УКП (например, в случае, когда УКП не подходит), можно использовать одно из следующих действий:
a) выбрать для использования внутри УКП биологический индикатор, имеющий большую популяцию и/или устойчивость;
b) предварительно обработать продукт перед стерилизацией с целью количественного уменьшения его бионагрузки;
c) подвергнуть оценке продукт, процесс или и то, и другое для определения, как количественно снизить бионагрузку или уменьшить ее устойчивость (например, за счет замены сырьевых материалов или изменения используемых производственных процессов, или за счет изменения производственной окружающей среды или модификации конструкции продукта);
d) разработать новое УКП.
Если сделано одно из перечисленных выше изменений, важно проверить его эффективность.
Конструкция продукта может не позволить расположить биологический индикатор в наиболее труднодоступном для стерилизации месте. В таких обстоятельствах может быть возможно помещение биологического индикатора в ином месте, для которого можно установить взаимосвязь с наиболее труднодоступным для стерилизации расположением. В дополнение к этому можно сказать, что наиболее трудные для стерилизации места многих медицинских изделий содержат меньшее количество микроорганизмов, и потому испытательная популяция может быть более тесно привязана к бионагрузке продукта.
Различные типы УКП описаны в D.7.1.6 (приложение D). Методы, схожие с теми, которые используются для определения пригодности биологического индикатора, могут быть использованы и для определения пригодности УКП. УКП, расположенное внутри границ продукта, в контейнере продукта или в упаковке грузоотправителя, является внутренним УКП, тогда как УКП, расположенное между упаковками или на внешних поверхностях стерилизационной загрузки, является внешним УКП. Внутренне УКП может использоваться в рутинном выпуске продукта; однако обычно используются внешние УКП, поскольку их легче восстанавливать после завершения стерилизационного процесса. Исследования, выполненные в разработочной камере, могут использоваться для демонстрации сравнительных испытаний летальности внутренних и внешних УКП; однако при выполнении таких исследований следует учитывать влияние объема загрузки и рабочих характеристик производственного стерилизатора. Если разработочная камера не способна продублировать производственный процесс, то сравнительные исследования следует выполнять в производственной камере.
Сравнительное исследование летальности "внутреннее УКП против внешнего УКП" можно оценивать, используя параллельные экспозиции во фракционных циклах. Полученные данные могут быть использованы в следующих действиях:
a) решение, какое внутреннее УКП наиболее подходит для валидации стерилизационного процесса;
b) оценка предлагаемых конструкций внешнего УКП (например, для рутинного мониторинга процесса);
c) оценка эквивалентности нового или модифицированного продукта или внутреннего УКП для его применения в валидированном стерилизационном процессе;
или
d) решение, станет ли новый или модифицированный продукт или внутренне УКП мастер-продуктом для семейства продуктов или обрабатываемой группы.
Могут встречаться случаи, когда желательно сравнить летальность нагрузки одного УКП с другим без сравнения обоих с летальностью нагрузки продукта. Это часто используется, когда внутреннее УКП доказало свою пригодность, а внешнее УКП внедряется для мониторинга рутинных производственных циклов для обычного выпуска, или когда желательно сменить одно внешнее УКП на другое. В таком случае методом оценки пригодности УКП является демонстрация эквивалентности или превышения летальности нагрузки внешнего УКП при его сравнении с внутренним УКП. В большинстве случаев это делается путем выполнения одного фракционного цикла, который сравнивает результаты фракционного негативного анализа данных от внешнего и внутреннего УКП. Если летальность внешнего УКП меньше, чем летальность внутреннего УКП (не более 20%, фармакопея США, "Биологические индикаторы для ЭО-стерилизации"), УКП может считаться эквивалентным, поскольку это является уровнем доверия биологического индикатора, использованного внутри УКП.
Примечание - Не является чем-то необычным обнаружение того, что внешнее УКП, расположенное в менее затруднительной для стерилизации конфигурации, показывает большую летальность нагрузки, чем внутреннее УКП, расположенное в наиболее трудной для стерилизации конфигурации. Имеется теория, объясняющая это явление тем, что ЭО выводится из внешнего УКП быстрее, чем из внутреннего, что приводит к меньшей экспозиции микробиологической нагрузки.
D.8.7 Дополнительное руководство не приводится.
D.8.8 Дополнительное руководство не приводится.
D.8.9 Дополнительное руководство не приводится.
D.9 Валидация
D.9.1 Общая информация
D.9.1.1 Целью валидации является документирование доказательств, требующихся для высокой степени уверенности в том, что специфический процесс будет постоянно выдавать продукт, обеспечивающий требуемый уровень обеспечения стерильности. Продукт, стерилизуемый валидированным процессом, должен быть продемонстрирован как соответствующий всем заранее определенным спецификациям и качественным характеристикам, относящимся к функциональности и безопасности продукта (например, с помощью исследования совместимости продукта).
Валидация стерилизационного процесса должна выполняться в соответствии с утвержденным письменным документом (например, протоколом), определяющим процедуры испытаний и критерии приемлемости еще до начала испытаний. Этот документ должен пересматриваться специалистами в области стерилизации.
Элементами валидации, как указывается в данной статье, являются:
a) аттестация установленного оборудования (IQ);
b) аттестация функционирующего оборудования (OQ);
c) аттестация эксплуатируемого оборудования (PQ).
В учреждении здравоохранения аттестация установленного оборудования и аттестация функционирующего оборудования обычно выполняется изготовителем стерилизатора, хотя эти действия могут быть выполнены и любым другим квалифицированным персоналом. Для типовых загрузок данные микробиологических исследований аттестации эксплуатируемого оборудования могут иметься у изготовителя стерилизатора.
Для учреждения здравоохранения это означает описание и документирование следующего:
a) необходимые для выполнения шаги валидации;
b) способы, какими эти шаги валидации будут выполняться, вместе с перечнем ответственных лиц, подразделений и/или внешних контрактных исполнителей;
c) критерии приемлемости успешной валидации.
Для учреждений здравоохранения: есть возможность заключения контракта с внешним исполнителем для проведения валидации; однако учреждение здравоохранения и в этом случае несет ответственность за обеспечение уверенности, что валидация соответствует требованиям настоящего стандарта.
D.9.1.2 Дополнительное руководство не приводится.
D.9.1.3 Дополнительное руководство не приводится.
D.9.1.4 Дополнительное руководство не приводится.
D.9.2 Аттестация установленного оборудования
D.9.2.1 Оборудование
D.9.2.1.1 Документация, поддерживающая аттестацию установленного оборудования, должна включать описания физических и эксплуатационных характеристик оборудования (включая вспомогательное оборудование). Примерами таких документов могут служить конструкторские спецификации, оригиналы заказов, спецификации требований пользователя (технические задания) и функциональные спецификации.
Далее перечисляются примеры компонентов оборудования, которые должны подвергаться аттестации для обеспечения уверенности, что оборудование было смонтировано в соответствии с применимыми спецификациями и иными требованиями:
a) конструкции камеры и двери;
b) уплотнительные прокладки и соединители конструкции камеры и трубной обвязки (т.е. способность поддерживать специфицированные максимальные давление и вакуум);
c) системы подачи газов и жидкостей (например, воздуха, азота, ЭО и воды), включая фильтры (если они используются);
d) системы подачи электроэнергии, которые должны адекватно и непрерывно подавать питание, необходимое для правильной работы оборудования и приборов;
e) системы газовой циркуляции, если таковые используются;
f) системы инжекции газа;
g) вакуумные системы, в том числе насосы, системы охлаждения насосов и трубная обвязка;
h) системы отвода, контроля выбросов и снижения концентрации;
i) прочие критические системы, способные влиять на условия выполнения процесса, такие как автоматизация процесса, системы безопасности и т.п.;
j) калибровка приборов (например, датчиков, самописцев/регистраторов, манометров и контрольных измерительных приборов), осуществляющих мониторинг, управление, индикацию и запись таких параметров как температура, влажность, давление и концентрация ЭО;
k) документированные процедуры аттестации установленного оборудования, которые должны описывать, как спланирован каждый элемент этой аттестации, и как он будет выполняться и пересматриваться.
D.9.2.1.2 Дополнительное руководство можно найти в документе МЭК 61010-2-40.
D.9.2.1.3 Дополнительное руководство не приводится.
D.9.2.2 Аттестация установленного оборудования
D.9.2.2.1 Место, на котором должно монтироваться оборудование, должно соответствовать требованиям всех имеющих к этому отношение национальных, региональных и местных нормативных документов.
D.9.2.2.2 Следует рассмотреть требования национальных и местных нормативных документов, применимых к охране здоровья и защиты окружающей среды в свете потенциальной экспозиции ЭО.
Для защиты здоровья и обеспечения безопасности персонала оборудование, определяющее атмосферные уровни ЭО или газовых смесей, должно устанавливаться рядом со стерилизатором и в любых иных местах, где может возникать угроза потенциальной экспозиции.
Безопасность в отношении ЭО достигается и поддерживается комбинацией факторов, включающих в себя:
a) правильную конструкцию, монтаж и обслуживание систем и оборудования;
b) соответствие требованиям применимых норм охраны здоровья, безопасности и защиты окружающей среды;
c) разработку и внедрение политик и процедур, поддерживающих безопасные рабочие (производственные) практики;
d) мониторинг атмосферы в зонах, в которых возможна экспозиция ЭО;
e) использование индивидуальных средств мониторинга при возможности;
f) обучение и вырабатывание навыков персонала;
g) периодический аудит оборудования, персонала и процессов для обеспечения уверенности в непрерывном соответствии техническим спецификациям и политикам и процедурам учреждения.
В учреждениях здравоохранения аттестация установленного оборудования обычно является ответственностью изготовителя стерилизатора, тогда как в промышленных организациях она часто выполняется собственным персоналом при участии представителя изготовителя. Если аттестация установленного оборудования выполняется изготовителем или третьей стороной, учреждение несет ответственность за поддержание системы управления документированием и за записи, относящиеся к закупке и монтажу оборудования.
D.9.2.2.3 Условия хранения ЭО должны соответствовать рекомендациям изготовителя ЭО и требованиям всех применимых норм и положений.
D.9.2.2.4 Дополнительное руководство не приводится.
D.9.2.2.5 Чертежи, описания процессов и схемы трубной обвязки и приборов должны проверяться на соответствие конфигурации на момент завершения монтажа и обновляться при необходимости.
Чертежи и перечни компонентов и частей оборудования должны включать в себя:
a) трубную обвязку и схемы приборной обвязки (т.е. схемы процессов и схемы их оснащения контрольно-измерительной аппаратурой);
b) перечни иных (относящихся к монтажу) механических чертежей и электрических схем с указанием их местонахождения;
c) перечень критических измерительных приборов и устройств, в особенности тех, которые имеют отношение к контролю процесса, чьи физические характеристики и заявленные изготовителем свойства (например, точность, повторяемость, размеры, модель) должны храниться в файлах;
d) документация автоматики или программного обеспечения контроля процесса, необходимая для поддержки валидации, в том числе схема системы управления, схемы управляющей логики и прикладное программное обеспечение (компьютеризированные системы измерения и управления), такое как листинги программ, схемы потоков, многоступенчатые логические схемы, если они применяются, и стратегические схемы.
D.9.2.2.6 Дополнительное руководство не приводится.
D.9.3 Аттестация функционирующего оборудования
D.9.3.1 В отношении всех приборов, используемых для мониторинга, управления, индикации и записи, должна быть документирована следующая информация:
a) идентификация прибора;
b) график калибровки:
c) фактическая дата каждой калибровки с информацией о том, кто ее выполнял;
d) дата следующей калибровки.
D.9.3.2 Аттестация функционирующего оборудования выполняется либо с пустой камерой стерилизатора, либо с использованием подходящих испытательных материалов для демонстрации способности оборудования обеспечивать ряд рабочих параметров в установленных пределах, указанных в спецификации процесса. Этот ряд параметров и их пределов должен включать в себя начальный стерилизационный процесс, ранее установленный в определении процесса (см. раздел 8).
Аттестация функционирующего оборудования должна также определять работу связанных вспомогательных систем. Например, способность испарителя ЭО обеспечивать минимальную входную температуру ЭО.
Системное программное обеспечение (например, компьютеризированных систем измерения и управления) в ходе выполнения аттестации функционирующего оборудования должно быть испытано во всех возможных условиях сбоя. Ответственность за валидацию программного обеспечения несет пользователь.
Аттестация функционирующего оборудования при использовании ранее определенного цикла может включать в себя следующее:
a) фаза предварительного кондиционирования:
1) должна быть определена модель циркуляции воздуха через зону, которую должна занимать стерилизационная загрузка. Это можно выполнить с помощью дымовых тестов в сочетании с расчетом скоростей воздухообмена и анемометрических измерений;
2) мониторинг температуры и влажности должен выполняться во всей зоне предварительного кондиционирования в течение периода времени достаточно длительного, чтобы продемонстрировать, что их значения поддерживаются в желаемых диапазонах. Должны быть определены температура и влажность в нескольких местах, распределенных по всей зоне предварительного кондиционирования;
Примечание - См. таблицу C.1 и таблицу C.2, где даны рекомендации о количестве датчиков температуры и влажности.
b) фаза стерилизации:
1) если вместо ЭО используются инертные газы, при оценке результатов следует брать в расчет разницу в относительной теплоемкости;
2) распределение температуры/влажности: датчики температуры/влажности должны располагаться в таких местах, которые с наибольшей вероятностью представляют максимальный дифференциал температуры, например, рядом с не нагреваемыми частями камеры или двери, и в местах рядом с входными портами пара или газа. Оставшиеся датчики температуры должны равномерно распределяться по всему полезному объему камеры;
Примечание - См. таблицу C.1, в которой даны рекомендации о количестве датчиков.
3) при исследованиях аттестации функционирующего оборудования с пустой камерой во время впуска ЭО или инертного газа по записям температуры после завершения периода уравновешивания в полезном объеме камеры должно быть достигнуто ±3% от средней записанной температуры камеры в каждой точке времени. Если исследования выполняются с загруженной камерой, тогда допуск ±3% может оказаться недостижимым;
4) скорость утечки камеры (измерение выполняется либо под вакуумом для субатмосферных циклов, либо под вакуумом или под давлением для суператмосферных циклов);
5) скорость роста давления при впуске пара во время выполнения фазы кондиционирования;
6) температура впущенного ЭО-газа должна быть в пределах спецификации испарителя или выше точки кипения ЭО (10,7°С при атмосферном давлении);
7) рост давления и скорость достижения (значения) при впуске ЭО и корреляция факторов, которые используются для мониторинга концентрации ЭО;
8) глубина и скорость достижения вакуума при удалении ЭО;
9) рост давления и скорость достижения (значения) при впуске воздуха (или иных газов);
10) число повторения этих последних стадий и любые отличия в успешных повторах;
11) надежность подачи фильтрованного воздуха, инертных газов, воды и пара;
12) для демонстрации повторяемости управления должны быть выполнены реплицирующие циклы;
13) должно быть выполнено исследование температуры стенки камеры для убеждения в том, что система нагрева рубашки обеспечивает единообразие температуры. Исследование должно характеризовать температурный профиль для последующего периодического сравнения с целью подтверждения продолжения эффективной работы системы.
c) фаза аэрации:
1) при выполнении аэрации должен быть определен температурный профиль зоны аэрации тем же способом, который рекомендован для зоны предварительного кондиционирования. Должны быть также определены скорости и модели воздушных потоков, проходящих через зону.
D.9.4 Аттестация эксплуатируемого оборудования
D.9.4.1 Общая информация
Аттестация эксплуатируемого оборудования состоит из тщательных микробиологических и физических испытаний, выходящих за рамки рутинного мониторинга, целью которых является демонстрация эффективности и воспроизводимости стерилизационного процесса. Аттестация эксплуатируемого оборудования обычно не начинается, пока не будут завершены и одобрены испытания аттестаций установленного и функционирующего оборудования. Критерии приемлемости должны включать соответствие спецификациям параметров стерилизационного процесса и микробиологической нагрузки. Действия в рамках аттестации эксплуатируемого оборудования должны быть четко и ясно определены в письменном документе (например, в протоколе). Там, где элементы аттестации эксплуатируемого оборудования выполняются третьими сторонами, эти стороны должны одобрять соответствующие документы. См. 4.1 и 4.2.
D.9.4.1.1 Дополнительное руководство не приводится.
D.9.4.1.2 См. AAMI TIR 28:2009 [26].
D.9.4.1.3 Дополнительное руководство не приводится.
D.9.4.1.4 В ходе специфицирования представления продукта должны рассматриваться как конфигурация загрузки (состав загрузки), так и размещение изделий внутри загрузки.
Типичные параметры загрузки, подлежащие определению, могут включать конфигурацию штабелирования, общую плотность, размеры, состав материалов, а также использование и тип обертки паллет. Конфигурация загрузки должна документироваться для каждого стерилизатора. Если рутинная стерилизация состоит из загрузок продукта меньших, чем полная камера, тогда микробиологические и физические исследования аттестации эксплуатируемого оборудования должны включать в себя и минимальные загрузки.
Размещение продукта также должно быть специфицировано. В больших промышленных стерилизаторах это относится к размещению паллет, коробок или больших пакетов. Для меньших стерилизаторов, которые используются в учреждениях здравоохранения, это относится к расположению корзин, упаковок и жестких контейнеров на стерилизационных тележках или других носителях.
Продукт и загрузка, используемые при аттестации эксплуатируемого оборудования, должны быть, по меньшей мере, настолько же трудно стерилизуемы, как наиболее трудная для стерилизации загрузка, которую предполагается обрабатывать в обычном производственном цикле. Загрузка может состоять из продуктов или материалов, имеющих характеристики, подобные характеристикам рутинно стерилизуемой загрузки. Изменения в конфигурации загрузки могут влиять на летальность стерилизационного процесса. Важно, чтобы приемлемые конфигурации загрузки были специфицированы. Если допускаются различные конфигурации загрузки, то конфигурация, используемая в ходе аттестации эксплуатируемого оборудования, должна представлять наиболее трудно стерилизуемую конфигурацию, или должна иметь хорошо известную взаимосвязь с трудно стерилизуемой конфигурацией. Некоторые небольшие изменения размера загрузки могут оцениваться как не имеющие заметного влияния.
В ходе аттестации эксплуатируемого оборудования могут быть выбраны два типа загрузки:
a) продаваемый продукт;
b) непродаваемый продукт или подходящий испытательный материал.
D.9.4.1.5 Когда загрузка составляется из продуктов, таких, как хирургические наборы, иглы различного размера и длины, разнообразные упаковки и различные физические массы, содержащие разные материалы (например, пластики, металлы, ткань и т.п.), важно верифицировать конфигурацию загрузки, поскольку эти материалы могут вести себя неодинаково при нагреве в ходе предварительного кондиционирования и кондиционирования.
D.9.4.1.6 В дополнение к рассмотрению максимального/минимального размера загрузки [см. D.9.4.1.4 (приложение (D)] и эффектов продукта [см. D.9.4.1.5 (приложение D)] валидация состава загрузки при разработке представителя наиболее трудной загрузки должна учитывать любые широко варьируемые характеристики рутинно стерилизуемых материалов/упаковок загрузки.
Продукт или имитирующие его материалы, используемые в валидационных загрузках, должны представлять те продукты и материалы, которые типичны для создания наиболее тяжелых условий для летальности (например, для проникновения тепла, влаги и диффузии газа ЭО; плотность). Внимание должно быть уделено включению материала загрузки с заметно изменяющимися характеристиками, такими как: абсорбирующие материалы, барьеры диффузии (твердые материалы), герметично упакованные жидкости, контейнеры и т.п.
D.9.4.1.7 Дополнительное руководство не приводится.
D.9.4.1.8 Если в ходе аттестации эксплуатируемого оборудования загрузка должна использоваться неоднократно, она должна быть аэрирована и уравновешена до условий окружающей среды до начала следующего цикла. После повторного использования пригодность загрузки к дальнейшему использованию должна быть оценена. Аэрация между экспозициями должна гарантировать, что остатки ЭО в загрузке не повлияют на биологический индикатор. Если время уравновешивания окажется недостаточным, загрузка может оказаться теплее нормальных показателей окружающей среды, или ее влажность может оказаться ниже нормальных условий. Любая из этих ситуаций выдаст данные, которые не будут представительными для нормального производства. Слишком высокая начальная температура приведет к нереалистично высокой скорости убивки. Слишком низкая влажность, при которой контрольные споры окажутся высохшими, приведет к нереалистично низкой скорости убивки. Слишком высокая влажность, возникающая там, где точка росы окружающей среды выше, чем у продукта и/или загрузки, в сочетании с повышенной температурой приведет к образованию в загрузке конденсата, что в свою очередь приведет к низкой и ошибочной степени убивки.
D.9.4.1.9 Дополнительное руководство не приводится.
D.9.4.1.10 Дополнительное руководство не приводится.
D.9.4.2 Аттестация эксплуатируемого оборудования - микробиологические исследования
D.9.4.2.1 Результаты, полученные в ходе определения процесса и (где применимо) в ходе аттестаций установленного и функционирующего оборудования, должны использоваться для задания параметров микробиологических исследований. Ключевым параметром является длительность экспозиции, которая варьируется в ходе микробиологической аттестации. Другие параметры могут настраиваться по необходимости для обеспечения уверенности в том, что микробиологическая аттестация обеспечивает меньшую летальность, чем нормальный производственный процесс. Например, температура, влажность и/или концентрация ЭО могут задаваться меньшие, чем нижние пределы диапазонов этих параметров для нормального процесса. Это обеспечит уверенность, что любые наблюдаемые значения в пределах специфицированного диапазона дадут приемлемую летальность.
Микробиологические исследования аттестации эксплуатируемого оборудования должны выполняться с продуктом, имеющим равную или более низкую температуру по сравнению с температурой, специфицированной для продукта при входе в зону предварительного кондиционирования. Если ожидается, что начальная температура продукта может изменяться, например, из-за транспортировки его для стерилизации в удаленное помещение, то расчеты аттестационных исследований должны учитывать эту возможность.
Для фракционных циклов (суб-летальный или половинный цикл) может оказаться необходимым укорочение по времени фаз, исполняемых после экспозиции, или извлекать биологические индикаторы до начала фазы аэрации, либо извлекать их после сокращенной фазы аэрации. Это делается для минимизации "остаточной убивки" биологических индикаторов тем ЭО, который остается в загрузке во время аэрации. При сокращении фаз, следующих после экспозиции, следует принимать во внимание такие факторы, как безопасность оператора. Параметры, выбранные для микробиологических исследований, за исключением длительности экспозиции, должны оставаться фиксированными на все время выполнения этих исследований.
Примечание - Следует уделить внимание наличию положений и нормативов, действующих в некоторых странах в отношении экспозиции персонала ЭО.
D.9.4.2.2 Микробиологическая нагрузка, определенная в микробиологических исследованиях, должна быть рассчитана на получение требуемого уровня обеспечения стерильности. Для достижения этой цели принято использовать УКП или продукт "худшего случая" для представления семейств продуктов ЭО.
УКП должны помещаться внутри коробки (упаковки) продукта и равномерно распределяться внутри стерилизационной загрузки, но при этом распределение должно охватывать те места, где условия стерилизации наиболее труднодостижимы. Места размещения должны включать и точки, выбранные для мониторинга температуры. Для загрузок, находящихся на паллетах, эти места размещения должны включать в себя верхние и нижние части паллет для убеждения, что оценены будут все возможные расслоения внутри камеры.
Руководство по выбору количества образцов см. в таблице C.3 (приложение C).
D.9.4.2.3 Дополнительное руководство не приводится.
D.9.4.2.4 Если для определения процесса была использована разработочная камера, следует рассмотреть установление взаимосвязи между данными исследований в разработочной камере и данными, полученными из производственной камеры. Разработка кривых микробиологической инактивации не всегда возможна в производственных камерах по причине их размеров и времени, затрачиваемого на инжекцию и откачку ЭО. Эти длительные инжекции и время откачки до вакуума ограничивают возможность достичь требуемого фракционного восстановления организмов-индикаторов. Такие кривые инактивации могут быть разработаны в разработочной камере, способной обеспечить эквивалентные параметры, в особенности концентрацию ЭО, используемую в производственной камере. Методы демонстрации взаимосвязи между данными от разработочной и производственной камер включают в себя сравнение физического профиля и сравнение плотности загрузки. Условия стерилизации, созданные в разработочной камере, должны сравниваться с физическим профилем, полученным в производственной камере. Сравнение летальности, достигнутой в разработочной и производственной камерах, должно принимать в расчет разницу в длительностях инжекций газообразного ЭО и в длительностях откачек для обеих камер.
D.9.4.2.5 См. AAMI TIR 16:2009 [25], пункт 4.3.2.
D.9.4.3 Аттестация эксплуатируемого оборудования - физические исследования
Примечание - Для идентификации факторов, необходимых для оценки в ходе аттестации эксплуатируемого оборудования, могут использоваться результаты, полученные из аттестации функционирующего оборудования.
D.9.4.3.1 Если в любом из этих циклов будут нарушены требования к стерильности или функциональности продукта, должно быть проведено расследование с целью определения необходимости выполнения дополнительных аттестационных циклов. Если процессные параметры не могут быть поддержаны в заданных пределах, также следует провести расследование причин. Если были внесены изменения/модификации, дополнительные циклы могут стать необходимыми.
D.9.4.3.2 Физические исследования должны проводиться с моделями загрузки и разделениями паллет, специфицированными в документированных процедурах. Для больших зон предварительного кондиционирования, где небольшая загрузка не оказывает заметного влияния на динамику в зоне, нет необходимости (и это даже может быть непрактично) выполнять исследования в зоне предварительного кондиционирования с разными состояниями загрузки.
Руководство по выполнению физических исследований аттестации эксплуатируемого оборудования в зоне предварительного кондиционирования относится также и к аттестации кондиционирования (например, во время стерилизации). Минимально необходимое количество датчиков приводится в таблицах C.1 и C.2 (приложение C):
a) дополнительное руководство не приводится;
b) важно установить и внести в отчет диапазоны температуры продукта и влажности стерилизационной загрузки после выдержки в течение специфицированного времени предварительного кондиционирования (если используется);
c) во время переноса продукта из зоны предварительного кондиционирования (если используется) в камеру стерилизатора могут быть нарушены условия температуры и влажности продукта. Важно, чтобы этот эффект был учтен во время аттестации эксплуатируемого оборудования путем обеспечения того, чтобы время переноса, специфицированное для аттестации, отражало максимальное специфицированное время переноса во время рутинной стерилизации;
d) датчики температуры и влажности должны располагаться внутри стерильной барьерной системы или между единичными упаковками в стерилизационной загрузке. Если используется предварительное кондиционирование, продукт должен ему подвергнуться в рамках специфицированного времени. Если предварительное кондиционирование не используется, температура и относительная влажности внутри загрузки должны оставаться в заданных пределах до окончания фазы кондиционирования цикла.
Профиль температуры и относительной влажности внутри стерилизационной загрузки должен быть оценен за время, потребное для достижения стерилизационной загрузкой минимальных заранее определенных значений температуры и влажности.
В отношении продукта следует рассмотреть размещение датчиков влажности в тех зонах загрузки, которые с наибольшей вероятностью могут испытывать колебания влажности, например, центры паллет, кромки паллет и поверхности. Во время аттестации эксплуатируемого оборудования датчики влажности должны размещаться внутри упаковок (где возможно), находящихся внутри загрузки. Этого можно достичь, размещая датчики внутри стерильной барьерной системы или между единичными упаковками в стерилизационной загрузке.
e) дополнительное руководство не приводится;
f) если применяется параметрический выпуск, должен быть оценен профиль концентрации ЭО для всего периода выдержки под воздействием газа с целью определения изменений концентрации на протяжении всей фазы;
g) дополнительное руководство не приводится;
h) дополнительное руководство не приводится;
i) датчики температуры внутри загрузки должны размещаться в местах, в которых наиболее вероятны наибольшие колебания температуры. В число этих мест должны входить горячие и холодные точки, определенные в ходе аттестации функционирующего оборудования. Координаты горячих и холодных точек внутри загрузки могут значительно отличаться от координат тех же точек в пустой камере.
Во время аттестации эксплуатируемого оборудования важно учесть взаимосвязь между температурой загрузки и температурой камеры с целью обеспечить адекватную температуру загрузки во время выполнения рутинного процесса. Если в стерилизационной камере устанавливаются датчики, и в то же время используется 100% ЭО или потенциально легковоспламеняющаяся смесь-стерилизующий агент, датчики температуры и влажности должны быть искро- и взрывобезопасными или иметь взрывозащищенную конструкцию. Эти датчики также должны быть функционально совместимыми с ЭО и любыми газами-разбавителями;
j) температура внутри стерилизационной загрузки во время процесса аэрации должна измеряться через период времени, необходимый для достижения стерилизационной загрузкой приемлемых остаточных уровней ЭО, или измеряться через период времени, необходимый для стабилизации температуры стерилизационной загрузки.
Примечание - Это может быть установлено в ходе дополнительных исследований после завершения аттестации эксплуатируемого оборудования.
D.9.5 Рассмотрение (анализ) и утверждение результатов валидации
D.9.5.1 Дополнительное руководство не приводится.
D.9.5.2 Любые несоответствия, обнаруженные в ходе процесса валидации, должны документироваться, а их воздействие на результаты валидации должно быть определено и задокументировано.
D.9.5.3 Обычно отчет о валидации одобряется (утверждается) назначенным ответственным лицом (лицами), которое указано в протоколе валидации.
D.9.5.4 Отчет о валидации должен также включать в себя (или давать ссылки на) следующее:
- спецификации стерилизатора и стерилизационного процесса:
a) данные аттестаций установленного и функционирующего оборудования;
b) записи данных (физических и микробиологических) по всем выполненным циклам аттестации эксплуатируемого оборудования;
c) доказательства того, что все измерительные приборы, регистраторы/самописцы и т.п. были откалиброваны и работали в пределах своих спецификаций;
d) положения о будущем пересмотре или повторной аттестации;
e) валидационные протоколы/процедуры;
f) документированные процедуры, которые использовались [для валидации];
g) документированные рабочие процедуры с указанием предельных значений для управления и контроля процесса;
h) если были отмечены сбои - описания проблем, предпринятые корректирующие действия и влияние произошедшего сбоя на цели валидации;
i) если произошло отклонение от протокола - подробности этого отклонения и оценка его влияния на валидацию и ее результаты.
D.9.5.5 Параметрический выпуск - это способ выпуска продукта, при котором продукт считается стерильным, если важнейшие физические параметры процесса соответствовали спецификациям, установленным во время валидации данного специфического продукта (продуктов) в определенной загрузке. Параметрический выпуск базируется больше на документированном пересмотре записей о процессах, чем на проверке биологических индикаторов или УКП.
Может понадобиться выработка значений и допусков для относительной влажности и концентрации ЭО после рассмотрения заранее определенного числа рутинных циклов. Во время этого оценочного периода биологические индикаторы могут использоваться как часть рутинного мониторинга и контроля обработанных загрузок. Обоснование выбранного числа циклов должно быть оценено и записано. На это могут влиять единообразие загрузок, уже имеющиеся данные, сезонные изменения или частота стерилизаций.
ЭО-стерилизаторы, используемые в учреждениях здравоохранения, могут быть недостаточно оборудованы для того, чтобы позволить применить параметрический выпуск продукта.
D.9.5.6 Дополнительное руководство не приводится.
D.10 Рутинный мониторинг и управление
D.10.1 Дополнительное руководство не приводится.
D.10.2 Руководство по отмеченным точками абзацам пункта 10.2 следующее:
a) температура продуктов, помещаемых в зону предварительного кондиционирования, должна быть равной или превышать минимальную специфицированную температуру, или должны быть соблюдены определенные условия хранения. Если продукт подвергался воздействию экстремально высоких температур, например, во время транспортировки, может оказаться необходимым выдержать его какое-то время до предварительного кондиционирования, или увеличить время предварительного кондиционирования, чтобы позволить температуре и влажности внутри продукта установиться в приемлемых пределах;
Примечание - Минимальная температура продукта, помещаемого в зону предварительного кондиционирования, или его условия хранения определяются в ходе аттестации эксплуатируемого оборудования.
b) контрольное положение для рутинного мониторинга температуры и относительной влажности во время предварительного кондиционирования должны коррелировать с местом, в котором достижение желаемых условий наиболее затруднительно. Мониторинг данных работы зоны предварительного кондиционирования должен проводиться в связке с остальными данными для выпуска продукта;
c) дополнительное руководство не приводится;
d) дополнительное руководство не приводится;
e) влажность обычно рассчитывается путем измерения изменений давления (см. также AAMI TIR15 [24]). Влажность в камере обычно рассчитывается по измерению парциального давления водяного пара, вводимого в камеру. Значение относительной влажности определяется с использованием паровых таблиц по соотношению парциального давления к давлению насыщенного пара для действующей температуры цикла. Это покажет значение относительной влажности в головной части камеры, и значение будет точным, пока загрузка или иные реакции не окажут воздействия на фактическое содержание водяного пара в головном пространстве. Следует учитывать объем влаги, введенной в камеру вместе с загрузкой из зоны предварительного кондиционирования;
f) дополнительное руководство не приводится;
g) усиленная циркуляция особенно важна, когда газовые смеси используются с целью обеспечения поддержания единообразных условий и для предотвращения расслоения газов, что могло бы воздействовать на микробиологическую летальность [см. D.6.3.2 (приложение D)];
h) дополнительное руководство не приводится;
i) рост давления из-за инжекции ЭО () обеспечивает косвенное измерение средней концентрации газообразного ЭО в доступном пространстве камеры стерилизатора. Поскольку концентрация ЭО является ключевой переменной, влияющей на эффективность стерилизационного процесса, считается необходимым, чтобы имелась отдельная вторичная система для документирования того, что рост давления произошел именно от впуска ЭО (см. AAMI TIR15 [25] для получения дополнительной информации). Во время инжекции ЭО и фазы экспозиции ЭО стерилизационного процесса ЭО абсорбируется продуктом и упаковочным материалом, что влияет на корреляцию между контрольным измерением (разность давлений) и вторичным измерением (например, массой впущенного ЭО или прямым измерением концентрации ЭО).
D.10.3 Наблюдения, показывающие рост в биологических индикаторах, не имеющий отношения к несоблюдению физических спецификаций процесса, должны быть проанализированы; это может приводить к необходимости модификаций процесса или оборудования и к повторению аттестации эксплуатируемого оборудования.
D.10.4 Для задач, выполняемых в учреждениях здравоохранения, предлагается следующее руководство:
Внешние химические индикаторы в учреждениях здравоохранения: Стерилизационные индикаторные ленты, индикаторные этикетки или индикаторные надпечатки должны быть закреплены или напечатаны на каждой упаковке, собранной в самом учреждении здравоохранения. Назначение внешних химических индикаторов - обеспечение видимого отличия обработанных изделий от необработанных. Они не являются доказательством достижения параметров стерилизации. Индикаторы должны иметь спецификации класса 1 согласно ISO 11140-1.
Внутренние химические индикаторы, используемые в учреждениях здравоохранения:
a) внутренний химический индикатор может быть использован внутри каждой упаковки, предназначенной для стерилизации. Такой химический индикатор должен располагаться в той зоне упаковки, которая считается наиболее труднодоступной для проникновения ЭО, тепла и влажности; это может быть (а может и не быть) центр упаковки. Хотя химические индикаторы и не свидетельствуют о стерильности, они позволяют обнаруживать несоответствия процесса и неправильное функционирование оборудования. Применение химических индикаторов, реагирующих на все параметры ЭО-процесса, является преимуществом;
b) внутренний химический индикатор извлекается на месте применения и интерпретируется пользователем. Пользователь должен быть адекватно обучен и иметь знания о рабочих характеристиках индикатора, чтобы принимать информированное решение, основанное на наблюдаемом результате;
c) если интерпретация индикатора предполагает неадекватную обработку ЭО, содержимое упаковки не подлежит дальнейшему использованию. Вся непригодная к употреблению упаковка, включая идентификатор загрузки и химический индикатор, подлежит возврату в отделение обработки для соответствующего расследования. Результаты физического мониторинга, химические индикаторы из всех мест в загрузке и результаты биологического мониторинга должны быть пересмотрены для получения заключения о том, должна или нет быть отозвана вся загрузка. Записи этого пересмотра подлежат хранению. Единичный не среагировавший или неправильно среагировавший индикатор не должен считаться доказательством того, что вся загрузка нестерильна. Химические индикаторы могут сигнализировать о проблемах, связанных с неправильным упаковыванием, неправильной загрузкой стерилизатора, перегрузкой камеры, неисправностью стерилизатора, неполным обеспечением стерилизационных параметров или с неправильным предварительным кондиционированием. "Хороший" результат химической индикации не доказывает, что изделие, в котором располагался индикатор, является стерильным;
d) химические индикаторы должны принадлежать к классам 3, 4, 5 или 6 в соответствии с ISO 11140-1.
D.10.5 Параметрический выпуск - это метод выпуска продукта из стерилизации как стерильного без использования биологических индикаторов, опираясь вместо этого на демонстрацию соответствия физических параметров процесса всем спецификациям. По этой причине собираются данные о дополнительных параметрах процесса, таких как прямой анализ относительной влажности в камере и концентрации ЭО, с целью убедиться, что стерилизационный процесс соответствовал всем спецификациям:
a) измерение температуры - требование об измерении температуры внутри стерилизатора как минимум в двух точках установлено с целью гарантировать, что необнаруженная неисправность одного датчика температуры не приведет к непреднамеренному выпуску неправильно обработанной загрузки. Если имеется разница между показаниями температуры в двух точках, то в процессных спецификациях должна быть определена допустимая разность этих температур. Если управляющий или контрольный датчик не соответствуют спецификациям, а расследование не может определить точность показаний, полученных из камеры, то загрузка бракуется;
b) измерение влажности - прямой анализ относительной влажности в головном пространстве может быть выполнен с применением электронных датчиков, газовой хроматографии, инфракрасного или иного доступного спектроскопического метода, используемого для измерения концентрации водяных паров и расчета значения относительной влажности. Преимуществом этих методов является индикация параметра в реальном времени на протяжении всей фазы кондиционирования. Электронные датчики требуют периодической калибровки для устранения эффекта воздействия на них ЭО, и могут потребовать замены после нескольких экспозиций ЭО по причине необратимого износа материалов, используемых в настоящее время в качестве чувствительных элементов;
с) измерение концентрации ЭО.
Частота анализов, требующихся для демонстрации того, что минимальная концентрация ЭО поддерживается на протяжении всей фазы экспозиции, должна быть установлена в ходе исследований аттестации эксплуатируемого оборудования. Мониторинг на протяжении фазы экспозиции ЭО также должен выполняться как часть валидации с целью определения изменений концентрации ЭО по времени. Результаты анализа будут специфичны для продукта и конфигурации загрузки, для которых он делался. Анализ, выполненный в ходе аттестации эксплуатируемого оборудования, должен завершиться документированными спецификациями частоты выполнения прямых анализов в ходе исполнения цикла. Если прямой анализ концентрации применяется, рекомендуется, чтобы он выполнялся как минимум во время первой и последней части фазы экспозиции ЭО.
Особое внимание следует уделить измерению и документированию влажности во время кондиционирования и концентрации ЭО во время экспозиции. Устройство для отбора проб ЭО, обеспечивающее прямой анализ концентрации с использованием газовой хроматографии, инфракрасного, микроволнового или иных схожих технологий, должно располагаться в месте, являющемся представительным для концентрации газа ЭО в камере стерилизатора. Однако важно понимать, что это измерение предоставляет значение концентрации ЭО именно в этом месте камеры на протяжении фазы экспозиции без учета любых ограничений, накладываемых реактивностью или воздействием загрузки. Воспроизводимость и точность результатов прямого анализа должна быть определена во время аттестации эксплуатируемого оборудования. Анализы, выполняемые в ходе рутинных циклов, должны быть в границах этого определенного диапазона, чтобы цикл считался приемлемым.
Может оказаться необходимым ввести определенное время уравновешивания в начале фазы экспозиции ЭО, чтобы обеспечить стабилизацию концентрации в камере по мере того, как газ распространяется по всей камере и проникает в полости загрузки.
Примечание 1 - Электронный датчик измеряет концентрацию газа ЭО только в одной точке пробоотбора, тогда как рассчитанная концентрация газа ЭО представляет среднюю концентрацию ЭО внутри объема (пространства), доступного для оседания молекул газа ЭО. Из-за нескольких факторов, таких, например, как динамические характеристики датчика ЭО, места размещения датчика внутри объема, занимаемого молекулами ЭО, потенциального расслоения внутри камеры, особенно если стерилизующий агент изготовлен из молекул ЭО и газа-разбавителя, селективной абсорбции и абсорбции ЭО в загрузке, объема, занимаемого загрузкой, значения, полученные из расчета средней концентрации газа ЭО, могут заметно отличаться от значений, полученных прямыми измерениями.
Примечание 2 - Учреждения здравоохранения не применяют в обычной практике рутинный параметрический выпуск.
D.11 Выпуск продукта после стерилизации
D.11.1 Это подтверждение должно включать формальный обзор документации процесса ответственным лицом (или с помощью валидированного автоматизированного процесса) для проверки и документирования того, что физические параметры цикла находятся в пределах допусков, определенных спецификациями процесса стерилизации. Если параметрический выпуск был одобрен и утвержден к использованию, продукт может быть выпущен на основе соответствия специфицированным параметрам процесса.
Рутинный выпуск продукта из стерилизации может базироваться на пересмотре электронных записей данных вместо бумажных записей. Подобным же образом необходимые подписи могут быть заменены электронными цифровыми подписями. Пользователи цифровых подписей и записей должны соблюдать национальные и/или международные требования к такому виду документирования. Пересмотр процессных записей и решение о выпуске должны выполняться квалифицированными лицами.
D.11.2 Дополнительное руководство не приводится.
D.11.3 Несоблюдение процессных спецификаций или обнаружение роста организмов-индикаторов из биологических индикаторов должно приводить к помещению стерилизационной загрузки в карантин и к расследованию причин неудачи. Это расследование должно быть документировано, а последующее обращение с продуктом должно соответствовать инструкциям, содержащимся в документированных процедурах.
Если вышел из строя датчик управления или мониторинга, цикл должен считаться забракованным, за исключением случаев, когда:
a) есть объяснимая причина неисправности,
b) данные от остальных датчиков находятся в пределах спецификаций.
Если принимается решение о повторной обработке загрузки, должна быть установлена пригодность к повторной обработке продукта и его упаковочной системы. Должен быть рассмотрен эффект от воздействия повторной экспозиции на функциональность продукта, а также учтены уровни остаточного ЭО и/или продуктов реакции. Записи первой стерилизации должны быть прослеживаемыми из записей повторной стерилизации (см. 7.2.2).
Если влияние повторной экспозиции на упаковочную систему неизвестно, продукт должен быть переупакован перед повторной стерилизацией.
D.11.4 Дополнительное руководство не приводится.
D.11.5 Дополнительное руководство не приводится.
D.12 Поддержание эффективности процесса
D.12.1 Общая информация
D.12.1.1 Для обеспечения непрерывности получения от стерилизационного процесса требуемого уровня обеспечения стерильности необходимо оценивать любые изменения продукта и упаковки, изменения процесса и оборудования. Рекомендуется использование комплексной системы управления изменениями продукта и процесса.
Одним из параметров, обычно наблюдаемых для убеждения в постоянной способности стерилизовать загрузку, является бионагрузка продукта. Бионагрузка подлежит мониторингу в соответствии с ISO 11737-1. Если наблюдаются значительные изменения в количестве и/или видах микроорганизмов, следует оценить их возможное влияние на способность стерилизационного процесса надлежащим образом стерилизовать нагрузку.
В учреждении здравоохранения рекомендуется установить периодический пересмотр данных об эффективности процесса очистки/деконтаминации с целью подтверждения того, что процесс остается эффективным обеспечивает адекватное снижение бионагрузки при подготовке продукта к последующему процессу стерилизации. Деконтаминированные медицинские изделия подлежат визуальному контролю чистоты перед окончательной стерилизацией. Загрязненные медицинские изделия не должны стерилизоваться. Должны иметься процедуры, гарантирующие, что медицинские изделия будут надлежащим образом деконтаминированы до их стерилизации (см. ISO 17664 и ISO 15883).
Для учреждений здравоохранения очень важно получение от изготовителей подробных инструкций по повторной обработке, специфических для каждого медицинского изделия, например, по их разборке. Должны иметься политики и процедуры, гарантирующие, что медицинские изделия надлежащим образом деконтаминированы.
D.12.1.2 Необходима документированная программа калибровки приборов, используемых для управления и мониторинга стерилизационного процесса, чтобы гарантировать, что процесс продолжает выдавать продукт с требуемым уровнем обеспечения стерильности и неизменными рабочими характеристиками.
D.12.2 Обслуживание оборудования
D.12.2.1 Для того чтобы быть эффективными, работы по профилактическому обслуживанию оборудования должны выполняться в соответствии с определенным графиком, основанном на рекомендациях изготовителя и наблюдениях за работой оборудования. Процедуры должны быть документированы, а обслуживающий персонал должен быть обучен.
Оборудование, которое должно обслуживаться и калиброваться на рутинной основе, может включать в себя (но не ограничиваться перечисленным) следующие узлы зоны предварительного кондиционирования, камеры и зоны аэрации:
a) прокладки и уплотнения;
b) приборы мониторинга;
c) устройства мониторинга ЭО (например, концентрация в окружающей среде и/или в камере);
d) блокировки безопасности дверей;
e) предохранительные клапаны или разрывные диски;
f) фильтры (для периодической замены);
g) испарители/выпариватели;
h) рециркуляционные системы рубашек камер;
i) системы рубашек камер;
j) звуковые и визуальные индикаторы тревоги;
k) оборудование, связанное с датчиками температуры и влажности;
I) бойлерные системы, обеспечивающие пар и тепло;
m) оборудование для откачки (вакуумные насосы);
n) весовое оборудование;
о) клапаны;
р) датчики/преобразователи давления;
q) таймеры;
r) записывающие устройства;
s) циркуляционные системы для газа/воздуха.
D.12.2.2 Стерилизационное оборудование, не прошедшее калибровку или неправильно обслуживаемое, может выдавать неверные записи процессных параметров во время выполнения стерилизационного цикла. Если эти данные используются для выпуска продукта, это может привести к выпуску загрузок, которые не были адекватно стерилизованы.
D.12.2.3 Дополнительное руководство не приводится.
D.12.2.4 Необходимо периодически пересматривать записи об обслуживании и выполнять необходимые настройки/регулировки, о которых сигнализируют данные.
D.12.3 Повторная аттестация
D.12.3.1 Пересмотр результатов аттестации установленного оборудования должен включать в себя подтверждение приемлемого статуса калибровки оборудования, управления и мониторинга. Программы контроля изменений и профилактического обслуживания должны показывать, что в стерилизационном оборудовании не было сделано никаких модификаций или значительных изменений, способных повлиять на процесс.
D.12.3.2 Пересмотр результатов аттестации функционирующего оборудования должен включать в себя оценку работы оборудования и инженерные изменения, выполненные в течение года, для обеспечения уверенности, что результаты первой аттестации функционирующего оборудования остаются действительными [см. рисунок D.1 (приложение D)].
Для достижения этой цели используется общая практика выполнения периодической повторной аттестации оборудования, которая должна включать в себя:
a) пересмотр состояния оборудования по данным аттестации установленного оборудования;
b) оценку трендов рабочих характеристик оборудования;
c) профили температуры и относительной влажности в зонах предварительного кондиционирования (если используются);
d) температурный профиль камеры;
e) температурные профили зон аэрации (если используются).
Эти действия при повторной аттестации должны показать отсутствие заметных изменений в работе предварительного кондиционирования (если используется), камеры или зон аэрации по сравнению с прошлой аттестацией.
Примечание - Для больших помещений предварительного кондиционирования или аэрации, содержащих много стерилизационных загрузок, охват повторной аттестации можно сузить, если в оборудовании не произошло никаких значительных изменений. Обоснование снижения охвата повторной аттестации должно быть документировано.
D.12.3.3 Пересмотр аттестации эксплуатируемого оборудования должен включать в себя оценку сохранения валидности стерилизационного процесса для определенного продукта (продуктов).
Подлежащие рассмотрению факторы должны включать в себя (но не ограничиваться перечисленными) следующее:
a) пересмотр статуса аттестации установленного оборудования;
b) пересмотр статуса аттестации функционирующего оборудования;
c) подтверждение, что в конструкции продукта, производственных и упаковочных материалах, в УКП, в перечне поставщиков, производственных площадок или заводов, в конфигурации загрузки или в производственных процессах не произошло значительных изменений, способных повлиять на стерильность продукта;
d) подтверждение, что не происходило значительных изменений в бионагрузке продукта и/или в сопротивляемости бионагрузки продукта процессу стерилизации, которые могли бы ухудшить способность процесса стерилизации стерилизовать продукт с требуемым уровнем обеспечения стерильности;
e) подтверждение, что индивидуальные процессы стерилизации работали в рамках спецификаций со времени последней аттестации;
f) подтверждение, что в стерилизационном процессе не произошло изменений, которые могли бы повлиять на стерильность продукта;
g) пересмотр неудачных стерилизаций биологических индикаторов или УКП, которые возникали при соблюдении процессных спецификаций, для определения необходимости в повторной аттестации.
Основываясь на этом пересмотре, специалист по стерилизации должен определить объем требуемой физической или микробиологической повторной аттестации. Пересмотр и решение должны быть документированы.
Существует три возможности повторной аттестации как результат пересмотра:
- полная аттестация - состоящая из физической и микробиологической аттестации. Это может потребоваться в определенных ситуациях, например, после внесения заметных изменений в конструкцию продукта/упаковки или в конфигурацию (создание нового условия "худшего случая"), в процесс или в оборудование и/или питающие среды;
- отсутствие необходимости в физической и микробиологической аттестации - в обстоятельствах, когда в конструкцию продукта/упаковки или в конфигурацию, в процесс, оборудование и/или питающие среды не вносилось никаких изменений, при наличии приемлемой работы камеры и приемлемого инженерного пересмотра, когда рутинный процесс стерилизации в прошедший период работал надежно, можно использовать профессиональное суждение для заключения о том, что до следующего пересмотра нет необходимости выполнять физическую и микробиологическую повторную аттестацию;
- уменьшенные физическая и микробиологическая аттестации - это может оказаться необходимым в определенных ситуациях, например, для перепроверки продолжающейся пригодности сопротивляемости внутреннего УКП в загрузке продукта по отношению к сопротивляемости бионагрузки продукта, или после определенного периода времени, для получения доказательств, что не произошло никаких нежелательных изменений по сравнению с предыдущими аттестационными исследованиями. Обычно это включает в себя как минимум один фракционный или половинный цикл экспозиции с измерениями температуры и влажности. Фракционные циклы в разработочной камере могут также использоваться для поддержки программы повторной аттестации, но повторная аттестация производственной камеры должна выполняться только в производственной камере;
Рекомендуется, чтобы цикл микробиологической аттестации эксплуатируемого оборудования и измерения температуры и влажности (микробиологическая плюс физическая аттестация эксплуатируемого оборудования) выполнялись по меньшей мере раз в два года для убеждения, что пересмотр документации выявил и зафиксировал любые изменения в продукте или в стерилизационном процессе.
Повторная аттестация может также включать в себя верификацию того, что если менялись спецификации стерилизационного процесса, то повторная аттестация процесса включала подтверждение, что продукт соответствует допустимым пределам по остаткам ЭО, указанным в ISO 10993-7.
Во всех вышеперечисленных случаях важно документировать принятые решения наряду с обоснованиями этих решений, и определять план последующих пересмотров или переаттестаций.
|
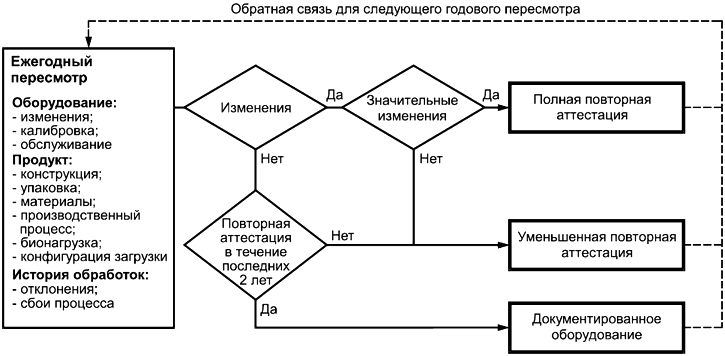
Примечание - Когда валидируется более одной конфигурации, это должно быть отражено в любом действии повторной аттестации
Рисунок D.1 - Дерево решений для повторной аттестации
D.12.3.4 Повторная аттестация выполняется для подтверждения того, что кумулятивный эффект от мелких изменений не ухудшил эффективность стерилизационного процесса.
Повторная аттестация может включать в себя верификацию того, что соблюдаются допустимые для продукта остатки ЭО согласно ISO 10993-7.
Важно выполнять формальную оценку необходимости в повторной аттестации стерилизационного процесса по меньшей мере ежегодно для обеспечения убеждения в том, что отсутствовали случайные изменения процесса и для демонстрации того, что первоначальная валидация остается валидной.
Программа повторной аттестации должна определить приемлемые диапазоны и уровни вариабельности характеристик, необходимые для поддержания валидности результатов первичной валидации из года в год.
D.12.3.5 Для определения причин несоответствий должно проводиться расследование. Влияние несоответствия на валидность повторной аттестации должно быть оценено, а обоснование принятого решения (решений) должно быть документировано. Дальнейшие действия по повторной аттестации должны выполняться под наблюдением надлежащей системы обеспечения качества.
D.12.4 Оценка изменений
D.12.4.1 События, которые могут потребовать повторной аттестации, включают в себя (но не ограничиваются перечисленными) следующее:
a) обширные ремонты стерилизатора и изменения (замена систем и органов управления, крупная реконструкция или установка новых важных компонентов);
b) неожиданные отрицательные показатели стерильности в рутинной стерилизации;
c) изменения в продукте;
d) изменения в упаковке;
e) модификация стерилизующего агента и/или его представления;
f) изменения в представлении продукта для стерилизации или конфигурации загрузки;
g) изменения плотности загрузки.
Важно убедиться, что контрольная загрузка, используемая в любой повторной аттестации, учитывает изменения, которые могли быть сделаны для обеспечения представительности контрольной загрузки по отношению к пересматриваемым продукту или конфигурации.
D.12.4.2 Могут понадобиться исследования при повторной аттестации, если были сделаны изменения в материалах, местах размещения производственных площадок или в методах обработки, которые могут повлиять на популяцию и/или устойчивость бионагрузки продукта. Исследования должны продемонстрировать, что популяция и устойчивость бионагрузки продукта не увеличились до уровня, который мог бы потенциально сделать недействительной пригодность внутреннего УКП или отрицательно повлиять на достижение требуемого уровня обеспечения стерильности.
D.12.4.3 Дополнительное руководство не приводится.
D.12.4.4 Дополнительное руководство не приводится.
D.12.4.5 Дополнительное руководство не приводится.
D.12.5 Оценка эквивалентности
D.12.5.1 Оценка эквивалентности процесса - это метод, используемый для демонстрации того, что двумя или более единицами или комплектами оборудования выполняется одинаковый валидированный процесс стерилизации. При этом не требуется, чтобы оборудование было физически идентичным. Даже если параметры, обеспечиваемые оборудованием, не идентичны статистически, выполняемый процесс все равно может быть эквивалентен, если все параметры способны обеспечивать процесс в определенных и валидированных процессных пределах (см. AAMI TIR 28 [26]).
Оценка эквивалентности процесса, выполняемого несколькими единицами оборудования, предназначена для уменьшения объема испытаний, необходимых для аттестации процесса. Процесс стерилизации должен быть валидирован в одной камере. Остальное оборудование может пройти сокращенную аттестацию эксплуатируемого оборудования, если это оборудование прошло аттестации установленного и функционирующего оборудования (9.2 и 9.3). Оборудование, выполняющее стерилизационный процесс, обычно состоит из камеры или комнаты и дополнительных систем управления. Оборудование стерилизационного процесса может находиться в пределах выделенного помещения для обработки или распределяться между несколькими помещениями. Это оборудование может использоваться независимо для создания одинаковых условий процесса и может иметь точно такую же конструкцию или отличаться по размерам или по объему вспомогательного оборудования.
Эквивалентность процесса может быть установлена с помощью анализа процессных данных в сочетании с микробиологической оценкой. Процессные данные должны продемонстрировать, что оборудование-кандидат работает в допустимом диапазоне контролируемых параметров (т.е. валидированные процессные параметры могут быть надежно применены к продукту). Анализ данных должен подтвердить, что процесс работает в пределах определенных для него допусков на валидированные параметры. Микробиологическая оценка должна подтвердить, что требуемый уровень обеспечения стерильности достигается.
D.12.5.2 Критерии для оценки эквивалентности процесса
Эквивалентность процесса может быть установлена вне зависимости от того, установлено ли оборудование в том же помещении или в иных помещениях. Критерии, которые должны быть соблюдены до установления программы оценки эквивалентности, следующие:
a) выполнение полной валидации стерилизационного процесса как минимум в одной существующей системе в соответствии с требованиями раздела 9;
b) выполнение аттестаций установленного и функционирующего оборудования, демонстрирующих документально, что все оборудование было установлено в соответствии с требованиями инженерных спецификаций, и что оно работает в соответствии с теми же требованиями;
c) выполнение определения процесса для включения разрешенных допусков и документирования всех фаз процесса;
d) выполнение анализа всех процессных данных с валидированными допусками для оборудования-кандидата и оригинального оборудования.
D.12.5.3 Определение эквивалентности процесса
Эквивалентность стерилизационного процесса, выполняемого одной единицей оборудования, процессу, выполняемому другой единицей, может быть установлена путем сравнения данных, полученных при выполнении одного и того же валидированного процесса на том и другом оборудовании. Это сравнение должно включать в себя оценку способности оборудования с хорошей воспроизводимостью соблюдать желаемые процессные параметры при обработке нормальной производственной загрузки. Данные, полученные в ходе аттестации эксплуатируемого оборудования, также могут быть использованы. Параметры и допуски должны быть теми, которые были ранее валидированы в ходе аттестации эксплуатируемого оборудования с выполнением процесса на оригинальном оборудовании. Оценка эквивалентности должна включать в себя анализ и оценку процесса наряду с микробиологической оценкой.
D.12.5.4 Анализ и оценка процесса
Выполняется анализ процессных данных, полученных из валидированного процесса, выполненного на оборудовании-кандидате и оригинальном оборудовании. Процессные данные должны быть получены от оборудования-кандидата. Эти данные должны быть сравнены с пределами параметров для данного специфического стерилизационного процесса и результатами, полученными из аттестации эксплуатируемого оригинального оборудования. Пределы параметров - это те пределы, которые были установлены в ходе первичной валидации стерилизационного процесса (включая все требования к процессу, описанные в настоящем стандарте), выполняемого в существующем оборудовании. Спецификации, критерии приемлемости и паллета с загрузкой в определенной конфигурации должны быть такими же, как те, что были определены в первичной аттестации эксплуатируемого оборудования. Фактические параметры, подлежащие оценке при определении эквивалентности, обычно представляют собой часть всей спецификации процесса. Выбранные параметры и обоснование их выбора должны быть документированы. В этой оценке могут быть использованы статистические методы, оценивающие как центральные тенденции данных испытаний, так и степень вариабельности данных. Примеры подхода с использованием статистического анализа представлены в AAMI TIR15 [24]. Примеры эти иллюстративны, и предназначены для обеспечения руководства по статистическим расчетам, требованиям к нормальности распределения, и по шагам, которые следует предпринимать, если данные не проходят проверку на нормальность. Если анализ процесса и оценка не отвечают установленным критериям приемлемости, то демонстрация эквивалентности процесса невозможна.
D.12.5.5 Оценка зон предварительного кондиционирования и аэрации
Критерии для установления эквивалентности процесса используются и для оценки зон предварительного кондиционирования и аэрации, за исключением того, что влажность для зон аэрации обычно не учитывается. Должна быть выполнена оценка, сравнивающая профили температуры загрузки, влажности в окружающей среде каждой зоны. Как минимум, должны оцениваться однородность температуры и влажности внутри загрузки и связь этого единообразия с соответствующими уставками и записанными контрольными переменными для зон. Если единицы оборудования используют разные уставки или имеют разные контрольные пределы, то доказать их эквивалентность может оказаться невозможно. Эквивалентность процесса для процессов предварительного кондиционирования или процессов аэрации может быть установлена, если после анализа рабочих данных можно сделать заключение что, условия внутри загрузки соответствуют пределам параметров (например, распределение температур, остаточные уровни и т.п.) в конце предварительного кондиционирования или в конце аэрации. Остаточные уровни ЭО в продукте после стерилизации должны верифицироваться в аэрационной комнате/помещении/ камере/ячейке-кандидате.
D.12.5.6 Оценка работы стерилизационной камеры
Это оценка, сравнивающая применение процессных параметров к загрузке в оборудовании-кандидате с данными, полученными из аттестации эксплуатируемого оригинального оборудования или из выполненных производственных циклов. Критические параметры процесса и загрузки, подлежащие сравнению, должны быть определены для стерилизационного процесса до выполнения оценки. Эти параметры индивидуальны для каждого стерилизационного процесса, но могут включать в себя следующее:
a) параметры загрузки:
1) температура продукта - достигнутые температуры и их распределение внутри загрузки во время выдержки ЭО;
2) влажность продукта - достигнутая влажность и ее распределение внутри загрузки к концу кондиционирования.
b) параметры процесса:
1) влажность в камере в выбранные периоды времени во время выполнения цикла (например, начало и/или конец кондиционирования). Этот параметр может быть измерен непосредственно или может базироваться на росте давления из-за инжекции пара;
2) температура в камере в выбранные периоды времени во время выполнения цикла (например, конец кондиционирования или во время экспозиции ЭО);
3) концентрация газа ЭО в камере в выбранные периоды времени во время выполнения цикла (если измеряется), или рост давления ЭО, или вес газа;
c) к другим параметрам процесса, которые можно рассматривать, относятся:
1) глубина вакуума и скорость откачки (/время) в выбранные периоды времени во время выполнения цикла;
2) время увлажнения и скорость впуска пара (/время);
3) температура инжекции ЭО и скорость (/время), а также объем использованного ЭО (вес, концентрация или давление);
4) скорость впуска воздуха или азота (/время).
Анализы процессных данных используются для того, чтобы показать, эквивалентны процессы или нет по своей способности соблюдать существующие пределы параметров и любые иные дополнительные критерии приемлемости. Полученные данные должны быть проанализированы и скомпилированы в формат, который позволит использовать их в дальнейших определениях эквивалентности процесса.
D.12.5.7 Микробиологическая оценка
В ходе микробиологической оценки выполняются фракционный или половинный цикл для демонстрации того, что стерилизационный процесс способен обеспечить определенный минимальный уровень обеспечения стерильности продукта во всех оцениваемых единицах или комплектах оборудования.
Примечание - Если цикл, используемый во время анализа процесса, был фракционным или половинным и включал микробиологический мониторинг, то его данные также могут использоваться для этой оценки.
В дополнение к обеспечению специфицированного уровня обеспечения стерильности продукта в число добавочных факторов, подлежащих оценке, включаются любые изменения мест расположения стерилизатора или мест расположения производственных мощностей, которые могли бы оказать влияние на уровень бионагрузки продукта, предоставляемого для стерилизации. Увеличение расстояний между предприятием-изготовителем и стерилизующим учреждением может привести к более высоким уровням бионагрузки, особенно если продукт сам способствует микробиологическому росту. Разница в окружающих средах различных производственных площадок тоже может приводить к изготовлению продуктов с более высокими уровнями бионагрузки или с более высокой ее устойчивостью, чем аттестованные ранее, даже если продукт не способствует микробиологическому росту. Еще одна проблема, подлежащая оценке при транспортировке продукта между площадками - это разница в условиях транспортировки, таких, как длительность перевозки и сезонные эффекты (температура, влажность и т.п.). Если нужно, следует выполнить имитацию доставки/транспортировки, выдерживая продукт при определенных условиях.
D.12.5.8 Оценка результатов
Результаты оценки должны определить, эквивалентно ли работают отдельные единицы или комплекты оборудования. Если они эквивалентны, тогда требование о сокращенных микробиологических исследованиях удовлетворяется за счет испытаний, которые уже были выполнены, и ни в какой дальнейшей аттестации нет необходимости. Если же выводы анализа процесса или микробиологической оценки говорят о том, что процессы неэквивалентны, то процесс должен быть объявлен "неэквивалентным", после чего должна быть выполнена полная аттестация эксплуатируемого оборудования.
D.12.5.9 Поддержание эквивалентности
Поддержка эквивалентности должна включать в себя обзор изменений для каждой единицы оборудования, каждого производственного процесса, загрузки продукта и стерилизационного процесса, чтобы убедиться, что эти изменения не повлияли отрицательно на общее определение эквивалентности. Этот обзор должен выполняться перед внесением изменений и должен быть частью процесса контроля изменений. Если какой-то процесс не проходит периодический обзор эквивалентности, он должен быть исключен из списка эквивалентности и заново аттестован.
D.12.5.10 Документация
Все решения, относящиеся к выходным данным анализа, определяющие, может ли оборудование-кандидат быть заявлено как "эквивалентное" в отношении существующего оборудования для стерилизационного процесса, должны быть документированы. Эта документация должна включать в себя как минимум следующее:
a) полная спецификация оборудования-кандидата, описывающая оборудование, его эксплуатационные спецификации с допусками, ссылающаяся или содержащая в себе перечень применимых рабочих процедур, процедур калибровки и графики обслуживания. Эта спецификация должна включать в себя или ссылаться на текущую аттестацию установленного оборудования, как указывает настоящий стандарт;
b) свидетельство или оценка способности оборудования выполнять рассматриваемый процесс. Свидетельство или оценка должны включать в себя или ссылаться на текущую аттестацию функционирующего оборудования;
c) результат сравнения оборудования-кандидата с существующим валидированным оборудованием процесса. Это сравнение должно четко продемонстрировать, что все основные системы и параметры были подвергнуты оценке, в том числе с помощью статистического анализа (если используется);
d) свидетельство или оценка условий продукта во время обработки в оборудовании-кандидате для демонстрации эквивалентности существующему процессу;
e) результат оценки любых дополнительных факторов, способных изменить летальность стерилизационного процесса, если это необходимо;
f) документированное заключение о том, что оборудование-кандидат эквивалентно оборудованию, указанному в текущих валидационных исследованиях, определяющих достижение специфицированного уровня обеспечения стерильности продукта. Это заключение должно включать в себя или ссылаться на все дополнительные испытания, выполненные для дополнения существующих валидационных исследований, и любые иные испытания, выполненные для подтверждения или аттестации рутинного выпуска продукта из существующего валидированного цикла (например, проверка остатков, функциональные испытания на трех первых лотах и т.п.);
g) одобрение специалистом по стерилизации и иными лицами, как этого требует нормальный контроль изменений или практики управления документацией, принятые в организации;
h) перечень применимых к стерилизатору рабочих процедур и спецификаций, выпущенных или измененных для авторизации применения оборудования-кандидата для рутинной обработки продукта.
D.12.5.11 Продукт
D.12.5.11.1 Семейство продукта
Семейство продукта - это набор продуктов, определенных как схожие или эквивалентные друг другу для целей валидации. Хотя семейства продуктов могут использоваться и по другим причинам (остатки ЭО, бионагрузка или биосовместимость), для ЭО-стерилизации семейство продукта обычно означает продукты, которые были сгруппированы вместе с целью определить, был ли достигнут требуемый уровень обеспечения стерильности в ходе микробиологических испытаний аттестации эксплуатируемого оборудования.
ЭО семейство продукта может состоять из различных комбинаций схожих продуктов. Например, семейство продукта может содержать набор катетеров, отличающихся только размерами, или это может быть набор продуктов, которые были изготовлены в одинаковой среде из одинаковых материалов. Когда продукты группируются в семейства, важно, чтобы они группировались на базе обоснования, подходящего для ЭО-процесса.
Использование семейств продуктов упрощает процесс валидации, поскольку все продукты в семействе могут быть определены как представляющие эквивалентную или меньшую нагрузку на стерилизационный процесс, чем продукт-представитель или внутреннее УКП. Семейство продуктов может быть представлено одним продуктом "худшего случая" (зачастую называемым "мастер-продуктом"); все семейство рассматривается как эквивалентная нагрузка на стерилизационный процесс, или представляется УКП продукта (внутренним УКП).
В дополнение к семействам продуктов, категории обработки также могут рутинно использоваться в ЭО-стерилизации после завершения аттестации эксплуатируемого оборудования. Категория обработки - это набор семейств ЭО-продуктов, которые могут быть несхожими в деталях, используемых для установления семейства продукта, таких, как материал, конструкция или упаковка, или изготовитель, но каждое семейство продуктов внутри категории обработки должно быть аттестовано в общем для них стерилизационном процессе. Как пример, набор продуктов (внутривенные наборы) может составлять семейство продукта и может войти в категорию обработки, которая включает в себя отдельный набор продуктов (например, семейство шприцев). Общностью внутри категории обработки может быть УКП, которое представляет микробиологическую нагрузку для продуктов в этой группе. Все продукты в этой категории обработки должны представлять эквивалентную или меньшую нагрузку на стерилизационный процесс при сравнении с продуктом "худшего случая", представительным членом, или при сравнении с УКП, располагаемым внутри стерильной барьерной системы продукта.
Пересмотр эквивалентности продукта может выполняться внутри каждого семейства продуктов или внутри категории обработки. В качестве альтернативы для аттестационного исследования может быть выбран продукт "худшего случая" или представительный член семейства. В последующих параграфах рассматриваются различные аспекты оценки продуктов.
D.12.5.11.2 Определение отрицательного влияния на продукт
Перед определением того, могут ли продукт-кандидат или упаковочная система быть приняты в семейство продуктов или в категорию обработки, следует определить, останутся ли продукт-кандидат или упаковочная система функциональными и эффективными. Система для оценки этих аспектов должна рассматриваться проектированием или процессом контроля изменений. Должно быть уделено внимание рассмотрению функциональности, целостности, стабильности, биосовместимости и остаткам, с особым упором на определение тех воздействий, которые процесс стерилизации может оказать на лекарственные средства, которые могут быть включены в состав изделий или их компонентов. Для продуктов, содержащих определенные типы законченных (готовых) компонентов (например, наборы с лекарствами), изготовитель должен рассмотреть нормативные требования в отношении безопасности и эффективности этих компонентов в дополнение к тому влиянию, которое стерилизационный процесс может оказать на срок годности рассматриваемого продукта.
ЭО-процесс, для которого продукт будет испытываться, должен составлять представительную нагрузку на продукт и его упаковочную систему. Документация должна отражать, насколько нагрузка этого процесса отличается от номинального процесса, а аттестация продукта должна демонстрировать, что все эти параметры подходят для приемки продукта.
Продукт-кандидат и его упаковка должны быть оценены для определения воздействия на продукт остаточных уровней ЭО и любые изменения в этом отношении должны оцениваться с точки зрения воздействия на выпуск продукта. В качестве руководства по выполнению этой оценки следует воспользоваться ISO 10993-7.
D.12.5.11.3 Определение влияния конструкции продукта
Конструкция продукта-кандидата должна быть тщательно рассмотрена на наличие любых изменений или отличий, которые могли бы представить собой большее препятствие для проникновения ЭО, тепла и/или влаги, чем существующий продукт или УКП. Примеры возможных изменений включают в себя более длинные каналы, добавление перегородок/крышек или увеличение количества матовых поверхностей или увеличение плотности продукта.
Пересмотр конструкции продукта по отношению к испытанию функциональности оригинального продукта необходим, чтобы убедиться, что изменения не имеют отрицательного влияния на функции продукта.
Примечание - Эта оценка обычно не включает в себя герметически закрытые зоны продукта, которые не могут быть оставлены незащищенными во время использования по назначению. Примерами могут служить такие изделия, как заваренные, полые, литые части или запаянные трубки.
D.12.5.11.4 Определение влияния материалов и характеристик продукта
Характеристики продукта-кандидата должны быть тщательно проверены на наличие любых отличий, которые могли бы оказать потенциальное влияние на бионагрузку продукта, например, на отличающиеся способы производства, различия в расположении производственных помещений, в самих помещениях, в сырьевых материалах и их источниках. Материалы конструкции должны быть пересмотрены для получения убеждения в том, что продукт не будет содержать уровней ЭО, превышающих нормативные предельные пороги.
D.12.5.11.5 Определение влияния стерильной барьерной системы
Стерильная барьерная система продукта-кандидата должна быть тщательно проверена на наличие любых факторов, которые могли бы представить собой большее препятствие для проникновения ЭО, тепла и/или влаги. В число этих факторов могут входить изменение пористости вентилирующего материала, уменьшение площади вентилирующей зоны, сжатие вентилирующей зоны или иные изменения, которые могут сделать продукт-кандидат большей нагрузкой для стерилизационного процесса по сравнению с существующим продуктом или внутренним УКП продукта. В дополнение к сказанному должно быть оценено влияние изменений стерильной барьерной системы на бионагрузку продукта и на остаточные уровни ЭО.
D.12.5.11.6 Определение влияния конфигурации загрузки
Конфигурация загрузки продукта-кандидата должна быть тщательно проверена на наличие любых изменений, способных повлиять на термодинамический отклик (термодинамическую реакцию) на процесс стерилизации. Эти изменения могут быть дополнительными слоями эластичной обертки или иной конфигурацией паллеты, изменением размера загрузки, изменениями общей плотности загрузки или любыми иными изменениями, которые могут сделать продукт-кандидат большей нагрузкой для стерилизационного процесса.
D.12.5.11.7 Заключения об оценке возможности принятия продукта
Если результаты письменного технического пересмотра показывают, что продукт-кандидат и существующие продукты или внутренние УКП схожи, а отличия между ними определены как несущественные или как представляющие нагрузку меньшую, чем имеющийся валидированный продукт или внутреннее УКП, то продукт-кандидат может быть включен в семейство продукта или в категорию обработки без дальнейших исследований. Если для пересмотра использовался AAMI TIR28:2009 [26] (приложение А), это решение должно быть виртуально поддержано ответами "нет" на все вопросы. Обоснование такого решения должно выдаваться специалистом по стерилизации и должно документироваться. Если технический пересмотр показывает, что продукт-кандидат имеет потенциал большей нагрузки для стерилизационного процесса, чем текущий валидированный продукт или внутреннее УКП, тогда требуются дальнейшие исследования. Если продукт-кандидат определен как представляющий большую нагрузку для стерилизационного процесса, это означает, что он не отвечает требованиям к включению в существующее семейство продукта или в категорию обработки и в этом случае необходимо выполнение полной аттестации эксплуатируемого оборудования. Эта аттестация может:
a) установить новое семейство продукта или категорию обработки с продуктом-кандидатом в качестве представительного продукта;
b) установить новое внутреннее УКП для процесса стерилизации;
c) установить, что продукт-кандидат эквивалентен текущему валидированному мастер-продукту;
d) установить новый стерилизационный процесс для продукта-кандидата.
D.13 Руководство к приложению А. Определение степени летальности стерилизационного процесса. Подход биологического индикатора/биологической нагрузки
D.13.1 [А.1] Общая информация
D.13.1.1 [A.1.1] Эта статья дает дополнение к информации, приведенной в приложениях A и D.8-D.9. Поскольку подход биологического индикатора/бионагрузки и подход "массовой гибели" используют много одинаковых процедур, часть текста в этой статье дублирует текст в D.14 (приложение D).
Комбинированный подход биологического индикатора/бионагрузки базируется на использовании биологического индикатора или иного внутреннего УКП с популяцией и устойчивостью равной или большей, чем устойчивость бионагрузки. Этот метод подходит тогда, когда доступны достаточные данные о бионагрузке из программы мониторинга бионагрузки, чтобы продемонстрировать, что устойчивость бионагрузки продукта наряду с популяцией могут быть достаточно полно представлены в ходе валидационных исследований по достижению уровня обеспечения стерильности продукта 10.
Примечание - Этот метод может потребовать использования биологических индикаторов или иных внутренних УКП с популяцией меньшей, чем 10.
Относительная устойчивость и популяция внутреннего УКП должны сравниваться с устойчивостью и популяцией бионагрузки продукта. Уменьшение логарифма внутреннего УКП может использоваться для расчета уровня обеспечения стерильности, достигнутого для бионагрузки продукта с наибольшим сопротивлением процессу стерилизации.
Если рассматривается такой случай, то для демонстрации эффективности процесса для продукта могут использоваться данные о снижении логарифма спор, полученные в исследовании летальности для биологического индикатора. Если данные сгенерированы с использованием метода перебора, снижение логарифма спор также может быть спрогнозировано из полученных данных кривой выживаемости. Пользователь должен помнить о том, что минимальное время цикла, вычисляемое при этом подходе, не является само по себе адекватным для валидации процесса стерилизации. Необходима демонстрация способности поддерживать параметры процесса в определенных пределах во время предложенного полного цикла.
Если бионагрузка продукта испытывается через короткие промежутки времени и показывает единообразие, то для определения процесса и/или микробиологической аттестации эксплуатируемого оборудования можно использовать комбинированный метод биологического индикатора/бионагрузки.
Определения летальности процесса: микробиологическая летальность, приложенная к продукту после его экспозиции отдельному процессу стерилизации, может быть рассчитана на основе величины специфицированного микроорганизма. Поскольку микроорганизмы в общем случае погибают со скоростью, приблизительно логарифмической для данного процесса, единица времени экспозиции газу ЭО может быть найдена как результирующая гибель 90% популяции микроорганизма вне зависимости от размера популяции. Каждая из этих единиц времени выражается как величина
для микробиологического контаминанта продукта при экспозиции специфицированному стерилизационному процессу.
Величина специфицированного микроорганизма и микробиологическая летальность, применяемая к продукту при экспозиции специфицированному процессу стерилизации, может быть рассчитана с использованием результатов от одного или двух широко применяемых методов. Первый метод (подсчет, перебор) состоит из перебора или физического подсчета выживших, а второй (фракционный негативный анализ) использует данные "рост/нет роста" во время выполнения фракционных циклов. Любой из этих методов может быть использован для приложений А или В. Величины
могут рассчитываться путем использования результатов от фракционных циклов и уравнений, описанных в ISO 11138-1 и ISO 14161.
Может потребоваться рассмотрение влияния времени инжекции ЭО и времени откачки после экспозиции для обеспечения большей точности в определении скорости достижения летальности. Это влияние может оказаться наиболее значительным, когда время инжекции ЭО и пост-экспозиции по длительности сравнимы с длительностью фазы экспозиции.
Вне зависимости от того, какой метод использовался, предполагается, что:
a) популяция микроорганизмов гомогенна;
b) параметры процесса постоянны от цикла к циклу;
c) существует полулогарифмическая связь с выжившими;
d) микроорганизмы, пережившие процесс и неэкспонированные микроорганизмы одинаково реагируют на восстановительную среду;
e) все методы микробиологических испытаний (проверка на стерильность, подсчет и т.д.) должны быть валидированы в соответствии с ISO 11737-1 и ISO 11737-2.
Подсчет: подсчет (перебор) состоит из экспонирования внутреннего УКП фракционному циклу, извлечения нагрузки и подсчета выживших на образцах или в биологическом индикаторе. Подсчет выживших может быть использован в разработке кривой выживания и величины . Величина
затем рассчитывается с помощью линейной регрессионной модели. См. также ISO 14161:2009.
Фракционный негативный анализ: фракционный негативный анализ предусматривает исполнение стерилизационных циклов, в которых инактивируются некоторые, но не все биологические индикаторы. Сюда входят:
a) процедура Холкомба-Спирмана-Карбера (HSK);
b) ограниченная процедура Холкомба-Спирмана-Карбера (LHSK);
c) процедура Стамбо-Мерфи-Кохрейна (SMC).
См. также ISO 14161:2009.
Количество образцов (проб): количество образцов зависит от используемого метода, а также от того, распределены ли образцы по всей загрузке или сосредоточены в одном месте. Использование одного места размещения может улучшить постоянство результатов по всем образцам; однако такой подход может не представить точку "худшего случая" в камере, если только не было выполнено экстенсивное картирование в каждой камере и при каждой возможной конфигурации загрузки.
При оценке результатов следует обратить внимание на то, было ли обеспечено условие, чтобы разночтения между количеством выживших микроорганизмов в повторяющихся контрольных заражениях имели причиной в большей степени случайный разброс внутри популяции, чем вариативность условий экспозиции.
Более детальные рекомендации по выбору количества биологических индикаторов см. в таблице С.З (приложение С). В дополнение к этому см. ISO 11138-1 и ISO 14161, чтобы убедиться, что соблюдено требование к минимально допустимому количеству образцов.
Для того чтобы получить желаемый результат, может понадобиться укоротить следующие после экспозиции фазы цикла.
D.13.1.2 [A.1.2] Информация относительно инкубационного периода для биологических индикаторов дается в ISO 14161:2009, пункт 12.3.
D.13.1.3 [A.1.3] Можно сочетать подходы подсчета и фракционного негативного анализа при определении летальности или величин . Два подхода основываются на разных методах расчета. Пользователи обычно выбирают один из них для определения летальности процесса.
D.13.2 [A.2] Процедура
Место внутри продукта, в котором труднее всего достичь стерильности, может быть не только среди зон, имеющих пониженную способность к проникновению стерилизующего агента, но также и среди тех зон, которые с наибольшей вероятностью могут иметь наиболее значительный объем бионагрузки. Для установления наиболее подходящего места размещения биологической нагрузки должен быть выполнен пересмотр продукта. Этот пересмотр должен быть документирован. См. также ISO 14161:2009, пункт 7.2.2.
Подлежащими рассмотрению аспектами являются:
a) длина и внутренний диаметр каналов, а также способность стенки медицинского изделия обеспечивать диффузию ЭО;
b) способность к абсорбции разных частей продукта и материала;
c) веса и плотности изделий;
d) конфигурация загрузки, особенно для загрузок со смешанными продуктами.
См. также ISO 11138-1 и ISO 14161, чтобы удовлетворить требование о минимальном количестве образцов.
ISO 14161:2009 (приложение A) обеспечивает дополнительное руководство по применению взаимосвязи между биологическим индикатором и бионагрузкой продукта в подходе "биологический индикатор/бионагрузка".
Важно, чтобы внутреннее УКП обеспечивало равную или большую нагрузку, чем бионагрузка, имеющаяся в наиболее труднодоступной части продукта. См. D.7.1.6 (приложение D) для получения информации о разработке УКП, и D.8.6 (приложение D) для получения информации об определении пригодности внутреннего УКП, размещаемого внутри стерильной барьерной системы продукта.
Параметры, в первую очередь влияющие на летальность - это время экспозиции, концентрация ЭО, влажность и температура. Если делается настройка остальных параметров, то кроме времени экспозиции, следует оценить общий эффект на работу цикла, поскольку такая настройка может не достичь желаемого результата из-за взаимосвязанности параметров. Например, результатом понижения температуры фактически может стать увеличение концентрации ЭО и относительной влажности, если при этом не были внесены изменения в параметры давления.
Данные, полученные из исследований летальности процесса, используются для установления минимального времени экспозиции газу ЭО, необходимого для процесса стерилизации. Если эти исследования выполняются в разработочной камере, то должны быть соблюдены предосторожности при прямом применении этого времени к стерилизационному процессу, потому что кривые гибели (скорости летальности или величины /логарифмической редукции спор) специфичны по отношению к параметрам процесса, конфигурации загрузки в камере и к месту размещения УКП внутри упакованного продукта внутри загрузки, использованной в исследованиях.
Для получения дополнительной информации о прямом подсчете и методах фракционного негативного анализа см. ISO 11138-1:2006 (приложение D), а также ISO 14161:2009 (приложение С).
D.14 Руководство к приложению В - консервативное определение скорости летальности стерилизационного процесса - подход "массовой гибели"
D.14.1 [B.1] Общая информация
D.14.1.1 [B.1.1] Эта статья дает дополнительное руководство по использованию информации, имеющейся в приложении В, а также дополнительное руководство к информации, содержащейся в разделах 8 и 9. Поскольку подход "биологического индикатора/бионагрузки" и подход "массовой гибели" используют много одинаковых процедур, часть текста в этом приложении дублирует текст в D.13 (приложение D). Однако при использовании подхода с расчетом цикла см. также D.13.1.1 (приложение D). Дополнительная информация относительно применения подхода "массовой гибели" приводится в стандарте ISO 14161:2009, пункт 7.2.
Пользователь должен помнить о том, что минимальное время цикла, вычисляемое при этом подходе, не является само по себе адекватным для валидации процесса стерилизации. Необходима демонстрация способности поддерживать параметры процесса в определенных пределах во время предложенного полного цикла.
D.14.1.2 [B.1.2] При этом подходе широко используются два метода.
Подход половинного цикла: по причине относительной легкости использования и получению традиционного уровня обеспечения стерильности изготовители медицинских изделий и учреждения здравоохранения обычно пользуются этим методом, который предназначен для демонстрации полной инактивации биологических индикаторов с нагрузкой 10 при времени экспозиции половинного цикла. Когда это время экспозиции удваивается, то в ходе экспозиции ЭО достигается уровень обеспечения стерильности 12. Этот подход приведет к процессу, создающему уровень обеспечения стерильности более 12.
Расчет цикла: этот метод состоит из экспозиции внутренних УКП экспериментальному циклу, выгрузке микробиологической нагрузки и испытанию ее на наличие выживших организмов. Эта проверка может выполняться с применением техники фракционного негативного анализа или путем подсчета жизнеспособных микробов на образцах или индикаторах с нагрузкой. Эта информация может быть использована для расчета цикла, необходимого для создания определенного уровня обеспечения стерильности для продукта. См. ISO 14161:2009. Если используется подход Стамбо-Мерфи-Кохрейна и подход расчета цикла массовой гибели, рекомендуемое количество биологических индикаторов/УКП может основываться на объеме продукта, подлежащего стерилизации, с минимальным количеством, равным 10 [см. [38] и С.3 (приложение С)]. Набор образцов, экспонированных за нулевое время, должен быть экспонирован всем фазам экспериментального цикла до инжекции стерилизующего агента.
D.14.1.3 [В.1.3] Информация относительно инкубационного периода для биологического индикатора приведена в ISO 14161:2009, пункт 12.3.
D.14.1.4 [B.1.4] Пригодность биологического индикатора в отношении времени инактивации бионагрузки может быть продемонстрирована проверкой на стерильность либо до, либо во время определения процесса путем использования фракционного цикла с подходящим временем экспозиции.
D.14.2 [B.2] Процедура
D.14.2.1 [B.2.1] Для этого метода могут быть использованы внутренние УКП, размещенные внутри стерильной барьерной системы продукта. Если они используются, то они должны обеспечивать по меньшей мере ту же нагрузку для стерилизационного процесса, какую создают представляемый ими продукт. Нагрузка, создаваемая внутренними УКП для стерилизационного процесса, должна быть по меньшей мере такой же, как бионагрузка, находящаяся в наиболее труднодоступной части продукта [см. D.7.1.6 и D.8.6 (приложение D)]. Информацию о разработке УКП см. в 7.1.6, а информацию об определении пригодности внутреннего УКП для микробиологической нагрузки продукта см. в 8.6 и D.8.6 (приложение D).
D.14.2.2 [В.2.2] Место внутри продукта, в котором достижение стерильности наиболее затруднено, может включать в себя не только зоны, которые имеют низкую проникающую способность для стерилизующего агента, но также и те зоны, которые с наибольшей вероятностью могут иметь значительное количество бионагрузки.
Аспектами, подлежащими рассмотрению, являются:
a) длина и внутренний диаметр каналов, а также способность стенок медицинского изделия позволять диффузию ЭО;
b) поглощающая способность отдельных частей продукта и материала;
c) вес и плотность изделий;
d) конфигурация загрузки, в особенности для загрузок со смешанными продуктами.
Соображения относительно учреждений здравоохранения: для демонстрации адекватного проникновения ЭО, влажности и тепла в продукт, должно быть выбрано УКП для рутинного мониторинга и валидации процесса ЭО-стерилизации. Устойчивость УКП воздействию ЭО должна быть доказана равной или превышающей устойчивость бионагрузки продукта, подлежащего стерилизации, в месте, наиболее трудном для стерилизации.
D.14.2.3 [B.2.3] Дополнительное руководство не приводится.
D.14.2.4 [B.2.4] Получение данных о подсчете микробов или данных об уничтожении в ходе фракционного цикла требует экспозиции микробиологической нагрузки меньшей летальности, чем обеспечивается нормальным производственным циклом. Это обычно достигается путем уменьшения времени экспозиции при удержании остальных параметров постоянными при номинальных условиях, или на выбранном минимально допустимом уровне процессных условий. Использование минимально допустимой температуры процесса для исследований с подсчетом обеспечивает получение требуемой летальности при работе в специфицированном температурном диапазоне.
Параметры, в первую очередь влияющие на летальность - это время экспозиции, концентрация ЭО, влажность и температура. Если делается настройка иных параметров, кроме температуры, должно быть оценено общее влияние изменений на цикл, поскольку настройка может не дать желаемого результата из-за взаимосвязи всех параметров. Например, результатом понижения температуры фактически может стать повышение концентрации ЭО и относительной влажности, если не будет сделано изменений давления впуска пара и роста давления инжекции ЭО.
D.14.2.5 [В.2.5] Логарифмическая редукция спор может быть рассчитана с использованием результатов фракционного цикла. Если нет выживших внутренних УКП, оценка "худшего случая" для логарифмической редукции спор может быть получена выполнением расчета с одним предполагаемым выжившим.
Независимо от применяемого метода предполагается, что:
a) популяция организмов гомогенна;
b) параметры процесса (за исключением времени экспозиции газа) постоянны от цикла к циклу;
c) имеется полулогарифмическое соотношение выживших;
d) экспонированные и неэкспонированные организмы одинаково реагируют на восстановительную среду.
Приложение E
(обязательное)
Выпуск единичного лота
E.1 Общая информация
Данное приложение оговаривает требования к выпуску продукта из стерилизационного процесса в тех случаях, когда загрузка состоит только из одного продукта, например, во время исследований и разработки нового продукта или во время клинических испытаний продукта.
Примечание - Следует рассмотреть возможность наличия национальных или региональных положений, относящихся к клиническим продуктам. Если такие положения имеются и действуют, следует выполнять их требования.
E.2 Процедура
E.2.1 Оцените упакованный продукт с целью определения возможности включения его в существующее семейство продуктов для целей стерилизации. Эта оценка должна рассматривать состав продукта, конструкцию, упаковку, бионагрузку и плотность загрузки. Результат оценки, включая обоснование принятых решений, должен документироваться.
E.2.2 Если упакованный продукт может быть включен в существующее семейство продукта, обратитесь к 12.5.2 и D.12.5.2 (приложение D).
E.2.3 Если нет существующего семейства (семейств) продукта или в случаях, когда упакованный продукт не может быть отнесен к существующему семейству:
a) выполните случайную выборку образцов из партии и определите среднюю бионагрузку партии в соответствии с ISO 11737-1;
b) распределите внутри стерилизационной загрузки образцы для проверки на стерильность и внутренние УКП, находящиеся внутри упакованного продукта, включая места, где наиболее трудно достичь условий стерилизации. Разместите внешние УКП (если они используются) на загрузке в определенных местах. УКП должны содержать биологические индикаторы в соответствии с ISO 11138-2:2006, раздел 5 и 9.5;
Примечание - Места размещения УКП должны включать в себя точки, используемые для мониторинга температуры.
c) подвергните стерилизационную загрузку экспонированию фракционному ЭО циклу с минимальными значениями параметров процесса, рассчитанными на достижение уровня обеспечения стерильности менее 10 для продукта и от 7 до 8
внутри УКП;
d) выньте внутренние УКП, внешние УКП (если использовались) и образцы продукта из загрузки и проведите проверку на стерильность в соответствии с ISO 11737-2;
Примечание - Если сравнительная устойчивость внутреннего УКП относительно бионагрузки продукта была ранее оценена с использованием фракционного цикла меньшей продолжительности, чем указано в E.2.3, перечисление b) (приложение E), и при этом не было обнаружено позитивных результатов проверки образцов продукта, нет необходимости выполнять проверку на стерильность образцов продукта, экспонированных во фракционном цикле, описанном в E.2.3, перечисление b) (приложение Е).
e) аэрируйте загрузку и доведите ее до условий окружающей среды. Аэрационный период достаточен для рассеивания остатков ЭО до уровня, который не окажет отрицательного влияния на новые УКП в стерилизационном цикле с полной экспозицией (см. f) и g) ниже);
f) распределите новые УКП, находящиеся внутри упаковок продукта, по всей стерилизационной загрузке, включая места, где наиболее трудно достичь условий стерилизации. Разместите внешние УКП (если они используются) на загрузке в определенных местах;
Примечание - Места размещения УКП должны включать в себя точки, используемые для мониторинга температуры.
g) обработайте ту же самую загрузку, подвергнув ее второму стерилизационному циклу с номинальными параметрами процесса, со специфицированным временем экспозиции по меньшей мере вдвое большим, чем время экспозиции фракционного цикла (см. b) выше) - это полный цикл;
h) выньте внешние УКП (если использовались) и внутренние УКП из заново обработанной загрузки и выполните проверку на стерильность.
E.2.4 Стерилизационная загрузка может быть выпущена из стерилизации при условии выполнения следующих требований:
a) бионагрузка продукта представляет собой меньшую нагрузку на процесс стерилизации, чем биологический индикатор, используемый во внешнем УКП (если используется) и во внутреннем УКП;
b) параметры процесса для фракционного цикла соответствуют процессным спецификациям;
c) загрузка была вторично экспонирована в полном стерилизационном цикле с номинальными процессными параметрами, со специфицированным временем экспозиции по меньшей мере вдвое большим, чем время экспозиции фракционного цикла, установленного в Е.2.3, перечисление b) (приложение E);
d) параметры процесса для полного стерилизационного цикла соответствуют процессным спецификациям;
e) имеется подтверждение отсутствия роста контрольных микроорганизмов из внешних УКП (если использовались) и внутренних УКП, экспонированных во фракционном цикле;
f) имеется подтверждение отсутствия позитивного роста при проверке на стерильность образцов продукта после экспонирования во фракционном цикле;
Примечание - Если сравнительная устойчивость внутреннего УКП относительно бионагрузки продукта была ранее оценена с использованием фракционного цикла меньшей продолжительности, чем указано в E.2.3, перечисление b) (приложение E), и при этом не было обнаружено позитивных результатов проверки образцов продукта, тогда нет необходимости выполнять проверку на стерильность образцов продукта, экспонированных во фракционном цикле, в соответствии с E.2.3, перечисление b) (приложение Е).
g) имеется подтверждение отсутствия роста контрольных микроорганизмов из УКП, экспонированных в полном стерилизационном цикле;
h) функциональность, стабильность продукта и целостность упаковки соответствуют требованиям после экспонирования в полном стерилизационном цикле;
i) имеется подтверждение, что остаточные уровни ЭО в продукте соответствуют ISO 10993-7 после экспонирования продукта как во фракционном, так и в полном стерилизационном цикле;
j) выполнены все требования к качеству и нормативные требования.
Примечание - Информация и данные, полученные при этом подходе, могут использоваться ретроспективно для поддержки последующей валидации стерилизационного процесса.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов ссылочным межгосударственным стандартам
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего межгосударственного стандарта |
ISO 10012 | - | * |
ISO 10993-7 | IDT | ГОСТ ISO 10993-7-2011 "Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации" |
ISO 11138-1:2006 | IDT | ГОСТ ISO 11138-1-2012 "Стерилизация медицинской продукции. Биологические индикаторы. Часть 1. Технические требования" |
ISO 11138-2:2009 | IDT | ГОСТ ISO 11138-2-2012 "Стерилизация медицинской продукции. Биологические индикаторы. Часть 2. Биологические индикаторы для стерилизации оксидом этилена" |
ISO 11140-1 | IDT | ГОСТ ISO 11140-1-2011 "Стерилизация медицинской продукции. Химические индикаторы. Часть 1. Общие требования" |
ISO 11737-1 | IDT | ГОСТ ISO 11737-1-2012 "Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукции" |
ISO 11737-2 | IDT | ГОСТ ISO 11737-2-2011 "Стерилизация медицинских изделий. Микробиологические методы. Часть 2. Испытания на стерильность, проводимые при валидации процессов стерилизации" |
ISO 13485:2003/Cor 1:2009 | IDT | ГОСТ ISO 13485-2011 "Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования" |
* Соответствующий межгосударственный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного стандарта. | ||
Библиография
[1] | ISO 9000:2005 | Quality management systems - Fundamentals and vocabulary |
[2] | ISO 9001 | Quality management systems - Requirements |
[3] | ISO 10993 (all parts) | Biological evaluation of medical devices |
[4] | ISO 11607-1 | Packaging for terminally sterilized medical devices - Part 1: Requirements for materials, sterile barrier systems and packaging systems |
[5] | ISO 11607-2 | Packaging for terminally sterilized medical devices - Part 2: Validation requirements for forming, sealing and assembly processes |
[6] | ISO 14001 | Environmental management systems - Requirements with guidance for use |
[7] | ISO 14040 | Environmental management - Life cycle assessment - Principles and framework |
[8] | ISO 14161:2009 | Sterilization of health care products - Biological indicators - Guidance for the selection, use and interpretation of results |
[9] | ISO 14937:2009 | Sterilization of health care products - General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process for medical devices |
[10] | ISO 14971 | Medical devices - Application of risk management to medical devices |
[11] | ISO 15883 (all parts) | Washer-disinfectors |
[12] | ISO 17664 | Sterilization of medical devices - Information to be provided by the device manufacturer for the processing of reusable medical devices |
[13] | ISO 22442-1 | Medical devices utilizing animal tissues and their derivatives - Part 1: Application of risk management |
[14] | ISO 22442-2 | Medical devices utilizing animal tissues and their derivatives - Part 2: Controls on sourcing, collection and handling |
[15] | ISO 22442-3 | Medical devices utilizing animal tissues and their derivatives - Part 3: Validation of the elimination and/or inactivation of viruses and transmissible spongiform encephalopathy (TSE) agents |
[16] | ISO/IEC Guide 99:2007 | International vocabulary of metrology - Basic and general concepts and associated terms (VIM) |
[17] | ISO/IEC 90003 | Software engineering - Guidelines for the application of ISO 9001:2000 to computer software |
[18] | ISO/TS 11139:2006 | Sterilization of health care products - Vocabulary |
[19] | ISO/TS 16775 | Packaging for terminally sterilized medical devices - Guidance on the application of ISO 11607-1 and ISO 11607-2 |
[20] | IEC 61010-1 | Safety requirements for electrical equipment for measurement, control and laboratory use - Part 1: General requirements |
[21] | IEC 61010-2-040 | Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-040: Particular requirements for sterilizers and washer-disinfectors used to treat medical materials |
[22] | ANSI/AAMI ST41:1999 | Ethylene oxide sterilization in health care facilities: Safety and effectiveness |
[23] | ANSI/AAMI ST67 Sterilization medical devices - Requirements for products labeled "STERILE". AAMI, Arlington, VA, 2006 | |
[24] | AAMI TIR15. Physical aspects of ethylene oxide. AAMI, Arlington, VA, 2009 | |
[25] | AAMI TIR16. 2009. Microbiological aspects of ethylene oxide sterilization. AAMI, Arlington, VA, 2009 | |
[26] | AAMI TIR28. Product adoption and process equivalence for ethylene oxide sterilization. AAMI, Arlington, VA, 2009 | |
[27] | AS/NZS 4187, Reprocessing of reusable medical devices in health service organizations | |
[28] | EN 556-1 | Sterilization of medical devices - Requirements for medical devices to be designated "STERILE" - Part 1: Requirements for terminally sterilized medical devices |
[29] | Manufacturers Directive ATEX 94/9/EC, European Parliament and Council, 1994, as amended, 1994 | |
[30] | Gillis J., & Schmidt W.C. Scanning electron microscopy of spores on inoculated product surfaces. Medical Device and Diagnostic Industry. 1983, 5 (6), pp.46-49 | |
[31] | Global Harmonization Taskforce (GHTF) Study Group 1 (SG1), Document No. N29R16:2005 Information Document Concerning the Definition of the Term "Medical Device" | |
[32] | Holcomb R.G, & Pflug I.J. The Spearman-Karber method of analyzing quantal assay microbial destruction data. In: Selected Papers on the Microbiology and Engineering of Sterilization Processes, (Pflug I.J. e.d.). Environmental Sterilization Laboratory, Minneapolis, Fifth Edition, 1988, pp.83-100 | |
[33] | Mosley G.A., Estimating the effects of EtO BIER-Vessel Operating Precision on D-value Calculations, Medical Device & Diagnostic Industry, April 2002 | |
[34] | Mosley G.A., & Gillis J.R. Factors Affecting Tailing in Ethylene Oxide Sterilization. Part 1: When Tailing is an Artifact and Scientific Deficiencies in ISO 11135 and EN 550. PDA J. Pharm. Sci. Technol. 2004, 58 (2), pp.81-95 | |
[35] | Mosley G.A., Gillis J.R., Krushefski G. Evaluating the formulae for integrated lethality in ethylene oxide sterilization using six different endospore forming strains of bacteria, and comparisons of integrated lethality for ethylene oxide and steam systems. PDA J. Pharm. Sci. Technol. 2005, 59 (1), pp.64-86 | |
[36] | Mosley G.A., Gillis J.R., Whitbourne J.E. Formulae for Calculations of Integrated Lethality for EtO Sterilization Processes, Refining the Concepts and Exploring the Applications. Pharm. Tech. 2002, 26 (10), pp.114-134 | |
[37] | Pflug I.J. Microbiology and Engineering of Sterilization Processes, Minneapolis. Environmental Sterilization Services, Minneapolis, Eleventh Edition, 2003 | |
[38] | Pflug I.J., Holcomb R.G., Gomez M.M. Thermal Destruction of Microorganisms. In: Disinfection, Sterilization, and Preservation, (Block S, ed.). Lippincott, Williams & Wilkins, Philadelphia, 2001, pp.79-129 | |
[39] | Rodriguez A.C., Young В., Caulk K., Zelewski J., Dwasnica S., Aguirre S. Calculating Accumulated Lethality and Survivorship in EtO Sterilization Processes. MD. 2001 September, p.1 | |
[40] | West K.L., Ethylene oxide sterilization: A study of resistance relationships. In: Sterilization of Medical Products, (Gaughran E., & Kereluk K. eds.). Johnson & Johnson, New Brunswick, NJ, 1977 | |
[41] | Shintani et al. Comparison of D10-value accuracy by the limited Spearman-Karber procedure (LSKP), the Stumbo-Murphy-Cochran procedure (SMCP), and the survival-curve method (EN). Biomed. Instrum. Technol. 1995, 29 (2), pp.113-124 | |
[42] | Stumbo C.R., Murphy J.R., Cochran J. Nature of Thermal Death Time Curves for P.A. 3679 and Clostridium Botulinum. Food Technol. 1950, 4, pp.321-326 | |
[43] | USP Monograph on Biological Indicator for Ethylene Oxide Sterilization, Paper Carrier, USP36-NF31 (2013), page 2659 | |
УДК 615.478.73:006.354 | МКС 11.080.01 | Р26 | IDT |
Ключевые слова: стерилизация, медицинская продукция, этиленоксид, разработка, управление процессом стерилизации, медицинские изделия | |||
Электронный текст документа
и сверен по:
, 2017

{kind=link}