ГОСТ Р 56033-2014/Руководство ИСО/МЭК 63:2012
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
РУКОВОДСТВО ПО РАЗРАБОТКЕ И ВКЛЮЧЕНИЮ АСПЕКТОВ БЕЗОПАСНОСТИ В МЕЖДУНАРОДНЫЕ СТАНДАРТЫ НА МЕДИЦИНСКИЕ ИЗДЕЛИЯ
Guide to the development and inclusion of safety aspects in international standards for medical devices
ОКС 01.080.20,
11.040.01
Дата введения 2015-06-01
Предисловие
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "МЕДИТЕСТ" (ЗАО "МЕДИТЕСТ") на основе собственного перевода на русский язык англоязычной версии документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 4 июня 2014 г. N 502-ст
4 Настоящий стандарт идентичен международному документу Руководство ИСО/МЭК 63:2012* "Руководство по разработке и включению аспектов безопасности в международные стандарты на медицинские изделия" (ISO/IEC Guide 63:2012 "Guide to the development and inclusion of safety aspects in International Standards for medical devices", IDT)
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
5 ВВЕДЕН ВПЕРВЫЕ
6 ПЕРЕИЗДАНИЕ. Март 2020 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт ИСО/МЭК 63 был разработан Техническим комитетом ИСО/ТК 210 "Менеджмент качества и соответствующие общие аспекты медицинских изделий" и подкомитетом МЭК/ПК 62А "Общие аспекты электрических изделий, применяемых в медицинской практике" в составе рабочей группы "Применение менеджмента риска к медицинским изделиям".
Настоящей стандарт отменяет и заменяет первое издание (ISO/IEC Guide 63:1999), которое было технически пересмотрено.
Концепция безопасности, включая связанные с безопасностью функциональные характеристики и эксплуатационную пригодность, близко связана с охраной целостности здоровья пациентов, являющихся объектами медицинского обслуживания, так же как медицинского персонала и любых других лиц. Поскольку медицинские изделия и медицинские системы становятся все более сложными, то возрастают и усилия, требуемые для обеспечения их безопасности.
Так как различные обстоятельства диктуют различные подходы к обеспечению безопасности, то невозможно предусмотреть четкие требования и руководства, которые будут иметь отношение к каждому конкретному случаю. В то же время, такие руководства, основываясь на принципе "используй, если применимо", могут помочь в разработке соответствующих стандартов.
1 Область применения
Настоящий стандарт предоставляет общие рекомендации по включению аспектов безопасности в стандарты на медицинские изделия, которые предназначены для использования в рамках менеджмента риска, установленного в ИСО 14971. Настоящий стандарт основывается на концепциях, установленных в Руководстве ИСО/МЭК 51, с целью включения в рассмотрение функциональных характеристик и эксплуатационной пригодности, связанных с безопасностью.
Настоящий стандарт предназначен для использования совместно с Руководством ИСО/МЭК 51 и ИСО 14971.
2 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
2.1 эксплуатационный документ (accompanying document): Документ, прилагаемый к медицинскому изделию и содержащий информацию для лиц, ответственных за сборку, применение и техническое обслуживание медицинского изделия, оператора или пользователя, особенно в отношении безопасности.
[ИСО 14971, определение 2.1]
2.2 вред (harm): Физическая травма или ущерб здоровью людей, имуществу или окружающей среде.
[ИСО/МЭК 51, определение 3.3]
2.3 опасность (hazard): Потенциальный источник вреда.
Примечание - Термин опасность может быть установлен для определения происхождения опасности или характера предполагаемого вреда (например, опасность поражения электрическим током, опасность разрушения конструкции, опасность повреждения ткани, токсикологическую опасность, опасность возгорания).
[ИСО/МЭК 51, определение 3.5]
2.4 опасная ситуация (hazardous situation): Обстоятельства, при которых люди, имущество или окружающая среда подвержены одной или нескольким опасностям.
[ИСО/МЭК 51, определение 3.6]
2.5 предусмотренное назначение/предусмотренное применение (intended use): Применение изделия, процесса или услуги по назначению в соответствии с техническими требованиями, инструкциями и информацией, предоставленными изготовителем.
[ИСО 14971, определение 2.5]
2.6 жизненный цикл (life-cycle): Все стадии существования медицинского изделия, от первоначальной концепции до вывода из эксплуатации и утилизации.
[ИСО 14971, определение 2.7]
2.7 изготовитель (manufacturer): Физическое или юридическое лицо, ответственное за проектирование, изготовление, упаковывание и/или маркировку медицинского изделия, установку/монтаж или модификацию медицинского изделия перед выпуском его в обращение или вводом в эксплуатацию, независимо от того, выполняет ли эти операции вышеупомянутое лицо или третья сторона от его имени.
Примечания
1 В определении термина "изготовитель" следует учитывать положения национальных и региональных регулирующих документов.
2 Определение термина "маркировка" - см. ИСО 13485, определение 3.6.
[ИСО 14971, определение 2.8]
2.8 медицинское изделие (medical device): Любой инструмент, аппарат, прибор, устройство, оборудование, имплантат, in vitro реагент или калибратор, программное обеспечение, материал или иные подобные или связанные с ними изделия, предназначенные изготовителем для применения к человеку по отдельности или в сочетании друг с другом в целях:
- диагностики, профилактики, мониторинга, лечения или облегчения заболеваний;
- диагностики, мониторинга, лечения, облегчения или компенсации последствий травмы;
- исследования, замещения или изменения анатомического строения или физиологических процессов;
- поддержания или сохранения жизни;
- управления зачатием;
- дезинфекции медицинских изделий;
- получения информации для медицинских целей посредством исследования in vitro проб, взятых из тела человека, при условии, что их функциональное воздействие на человеческий организм не реализуется за счет фармакологических, иммунологических или метаболических средств, но может поддерживаться такими средствами.
Примечания
1 Данное определение разработано Целевой группой по глобальной гармонизации (Global Harmonization Task Force - GHTF).
2 Следующие изделия могут рассматриваться в некоторых странах как медицинские, но в их отношении еще не выработан единый подход:
- вспомогательные средства для лиц с ограниченными возможностями или с физическими и умственными недостатками;
- изделия для лечения/диагностики заболеваний и травм у животных;
- принадлежности медицинских изделий;
- дезинфицирующие вещества;
- изделия, включающие ткани животных или человека, которые могут подпадать под определение медицинского изделия, но являются предметом другого регулирования.
3 Адаптировано из ИСО 13485, определение 3.7, и ИСО 14971, определение 2.9.
2.9 остаточный риск (residual risk): Риск, остающийся после выполнения мер по управлению риском.
Примечания
1 ИСО/МЭК 51, определение 3.9, использует термин "защитные меры" вместо термина "меры по управлению риском".
2 Адаптировано из ИСО 14971, определение 2.15.
2.10 риск (risk): Сочетание вероятности причинения вреда и тяжести этого вреда.
[ИСО/МЭК 51, определение 3.2]
2.11 анализ риска (risk analysis): Систематическое использование имеющейся информации для выявления опасностей и определения риска.
[ИСО/МЭК 51, определение 3.10]
Примечание - Анализ риска включает в себя изучение последовательности событий, которые могут привести к опасным ситуациям и причинению вреда.
2.12 управление риском (risk control): Процесс принятия решений и выполнения мер по уменьшению рисков до установленных уровней или поддержания рисков на установленных уровнях.
[ИСО 14971, определение 2.19]
2.13 определение риска (risk estimation): Процесс, применяемый для присвоения значений вероятности наступления вреда и тяжести этого вреда.
[ИСО 14971, определение 2.20]
2.14 оценивание риска (risk evaluation): Процесс сравнения риска, который уже определен, с установленными критериями риска для определения допустимости риска.
[ИСО 14971, определение 2.21]
2.15 менеджмент риска (risk management): Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания, управления и мониторинга риска.
[ИСО 14971, определение 2.22]
2.16 безопасность (safety): Отсутствие недопустимого риска.
[ИСО/МЭК 51, определение 3.1]
2.17 тяжесть (severity): Мера возможных последствий опасности.
[ИСО 14971, определение 2.25]
2.18 эксплуатационная пригодность (usability): Характеристика пользовательского интерфейса, которая определяет результативность, эффективность, простоту обучения пользователя и степень удовлетворенности пользователя.
[МЭК 62366, определение 3.17]
2.19 ошибка эксплуатации/применения (use error): Выполнение или невыполнение действия, приводящее к функционированию медицинского изделия, отличающемуся от предусмотренного изготовителем или ожидаемого пользователем.
Примечание 1 - К ошибкам эксплуатации/применения относят промахи, упущения и заблуждения.
Примечание 2 - См. также приложения С и D, D.1.3.
Примечание 3 - Неадекватную физиологическую реакцию пациента саму по себе не относят к ошибке эксплуатации/применения.
[МЭК 62366, определение 3.21]
2.20 верификация (verification): Подтверждение посредством представления объективных свидетельств того, что установленные требования были выполнены.
Примечание 1 - Термин "верифицирован" используют для обозначения соответствующего статуса.
Примечание 2 - Деятельность по подтверждению требованиям может включать в себя:
- осуществление альтернативных расчетов;
- сравнение спецификации на новый проект с аналогичной документацией на апробированный проект;
- проведение испытаний и демонстраций;
- анализ документов до их выпуска.
[ИСО 9000, определение 3.8.4]
3 Принципы подготовки стандартов по безопасности медицинских изделий
3.1 Общие положения
Целью стандартов по безопасности медицинских изделий является поддержка разработки и производства медицинских изделий с предсказуемым, последовательным уровнем безопасности.
Для достижения этой цели стандарты по безопасности медицинских изделий должны:
a) содействовать изготовителям в проектировании и производстве безопасных и результативных медицинских изделий;
b) содействовать изготовителям, органам по сертификации, испытательным лабораториям, регулирующим органам власти в оценке соответствия установленным требованиям;
c) содействовать поставщикам услуг здравоохранения в осуществлении менеджмента рисков, связанных с использованием медицинских изделий.
Для создания стандартов по безопасности медицинских изделий, которые будут способны должным образом реализовывать поставленные задачи, разработчикам стандартов следует использовать структуру на основе концепции риска (см. раздел 4).
3.2 Область применения стандартов по безопасности
Планирование и разработка стандартов по безопасности медицинских изделий требует глобального подхода, который включает вовлечение в эти процессы изготовителей, пользователей, регулирующие органы власти и другие заинтересованные стороны. Тесное взаимодействие внутри и между комитетами (технические комитеты по стандартизации), ответственными за различные медицинские изделия, является необходимым элементом в создании последовательного подхода к трактовке безопасности при подготовке стандартов. Четкое определение области применения стандартов по безопасности должно обеспечивать ограничение каждого стандарта определенными аспектами и давать ссылку на стандарты более широкого применения для всех других уместных аспектов.
На такой иерархии основаны:
- базовые стандарты безопасности, содержащие фундаментальные понятия и концепции, принципы и требования в отношении общих аспектов безопасности, применимых ко всем видам или широкому диапазону продукции, процессов и услуг (базовые стандарты безопасности иногда называют горизонтальными стандартами);
- групповые стандарты безопасности, содержащие аспекты безопасности, применимые к некоторым видам или к однородной продукции, процессам и услугам, которые находятся в ведении двух или более технических комитетов или рабочих групп и в которых, по возможности, делается ссылка на базовые стандарты безопасности;
- стандарты безопасности продукции, содержащие все необходимые аспекты безопасности, применимые к конкретной продукции или к однородной продукции; продукции, процессам и услугам, которые находятся в ведении одного технического комитета или рабочей группы и в которых, по возможности, делается ссылка на базовые стандарты безопасности и групповые стандарты безопасности (стандарты безопасности продукции иногда называют вертикальными стандартами).
Такая иерархия изложена в ИСО/МЭК 51, подраздел 7.1.
Требования безопасности для медицинских изделий могут быть включены в стандарты различных типов (см. 3.3), которые могут быть найдены на любом соответствующем уровне в иерархии, описанной выше.
3.3 Типы стандартов
3.3.1 Стандарты на продукцию
Эти стандарты могут быть:
- стандартами, которые устанавливают безопасность или параметры функциональных характеристик, а также содержат ссылки на методы испытаний, которые могут быть использованы для демонстрации соответствия этим параметрам, или
- стандартами на методы и методики испытаний, когда строгое соблюдение заявленных критериев сдачи-приемки является необходимым для обеспечения безопасности и функциональных характеристик.
См. А.1 приложения А для информации о том, как стандарты на продукцию могут внести вклад в безопасность и результативность медицинских изделий.
3.3.2 Стандарты на процессы
Эти стандарты могут быть:
a) стандартами на системы качества, которые устанавливают структуру, в рамках которой изготовитель становится способным проектировать, разрабатывать и производить медицинские изделия, соответствующие спецификациям, или
b) стандартами, которые устанавливают структуру, в рамках которой изготовитель становится способным проектировать и разрабатывать медицинские изделия, соответствующие требованиям безопасности и результативности, или
c) стандартами на процессы, которые могут быть применимы для проектирования, разработки или производства безопасных и результативных медицинских изделий (например, стерилизация, оценивание биологического действия, клиническое исследование).
См. А.2 приложения А для информации о том, как стандарты на процессы могут внести вклад в безопасность и результативность медицинских изделий.
Некоторые стандарты не могут быть однозначно отнесены к одной из этих категорий, так как имеют общие признаки стандартов на продукцию и стандартов на процессы. Примеры приведены в 3.3.3 и 3.3.4.
3.3.3 Стандарты на установку/монтаж и стандарты, относящиеся к окружающей среде
Такие стандарты являются общеприменимыми для больших систем и активных медицинских изделий. Эти стандарты могут быть:
a) стандартами в отношении конструкции и установки/монтажа (например, защитное ограждение/экранирование рентгена, прокладка электропроводки);
b) стандартами на системы, которые могут быть применены в отношении надлежащего обращения при объединении отдельных изделий и устройств в единую систему, а также используемых при этом процедур;
c) стандартами на ввод в эксплуатацию, которые могут быть применены в отношении испытаний и проверочных процедур для применения к стационарно устанавливаемому оборудованию и системам, до их первоначального использования, или
d) стандартами, относящимися к окружающей среде, которые могут быть применены в отношении надлежащего проведения испытаний для обеспечения уверенности в том, что медицинское изделие не оказывает отрицательного влияния на окружающую среду и что окружающая среда не ухудшает или как-либо иначе негативно сказывается на функциональных характеристиках медицинского изделия (например, стандарты на электромагнитную совместимость).
3.3.4 Стандарты, используемые в процессе эксплуатации
Эти стандарты могут быть:
a) стандартами на рутинные штатные испытания, проводимые для обеспечения уверенности в том, что безопасность активных медицинских изделий поддерживается в течение всего срока эксплуатации оборудования, или
b) стандартами по обеспечению качества и калибровочными стандартами, которые могут быть применены для обеспечения постоянной надлежащей функциональности и метрологической точности медицинских изделий и, где уместно, безопасности.
3.4 Восприятие практического суждения о безопасности
Риск должен быть сбалансирован относительно других требований к продукции, процессу или обслуживанию. Такие требования включают выгоду, пригодность и стоимость. Разработчики стандартов должны помнить, что уровень требуемых усилий и затрат изготовителя (например, для ведения регуляторной документации или проведения испытаний) должен быть соразмерен с уровнем риска.
Поскольку нулевой риск недосягаем, безопасность определяется как отсутствие недопустимого риска. Хотя нулевой риск является наиболее желательным, достижение этого уровня не должно ожидаться на практике. Реалистическое ожидание состоит в том, чтобы выбрать такие критерии допустимости риска, которые основываются на доступной информации о современном уровне научно-технического развития и потребностях заинтересованных сторон, а также существующем положении дел в повышении уровня безопасности и защиты здоровья.
При оценивании безопасности медицинских изделий необходимо учитывать, что в силу своего способа действия, состава или обстоятельств применения, некоторые изделия содержат присущий им риск, который не может быть устранен без ухудшения их результативности.
Как технологии и общественные ценности подчинены влиянию времени и среди различных культур существуют определенные различия в практическом здравоохранении, так и суждение о безопасности медицинских изделий и медицинских процедур изменяется с течением времени и различается в разных странах. Другими словами, оценивание риска субъективно и не является объективной задачей. С такими спорными вопросами можно столкнуться при идентификации определенных условий, при которых применяется техническое требование. Разработчики стандартов должны сосредоточиться на требованиях, связанных с безопасностью изделия или с безопасным использованием изделия, избегая при этом особенностей или функций, которые не являются существенными с точки зрения безопасности.
3.5 Менеджмент рисков
Процесс менеджмента риска для медицинских изделий включает следующее:
a) установление критериев допустимости риска;
b) идентификацию опасностей и сценариев, которые приводят к опасным ситуациям;
c) определение связанных рисков;
d) идентификацию мер по управлению риском с целью снижения каждого риска для необходимого соответствия критериям допустимости риска;
e) осуществление мер по управлению риском;
f) верификацию результативности мер по управлению риском.
Процесс менеджмента риска более подробно изложен в ИСО 14971.
Разработка стандартов должна базироваться на идентификации опасности. Цель разработки стандартов состоит в создании таких стандартов, которые устанавливают методы идентификации опасности или меры по управлению риском, способные привести к соответствию критериям допустимости риска (см. 3.6).
Это должно быть обязательно принято во внимание, поскольку разработчики стандартов устанавливают соответствующие требования, которые будут включены в стандарт.
В разделе 5 приведен краткий обзор того, как стандарты по безопасности медицинских изделий могут облегчить внедрение системы менеджмента риска в соответствии с ИСО 14971.
3.6 Критерии допустимости риска
Целью стандартов по безопасности медицинских изделий является содействие и поддержание разработки медицинских изделий, имеющих допустимый уровень риска. При описании критериев допустимости стандарты по безопасности медицинских изделий должны предоставлять практические критерии, такие как предельные значения для проектирования и критерии в отношении функциональных характеристик.
3.7 Методы и методология управления риском
Стандарты по безопасности медицинских изделий должны устанавливать методы управления рисками или методологию применения мер по управлению рисками, которые связаны с соответствующими медицинскими изделиями. Они могут включать аспекты в отношении функциональных характеристик, проектирования, процесса производства, монтажа, обслуживания и т.д. и базироваться на подходе менеджмента риска в отношении всего жизненного цикла изделия.
3.8 Координация стандартов по безопасности медицинских изделий
Разработка каждого нового стандарта по безопасности медицинских изделий должна рассматриваться в контексте существующих медицинских изделий и стандартов, а также в контексте национального, регионального и международного законодательства. По возможности, где это уместно, новые стандарты должны использовать как основу существующие стандарты, содержать их текст или ссылаться на них, исходя из удобства изложения (см. Директиву ИСО/МЭК, Часть 2, 2011, раздел А.7).
3.9 Влияние регулирования
Безопасность и результативность медицинских изделий, выпуск в обращение и применение которых регулируется во многих странах, являются предметом пристального внимания со стороны регулирующих органов власти.
На стандарты часто ссылаются в регулирующих документах и в законодательстве. В этом случае такие стандарты становятся юридически закрепленными. В то же время существуют альтернативные системы регулирования, в которых медицинское изделие, удовлетворяющее конкретному стандарту, считают соответствующим действующему регулированию.
На стандарты могут также ссылаться при судебных разбирательствах в случаях, когда это обоснованно ожидается обществом.
Разработчики должны быть осведомлены о возможном влиянии законодательства и регулирования на разрабатываемый стандарт.
4 Структура для разработки стандарта по безопасности медицинских изделий, основанная на риске
4.1 Общие положения
При применении ИСО 14971 к рискам медицинского изделия выбранные меры по управлению риском могут включать как особенности проектирования продукции, так и управления процессами. Выбор из доступных мер по управлению риском зависит от их результативности и выполнимости. Точно так же при применении принципов ИСО 14971 к разработке стандартов стандарт может являться стандартом на продукцию, стандартом на процесс или их комбинацией (см. 3.3). Выбор зависит от результата анализа идентифицированных рисков медицинского изделия или процесса. Область применения стандарта должна отражать такой выбор.
Настоящий раздел соответствует процедурным этапам ИСО 14971. Это позволяет разработчикам стандартов использовать систематический подход при идентификации потребности в стандарте по безопасности и управлению риском, который должен быть включен в стандарт, для осуществления менеджмента риска. Также эти этапы могут помочь в подготовке входных данных для нового проекта. Стандарты на продукцию и стандарты на процесс могут быть использованы как различные подходы к управлению рисками продукции.
Хотя положения раздела 4 ориентированы на продукцию, они могут также применяется в отношении результата, приводящего к выбору разработки стандарта на процесс.
Для этого имеются две причины:
- так как менеджмент рисков различных типов может повлечь за собой необходимость управления процессами, используя при их управлении различные процедуры, модели или методы, разработчикам стандартов на процессы необходимо обеспечить, чтобы цели менеджмента риска в отношении продукции были достигнуты;
- ценность стандартов на процессы определяется их полноценностью в достижении целей менеджмента риска продукции.
Таким образом, 4.2-4.8 могут рассматриваться как помощь на этапе идентификации проблемы, 4.9 - как помощь при решении потребности в менеджменте рисков, 4.10 - как помощь в решениях относительно методов управления рисками, например, безопасная конструкция, управление процессами или параметрами процессов, информация по безопасности и т.д.
4.2 Менеджмент структуры стандарта, основанной на риске
Если принято решение о потребности в стандарте по безопасности медицинских изделий, то независимо от типа стандарта начальная задача должна быть сведена к следующему:
- определить область применения - медицинское изделие или процесс;
- идентифицировать характеристики, связанные с безопасностью медицинского изделия или связанного с ним процесса;
- идентифицировать опасности и опасные ситуации, связанные с использованием медицинских изделий и их жизненным циклом;
- определить, должны ли управляться риски, обусловленные этими опасностями;
- определить риски, которые могут управляться с помощью применения разрабатываемого стандарта.
4.3 Применение и характеристики
Установление области применения (медицинское изделие или процесс), включает следующее:
a) предполагаемый медицинский показатель (например, состояние или болезнь, которая будет обнаружена, наблюдаться, изучаться, диагностироваться или предотвращаться);
b) предполагаемая группа пациентов (например, возраст, масса, здоровье, состояние);
c) предполагаемая часть тела, к которой применяется изделие, или тип ткани, с которым оно взаимодействует;
d) предполагаемый профиль пользователя;
e) предполагаемая окружающая среда применения, состояние и методы использования;
f) предварительные процедуры и процедуры обслуживания до использования (например, стерилизация, сборка, калибровка).
При рассмотрении потребности в разработке стандарта должны быть приняты во внимание характеристики медицинского изделия или процесса, которые могут оказать влияние на безопасность пациентов и пользователей. Типичные характеристики включают следующее:
- стерильность;
- биологическая совместимость;
- пирогенность;
- надежность;
- эксплуатационная пригодность;
- функциональные возможности;
- чувствительность и специфичность;
- электрическая безопасность;
- механическое сопротивление;
- радиационная безопасность.
Для систематической идентификации опасности и опасных ситуаций, важно определить глубину анализа рассматриваемого медицинского изделия или процесса, чтобы установить, где могут возникнуть проблемы с безопасностью.
4.4 Идентификации опасности и опасных ситуаций
Идентификация опасности обычно включает в себя следующие этапы:
- идентификация предполагаемых опасностей, связанных с обычным использованием медицинского изделия или с обоснованно прогнозируемым неправильным применением;
- идентификация последовательности событий, которые могли бы привести к опасной ситуации.
После того как идентифицированы известные и предполагаемые опасности, связанные с медицинским изделием, как при работе в нормальном состоянии, так и в состоянии неисправности, должна быть рассмотрена прогнозируемая последовательность событий, которые могут привести к опасной ситуации и причинить вред. Эти отношения показаны на рисунке 1.
Опасность не может привести к причинению вреда до тех пор, пока последовательность событий или другие обстоятельства (в том числе условия нормального применения) не приведут к возникновению опасной ситуации. На данном этапе риск может быть оценен путем определения тяжести и вероятности причинения вреда, которым могла бы закончиться опасная ситуация. Следует обратить внимание на то, что прогнозируемая последовательность событий, связанная с состояниями неисправности может включать действия, связанные с производством, установкой или обслуживанием медицинского изделия.
|
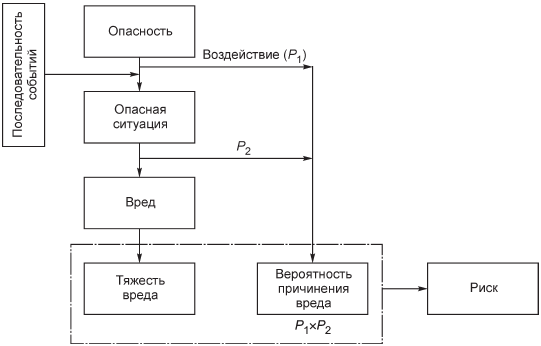
- вероятность возникновения опасной ситуации;
- вероятность возникновения опасной ситуации, ведущей к причинению вреда
Рисунок 1 - Схема взаимосвязи между опасностью, последовательностью событий, опасной ситуацией и вредом
4.5 Типы опасностей и опасных ситуаций
4.5.1 Общие положения
Для подробного рассмотрения примеров опасностей и опасных ситуаций см. ИСО 14971, приложение Е.
4.5.2 Опасности и опасные ситуации, связанные с медицинским изделием
В значительной степени возможные опасности, связанные с медицинским изделием, зависят от характера этого изделия.
Так как невозможно обеспечить, чтобы медицинское изделие никогда не вышло из строя, стандарт должен распознать опасности, присущие медицинским изделиям, поскольку они имеют отношение к состоянию, здоровью и безопасности пациента, а также здоровью и безопасности пользователя. Предпочтительным результатом в случае возникновения отказа должен являться отказ по безопасному сценарию.
В то же время в качестве альтернативы, когда это возможно или целесообразно, медицинское изделие должно сгенерировать предупреждающий сигнал до отказа или во время отказа. Хотя функция генерирования сигнала предупреждения и тревоги более легко может быть реализована в активных медицинских изделиях, она может быть включена во многие неактивные медицинские изделия.
Некоторые примеры опасностей и опасных ситуаций, связанных с медицинским изделием, приведены ниже:
a) отказ медицинского изделия, приводящий к невыполнению его предназначенной функции, например, отказ дефибриллятора произвести надлежащий разряд, механический отказ протеза клапана сердца;
b) неправильное срабатывание медицинского изделия, например, автоматический внешний кардиостимулятор произвел сигнал в несоответствующее время;
c) для активных медицинских изделий - опасности, возникающие из-за энергий, используемых для надлежащего функционирования медицинского изделия, например, короткое замыкание электрического тока на пациента или пользователя, чрезмерное или неадекватное нагревание или охлаждение пациента или части его тела;
d) микроорганизмы, занесенные нестерильным медицинским изделием, например, из-за неправильно проведенной стерилизации или отказа барьера для обеспечения стерильности;
e) опасности, непредвиденные для функции медицинских изделий, например, вызванные воспламенением огнеопасных материалов около медицинского изделия;
f) опасные ситуации, являющиеся результатом неправильной интерпретации результатов, полученных от медицинского изделия, например, интерпретация результатов in vitro диагностики или других диагностических результатов, интерпретация полученных данных, сигнала тревоги от устройств мониторинга жизненных функций;
g) опасные ситуации, являющиеся результатом механического отказа медицинского изделия или его вспомогательного оборудования, например, разрушение части стола рентгеновской установки, поломка салазок больничной кровати;
h) опасности, связанные с токсичностью материалов, используемых в медицинском изделии или выщелачиваемых из медицинского изделия, отсутствие должной биологической совместимости составляющих материалов медицинского изделия; эти опасности могут быть непосредственными (проявляются вскоре после первоначального использования медицинского изделия) или отдаленными (проявляются только после длительного применения медицинского изделия);
i) опасные ситуации, являющиеся результатом взаимодействия несовместимых изделий.
4.5.3 Опасности и опасные ситуации, связанные с пациентом
Некоторые опасности и опасные ситуации напрямую связаны с пациентом, а также с состоянием окружающей среды в зоне нахождения пациента.
Некоторые примеры приведены ниже:
a) неспособность пациента или пользователя обнаружить присутствие некоторых опасностей, таких как ионизирующее или высокочастотное излучение;
b) отсутствие у пациента нормальной реакции, которая должна проявляться исходя из его физического состояния, например, по мнению пациента, на него не действует анестезия;
c) пониженный иммунитет пациента не способен противостоять инфекции;
d) состояние окружающей среды в зоне нахождения пациента, которое может являться сочетанием температуры и влажности, а также опасностей взрыва, связанных с присутствием чистого кислорода, окиси азота и других различных агентов, обезболивающих или чистящих средств;
е) аллергия у пациента или другая гиперчувствительность к материалу, используемому в медицинском изделии, например, натуральный латекс.
4.5.4 Опасности и опасные ситуации, связанные с применением
Некоторые опасности и опасные ситуации напрямую связаны с применением медицинского изделия. Некоторые примеры приведены ниже:
a) неправильные процедуры предварительной подготовки и обслуживания, проводимые до использования медицинского изделия;
b) использование медицинского изделия по назначению, но в условиях, которые не предусмотрены изготовителем;
c) использование медицинского изделия, когда пользователь в замешательстве, отвлечен или утомлен;
d) использование медицинского изделия непрофессиональным пользователем или необученным человеком, например, изделия для самостоятельной диагностики.
4.5.5 Опасности и опасные ситуации, связанные с исполнением профессиональных обязанностей
При использовании, обслуживании или ремонте некоторых медицинских изделий пользователь или другие лица могут быть подвергнуты инфекционному заражению, воздействию опасных химических веществ, радиации или другим опасностям, связанным с их профессией. Для пользователя или других лиц риск от таких опасностей должен быть допустимым.
4.6 Систематический или случайный характер рисков
4.6.1 Общие положения
Наличие опасностей и возникновение опасных ситуаций может носить систематический или случайный характер; поэтому риски могут быть систематическими или случайными.
Разработчики стандартов должны учитывать как систематические, так и случайные риски.
Разработчики стандартов должны также знать, что некоторые опасности и возникающие опасные ситуации могут быть связаны как с систематическими, так и со случайными событиями. Например, токсичное вещество могло попасть на медицинское изделие в результате систематических несоответствий в производственном процессе (например, прохождение не всех этапов очистки, недостаточное проветривание после стерилизации). С другой стороны, случайные изменения качества сырья или иные вещества, используемые в течение обработки, могут также привести к присутствию токсичных веществ.
4.6.2 Риски, являющиеся результатом систематических событий
Достоверность определения риска возрастает, когда на основании точных и надежных данных может быть установлено количественное определение вероятности возникновения вреда, или имеется возможность выполнить обоснованное качественное определение. К сожалению, это не всегда возможно. Например, вероятность систематических неисправностей чрезвычайно трудно определить. Когда точность определения вероятности вызывает сомнение, необходимо или установить стопроцентную вероятность, или принять решение и обосновать приоритет некой специфической ценности (например, важной функциональной характеристики).
Примеры, где определение вероятности затруднительно:
a) сбой программного обеспечения;
b) поведение человека, например, ошибки эксплуатации, вызванные неадекватным проектированием или ошибками в инструкциях по применению;
c) сложные ситуации;
d) новые медицинские назначения, в отношении которых недостаточно клинических данных.
Если отсутствует достаточное количество данных относительно вероятности возникновения вреда, то любое определение риска невозможно. В таких случаях риск необходимо оценивать только на основании характера вреда. Должное управление риском может принять в качестве приоритета уровень тяжести. Часто следует установить требуемую строгость управления риском, рассматривая тяжесть последствия различных видов систематических случаев, а также результативность внешних мер по управлению риском. Чем больше последствие и менее результативны внешние меры по управлению риском, тем выше требуемая строгость управления риском.
Примеры мер по управлению риском могут включать следующее:
- строгость применяемых процессов проектирования, разработки и производства: обычно предполагается, что чем строже процессы, использованные в проектировании и разработке или производстве, тем ниже вероятность возникновения систематических неисправностей, которые будут или не будут обнаружены;
- применение принципа избыточности в отношении мер по управлению риском: хотя иногда определение вероятности является невозможным в отношении любой отдельной меры по управлению риском, использование одной или более независимых дополнительных мер по управлению риском, как правило, увеличивает общую защиту;
- сокращение промежутка времени, в пределах которого должны произойти два или больше независимых события, способствующих возникновению опасной ситуации, приводящей к вреду: детальные меры могут быть разработаны в диапазоне от периодических самопроверок до периодического обслуживания;
- применение процессов и механизмов для непрерывного мониторинга критических параметров, а также последующего оценивания и корректирующих действий.
4.6.3 Риски, являющиеся результатом случайных событий
Для многих событий, ведущих к появлению неисправности, может быть проведена числовая оценка вероятности. Количественное определение может быть использовано только тогда, когда имеется достаточная информация об опасности и обстоятельствах, влияющих на вероятность возникновения опасной ситуации.
Некоторые примеры случайных неисправностей приведены ниже:
- отказ составной части, например, интегральной микросхемы на печатной плате;
- загрязнение реактива для in vitro диагностики, которое ведет к ухудшению его характеристик и приводит к неправильным результатам;
- внешнее или внутреннее инфекционное загрязнение медицинского изделия;
- внешнее или внутреннее загрязнение медицинского изделия токсичным веществом. Риск, являющийся результатом вымывания токсичного вещества из изделия, может быть определен и оценен с помощью ИСО 10993-17. В этом случае может быть обеспечена уверенность в том, что степень воздействия, которое ожидается от применения изделия, является незначительной для причинения вреда здоровью;
- ошибка эксплуатации, являющаяся результатом промахов, упущений и заблуждений.
4.7 Определение риска
4.7.1 Общие положения
Настоящий стандарт не предлагает использование разработчиками стандартов специфического инструмента или метода определения риска. В принципе, не следует отдавать предпочтение только количественному или только качественному определению вероятности возникновения вреда.
Для установления в стандартах необходимого и достаточного перечня требований разработчики стандартов должны предложить такой способ определения риска, который был бы основан на доступных данных, представляющих последние достижения в этой области. Предпочтение следует отдавать опубликованным проверенным данным, специфичным для рассматриваемого медицинского изделия. Ссылка на такие данные должна быть включена в стандарт.
Результаты деятельности по определению риска необходимо регистрировать с такой степенью детализации, чтобы была установлена прослеживаемость к используемым источникам.
Там, где разработчики стандартов полагают, что существует проблема, связанная с безопасностью, и ее необходимо отразить в стандарте, они должны документировать такие предположения и разъяснять их.
4.7.2 Компоненты риска
Концепция риска предполагает наличие двух составляющих, которые должны быть проанализированы отдельно:
a) вероятность возникновения вреда;
b) тяжесть этого вреда.
Определение риска должно включать рассмотрение, например, следующего:
- характер и тяжесть вреда, который мог быть причинен;
- последовательность событий, которые могли бы привести к возникновению опасной ситуации;
- вероятность того, что опасная ситуация приведет к вреду (см. рисунок 1);
- инициирующее событие или обстоятельство.
4.7.3 Сравнение количественного и качественного методов
В основном регулирование медицинских изделий обозначает тяжесть вреда качественно, например, когда устанавливаются критерии для сообщения об инциденте или отзыва продукции. Поскольку для изготовителей и регулирующих органов стандарты по безопасности медицинских изделий могут быть ссылочными, использование сопоставимых качественных уровней может увеличить ценность стандартов по безопасности медицинских изделий в отношении проектирования и процедур сообщения об инцидентах или отзыва продукции.
Количественное определение вероятности возникновения вреда может быть затруднено факторами, способствующими определению полной вероятности, которые имеют случайный характер и могут быть подтверждены достоверными статистическими данными только в перспективе. Необходимо понимать, что вероятность систематических событий (см. 4.6.2) не всегда может быть подтверждена количественными результатами.
4.8 Критерии допустимости риска
4.8.1 Общие положения
Для разработчиков стандартов важно установить критерии допустимости, подходящие для использования при оценивании применимости требований в стандарте.
4.8.2 Установление критериев
Методы установления допустимости риска включают следующие, не ограничиваясь ими:
- использование применимых стандартов, представляющих собой последние достижения в соответствующей области или лучшую признанную практику, а также требования, достаточные для демонстрации допустимости риска;
- сравнение уровней риска с уже находящимися в применении медицинскими изделиями может считаться как использование последних достижений или лучшей признанной практики;
- использование мнения экспертов;
- использование результатов научных исследований, включая клинические данные.
4.8.3 Последние достижения или лучшая признанная практика
В настоящем стандарте понятие "Последние достижения или лучшая признанная практика" используется для обозначения того, что в настоящее время является общепринятым и обычно расценивается как надлежащая практика. "Последние достижения или лучшая признанная практика" не обязательно подразумевает наиболее технологически продвинутое решение.
4.8.4 Общественные ценности
Общеизвестно, что субъективное восприятие риска часто отличается от значения риска, определенного опытным путем. При решении того, какой риск является допустимым, должно быть принято во внимание восприятие этого риска широкой общественностью. Для согласованности с ожиданиями общества и общественным мнением необходимо к некоторым рискам сделать дополнительный "запас".
4.8.5 Социальные ожидания
Восприятие риска может отличаться в различных социальных сообществах/популяциях, и это должно быть принято во внимание. Процесс разработки и утверждения стандарта по безопасности медицинского изделия должен обеспечивать, что различные социальные ожидания будут учтены до принятия стандарта.
4.9 Оценивание риска
Используя критерии допустимости риска, установленные в 4.7, разработчики стандартов должны принять решение относительно необходимости управления рассматриваемыми рисками. Если риск может быть управляемым, то разработчики должны установить соответствующее требование по управлению риском в стандарте на продукцию или на процесс.
4.10 Управление риском
4.10.1 Общие положения
Меры по управлению риском могут уменьшить тяжесть вреда, вероятность его возникновения или того и другого. При выборе мер по управлению риском в отношении рисков, идентифицированных в 4.4, которые могут быть включены в стандарт, рекомендуется, чтобы сначала были рассмотрены общие методы, пригодные для уменьшения риска.
Такие методы включают:
a) надлежащее проектирование, принимающее во внимание предполагаемый риск, а также способы его исключения или снижения;
b) производство изделий в соответствии с подходящей системой или процессом, например управляемый производственный процесс или система качества, которые ориентированы на риск;
c) выбор пользователем соответствующего изделия, который, в свою очередь, зависит от предоставления изготовителем ясного описания предусмотренного назначения изделия;
d) понимание изделия пользователем, которое может зависеть от обучения или от полноты и ясности инструкций, а также предупреждений, которые установлены изготовителем (это особенно важно в случае изделий, предназначенных для использования в домашних условиях);
e) идентификация и использование совместимых принадлежностей;
f) повторная стерилизация медицинского изделия или его части, необходимая для использования медицинского изделия;
g) выполнение предписанного предупредительного ремонта и обслуживания, включая компоненты изделия.
Обсуждение таких методов должно привести к разработке требований, устанавливаемых к продукции или к процессу, в зависимости от области применения стандарта, способных внести вклад в снижение рисков, связанных с этим изделием или процессом.
4.10.2 Иерархический подход к управлению риском
При выборе мер по управлению риском, способных уменьшить риск(и) до допустимого уровня, в порядке приоритета должны быть применены следующие варианты управления риском:
a) безопасность, обеспечиваемая проектом;
b) защитные меры, включая:
- наличие сигнала тревоги, встроенного в медицинское изделие, или
- защитные меры, предусмотренные в процессе производства;
c) информация по безопасности.
4.10.3 Остаточные риски
4.10.3.1 Общие положения
После разработки перечня мер по управлению риском должно быть установлено, сможет ли осуществление этих мер сделать риск допустимым. Если в отношении допустимости риска имеются обоснованные сомнения, следует рассмотреть дополнительные меры по управлению риском. Такая повторяющаяся процедура должна продолжаться до тех пор, пока риск не будет уменьшен до допустимых уровней.
Пользователь стандарта должен быть обеспечен уместной информацией относительно остаточных рисков. Такое информирование должно позволять пользователю принимать обоснованное решение относительно достаточности стандарта для конкретной продукции или процесса.
Все требования стандарта должны базироваться на объективной оценке риска, которая демонстрирует, что остаточные риски являются допустимыми.
4.10.3.2 Верификация результативности
Меры по управлению риском и связанные с ними методы испытаний, установленные в стандартах, должны быть верифицированы относительно их результативности, причем до издания стандарта. Верификация частично может быть проведена на основе анализа научной литературы или другим способом. В некоторых случаях разработчики стандартов могут принять решение установить систему межлабораторных исследований.
4.10.3.3 Валидация того, что потребности пользователя выполнены
Требование проводить валидацию того, что потребности пользователя выполнены, было выдвинуто в результате международного процесса согласования, при котором стандарт рассылают всем заинтересованным сторонам для нескольких раундов голосования и комментариев.
4.10.3.4 Влияние вводимых мер по управлению риском
Разработчики стандартов должны рассмотреть воздействие введенных мер по управлению риском на медицинское изделие, а также возможность возникновения из-за этих мер новых рисков, которые могли бы изменить первоначальную оценку риска. Например, разработчики стандартов должны оценить воздействие меры по управлению риском на эксплуатационную пригодность медицинского изделия.
4.11 Заключение
Выполнение вышеуказанных этапов облегчит разработку полноценного, основанного на концепции риска, стандарта на продукцию или процесс. Кроме того, такой подход облегчает обеспечение выполнения системы менеджмента риска, совместимой с ИСО 14971.
5 Практическая помощь по выполнению требований ИСО 14971 посредством стандартов на продукцию или процессы
5.1 Стандарты на продукцию
Если стандарт на продукцию устанавливает требования, которые предназначены для управления риском, связанным с медицинским изделием, то должна быть приведена следующая информация:
a) обоснование выбора предельных значений параметров, деталей конструкции, перечня опасных обстоятельств, а также обоснование вывода о том, что принятые решения можно счесть безопасными (например, в отношении научной литературы);
b) область применения, которая четко определяет продукцию, а также предусмотренное назначение этой продукции;
с) методы испытаний и предельные значения параметров, которые обеспечивают уверенность в том, что риски уменьшены до допустимых уровней, информация о которых должна содержать, по крайней мере, следующее:
- предполагаемый медицинский признак (например, состояние или болезнь, которая будет обнаружена, наблюдаться, изучаться, диагностироваться или предотвращаться);
- предполагаемая группа пациентов (например, возраст, масса, здоровье, состояние);
- предполагаемая часть тела, к которой применяется изделие, или тип ткани, с которым оно взаимодействует;
- предполагаемый профиль пользователя;
- предполагаемая окружающая среда применения, состояния и методы использования;
- предварительные процедуры и процедуры обслуживания до использования (например, стерилизация, сборка, калибровка);
- процедуры ремонта и обслуживания в течение всего жизненного цикла медицинского изделия.
Приведенная выше информация позволит изготовителям устанавливать степень вклада выполнения требований стандарта в удовлетворение их потребности в управлении риском.
5.2 Стандарты на процессы
Если стандарт на процесс устанавливает требования, которые предназначены для управления риском(ами), связанным(и) с медицинским изделием, то должна быть приведена следующая информация:
- обоснование вывода о том, что применение стандарта уменьшит риск до допустимого уровня, как того требует ИСО 14971;
- методы снижения риска, а также методы получения поддающихся проверке результатов, позволяющих сделать обоснованный вывод о том, что остаточный риск является допустимым, и
- методы для установления целей (например, цели эксплуатационной пригодности, описанные в МЭК 62366), достижение которых, как предполагается, приводит к достижению допустимого остаточного риска.
Стандарт на процесс должен предоставить такую информацию, которая способствовала бы выполнению требований ИСО 14971 в отношении определения риска, управления риском, а также верификации мер по управлению риском. Хотя валидация процесса не является требованием ИСО 14971, некоторые стандарты на процессы включают этап валидации для производственных процессов. Валидация процесса может демонстрировать, что с целью выполнения критериев допустимости, было достигнуто достаточное уменьшение риска.
Приведенная выше информация позволит изготовителям устанавливать степень вклада выполнения требований стандарта в удовлетворение их потребности в управлении риском.
5.3 Краткий обзор применения стандартов по безопасности медицинских изделий в рамках ИСО 14971
Рекомендуется указывать в виде ссылки, как стандарты по безопасности на процессы и продукцию могут облегчить выполнение процесса менеджмента риска в соответствии с требованиями ИСО 14971 (например, это может быть сделано, как приведено в таблице 1). Если стандарты по безопасности на процессы или продукцию не устанавливают измеримые параметры и допустимые предельные значения, то для изготовителя следует установить метод для разработки таких допустимых предельных значений. В то же время установление соответствующих критериев допустимости должно повлечь использование ИСО 14971. Разработчики стандартов должны заботиться, в первую очередь, о том, чтобы побуждать изготовителей использовать процесс, установленный в ИСО 14971, с целью обеспечения как большей результативности, так и безопасности.
Таблица 1 - Руководство по применению стандартов по безопасности на процессы и продукцию для облегчения выполнения процесса менеджмента риска
ИСО 14971 раздел/пункт | Стандарт по безопасности продукции | Стандарты на процесс |
3.2 Ответственность высшего руководства | Может быть применим частично | Может определить дополнительную ответственность и задачи для менеджмента |
3.4 План менеджмента риска | Может помочь устанавливать критерии допустимости риска | Может предусматривать процедуры для планирования менеджмента риска |
3.5 Файл менеджмента риска | Может определить некоторые сведения, которые будут включены в файл менеджмента риска | |
4.2 Предусмотренное применение | Может идентифицировать предусмотренное применение и обоснованно прогнозируемое неправильное применение продукции и процессов, на которые распространяется стандарт, а также характеристики, связанные с безопасностью, к которым обращается стандарт | |
4.3 Идентификация опасностей | Может идентифицировать известные и прогнозируемые опасности, связанные с медицинским изделием, которые будут рассмотрены при анализе риска этого изделия | Может предоставлять формальные процедуры идентификации известных и прогнозируемых опасностей, связанных с медицинским изделием |
4.4 Определение риска(ов) от опасной ситуации | Может предоставлять информацию о степени тяжести вреда или вероятности возникновения вреда при возникновении опасных ситуаций, которая может использоваться изготовителем при анализе риска медицинского изделия | Может предоставлять конкретную информацию или процедуры для систематического сбора информации в отношении степени тяжести вреда или вероятности возникновения вреда при возникновении опасных ситуаций |
5 Оценивание риска | Предельные значения, которые устанавливаются в стандарте, обычно отражают лучшую общепринятую практику | Может предоставлять методы или процедуры по оцениванию и установлению допустимости риска |
6.2 Анализ возможностей управления риском | Может предоставлять возможные варианты управления риском, предположительно являющиеся результативными для снижения риска, которые могут быть осуществлены при проектировании изготовителем медицинского изделия | Может предоставлять возможные варианты управления риском, предположительно являющиеся результативными для снижения риска, которые могут быть осуществлены изготовителем |
6.3 Выполнение мер по управлению риском | Может предусматривать испытания для верификации результативности мер по управлению риском | Может предоставлять возможные меры по управлению риском, предположительно являющиеся результативными для снижения риска, которые могут быть осуществлены и верифицированы изготовителем |
6.4 Оценивание остаточного риска | Предельные значения, которые устанавливаются в стандарте, обычно отражают лучшую общепринятую практику при управлении риском | Может предоставлять методы или процедуры по оцениванию и установлению допустимости риска (см. раздел 5 "Оценивание риска") |
9 Производственная и постпроизводственная информация | Может предусматривать определенные требования для механизмов сбора информации на стадиях производства и постпроизводства | Может предоставлять процедуры для выбора и оценивания постпроизводственной информации для последующих корректирующих действий |
Приложение А
(справочное)
Стандарты по безопасности на продукцию и процессы
А.1 Стандарты на продукцию
Как правило, с целью содействия обеспечению безопасности и результативности медицинских изделий, стандарты на продукцию должны базироваться на научных данных. Такие данные могут быть получены от исследований в условиях лаборатории или клиники, при использовании таких последних достижений в области научно обоснованных методов, как статистическая обработка результатов, метод сравнения или лучших практических решений, которые используются в таких же изделиях или изделиях подобного типа.
С целью управления риском до допустимого уровня стандарты на продукцию могут использовать разнообразные прикладные методы, например:
- установление общепринятых в настоящее время предельных значений таких показателей безопасности, как физическое, химическое или биологическое воздействие на человека (доза рентгена, предельные значения электрического тока, ограничения температуры поверхности, предел бионагрузки, предельные значения для вымываемых веществ);
- стандартизированные состояния окружающей среды в зоне нахождения пациента (диапазоны температуры и влажности, электромагнитные поля, особо управляемые условия в операционных);
- стандартизированные интерфейсы человек - изделие (индикаторы, различные цвета, символы, тревожная сигнализация, требования к документации);
- установление общепринятых в настоящее время конструктивных деталей (электрическая изоляция, кабельные соединения);
- стандартизированные испытания для демонстрации соответствия (испытания на электромагнитную совместимость, испытания на биосовместимость).
А.2 Стандарты на процессы
Общепризнано, что стандарты на процессы способны внести вклад в обеспечение безопасности и результативности, но при этом весьма трудно определить степень этого вклада количественно. Тем не менее, стандарты могут отражать общепринятую в настоящее время практику, главным образом фокусируясь на тех аспектах, которые приводят к достижению желаемого результата управляемым способом (например, соответствие спецификации по безопасности). Стандарты на процессы также вносят вклад в обеспечение уверенности в том, что стандарты на продукцию последовательно осуществлены.
Стандарты на процессы, включая процессы управления проектированием, валидации, производства и обслуживания, могут внести вклад в безопасность медицинских изделий посредством стандартизации этих процессов, которая является существенной для создания безопасных медицинских изделий. Ниже приведены примеры стандартов на процессы, которые облегчают менеджмент риска, хотя они и не разработаны специально для целей менеджмента риска:
- общепринятое в настоящее время управление документацией, которое является предпосылкой для любого процесса создания продукции; обычно является элементом системы менеджмента качества (например, ИСО 13485);
- согласованные рамки для выбора биологических испытаний (например, серия стандартов ИСО 10993) и согласованные методы на процессы стерилизации медицинских изделий (например, ИСО 11135-1 или ИСО 11137-1).
Приложение В
(справочное)
Информация по риску
Разработчики стандартов должны определить источники или методы получения информации о риске. К такой информации, например, относится вред, который мог бы следовать от некоторых опасностей, вероятность возникновения вреда в результате создания опасной ситуации или данных надежности, которые могут иметь отношение к связанным рискам. Эта информация может быть получена от внешних источников или экспериментов, проведенных внутри организации (система сбора и анализа данных).
Возможные источники данных для информации по риску включают:
- систематический анализ рассмотренной литературы, с использованием медицинских и парамедицинских баз данных;
- технические документы от соответствующих комитетов по стандартизации;
- научную литературу и внутренние документы промышленных ассоциаций;
- другие неопубликованные источники, известные экспертам в этой области;
- необработанные данные из открытых источников;
- базы данных компетентных органов власти;
- основные данные надежности;
- ссылки в других стандартах;
- журналы и непроверенные источники из сети Интернет.
Необходимо с осторожностью подходить к выбору информационных источников.
Библиография
[1] | ИСО 5840:2005 | Сердечно-сосудистые имплантаты. Протезы клапанов сердца |
ISO 5840:2005 | Cardiovascular implants - Cardiac valve prostheses | |
[2] | ИСО 9000:2005 | Системы менеджмента качества. Основные положения и словарь |
ISO 9000:2005 | Quality management systems - Fundamentals and vocabulary | |
[3] | ИСО 10993 (все части) | Изделия медицинские. Оценка биологического действия медицинских изделий |
ISO 10993 (all parts) | Biological evaluation of medical devices | |
[4] | ИСО 10993-17 | Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 17. Установление пороговых значений для вымываемых веществ |
ISO 10993-17 | Biological evaluation of medical devices - Part 17: Establishment of allowable limits for leachable substances | |
[5] | ИСО 11135-1 | Стерилизация медицинской продукции. Стерилизация оксидом этилена. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий |
ISO 11135-1 | Sterilization of health care products - Ethylene oxide - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices | |
[6] | ИСО 11137-1 | Стерилизация медицинской продукции. Радиационная стерилизация. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий |
ISO 11137-1 | Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices | |
[7] | ИСО 13485:2003 | Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования |
ISO 13485:2003 | Medical devices - Quality management systems - Requirements for regulatory purposes | |
[8] | ИСО 14971:2007 | Изделия медицинские. Применение менеджмента риска к медицинским изделиям |
ISO 14971:2007 | Medical devices - Application of risk management to medical devices | |
[9] | Руководство | Аспекты безопасности. Руководящие указания по включению их в стандарты |
ISO/IEC Guide 51:1999 | Safety aspects - Guidelines for their inclusion in standards | |
[10] | МЭК 62366:2007 | Медицинские изделия. Проектирование медицинских изделий с учетом эксплуатационной пригодности |
IEC 62366:2007 | Medical devices - Application of usability engineering to medical devices | |
[11] | Директива ИСО/МЭК, Часть 2, 2011 | Правила построения и разработки международных стандартов |
ISO/IEC Directives, Part 2, 2011 | Rules for the structure and drafting of International Standard |
УДК 006.83:006.354 | ОКС 01.080.20, |
11.040.01 | |
Ключевые слова: аспекты безопасности, медицинские изделия, международные стандарты, руководство | |
Электронный текст документа
и сверен по:
, 2020

{kind=link}