ГОСТ 7636-85
Группа Н29
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
РЫБА, МОРСКИЕ МЛЕКОПИТАЮЩИЕ, МОРСКИЕ БЕСПОЗВОНОЧНЫЕ И ПРОДУКТЫ ИХ ПЕРЕРАБОТКИ
Методы анализа
Fish, marine mammals, invertebrates and products of their processing. Methods of analysis
МКС 67.120.30
ОКСТУ 9109
Дата введения 1986-01-01
Постановлением Государственного комитета СССР по стандартам от 27 марта 1985 г. N 898 дата введения установлена 01.01.86
Ограничение срока действия снято по протоколу N 5-94 Межгосударственного совета по стандартизации, метрологии и сертификации (ИУС 11-12-94)
ВЗАМЕН ГОСТ 7636-55; ГОСТ 13893-68; ГОСТ 13929-68, кроме определения хлористого натрия в водорослях и продуктах их переработки; ГОСТ 13930-68, кроме разд.2 и 3 в части определения влажности рыбной муки, муки из морских млекопитающих и ракообразных и определения влаги в водорослях и продуктах их переработки; ГОСТ 17258-71, ГОСТ 17259-71, ГОСТ 18657-73, ГОСТ 8714-72 в части разд.3, пп.3.3-3.8; ГОСТ 18170-72 в части разд.3, пп.3.3-3.5; ГОСТ 7047-55 разд.II в части определения витамина А в печени рыб, морских млекопитающих и морских беспозвоночных; ГОСТ 18173-72 в части разд.3, п.3.2.
ИЗДАНИЕ (июнь 2010 г.) с Поправкой (ИУС 12-2005)
ВНЕСЕНО Изменение N 1, принятое Евразийским советом по стандартизации, метрологии и сертификации (протокол N 67-П от 30.05.2014). Государство-разработчик Россия. Приказом Федерального агентства по техническому регулированию и метрологии от 25.06.2014 г. N 667-ст вводится в действие на территории РФ с 01.01.2015
Изменение N 1 внесено изготовителем базы данных по тексту ИУС N 11, 2014 год
Настоящий стандарт распространяется на рыбу, морских млекопитающих, морских беспозвоночных и продукты их переработки и устанавливает методы физического и химического анализа.
Стандарт не распространяется на рыбные консервы и пресервы.
1. МЕТОДЫ ОТБОРА ПРОБ
1.1. Отбор проб - по ГОСТ 7631-2008, ГОСТ 31339-2006 кормовой муки из рыбы, морских млекопитающих и ракообразных - по ГОСТ 13496.0-80.
2. ПОДГОТОВКА К АНАЛИЗУ СРЕДНЕЙ ПРОБЫ
2.1. Подготовка к анализу средней пробы свежей, охлажденной, мороженой, соленой, пряной, маринованной, вяленой, сушеной и копченой рыбы, сырья морских млекопитающих и пищевого рыбного фарша
2.1.1. Рыбу, отобранную для анализа, очищают от механических загрязнений, целых и крупнодробленых пряностей и чешуи. Обмывать рыбу не допускается. Мороженую рыбу предварительно размораживают до температуры в толще рыбы минус 1 °С.
2.1.2. Среднюю пробу, составленную из мелкой рыбы массой экземпляра 0,1 кг и менее (кроме бычка, мойвы, черноморской ставриды всех размеров и салаки длиной свыше 15 см), размалывают без разделки. У салаки длиной более 15 см, у бычка, черноморской ставриды перед размалыванием удаляют голову, внутренности вместе с икрой или молоками и хвостовой плавник. У мойвы удаляют голову вместе с пучком внутренностей, не разрезая брюшко, и хвостовой плавник.
2.1.3. При подготовке средней пробы, составленной из рыбы массой экземпляра от 0,1 до 1 кг, рыбу разделывают на филе: отделяют голову и плавники, разрезают тушку по брюшку и удаляют все внутренности вместе с икрой или молоками; разрезают вдоль спинки, удаляют позвоночник и, по возможности, все ребра и кожу.
У рыбы свежей, охлажденной, мороженой (за исключением рыбы с плотной кожей: акулы, макруруса, осетровых, пинагора, сома, ставриды, угря, лососевых и др.) удаляют чешую, не удаляя кожу.
2.1.4. Среднюю пробу в виде кусков, отобранную от крупной рыбы массой экземпляра более 1 кг, измельчают после обесшкуривания и удаления костей.
2.1.5. Среднюю пробу мелкой неразделанной рыбы или крупной рыбы дважды пропускают через ручную мясорубку или один раз через электрическую мясорубку. Фарш тщательно перемешивают, квартуют и часть его в количестве 100-200 г переносят в широкогорлую банку с плотно закрывающейся крышкой.
2.1.6. Средние пробы мороженого китового мяса и печени измельчают мясорубкой. При поступлении средней пробы в мороженом виде ее размораживают на воздухе до температуры от 0 до минус 1 °С.
2.1.7. Среднюю пробу соленой китовой печени перед измельчением оставляют для стекания тузлука на 20-30 мин.
2.1.8. Китовое мясо измельчают трехкратным, а печень двукратным пропусканием через мясорубку. Фарш тщательно перемешивают, отбирают 250-300 г для анализа и переносят в широкогорлую банку с плотно закрывающейся крышкой.
2.1.9. Среднюю пробу мороженого рыбного фарша размораживают на воздухе до температуры 0-2 °С, тщательно перемешивают и переносят в широкогорлую банку с притертой пробкой.
2.1.10. При определении водоудерживающей способности среднюю пробу делят на две равные части. Одну часть пробы, предназначенную для определения водоудерживающей способности, размораживают до 3-4 °С, разделывают на филе, удаляя кости, и при необходимости пропускают через мясорубку. Полученный фарш тщательно перемешивают и помещают в широкогорлую банку с плотно закрывающейся крышкой.
2.2. Подготовка к анализу средней пробы полуфабрикатов и кулинарных изделий
2.2.1. Среднюю пробу, доставленную в лабораторию, направляют на анализ не позднее чем через 30 мин. Замороженную пробу предварительно размораживают при комнатной температуре в плотно закрытой банке.
2.2.2. После определения физических показателей (длины, массы нетто, составных частей) и органолептической оценки по ГОСТ 7631-2008 пробу освобождают от несъедобных частей (кости, целые и крупнодробленые пряности и др.), плотную часть пропускают через мясорубку, смешивают с жидкой фракцией (при ее наличии) и растирают в ступке до однородной массы.
2.2.3. Рыбомучные изделия после определения соотношения составных частей (в случае необходимости) измельчают.
Начинку пропускают через мясорубку и растирают в ступке до однородной массы, а мучную часть или целые кулинарные изделия измельчают вместе с корочкой ножом или пропускают дважды через мясорубку.
При необходимости анализа кулинарных изделий с начинкой целиком составные части их смешивают.
2.2.4. Пробу, отобранную из кулинарных изделий или полуфабрикатов, приготовленных из измельченного сырья (фарш, паста и др.), перед анализом разрезают на кусочки, тщательно перемешивают и растирают в ступке до однородной массы.
2.3. Подготовка к анализу средней пробы икры
2.3.1. Пробу зернистой икры осетровых и лососевых рыб, а также пробойной икры различных видов рыб измельчают в гомогенизаторе или растирают в ступке до получения однородной массы.
2.3.2. Паюсную икру осетровых рыб не измельчают. Навески отбирают из разных мест средней пробы.
2.3.3. Среднюю пробу ястычной икры всех видов рыб дважды пропускают через мясорубку для измельчения пленок, а затем растирают в ступке до получения однородной массы.
С обвощенных ястыков предварительно удаляют слой воска.
2.4. Подготовка к анализу средней пробы концентратов, белковой массы, бульонов, гидролизатов и паст
2.4.1. Среднюю пробу концентрата рыбного белка помещают в чистую сухую банку вместимостью 500 см, перемешивают и направляют на анализ без предварительной обработки.
2.4.2. 500 г белковой массы размораживают на воздухе до температуры 0 минус 2 °С, пропускают через мясорубку, тщательно перемешивают, квартируют и часть ее переносят в широкогорлую банку с плотно закрывающейся крышкой.
2.4.3. Среднюю пробу гидролизата и рыбного бульона помещают в стеклянную банку с притертой пробкой вместимостью 500 см. Перед анализом содержимое банки тщательно перемешивают.
2.4.4. При подготовке средней пробы пасты содержимое вскрытых банок тщательно перемешивают и растирают в ступке до получения однородной массы.
2.5. Подготовка к анализу средней пробы жиров и жидких витаминных препаратов
2.5.1. Среднюю пробу исследуемого жира тщательно взбалтывают в течение 3-5 мин и делят на две части.
Одну часть фильтруют через бумажный складчатый фильтр (при температуре, указанной в стандарте на данный жир для определения прозрачности) и затем используют для определения цвета, плотности, кислотного числа, йодного числа, числа омыления, неомыляемых веществ и др.
Нефильтрованную часть средней пробы используют для определения прозрачности, содержания воды и примесей нежирового характера.
2.5.2. Среднюю пробу жидких витаминных препаратов фильтруют при температуре, указанной в стандарте для определения прозрачности, и направляют на анализ.
2.6. Подготовка к анализу средней пробы кормовой муки из рыбы, морских млекопитающих и ракообразных
2.6.1. Поступившую в лабораторию среднюю пробу муки в количестве 500 г делят методом квартования на две части.
Одну часть просеивают через металлическое сито со стороной отверстий 1 мм. Не прошедшую через сито муку растирают в фарфоровой ступке или на лабораторной мельнице и снова просеивают. Растирание и просеивание продолжают до тех пор, пока вся мука не будет просеяна через сито. Просеянную муку тщательно перемешивают и помещают в банку с притертой пробкой. Непросеянную часть муки оставляют для определения крупности помола, наличия песка, частиц железа (металлических примесей) и обнаружения примеси стекла.
2.7. Подготовка к анализу средней пробы рыбного клея
2.7.1. Среднюю пробу жидкого технического клея, поступившую в лабораторию, хранят в широкогорлой банке вместимостью 500 см с притертой пробкой; перед анализом содержимое банки тщательно перемешивают.
2.7.2. Среднюю пробу пищевого клея в количестве 0,3 кг предварительно нагревают на водяной бане при температуре 85-90 °С до полного расплавления клея, а затем после перемешивания отбирают часть его и помещают в чистую сухую широкогорлую банку.
2.7.3. При подготовке пробы для определения вязкости и стойкости клеевого раствора при хранении готовят 300 г раствора клея 18%-ной концентрации по товарно-сухому клею или 15%-ной концентрации по беззольному и безводному клею. Навеску клея () в граммах для приготовления раствора клея вычисляют по формуле
![]() ,
,
где 15 - массовая доля безводного и беззольного клея в растворе в процентах;
- массовая доля золы в клее в процентах;
- массовая доля воды в клее в процентах.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений.
Необходимое количество дистиллированной воды для приготовления раствора 15%-ной концентрации клея вычисляют по разности между 300 г и навеской клея. Навеску клея, взвешенную с абсолютной погрешностью не более 0,01 г, помещают в стакан, приливают необходимое количество дистиллированной воды (300 г) и, накрыв стакан часовым стеклом, нагревают при помешивании на водяной бане до полного растворения клея. Концентрацию клеевого раствора проверяют клеемером по ГОСТ 2067-93.
2.8. Подготовка к анализу средней пробы кормовых продуктов, консервированных пиросульфитом натрия и кислотами
2.8.1. Среднюю пробу растирают до однородной массы и тщательно перемешивают.
2.9. Подготовка к анализу средней пробы морских беспозвоночных и продуктов их переработки
2.9.1. Средние пробы водных беспозвоночных очищают от загрязнений и при наличии излишней воды обсушивают фильтровальной бумагой или марлей.
Разделку беспозвоночных для подготовки пробы проводят аккуратно и по возможности быстро во избежание подсыхания, а мороженых - во избежание потери дефростационной воды.
Съедобные части собирают в чистую сухую посуду (кюветы, противни) и немедленно измельчают мясорубкой. Фарш тщательно перемешивают и часть его в количестве 250-300 г переносят в широкогорлую склянку с пробкой.
А. Свежие и охлажденные беспозвоночные
2.9.2. Двустворчатые моллюски. Для раскрытия раковины тонкий нож или скальпель вводят между створками и разрезают мускул-замыкатель. Из открытой раковины, надрезав мантию в передней ее части, сливают межстворчатую жидкость. Для более полного удаления жидкости раковины можно ставить на 5-10 мин вертикально на сетке, замком вверх. После этого из раковины тщательно извлекают все мясо (тело моллюска). У черноморских мидий и устриц для пробы берут всю массу заключенного в раковине тела (без биссуса).
При разделке тихоокеанской мидии, гребешка и других крупных моллюсков для средней пробы берут только съедобные части (мускул-замыкатель, мантию и половые железы). Для этого тело моллюска дополнительно разделывают, отделяя несъедобные части (желудок, кишечник, жабры и биссус). При наличии песка на поверхности съедобных частей последние тщательно очищают от него. Допускается быстрая промывка в проточной воде с последующей подсушкой поверхности фильтровальной бумагой.
Выделенное мясо дважды измельчают мясорубкой. Остаток из мясорубки тщательно измельчают ножницами и добавляют к фаршу.
2.9.3. Головоногие моллюски. При разделке целого кальмара острым ножом делают неглубокий разрез туловища от края мантии до основания плавника, стараясь не повредить мешочек с сепией. Отгибают стенки мантии и удаляют внутренности и хитиновую пластинку (раковину). Брюшную полость зачищают тупой стороной ножа. После этого разрезают голову, удаляют глаза и клюв.
У разделанного кальмара с мантии и конечностей снимают вручную с тонкого края предварительно надрезанную наружную пленку с присосками. Мясо измельчают в мясорубке.
При разделке осьминога удаляют внутренности, пищевод, ротовой аппарат, глаза и кожу вместе с присосками. Для этого осьминога кладут на спину и делают разрез вдоль головы-туловища от клюва до конца брюшной полости. Через разрез удаляют внутренности. Операцию проводят осторожно, чтобы не раздавить мешочек с сепией; удаляют глаза, клюв, выворачивают разделанного осьминога наизнанку и тщательно зачищают от остатков внутренностей, песка и крови. С туловища и конечностей снимают кожу вместе с присосками, после чего измельчают мясорубкой.
2.9.4. Ракообразные. При разделке ракообразных берут: у краба - мясо клешненосных и ходильных конечностей, у креветок и лангустов - мясо абдомена (шейки), у омаров и раков - мясо клешней и абдомена (шейки).
Отделение конечностей у крабов, клешней и абдомена у омаров, абдомена у креветок и лангустов проводят как при промысловой разделке. С ходильных конечностей краба осторожно, не нарушая кожистой пленки, прикрывающей мясо плечевого сустава, срезают жабры.
Отделенные части ракообразных очищают от остатков внутренностей; при необходимости панцирное покрытие осушают фильтровальной бумагой, после чего отделенные части помещают в чистые сухие кюветы или противни, на которых проводят дальнейшую разделку во избежание потерь студнеобразного мяса.
У краба перерезают перегородки, соединяющие конечности. Ножницами разрезают конечности на части (поперек) вблизи кожистых суставов, одновременно разрезая хитиновую пластинку, прикрепленную к суставу. Панцирные трубки разрезают вдоль и тщательно (шпателем или ложкой) извлекают мясо вместе с пигментной пленкой. Клешню разбивают резким и сильным ударом деревянного молотка, укладывая ее на чистую сухую поверхность выпуклой стороной кверху.
Пинцетом из мяса тщательно удаляют остатки кусочков панциря и хитиновые пластинки.
Для выделения мяса из абдомена (у креветок, раков и лангустов) ножницами разрезают панцирь от верхнего края шейки до тельсона, после чего аккуратно извлекают мясо вместе с пигментной пленкой.
Выделенное мясо ракообразных, полностью очищенное от панциря и хитиновых пластинок, измельчают мясорубкой.
2.9.5. Иглокожие. При разделке голотурий (трепанг, кукумария) берут оболочку с венчиком щупальцев. Тело голотурий разрезают по брюшку и спинке через анальное отверстие. Перед разделкой предварительно выпускают полостную жидкость, сделав прокол острием ножа в оболочке. Через разрез удаляют внутренности и тщательно зачищают брюшную полость от остатков внутренностей и песка. Очищенные оболочки измельчают. Для лучшего измельчения оболочки голотурий предварительно замораживают, помещая их в морозильную камеру бытового холодильника, после чего быстро пропускают через охлажденную там же мясорубку.
При отсутствии условий для замораживания оболочки разрезают на кусочки и постепенно пропускают через мясорубку. Остаток в мясорубке тщательно измельчают ножницами в узком небольшом стаканчике и добавляют к основной массе, после чего тщательно растирают в фарфоровой ступке.
При разделке морских ежей берут икру, которая расположена внутри известковой скорлупы в виде пяти желез желто-оранжевой окраски.
Для извлечения икры скорлупу ежей раскалывают при помощи ножа или щипцов на две части и ястыки осторожно извлекают из панциря деревянной лопаточкой. Можно предварительно встряхнуть расколотые половинки скорлупы для удаления основной части внутренностей, после чего лопаточкой извлечь икру. Выделенную икру тщательно растирают в фарфоровой ступке.
Б. Сыро-мороженые и варено-мороженые беспозвоночные
2.9.6. Во избежание потери воды мороженых беспозвоночных размораживают при комнатной температуре, до температуры в толще тела (блока) от 0 - до минус 1 °С.
Для размораживания беспозвоночных кладут в чистые кюветы, противни и накрывают сверху влажной тканью или бумагой во избежание подсыхания.
Жидкость, образующуюся в результате таяния ледяной глазури или налета снега, удаляют, а поверхность обсушивают фильтровальной бумагой.
Образцы беспозвоночных, разделанные без замораживания, сразу измельчают мясорубкой. Неразделанные или частично разделанные образцы дополнительно разделывают способами, изложенными в разд.А, после чего дважды измельчают мясорубкой и растирают в фарфоровой ступке.
В. Сушеная продукция из беспозвоночных
2.9.7. Среднюю пробу сушеного кальмара, мидии, гребешка, мактры и мяса краба и креветок предварительно разрезают ножницами, после чего измельчают лабораторной мельницей (типа кофейной), или пропускают через мясорубку. Сушеное мясо краба и креветок можно измельчать в ступке.
Сушеных трепанга и кукумарию разрезают на кусочки острым ножом на сухой чистой доске, после чего измельчают мясорубкой. Если оболочки кукумарии или трепанга не могут быть разрезаны ножом, их сначала дробят в металлической ступке на куски, которые затем измельчают в однородный порошок в лабораторной мельнице.
Г. Подготовка к анализу средней пробы белковой пасты "Океан"
2.9.8. Среднюю пробу мороженой пасты "Океан" размораживают на воздухе при температуре не выше 20 °С до температуры в толще блока (куска, брикета) от 0 до минус 1 °С.
Для размораживания пасту укладывают в чистые кюветы, противни и накрывают сверху влажной тканью или бумагой во избежание подсыхания.
Размороженную пасту дважды пропускают через мясорубку и растирают в фарфоровой ступке.
2.10. Подготовка к анализу средней пробы натуральной амбры
2.10.1. Пробу хрупкой амбры растирают в фарфоровой ступке до порошкообразного состояния, а воскообразной - до мелкой крупки.
При невозможности измельчения воскообразной амбры в ступке ее режут ножом или ланцетом на мелкие, тонкие ломтики или кусочки размером 2-4 мм, которые затем тщательно перемешивают.
2.11. Подготовка к анализу средней пробы жемчужного пата и перламутрового препарата
2.11.1. Среднюю пробу тщательно перемешивают и часть ее массой от 150 до 200 г помещают в чистую сухую стеклянную банку с притертой пробкой.
2.12. Подготовка к анализу средней пробы кристаллического спермацета
2.12.1. Среднюю пробу спермацета расплавляют и после тщательного перемешивания часть ее в количестве от 150 до 200 г помещают в чистую сухую стеклянную банку с притертой пробкой.
3. МЕТОДЫ АНАЛИЗА РЫБЫ, МОРСКИХ МЛЕКОПИТАЮЩИХ И БЕСПОЗВОНОЧНЫХ
[свежей, охлажденной, мороженой, соленой, пряной, маринованной, вяленой, сушеной и копченой рыбы, сырья морских млекопитающих, пищевого рыбного фарша, печени рыб]
3.1. Подготовка средней пробы к анализу - по п.2.1.
3.2. Методы определения азота летучих оснований, аммиака и сероводорода
3.2.1. Определение азота летучих оснований титриметрическим методом
3.2.1.1. Сущность метода
Свободные и связанные летучие основания отгоняют с паром. Образующий аммиак взаимодействует с серной кислотой. Избыток серной кислоты оттитровывают щелочью.
3.2.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт*.
______________
* Изменением N 1 по всему тексту стандарта заменены слова: "Весы аналитические класса 2 с пределами измерений от 0 до 200 г по ГОСТ 24104-88" на "Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт". - .
Вода дистиллированная по ГОСТ 6709-72.
Часы механические по ГОСТ 10733-98.
Аппарат для отгонки вместимостью 0,7-1,0 дм (черт.1).
Колба коническая или плоскодонная по ГОСТ 25336-82, вместимостью 500 см.
Бюретка по ГОСТ 29251-91, вместимостью 25 или 50 см с делениями на 0,1 см
.
Электроплитка бытовая по ГОСТ 14919-83.
Капельница по ГОСТ 25336-82.
Кислота серная по ГОСТ 4204-77, раствор 0,05 моль/дм (0,1 н).
Натрия гидроокись по ГОСТ 4328-77, раствор 0,1 моль/дм (0,1 н).
Магния окись по ГОСТ 4526-75.
Парафин по ГОСТ 23683-89.
Метиловый красный, раствор 0,2 г/дм (0,02%-ный): растворяют 0,02 г метилового красного в 100 см
спирта 600 г/дм
(60%-ного).
(Измененная редакция, Изм. N 1).
3.2.1.3. Проведение анализа
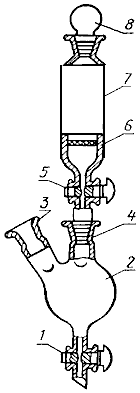
Собирают аппарат (черт.1), состоящий из отгонной колбы 4, каплеуловителя 3, парообразователя 2, холодильника 5, нагревательного элемента 1, приемника 6. Всю систему предварительно пропаривают в течение 10-15 мин.
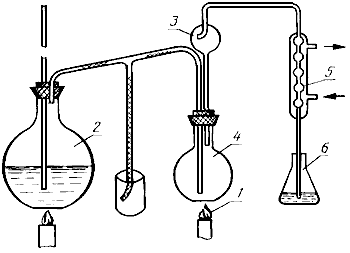
1 - нагревательный элемент; 2 - парообразователь; 3 - каплеуловитель; 4 - отгонная колба; 5 - холодильник; 6 - приемная колба
Черт.1
Навеску исследуемого продукта массой от 9 до 10 г, взвешенную с абсолютной погрешностью не более 0,01 г, количественно переносят 250 см, дистиллированной воды в отогнанную колбу 4, туда же добавляют 1 г окиси магния и, во избежание вспенивания, кусочек чистого парафина. Колбу закрывают пробкой с каплеуловителем 3, соединяют с холодильником 5 и парообразователем 2.
Подогревая колбу на слабом огне, пропускают в нее пар и проводят отгонку в течение 30 мин, считая с момента появления капли дистиллята в холодильнике. Дистиллят собирают в приемник 6, в который предварительно внесено 15-25 см 0,05 моль/дм
раствора серной кислоты. Конец трубки холодильника должен быть погружен в серную кислоту.
За 5-7 мин до окончания отгонки конец холодильника вынимают из раствора.
По окончании отгонки конец трубки холодильника обмывают водой в приемную колбу и избыток кислоты в ней оттитровывают раствором гидроокиси натрия 0,1 моль/дм в присутствии 5 капель метилового красного до перехода окраски от розовой до слабо-желтой.
Параллельно с рабочим проводят контрольный анализ без навески исследуемого образца.
3.2.1.4. Обработка результатов
Массовую долю азота летучих оснований () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора 0,1 моль/дм
гидроокиси натрия, израсходованный на титрование серной кислоты в контрольном анализе, см
;
- объем раствора 0,1 моль/дм
гидроокиси натрия, израсходованный на титрование серной кислоты в рабочем анализе, см
;
0,0014 - количество азота, эквивалентное 1 см раствора 0,1 моль/дм
гидроокиси натрия, г;
- коэффициент пересчета на точный раствор 0,1 моль/дм
гидроокиси натрия;
- масса исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,001%.
Вычисления проводят до третьего десятичного з
нака.
3.2.2. Определение азота летучих оснований колориметрическим методом (с реактивом Несслера)
3.2.2.1. Сущность метода
Свободные и связанные летучие основания отгоняют с паром. Аммиак определяют после обработки дистиллята реактивом Несслера.
3.2.2.2. Аппаратура, материалы и реактивы
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Аппарат для встряхивания.
Аппарат для отгонки.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Цилиндр мерный с пришлифованной пробкой по ГОСТ 1770-74, вместимостью 200 см или коническая колба по ГОСТ 25336-82, вместимостью 250-300 см
.
Пробирка по ГОСТ 25336-82, диаметром 20-30 мм.
Воронка химическая по ГОСТ 25336-82, диаметром 60-80 мм.
Колбы мерные по ГОСТ 1770-74, вместимостью 50, 500 и 1000 см.
Пипетки по ГОСТ 29227-91, вместимостью 1, 2, 5, 10 и 25 см.
Бюретка по ГОСТ 29251-91, вместимостью 25 см с делениями на 0,1 см
.
Аммоний хлористый по ГОСТ 26600-98.
Натрия гидроокись по ГОСТ 4328-77, раствор 100 г/дм (10%-ный).
Кислота серная по ГОСТ 4204-77, раствор 0,05 моль/дм (0,1 н).
Калий-натрий виннокислый 4-водный (сегнетова соль) по ГОСТ 5845-79, раствор 500 г/дм (50%-ный).
Магния окись по ГОСТ 4526-75, водная суспензия 50 г/дм (5%-ная) - магнезиальное молоко.
Йод по ГОСТ 4159-79.
Калий йодистый по ГОСТ 4232-74.
Ртуть по ГОСТ 4658-73.
Вода дистиллированная, безаммиачная, приготовленная по ГОСТ 4517
-87.
3.2.2.3. Подготовка к анализу
Приготовление реактива Несслера
22,5 г йода растворяют в 20 см воды, содержащей 30 г йодистого калия. К раствору йода прибавляют 30 г металлической ртути и сильно встряхивают до исчезновения окраски от йода. Если после этого раствор не будет давать с крахмалом реакцию на йод, к нему прибавляют по каплям раствор йода в йодистом калии до положительной реакции. После этого раствор разбавляют водой до 200 см
, перемешивают и прибавляют к нему 375 см
раствора гидроокиси натрия 100 г/дм
(10%-ного); дают раствору отстояться и сливают сифоном в склянку оранжевого стекла.
Реактив хранят в темноте.
Приготовление раствора хлористого аммония
1,9095 г дважды перекристаллизованного и высушенного при комнатной температуре до постоянной массы хлористого аммония растворяют в мерной колбе вместимостью 1 дм (основной раствор). Этот раствор, содержащий 0,5 мг азота в 1 см
, может сохраняться в течение нескольких месяцев в темном прохладном месте. Перед началом работы 5 см
основного раствора отбирают пипеткой в мерную колбу вместимостью 500 см
и доводят объем дистиллированной водой до метки (рабочий раствор). Этот раствор содержит 0,005 мг азота в 1 см
.
Магнезиальное молоко
5 г окиси магния, отвешенных с абсолютной погрешностью не более 0,01 г, помещают в фарфоровую ступку и, отмерив цилиндром 95 см дистиллированной воды, приливают ее небольшими порциями, тщательно растирая густую массу пестиком. Гомогенизированную массу переносят остатком воды через воронку в склянку или колбу с пришлифованной пробкой. Взвесь перед использованием тщательно взбалтывают и, не давая осесть осадку, отбирают пипеткой с широким отверстием.
3.2.2.4. Проведение анализа
От 9 до 10 г исследуемого продукта, взвешенных с абсолютной погрешностью не более 0,01 г, переносят в мерный цилиндр с пришлифованной пробкой вместимостью 200 см или колбу вместимостью 250-300 см
, заливают дистиллированной водой до объема 100 см
(при применении колбы приливают 90 см
воды) и встряхивают на аппарате в течение 10 мин со скоростью 40-50 качаний в минуту.
Примечание. Встряхивание можно заменить настаиванием в течение 20 мин при периодическом взбалтывании.
Взвесь фильтруют через марлю, уложенную на воронку диаметром 60-80 мм.
Отгонку летучих оснований проводят в аппарате, аналогичном приведенному в пп.3.2.1.3 (черт.1), но меньшей вместимости (100 см).
Предварительно парообразователь доводят до кипения и собранный аппарат пропаривают в течение 5-10 мин.
В дистиллированную колбу вносят 5 см профильтрованной вытяжки, добавляют 2 см
магнезиального молока 50 г/дм
и проводят отгонку с паром в течение 10 мин, считая с момента появления капли дистиллята в холодильнике. Дистиллят собирают в приемник (широкая пробирка). Предварительно в приемник вносят 1-2 см
раствора 0,05 моль/дм
серной кислоты, в которую опускают конец трубки холодильника. По окончании отгонки последний обмывают водой в приемную пробирку.
Дистиллят переносят в мерную колбу вместимостью 50 см, смывают приемную пробирку и разбавляют дистиллированной водой до
объема колбы. Одновременно в три мерные колбы вместимостью 50 см
отбирают 5, 10 и 25 см
рабочего стандартного раствора хлористого аммония и добавляют безаммиачной воды до
их объема.
Во все колбы добавляют по 1 см раствора сегнетовой соли 500 г/дм
, взбалтывают, добавляют по 2 см
реактива Несслера, доводят объемы до метки безаммиачной водой и перемешивают взбалтыванием.
Растворы оставляют в покое на 10-15 мин, после чего определяют оптическую плотность фотоэлектроколориметром при длине волны 400 нм в кюветах с рабочей длиной 10 мм по отношению к контрольному раствору.
Количество азота, соответствующее определенной оптической плотности, рассчитывают по градуировочн
ому графику.
3.2.2.5. Построение градуировочного графика
Готовят ряд разведений раствора хлористого аммония с известной концентрацией. Для этого в мерные колбы вместимостью 50 см из бюретки последовательно вносят указанные в табл.1 количества рабочего раствора хлористого аммония и доводят объем дистиллированной водой до метки.
Таблица 1
Номер колбы | Количество рабочего раствора хлористого аммония, см | Количество азота в колбе, г |
1 | 10 | 0,000050 |
2 | 15 | 0,000075 |
3 | 20 | 0,000100 |
4 | 25 | 0,000125 |
5 | 30 | 0,000150 |
6 | 35 | 0,000175 |
7 | 40 | 0,000200 |
8 | 45 | 0,000225 |
Оптическую плотность растворов определяют при длине волны 400 нм в кюветах с рабочей длиной 10 мм.
По полученным данным строят градуировочный график, откладывая на оси абсцисс содержание азота (г в 50 см), на оси ординат - соответствующую оптическую плотность.
3.2.2.6. Обработка результатов
Массовую долю азота летучих оснований () в процентах вычисляют по формуле
![]() ,
,
где - масса образца, взятая для приготовления вытяжки, г;
- массовая доля азота, найденная по градуировочному графику, г;
- объем смеси, полученный при приготовлении вытяжки из навески, см
;
- объем профильтрованной вытяжки, взятый для отгонки летучих оснований, см
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,001%. Вычисления проводят до третьего десятичного знака.
3.2.3. Определение аммиака (качественная реакция)
3.2.3.1. Сущность метода
Метод основан на взаимодействии аммиака, образующегося при порче рыбы, с соляной кислотой и появлении при этом облачка хлористого аммония.
3.2.3.2. Аппаратура, материалы и реактивы
Пробирка химическая по ГОСТ 25336-82, диаметром не менее 20 мм.
Кислота соляная по ГОСТ 3118-77, раствор 250 г/дм (25%-ный).
Спирт этиловый питьевой по ГОСТ 5963-67*, раствор 950 г/дм (95%-ный).
________________
* На территории Российской Федерации действует ГОСТ Р 51723-2001 (здесь и далее).
Эфир медицинский по Госфармакопее СССР.
3.2.3.3. Подготовка к анализу
Приготовление реактива Эбера
Смешивают одну часть соляной кислоты 250 г/дм (плотность 1120 кг/м
), три части этилового спирта 950 г/дм
и одну часть серного эфира.
3.2.3.4. Проведение анализа
В широкую пробирку наливают 2-3 см смеси Эбера, закрывают ее пробкой и встряхивают 2-3 раза.
Вынимают пробку из пробирки и сразу же закрывают ее другой пробкой, через которую продета тонкая стеклянная палочка с загнутым концом. На конец палочки должен быть прикреплен кусочек исследуемого мяса рыбы. Исследуемый объект должен иметь температуру, наиболее близкую к температуре воздуха лаборатории в момент проведения анализа. Мясо вводят в пробирку так, чтобы не запачкать стенок пробирки и чтобы оно находилось на расстоянии 1-2 см от уровня жидкости.
3.2.3.5. Обработка результатов
Через нескольку секунд в результате реакции аммиака с соляной кислотой образуется облачко хлористого аммония.
Интенсивность реакции обозначается следующим образом:
- реакция отрицательная;
+ реакция слабоположительная (быстро исчезающее расплывчатое облачко);
++ реакция положительная (устойчивое облачко, появляющееся через несколько секунд после внесения мяса в пробирку с реактивом);
+++ реакция резко положительная (облачко появляется сразу после внесения мяса в пробирку с реактивом).
3.2.4. Определение сероводорода (качественная реакция)
3.2.4.1. Сущность метода
Метод основан на взаимодействии сероводорода, образующегося при порче рыбы, со свинцовой солью с появлением темного окрашивания вследствие образования сернистого свинца.
3.2.4.2. Аппаратура, реактивы и материалы
Бюксы по ГОСТ 25336-82, вместимостью 40-50 см.
Натрия гидроксид по ГОСТ 4328-77, раствор 330 г/дм (33%-ный).
Свинец уксуснокислый по ГОСТ 1027-67, раствор 40 г/дм (4%-ный).
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Весы лабораторные технические класса 1 с пределами измерений от 0 до 1000 г.
3.2.4.3. Подготовка к анализу
Приготовление раствора свинцовой соли
К раствору уксуснокислого свинца 40 г/дм добавляют раствор гидроксида натрия 300 г/дм
до растворения образующегося вначале осадка гидрата оксида свинца (необходимо избегать большого избытка щелочи). Полученный раствор фильтруют через бумажный фильтр.
3.2.4.4. Проведение анализа
15-25 г исследуемого фарша помещают рыхлым слоем в бюксу вместимостью 40-50 см. В бюксу подвешивают горизонтально над фаршем полоску плотной фильтровальной бумаги, на поверхность которой, обращенной к фаршу, нанесены 3-4 капли раствора свинцовой соли. Диаметр капли 2-3 мм. Расстояние между бумагой и поверхностью фарша должно быть 1 см.
Бюксу закрывают сверху крышкой, зажимая фильтровальную бумагу между крышкой и корпусом бюксы, и оставляют стоять при комнатной температуре.
Параллельно проводят контрольный анализ без навески продукта.
По истечении 15 мин бумагу снимают и сравнивают ее окраску с окраской бумаги, смоченной тем же раствором свинцовой соли (контрольный анализ).
При наличии в исследуемом образце свободного сероводорода происходит побурение или почернение участков бумаги, смоченных раствором свинцовой соли.
Интенсивность реакции обозначают следующим образом:
- реакция отрицательная;
± следы окрашивания капли;
+ реакция слабоположительная (бурое окрашивание по краям капли);
++ реакция положительная (бурое окрашивание всей капли, более интенсивное по краям);
+++ реакция резко положительная (интенсивное темно-бурое окрашивание всей капли).
3.3. Методы определения воды
3.3.1. Определение массовой доли воды высушиванием при 100-105 °С
3.3.1.1. Сущность метода
Метод основан на выделении (испарении) воды из продукта при тепловой обработке и определении изменения массы его взвешиванием.
Метод применяется для анализа рыбы, морских млекопитающих, морских беспозвоночных и продуктов их переработки.
3.3.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Шкаф сушильный лабораторный.
Эксикатор по ГОСТ 25336-82.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Стаканчики для взвешивания (бюксы) стеклянные по ГОСТ 25336-82 или металлические.
Песок силикатный речной или морской очищенный и прокаленный.
3.3.1.3. Проведение анализа
Навеску анализируемой пробы от 1,5 до 2 г (3-4 г для паюсной икры), взвешенную с абсолютной погрешностью не более 0,001 г, помещают в чистую высушенную и тарированную бюксу со стеклянной палочкой, при помощи которой распределяют навеску продукта в бюксе ровным тонким слоем. Навеска исследуемого продукта может быть увеличена до 5 г при использовании ее после высушивания для определения содержания жира. Бюксу закрывают притертой крышкой, взвешивают на аналитических весах и высушивают в сушильном шкафу при 100-105 °С до постоянной массы.
Навески продуктов, за исключением сушеных, вялых и обработанных холодным копчением, первые 2 ч сушат при 60-80 °С. Навески продуктов с массовой долей жира более 20% необходимо первые 2 ч сушить при температуре 60-65 °С, а с массовой долей жира более 40% (печень тресковых рыб и т.д.) - 2 ч при 60-65 °С в токе инертного газа.
Первое взвешивание проводят через 3 ч после начала сушки, последующие - через 30-40 мин.
Постоянная масса считается достигнутой, если разница между двумя взвешиваниями не превышает 0,001 г.
Перед каждым взвешиванием бюксу с пробой закрывают крышкой и охлаждают 30 мин в эксикаторе.
Для рыбы и других продуктов, способных при высушивании спекаться в плотную массу, в бюксу предварительно вносят 5-10 г песка и навеску продукта тщательно перемешивают. Кварцевый песок предварительно очищают следующим образом: промывают водопроводной чистой водой, заливают раствором соляной кислоты (1:1) на сутки, тщательно промывают водопроводной, а затем дистиллированной водой до исчезновения кислой реакции на лакмус, высушивают, прокаливают и просеивают.
Очистку песка описанным выше способом проводят во всех случаях, где требуется использование песка.
3.3.1.4. Обработка результатов
Массовую долю воды () в процентах вычисляют по формуле
![]() ,
,
где - масса бюксы с песком, г;
- масса бюксы с навеской и песком до высушивания, г;
- масса бюксы с навеской и песком после высушивания, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного знака.
3.3.2. Определение массовой доли воды высушиванием при 130 °С (рыба соленая, вяленая, сушеная, холодного копчения, мука)
3.3.2.1. Сущность метода - по п.3.3.1.1.
3.3.2.2. Аппаратура, материалы и реактивы - по п.3.3.1.2.
3.3.2.3. Проведение анализа
Навеску рыбы подсушивают в течение 30 мин при 60-80 °С, затем окончательно высушивают в течение 1 ч при температуре 130 °С. Навеску жирных видов рыб берут с песком.
По истечении указанного времени бюксу вынимают, охлаждают 30 мин в эксикаторе и взвешивают с абсолютной погрешностью не более 0,001 г.
3.3.2.4. Обработка результатов - по п.3.3.1.4.
3.3.3. Определение массовой доли воды отгонкой в жирах и витаминных препаратах
3.3.3.1. Сущность метода
Метод основан на выделении воды из продукта отгонкой с парами растворителя жира и определении ее объема.
3.3.3.2. Аппаратура, материалы и реактивы
Аппарат количественного определения воды АКОВ (типа Дина и Старка) (черт. 2).
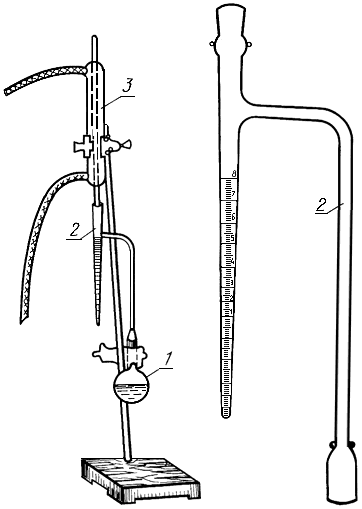
1 - отгонная колба; 2 - приемник; 3 - холодильник
Черт.2
Штатив с кольцом и зажимом.
Цилиндр по ГОСТ 1770-74, вместимостью 100 см, с делениями на 1 см
.
Баня песочная.
Стеклянная палочка диаметром 3-4 мм с резиновым наконечником или медная проволочка, согнутая полукольцом.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Толуол по ГОСТ 5789-78, или ксилол каменноугольный по ГОСТ 9949-76, или бензин.
3.3.3.3. Проведение анализа
В стеклянную колбу 1 аппарата отвешивают с абсолютной погрешностью не более 0,1 г 10-15 г тщательно измельченного продукта или 50-200 г жира (в зависимости от предполагаемого содержания воды) с таким расчетом, чтобы отогнанная вода составляла не более 10 см, т.е. не более вместимости приемника-ловушки 2, приливают 80-100 см
одного из перечисленных растворителей и тщательно перемешивают содержимое колбы. В колбу помещают несколько кусочков неглазурованного фаянса, пемзы или фарфора и с помощью шлифа присоединяют ее к отводной трубке приемника, соединенного с холодильником 3.
Колбу нагревают, доводят содержимое до интенсивного кипения, и поддерживают его до окончания анализа.
Если на стенках холодильника или приемника остаются капли воды, их осторожно сталкивают со стенки в нижнюю часть приемника стеклянной палочкой с резиновым наконечником или медной проволокой.
Отгонку прекращают, когда объем воды в приемнике перестает увеличиваться и верхний слой растворителя в ловушке-приемнике станет совершенно прозрачным.
Содержимое колбы охлаждают до комнатной температуры и проводят отсчет объема воды в приемнике.
3.3.3.4. Обработка результатов
Массовую долю воды () в процентах вычисляют по формуле
![]() ,
,
где - масса пробы, г;
- масса воды в приемнике, г (1 см
воды принимают равным 1 г).
Вычисление проводят до первого десятичного знака.
3.3.4. Определение массовой доли воды высушиванием на приборе ВЧМ (прибор Чижовой)
3.3.4.1. Сущность метода
Метод основан на выделении воды из продукта при нагревании инфракрасными лучами и определении изменения его массы взвешиванием.
Метод применяют для определения воды в вяленой рыбе и рыбе холодного копчения.
3.3.4.2. Аппаратура, материалы и реактивы
Прибор ВЧМ Чижовой (черт 3).
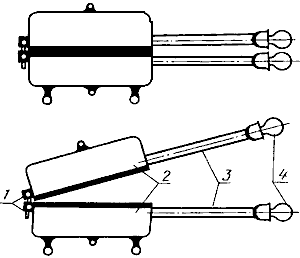
1 - шарниры; 2 - металлические плиты; 3 - ручка; 4 - термометры
Черт.3
Автотрансформатор (типа ЛАТР-1).
Эксикатор по ГОСТ 25336-82.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
3.3.4.3. Подготовка к анализу
Прибор Чижовой нагревают до температуры обезвоживания исследуемого продукта (125-180 °С) в соответствии с установленным режимом.
Для изготовления бумажных пакетов лист бумаги размером 15x15 см складывают по диагонали пополам и края загибают в одну сторону на 1 см. При определении воды в жирных пробах (сельдь и др.) в бумажный пакет помещают дополнительно лист фильтровальной бумаги.
Заготовительные пакеты просушивают 1-3,5 мин между нагретыми плитами прибора при температуре, при которой будет высушиваться навеска, и переносят на 5 мин в эксикатор для охлаждения. После этого пакеты взвешивают с абсолютной погрешностью не более 0,01 г.
3.3.4.4. Проведение анализа
Навеску анализируемой пробы 2-3 г, взвешенную с абсолютной погрешностью не более 0,01 г, помещают в предварительно высушенный и взвешенный пакет и распределяют ее шпателем равномерным тонким слоем по внутренней поверхности пакета. Шпатель вытирают о внутреннюю сторону пакета. Пакет с навеской складывают, помещают в прибор между плитами и выдерживают 1-3,5 мин в соответствии с режимом обезвоживания, указанным в табл.2.
Таблица 2
Наименование продукта | Масса анализируемой пробы, г | Температура высушивания, °С | Продолжительность высушивания, мин |
Рыба вяленая и холодного копчения | 2 | 135 | 3,0 |
То же | 3 | 145 | 3,5 |
" | 3 | 155 | 3,0 |
" | 3 | 180 | 1,0 |
3.3.4.5. Обработка результатов
Массовую долю воды () в процентах вычисляют по формуле
![]() ,
,
где - масса пакета, г;
- масса пакета с навеской до обезвоживания, г;
- масса пакета с навеской после обезвоживания, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного знака.
3.4. Методы определения белковых веществ
3.4.1. Определение массовой доли белковых веществ макрометодом - по п.8.9.1.
3.4.2. Определение массовой доли белковых веществ макрометодом с селеновой смесью - по п.8.9.2.
3.4.3. Определение массовой доли белковых веществ макрометодом с перекисью водорода без отгонки - по п.8.9.3.
3.4.4. Определение массовой доли белковых веществ полумикрометодом - по п.8.9.4.
3.5. Методы определения хлористого натрия (поваренной соли)
3.5.1. Определение массовой доли хлористого натрия аргентометрическим методом (используется при разногласиях в оценке качества продукции)
3.5.1.1. Сущность метода
Метод основан на взаимодействии хлористого натрия с азотнокислым серебром в присутствии хромовокислого калия с образованием красного осадка - хромовокислого серебра.
3.5.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Бюретки по ГОСТ 29251-91, вместимостью 10, 25, 50 см.
Колбы мерные по ГОСТ 1770-74, вместимостью 200, 250 см.
Палочки стеклянные по ГОСТ 25336-82.
Стекло часовое.
Электропечь сопротивления лабораторная.
Бумага фильтровальная по ГОСТ 12026-76 или фильтры бумажные.
Капельница лабораторная по ГОСТ 25336-82.
Пипетки по ГОСТ 29227-91, вместимостью 10, 25, 50 см.
Воронка по ГОСТ 25336-82.
Тигли фарфоровые по ГОСТ 9147-80.
Марля медицинская по ГОСТ 9412-93.
Вата медицинская гигроскопическая по ГОСТ 5556-81.
Натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм (0,1 н).
Серебро азотнокислое по ГОСТ 1277-75, раствор 0,1 моль/дм (0,1 н).
Калий хромовокислый по ГОСТ 4459-75, 100 г/дм (10%-ный) или насыщенный раствор.
Натрий двууглекислый по ГОСТ 4201-79, раствор 0,01 моль/дм (0,01 н).
Кислота уксусная ледяная по ГОСТ 61-75 и раствор 0,01 моль/дм (0,01 н).
Фенолфталеин, раствор 10 г/дм (1%-ный).
Паранитрофенол, раствор 0,5 г/дм (0,05%-ный).
Вода дистиллированная по ГОСТ 6709-72
.
3.5.1.3. Проведение анализа
Навеску исследуемого образца 2-5 г (для рыбного клея 5-10 г), взвешенную с абсолютной погрешностью не более 0,01 г, помещают в мерную колбу вместимостью 200-250 см и заливают на
объема дистиллированной водой, нагретой до 60 °С. Содержимое колбы настаивают в течение 15-20 мин, периодически сильно взбалтывая. Допускается экстрагирование хлористого натрия из фарша водой комнатной температуры, при этом время настаивания увеличивают до 25-30 мин. По окончании настаивания жидкость в колбе охлаждают до комнатной температуры, объем доводят водой до метки.
При определении хлористого натрия в пробах жирной рыбы (массовая доля жира более 20%) и кормовой муки навеску средней пробы от 2 до 2,5 г подсушивают (при анализе рыбы), а затем осторожно обугливают в фарфоровом тигле на пламени газовой горелки или в муфельной печи до прекращения выделения дыма.
Уголь измельчают, смывают горячей дистиллированной водой в мерную колбу вместимостью 200-250 см и после охлаждения до комнатной температуры объем доводят водой до метки.
Содержимое мерной колбы в обоих случаях тщательно взбалтывают и фильтруют через сухой бумажный фильтр, вату или двойной слой марли, причем первые 20-30 см фильтрата отбрасывают.
Для устранения испарения жидкости во время фильтрования воронку с фильтром накрывают часовым стеклом.
В две конические колбы отбирают по 10-25 см фильтрата (50 см
- при анализе кормовой муки) и титруют раствором азотнокислого серебра 0,1 моль/дм
в присутствии 3-4 капель раствора хромовокислого калия 100 г/дм
(10%-ного) или 1 капли насыщенного раствора до получения неисчезающей красновато-бурой окраски.
При исследовании средне- или крепкосоленой рыбы отбирают для титрования меньшее количество фильтрата (но не менее 10 см).
В случае исследования продуктов, имеющих кислую или щелочную реакцию (маринады, испорченная соленая рыба), перед титрованием раствором азотнокислого серебра отобранную порцию фильтрата нейтрализуют раствором двууглекислого натрия 0,01 моль/дм или раствором 0,01 моль/дм
уксусной кислоты в присутствии индикаторов фенолфталеина или паранитрофенола. После нейтрализации двууглекислым натрием фенолфталеин должен оставаться бесцветным, в случае использования гидроксида натрия - окраситься в бледно-розовый цвет. Паранитрофенол после нейтрализации приобретает слабо-желтую окр
аску.
3.5.1.4. Обработка результатов
Массовую долю хлористого натрия () в процентах вычисляют по формуле
![]() ,
,
где - объем водной вытяжки в мерной колбе, см
;
- объем раствора азотнокислого серебра 0,1 моль/дм
, израсходованный на титрование исследуемого раствора, см
;
- объем водной вытяжки, взятый для титрования, см
;
- навеска исследуемого образца, г;
0,00585 - количество хлористого натрия, соответствующее 1 см раствора 0,1 моль/дм
азотнокислого серебра, г;
- коэффициент пересчета на точный раствор 0,1 моль/дм
азотнокислого серебра.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до первого десятичного
знака.
3.5.2. Упрощенный аргентометрический метод
3.5.2.1. Сущность метода - по п.3.5.1.1.
3.5.2.2. Аппаратура, материалы и реактивы - по п.3.5.1.2.
3.5.2.3. Проведение анализа
Навеску фарша 2-5 г, взвешенную с абсолютной погрешностью не более 0,01 г, помещают в химический стакан и приливают в него мерным цилиндром соответственно 98-95 см или 248-245 см
дистиллированной воды, размешивают стеклянной палочкой с резиновым наконечником. Через 25-30 мин настаивания содержимое стакана фильтруют через бумажный фильтр, вату или двойной слой марли.
В две колбы для титрования отбирают пипеткой 10-25 см фильтрата, добавляют 3-4 капли раствора хромовокислого калия и титруют из бюретки раствором азотнокислого серебра до неисчезающей красновато-бурой окраски.
3.5.2.4. Обработка результатов - по п.3.5.1.4.
3.5.3. Меркурометрический метод
3.5.3.1. Сущность метода
Метод основан на взаимодействии хлористого натрия с азотнокислой ртутью (II) или азотнокислой ртутью (I) в присутствии дифенилкарбазида или дифенилкарбазона с образованием комплекса, окрашивающего раствор в фиолетовый цвет, и титриметрическом определении его.
Раствор азотнокислой ртути (I) используют при исследовании продуктов с массовой долей соли 50 г/дм (5%) и более.
3.5.3.2. Аппаратура, материалы и реактивы - по п.3.5.1.2 со следующими дополнениями:
Баня водяная.
Склянка с притертой пробкой вместимостью 1000 см.
Спирт этиловый питьевой по ГОСТ 5963-67*, раствор 950 г/дм (95%-ный).
_________________
* На территории Российской Федерации действует ГОСТ Р 51723-2001.
Дифенилкарбазид, насыщенный раствор.
Бромфеноловый синий, раствор 1 г/дм (0,1%-ный).
Кислота азотная по ГОСТ 4461-77, концентрированная.
Ртуть (I) азотнокислая двуводная по ГОСТ 4521-78, раствор 0,1 моль/дм.
Вода дистиллированная по ГОСТ 6709-72.
Ртуть (II) азотнокислая одноводная по ГОСТ 4520-78, раствор 0,025 моль/дм (0,05 н).
Дифенилкарбазон, спиртовой раствор 10 г/дм (1%-ный).
3.5.3.3. Подготовка к анализу
Приготовление раствора азотнокислой ртути (II) 0,025 моль/дм (0,05 н)
5,5 г желтой окиси ртути растворяют в 7-10 см концентрированной азотной кислоты. Раствор количественно переносят в мерную колбу вместимостью 1 дм
и доводят объем дистиллированной водой до метки, хорошо перемешивают и затем фильтруют через бумажный складчатый фильтр. Профильтрованный раствор хранят в склянке с притертой пробкой.
Приготовление раствора азотнокислой ртути (I) 0,1 моль/дм (0,1 н)
30 г азотнокислой ртути (I) растворяют в 1 дм азотной кислоты 0,2 моль/дм
при легком нагревании на водяной бане. При наличии в растворе нерастворившегося осадка его фильтруют. Титр рабочего раствора азотнокислой ртути (I) устанавливают через сутки после приготовления раствора по фиксаналам или навескам х.ч. хлористого натрия (или калия).
Приготовление раствора индикатора бромфенолового синего (бромфенолблау) 1 г/дм (0,1%-ного)
В мерной колбе вместимостью 100 см растворяют 100 мг индикатора в небольшом количестве дистиллированной воды, добавляют 1,5 см
раствора гидроокиси натрия 0,1 моль/дм
и доводят объем дистиллированной водой до метки.
Приготовление раствора индикатора дифенилкарбазона 10 г/дм (1%-ного)
1 г дифенилкарбазона растворяют в 100 см спирта 950 г/дм
(95%). Раствор хранят в склянке из темного стекла.
При установлении титра пользуются тем индикатором, который будет применяться при анализах проб.
3.5.3.4. Проведение анализа
Навеску средней пробы массой от 2 до 5 г, отвешенную с абсолютной погрешностью не более 0,001 г (в зависимости от предполагаемого содержания соли), помещают в мерную колбу вместимостью 200-250 см и заливают на
объема дистиллированной водой комнатной температуры. Содержимое колбы настаивают 25-30 мин, периодически сильно взбалтывая. Объем жидкости в колбе доводят до метки и фильтруют через сухой бумажный фильтр, вату или двойной слой марли.
Титрование проводят одним из следующих способов:
- титрование азотнокислой ртутью (I) с индикатором бромфеноловым синим.
К 25 см фильтрата прибавляют 15 капель раствора бромфенолблау 1 г/дм
и титруют раствором азотнокислой ртути (I) 0,1 моль/дм
до перехода окраски из зеленовато-синей через светло-серую в сиреневую;
- титрование азотнокислой ртутью (I) с индикатором дифенилкарбазоном.
25 см фильтрата подкисляют 1 см
концентрированной азотной кислоты (свободной от окислов азота), добавляют 8-10 капель спиртового раствора дифенилкарбазона 10 г/дм
(1%-ного) (реактив хранят в склянке из темного стекла) и титруют раствором 0,1 моль/дм
азотнокислой ртути (I) до резкого изменения окраски в голубой или сине-фиолетовый цвет;
- титрование азотнокислой ртутью (II).
К 25 см фильтрата прибавляют 1-2 капли концентрированной азотной кислоты (оптимальное значение рН 4) и затем 3-5 капель насыщенного раствора дифенилкарбазида в спирте.
Смесь хорошо взбалтывают и титруют раствором азотнокислой ртути (II) 0,025 моль/дм до появления слабой фиолетовой окр
аски.
3.5.3.5. Обработка результатов
Массовую долю хлористого натрия () в процентах вычисляют по формуле
![]() ,
,
где - титры растворов азотнокислой ртути:
0,0029 - для 0,025 моль/дм раствора азотнокислой ртути (II);
0,0058 - для 0,1 моль/дм раствора азотнокислой ртути (I);
- общий объем вытяжки в мерной колбе, см
;
- объем 0,1 моль/дм
раствора азотнокислой ртути (I) или 0,025 моль/дм
раствора азотнокислой ртути (II), израсходованный на титрование, см
;
- объем водной вытяжки, взятый для титрования, см
;
- масса исследуемого образца, г;
- коэффициент пересчета на точный раствор 0,1 моль/дм
раствор азотнокислой ртути (I).
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%. Вычисление проводят до первого десятичног
о знака.
3.6. Методы определения кислотности
3.6.1. Определение общей кислотности титриметрическим методом - по ГОСТ 27082-89
3.6.2. Определение свободной уксусной кислоты маринадов титриметрическим методом
3.6.2.1. Сущность метода
Метод основан на выделении (отгоне) уксусной кислоты из водной вытяжки рыбы или разбавленной заливки и количественном определении ее титрованием.
3.6.2.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Аппарат для отгонки (черт.4).
Аппарат для отгонки
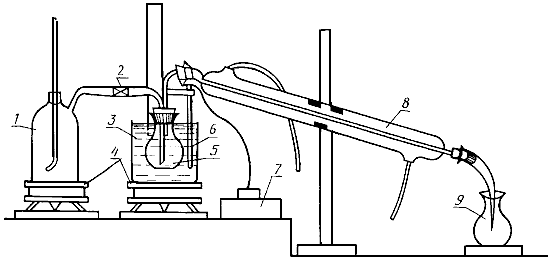
1 - парообразователь; 2 - кран; 3 - глицериновая баня; 4 - нагревательный прибор; 5 - отгонная колба; 6 - термометр; 7 - терморегулирующее устройство; 8 - холодильник; 9 - приемная колба
Черт.4
Глицерин по ГОСТ 6259-75.
Натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм или калия гидроксид по ГОСТ 24363-80, раствор 0,1 моль/дм
.
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный).
Вода дистиллированная по ГОСТ 6709-72.
3.6.2.3. Подготовка к анализу
При подготовке прибора к работе температуру глицериновой (масляной) бани доводят до 150 °С, а воду в парообразователе - до кипения. Для парообразователя необходимо брать дистиллированную воду, проверенную на полное отсутствие углекислоты. Новый прибор с замененными частями или прибор, не эксплуатировавшийся несколько суток, необходимо до начала работы пропарить вначале с холодильником, свободным от воды в течение 10 мин, а затем с холодильником, заполненным водой, до нейтральной реакции дистиллята.
Отгонную колбу 5 помещают в глицериновую баню до пробки или шлифа во избежание конденсации паров при выходе из колбы.
3.6.2.4. Проведение анализа
Фильтрат вытяжки рыбы или разбавленной заливки готовят по ГОСТ 27082-89.
10 см фильтрата помещают в отгонную колбу 5, которую соединяют с холодильником 8 и при закрытом кране 2 помещают в глицериновую баню 3. Пробу нагревают в течение 5 мин, после чего открывают кран и пропускают пар через пробу в течение 40 мин. За это время в приемной колбе 9 образуется 200-250 см
дистиллята.
Перед отгонкой в приемную колбу наливают от 10 до 15 см дистиллированной воды, свободной от углекислоты, и погружают в нее конец трубки холодильника. За 15 мин до окончания отгонки конец трубки холодильника вынимают из дистиллята и смывают дистиллированной водой (от 3 до 5 см
) в приемную колбу (в процессе отгонки температура глицериновой бани должна поддерживаться на уровне 145-160 °С). Включение в схему терморегулирующего устройства не обязательно, однако наличие его значительно облегчает проведение анализа. Собранный дистиллят титруют раствором 0,1 г/дм
гидроксида натрия в присутствии нескольких капель фенолфталеина.
Одновременно готовят контрольную пробу - 200-250 см дистиллированной воды, свободной от углекислоты, и титруют в присутствии нескольких капель фенолфталеина раствором 0,1 моль/дм
гидроксида натрия
.
3.6.2.5. Обработка результатов
Массовую долю свободной уксусной кислоты () в процентах вычисляют по формуле
![]() ,
,
где - объем фильтрата, взятый для отгонки, см
;
- общий объем вытяжки, см
;
- объем раствора 0,1 моль/дм
гидроксида натрия, израсходованный на титрование дистиллята, см
;
- объем раствора 0,1 моль/дм
гидроксида натрия, израсходованный на титрование контрольной пробы, см
(при объеме дистиллята 200-250 см
- 0,1 см
);
- масса исследуемого образца, г;
- коэффициент пересчета на точный раствор 0,1 моль/дм
гидроксида натрия;
- коэффициент пересчета на соответствующую кислоту (уксусная кислота - 0,0060; яблочная кислота - 0,0067; молочная кислота - 0,0090; винная кислота - 0,0075).
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,03%.
Вычисление проводят до второго десятичн
ого знака.
3.6.3. Определение активной кислотности потенциометрическим методом - по ГОСТ 28972-91.
3.6.4. Определение кислотности рыбной печени титриметрическим методом
3.6.4.1. Сущность метода
Метод основан на извлечении спиртоэфирной смесью продуктов, обладающих кислотными свойствами, и количественном определении их титрованием.
3.6.4.2. Аппаратура, материалы и реактивы
Мясорубка бытовая по ГОСТ 4025-95.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Ступка фарфоровая по ГОСТ 9147-80.
Чашка фарфоровая по ГОСТ 9147-80.
Колба лабораторная по ГОСТ 25336-82, вместимостью 100 см.
Палочка стеклянная.
Спирт этиловый технический по ГОСТ 17299-78.
Эфир серный по Госфармокопее СССР.
Натрий сернокислый безводный по ГОСТ 4166-76.
Калия гидроксид по ГОСТ 24363-80, раствор 0,1 моль/дм или натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм
(0,1 н).
Фенолфталеин, спиртовой раствор 10 г/дм (0,1%-ный).
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
3.6.4.3. Проведение анализа
От 1,5 до 2 г дважды измельченной в мясорубке и растертой в ступке рыбной печени отвешивают в фарфоровую чашку с погрешностью не более 0,01 г, добавляют 4,0 г безводного сернокислого натрия, перемешивают стеклянной палочкой, переносят в колбу вместимостью 100 см, прибавляют 40 см
спиртоэфирной смеси (1 объем спирта и 2 объема эфира), закрывают пробкой, взбалтывают и выдерживают при периодическом перемешивании в течение 1 ч. Отстоявшийся раствор сливают в другую колбу через бумажный фильтр, остаток в колбе промывают небольшими порциями спиртоэфирной смеси, собирая промывную жидкость в ту же колбу так, чтобы общий объем фильтрата не превысил 50 см
.
Для титрования берут весь объем фильтрата, добавляют 1 см раствора фенолфталеина 10 г/дм
и титруют раствором 0,1 моль/дм
гидроксида натрия.
3.6.4.4. Обработка результатов
Кислотность печени () в мг КОН на 1 г печени вычисляют по формуле
![]() ,
,
где 5,61 - количество гидроксида калия в 1 см раствора 0,1 моль/дм
, мг;
- объем гидроксида калия, израсходованный на титрование, см
;
- навеска печени, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1 мг.
Вычисление проводят до первого десятичного знака.
3.7. Методы определения жира
3.7.1. Определение массовой доли жира экстракционным методом в аппарате Сокслета*
________________
* В части определения сырого жира в кормовой рыбной муке и из морских млекопитающих и ракообразных, предназначенной для производства комбикормов, - ГОСТ 13496.15-97.
(Поправка).
3.7.1.1. Сущность метода
Метод основан на экстракции жира органическим растворителем из сухой навески и определении его массы взвешиванием.
Метод применяется при разногласиях в оценке качества продукта.
3.7.1.2. Аппаратура, материалы и реактивы
Аппарат экстракционный (аппарат Сокслета).
Баня водяная.
Шкаф сушильный лабораторный.
Стекло часовое.
Эксикатор по ГОСТ 25336-82.
Холодильник стеклянный лабораторный по ГОСТ 25336-82.
Ступка фарфоровая по ГОСТ 9147-80.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Стаканчики для взвешивания (бюксы) стеклянные по ГОСТ 25336-82 или металлические.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Воронка стеклянная делительная по ГОСТ 25336-82.
Вата медицинская гигроскопическая по ГОСТ 5556-81.
Натрий сернокислый безводный по ГОСТ 4166-76 или натрий фосфорнокислый двузамещенный 12-водный по ГОСТ 4172-76.
Эфир петролейный температурой кипения 40-60 °С или эфир этиловый по Госфармакопее СССР, не содержащий перекисей, высушенный над хлористым кальцием или сернокислым натрием.
Песок силикатный речной или морской очищенный и прокаленный.
3.7.1.3. Проведение анализа
Навеску исследуемого продукта 5-10 г, взвешенную с абсолютной погрешностью не более 0,01 г, помещают в фарфоровую ступку. Туда же добавляют двойное-тройное по массе количество безводного сернокислого (или фосфорнокислого) натрия и смесь хорошо растирают пестиком. Обезвоженный продукт количественно переносят в пакет или специальный патрон (см. п.3.7.2.3). Ступку протирают ваткой, смоченной эфиром, которую затем присоединяют к сухой навеске и помещают в экстрактор аппарата Сокслета. Экстрактор соединяют с предварительно высушенной при 105 °С и взвешенной колбой и наливают эфир с таким расчетом, чтобы количество его в 1,5 раза превышало объем экстрактора. Экстрактор с помощью пришлифованной пробки соединяют с холодильником. Воду пропускают в холодильник аппарата, и колбу слабо нагревают на водяной бане.
Экстракцию жира проводят в течение 10-12 ч. Интенсивность нагрева должна быть такой, чтобы в течение 1 ч происходило не менее 5-6 и не более 8-10 сливаний эфира. Полноту извлечения жира проверяют нанесением капли стекающего из экстрактора растворителя на часовое стекло. После испарения растворителя на стекле не должно оставаться жирного пятна.
При перерыве в работе в экстракторе оставляют эфир в таком количестве, чтобы патрон с навеской был погружен в него и извлечение жира из навески продолжалось настаиванием в течение времени перерыва.
По окончании экстракции жира эфир из колбочки отгоняют, а колбу с жиром помещают в сушильный шкаф, высушивают при 100-105 °С до постоянной массы, охлаждают в эксикаторе и взвешивают с абсолютной погрешностью не более 0,001 г. Колбу с жиром лучше сушить в атмосфере инертного газа или под вакуумом.
3.7.1.4. Обработка результатов
Массовую долю жира () в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- масса колбочки с жиром, г;
- масса пустой колбочки, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного знака.
3.7.2. Определение массовой доли жира экстракционным методом по обезжиренному остатку
3.7.2.1. Сущность метода
Метод основан на определении изменения массы образца после экстракции жира растворителем.
3.7.2.2. Аппаратура, реактивы и материалы - по п.3.7.1.2.
3.7.2.3. Проведение анализа
2-5 г исследуемого образца, отвешенных с абсолютной погрешностью не более 0,001 г, высушивают в бюксе в сушильном шкафу при температуре 100-105 °С.
Высушенную навеску количественно переносят в пакеты из фильтровальной бумаги размером 8x9 см. Стенки бюксы протирают небольшим кусочком ваты, смоченным в эфире, вату присоединяют к навеске в пакет из фильтровальной бумаги.
Пакет с навеской вкладывают в другой пакет из фильтровальной бумаги размером 9х10 см так, чтобы линии загиба обоих пакетов не совпадали. Пакеты можно перевязать ниткой. Наружный пакет нумеруют графитовым карандашом. Пакет с навеской помещают в ту же бюксу и высушивают до постоянной массы в сушильном шкафу при 100-105 °С. Допускается сушить пробы для нежирных продуктов при 100-105 °С непосредственно в пакетах. Высушенный до постоянной массы пакет с навеской помещают в экстрактор аппарата Сокслета.
В аппарат Сокслета можно поместить несколько пакетов при условии, что в процессе экстракции все пакеты будут погружены в эфир и хорошо им омыты.
Экстракцию эфиром продолжают в течение 10-12 ч. Окончание экстракции проверяют нанесением капли стекающего из экстрактора растворителя на часовое стекло. После испарения растворителя на стекле не должно оставаться жирного пятна. По окончании экстракции пакет помещают в ту же бюксу и в течение 20-30 мин выдерживают в вытяжном шкафу для удаления эфира, затем высушивают в шкафу при температуре 100-105 °С до постоянной массы в течение 1-3 ч, охлаждают в эксикаторе и взвешивают с абсолютной погрешностью не более 0,001 г.
3.7.2.4. Обработка результатов
Массовую долю жира () в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- масса высушенных бюксы, пакета и образца до экстракции, г;
- масса высушенных бюксы, пакета и образца после экстракции, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного знака.
3.7.3. Определение массовой доли жира экстракционно-весовым методом ВНИИКОП (ускоренным) - по ГОСТ 8756.21-89.
3.7.4. Определение массовой доли жира рефрактометрическим методом (ускоренным)
3.7.4.1. Сущность метода
Метод основан на измерении разности коэффициентов преломления чистого растворителя и мисцеллы.
3.7.4.2. Аппаратура, материалы и реактивы
Рефрактометр, имеющий шкалу показателя преломления в интервале 1,3-1,7.
Ступка фарфоровая по ГОСТ 9147-80.
Пробирки химические стеклянные по ГОСТ 25336-82 (14х120 мм).
Пипетки с делениями по ГОСТ 29227-91, вместимостью 10 см.
Палочки стеклянные по ГОСТ 25336-82.
Воронки стеклянные по ГОСТ 25336-82, диаметром 50 мм.
Фильтры обеззоленные.
Вата медицинская гигроскопическая по ГОСТ 5556-81.
Спирт этиловый ректификованный по ГОСТ 5962-67*.
________________
* На территории Российской Федерации действует ГОСТ Р 51652-2000 (здесь и далее).
-хлорнафталин.
-бромнафталин.
Трикрезилортофосфат.
3.7.4.3. Проведение анализа
2 г исследуемого образца взвешивают с абсолютной погрешностью не более 0,001 г, помещают в фарфоровую ступку*. Туда же градуированной пипеткой вместимостью 10 см приливают 5 см
растворителя (
-бромнафталина,
-хлорнафталина или трикрезилортофосфата). Навеску с растворителем тщательно растирают пестиком в течение 5 мин. Полученную массу фильтруют через бумажный складчатый фильтр в чистую сухую пробирку.
________________
* Навеску икры предварительно подсушивают при температуре 60-70 °С в течение 15-30 мин, а затем помещают в фарфоровую ступку.
1-2 капли прозрачного фильтрата наносят стеклянной палочкой на нижнюю призму рефрактометра. После этого осторожно (не допуская удара) закрывают верхнюю призму и через 1-2 мин определяют показатель рефракции мисцеллы. Определение проводят трижды. Из трех определений берут среднеарифметическое значение. По окончании каждого определения мисцеллу удаляют с поверхности фильтровальной бумагой или ватой, смоченной спиртом.
Преломление чистого растворителя определяют один раз перед началом работы, а плотность его - не реже одного раза в месяц и при получении новой партии.
3.7.4.4. Обработка результатов
Массовую долю жира () в процентах вычисляют по формуле
![]() , (3)*
, (3)*
_______________
* Нумерация соответствует оригиналу. - .
где - масса исследуемого образца, г;
- масса растворителя, г;
- показатель преломления мисцеллы;
- показатель преломления чистого растворителя;
- показатель отношения массовой доли жира в растворителе к разности между показателями преломления растворителя и мисцеллы (определяют экспериментально).
Так как ,
,
являются постоянными величинами для растворителя, с которыми проводятся работы, выражение
![]() в формуле (3) можно заменить обозначением
в формуле (3) можно заменить обозначением (постоянная величина). Тогда расчет количества жира в анализируемом продукте сводится к умножению постоянной величины
на разность показателей преломления чистого растворителя и мисцеллы.
![]() . (4)
. (4)
Значения коэффициента и постоянной величины
при применении вышеуказанных растворителей приведены в табл.3.
Таблица 3
Растворитель | ||
| 0,0407 | 1514 |
| 0,0612 | 1840 |
Трикрезилортофосфат | 0,1212 | 3514 |
Формулы для вычисления жира рефрактометрическим методом даны применительно к температуре 20 °С.
Измеряя рефракцию мисцеллы и растворителя при другой температуре (выше или ниже 20 °С), пользуются температурными поправками, приведенными в табл.4.
Таблица 4
Изменение показателя рефракции при изменении температуры на 1 °С
Наименование растворителя, используемого в работе | При повышении температуры на 1 °С | При понижении температуры на 1 °С |
| -0,00043 | 0,00043 |
| -0,00045 | 0,00045 |
Трикрезилортофосфат | -0,00039 | 0,00039 |
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного знака.
3.7.5. Определение массовой доли жира и воды отгонкой
3.7.5.1. Сущность метода
Метод основан на одновременном извлечении жира и воды из навески растворителем и последующем раздельном количественном их определении взвешиванием.
3.7.5.2. Аппаратура, материалы и реактивы - по п.3.3.3.2 со следующими дополнениями:
Бумага фильтровальная по ГОСТ 12026-76.
Воронка стеклянная по ГОСТ 25336-82.
Колба мерная по ГОСТ 1770-74, вместимостью 100 см.
Пипетка по ГОСТ 29227-91, вместимостью 20 см.
Бюксы по ГОСТ 25336-82, диаметром 40-50 мм.
Баня водяная.
Электроплитка по ГОСТ 14919-83.
3.7.5.3. Проведение анализа
Определение массовой доли воды проводят по п.3.3.3.
По окончании отгонки воды и охлаждения раствора в отгонной колбе раствор фильтруют через бумажный фильтр или вату в мерную колбу вместимостью 100 см. Осадок переносят на фильтр, смывают колбу и промывают осадок на фильтре несколькими порциями свежего растворителя. Объем в колбе доводят до метки растворителем и перемешивают взбалтыванием.
Отбирают пипеткой 20-30 см раствора, переносят в бюксу диаметром 40-50 мм, предварительно взвешенную с погрешностью не более 0,001 г, и растворитель выпаривают на водяной бане до полного исчезновения запаха. Бюксу тщательно вытирают снаружи фильтровальной бумагой и взвешивают с абсолютной погрешностью не более 0,001 г.
3.7.5.4. Обработка результатов
Массовую долю жира () в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- масса бюксы с жиром, г;
- масса пустой бюксы, г;
- общий объем раствора, см
;
- объем раствора, отобранный для определения жира, см
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,3%.
Вычисление проводят до первого десятичного знака
.
3.7.6. Определение массовой доли жира ускоренным экстракционно-весовым методом института питания АМН СССР
3.7.6.1. Сущность метода
Метод основан на растворении липидов бинарной смесью органических растворителей, отделении (отгонке) растворителей и весовом определении массы липидов.
3.7.6.2. Аппаратура, материалы, реактивы
Шкаф сушильный.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Насос водоструйный лабораторный стеклянный по ГОСТ 25336-82.
Мясорубка бытовая по ГОСТ 4025-95.
Часы песочные на 1 мин или секундомеры механические.
Экстрактор Клинского завода "Лабприбор".
Тигли, вместимостью 50 см или цилиндры мерные, вместимостью 100 см
.
Бюксы по ГОСТ 25336-82 или стаканчики химические по ГОСТ 25336-82.
Эксикатор по ГОСТ 25336-82.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Хлороформ технический по ГОСТ 20015-88.
Бензол по ГОСТ 5955-75.
3.7.6.3. Проведение анализа
3-5 г анализируемого продукта, отвешенных с абсолютной погрешностью не более 0,001 г, помещают в фильтрующую делительную воронку 7 экстрактора (черт.5), приливают 20 см 96° этилового спирта, закрывают ее притертой пробкой, оставляют на 5 мин, после чего интенсивно встряхивают, переворачивая в течение 1-2 мин.
1 - сливной кран; 2 - приемник экстракта; 3 - шлифовое соединение отвода к вакуумному насосу; 4 - шлифовое соединение; 5 - кран для слива экстракта в приемник; 6 - впаянная пористая пластина 2 или 3; 7 - фильтрующая делительная воронка; 8 - притертая пробка
Черт.5
Воронку устанавливают на приемке экстрактора 2, отвод 3 которого соединяют с вакуумным насосом, открывают сливной кран 5 делительной воронки, включают вакуумный насос и отсасывают экстракт в приемник. В воронку приливают 20 см экстрагирующей бинарной смеси: хлороформ-эталон (2:1), проводят одноминутную экстракцию, как описано выше, и отсасывают экстракт в приемник.
Экстракцию бинарной смесью хлороформ-этанол повторяют еще 3 раза. Экстракты из приемника переносят в мерный цилиндр с притертой пробкой и тщательно перемешивают. Отбирают пипеткой 50 см экстракта и переносят в предварительно высушенный и взвешенный стаканчик или бюксу.
Растворители отгоняют на кипящей водяной бане до исчезновения запаха.
Оставшийся в бюксе так называемый "сырой" жир сушат в сушильном шкафу 5 мин при температуре 105 °С, охлаждают до комнатной температуры в эксикаторе над хлористым кальцием и взвешивают с абсолютной погрешностью не более 0,001 г.
Для отделения нелипидных компонентов в бюксу с "сырым" жиром приливают 10 см хлороформа и через 5 мин хлороформный экстракт сливают. Операцию повторяют дважды, после чего в бюксе остаются только нелипидные компоненты.
Бюксу с остатком подсушивают в сушильном шкафу 5 мин при 105 °С, охлаждают в эксикаторе и взвешивают. Содержание липидов находят по разности значений масс "сырого" жира и нелипидных компонентов.
Примечание. Метод распространяется на все продукты, кроме рыбной муки. Для рыбной муки используется смесь хлороформ-бензол (1:1) или хлороформ-этанол (2:1).
3.7.6.4. Обработка результатов
Массовую долю липидов () в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- концентрация "сырого" жира в 50 см
экстракта, г/см
;
- концентрация нелипидных компонентов в 50 см
экстракта, г/см
;
- общий объем экстракта, см
;
- объем экстракта, взятый для анализа, см
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до первого десятичного з
нака.
3.8. Определение общей ртути по ГОСТ 26927-86.
3.9. Определение водоудерживающей способности свежего, охлажденного, сыро-, вареномороженого мяса (фарша) рыбы, морских млекопитающих, морских беспозвоночных и кулинарных изделий
3.9.1. Сущность метода
Метод основан на выделении воды из анализируемой пробы прессованием и определении ее по массе или площади "влажного" пятна.
3.9.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Мясорубка бытовая по ГОСТ 4025-95.
Равновес (гиря) массой 1 кг:
Пластина (стеклянная, плексигласовая), 100x100 мм или круг диаметром 100-150 мм.
Холодильник бытовой электрический по ГОСТ 16317-87.
Планиметр.
Стаканчики для взвешивания (бюксы) по ГОСТ 25336-82.
Чашка фарфоровая по ГОСТ 9147-80.
Эксикатор по ГОСТ 25336-82.
Часы песочные на 10 мин.
Калий хлористый по ГОСТ 4234-77, насыщенный раствор.
Бумага фильтровальная по ГОСТ 12026-76.
Фильтры (средней плотности).
3.9.3. Проведение анализа (весовой метод)
Мясо или размороженный до температуры 3-4 °С фарш массой 0,1-0,3 кг пропускают через мясорубку с решеткой диаметром отверстий 3 мм, не допуская потери сока. После тщательного перемешивания всей массы часть ее помещают в бюксу с притертой крышкой.
0,3 кг фарша взвешивают с абсолютной погрешностью не более 0,001 г, помещают на полиэтиленовый кружок, который переносят на кружок фильтровальной бумаги, расположенный на стеклянной или плексигласовой пластинке (кругу) так, чтобы навеска фарша лежала на фильтровальной бумаге. Сверху полиэтиленовый кружок закрывают стеклянной или плексигласовой пластиной (кругом), на которую ставят груз (гирю) массой 1 кг. Продолжительность прессования 10 мин.
По окончании прессования массу освобождают от фильтровальной бумаги и полиэтиленового кружка, помещают в предварительно тарированную бюксу, взвешивают и высушивают при температуре 105 °С. Массовую долю воды в отпрессованной навеске определяют по п.3.3.
3.9.4. Обработка результатов
Количество отпрессованной воды () в г вычисляют по формуле
![]() ,
,
где - масса исследуемого образца до прессования, г;
- масса исследуемого образца после прессования, г.
Водоудерживающую способность () исследуемого продукта в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- массовая доля воды в отпрессованной навеске, %;
- масса воды, отпрессованной из навески, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 1%.
Вычисление проводят до единицы.
3.9.5. Проведение анализа (по площади влажного пятна)
Метод применяют для исследования рыбы тощей и средней жирности.
Прессование проводят по п.3.9.3, используя фильтры средней плотности, предварительно выдержанные 3 сут в эксикаторе над насыщенным раствором хлористого кальция (подготовленные фильтры должны храниться в полиэтиленовом пакете в холодильнике).
По окончании прессования фильтр освобождают от навески, очерчивают карандашом контур пятна вокруг прессованного мяса и контур общего пятна по границе распространения воды. Площадь пятен определяют планиметром.
Площадь "влажного" пятна находят по разности между площадью общего пятна и площадью пятна, образованного отпрессованной массой.
Одновременно определяют массовую долю воды в исследуемом образце по п.3.3.
3.9.6. Обработка результатов
Водоудерживающую способность исследуемого продукта () в процентах вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- массовая доля воды в навеске, г;
- площадь "влажного" пятна, см
;
0,0084 - количество воды в 1 см "влажного" пятна, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 1.
Вычисление проводят до первого десятичного знака.
3.10 Определение сорбиновой кислоты - по 5.7.
(Введен дополнительно, Изм. N 1).
4. МЕТОДЫ АНАЛИЗА ПОЛУФАБРИКАТОВ И КУЛИНАРНЫХ ИЗДЕЛИЙ
4.1. Подготовка средней пробы к анализу - по п.2.2.
4.2. Определение воды - по п.3.3.
4.3. Определение хлористого натрия (поваренной соли) - по пп.3.5.1 и 3.5.2.
4.4. Определение жира - по пп.3.7.1; 3.7.2; 3.7.4.
4.5. Определение соотношения отдельных частей продукта
4.5.1. Сущность метода
Метод основан на определении отдельных частей продукта взвешиванием.
4.5.2. Аппаратура и материалы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Чашки фарфоровые по ГОСТ 9147-80.
Стаканы химические по ГОСТ 25336-82.
Пинцет по ГОСТ 21241-89.
Шпатель.
4.5.3. Определение соотношения рыбы (морских беспозвоночных) и овощей
Из навески исследуемого образца массой 100 г выбирают отдельно в две тарированные фарфоровые чашки рыбу и нерыбные объекты и овощи.
Отобранные продукты взвешивают с абсолютной погрешностью не более 0,5 г и выражают соотношение в процентах от взятой навески.
4.5.4. Определение соотношения рыбы (морских беспозвоночных) и желе (или желе с другими компонентами и т.д.).
Навеску средней пробы массой 100-200 г взвешивают в фарфоровой чашке с абсолютной погрешностью не более 0,1 г и отделяют рыбу, тщательно счищая желе или желе с другими компонентами в тарированную чашку или стакан. Чашку с содержимым взвешивают и массу желе (или желе с другими компонентами) выражают в процентах. Процентное содержание рыбы находят по разности между массой исследуемого образца и массой желе (или желе с другими компонентами).
4.5.5. Определение соотношения рыбы (морских беспозвоночных) и заливки (томатный соус, маринад и т.д.).
Соотношение в продукте рыбы и заливки определяют отдельно для каждой упаковочной единицы из числа выделенных для средней пробы. Результаты определений вычисляют для каждой упаковочной единицы в отдельности.
Тщательно вытертую снаружи единицу упаковки (банку, пакет и т.д.) взвешивают с абсолютной погрешностью не более 0,5 г - для мелкой фасовки и до 1 г - для крупной упаковочной единицы (более 350 г).
Упаковку вскрывают и осторожно, так, чтобы твердая часть продукта не проходила, сливают жидкую часть в фарфоровую чашку или стакан. Слив продолжают 15 мин, причем каждые 5 мин упаковку осторожно переворачивают. По окончании стекания упаковку с твердой частью продукта взвешивают и по разности рассчитывают массу жидкой части. Содержимое упаковки (плотную часть) переносят в фарфоровую чашку.
Упаковку моют, высушивают и после взвешивания по разности между массой брутто и упаковки определяют массу нетто.
Примечание. При необходимости проводят:
1) Определение соотношения других компонентов продукта (например, овощей, заливки и т.п.).
2) Подогревание продукции на водяной бане до 50-60 °С перед сливом жидкой части для лучшего разделения компонентов.
Массу рыбы находят по разности между массой нетто и массой жидкой части.
Массовую долю рыбы и жидкой части вычисляют в процентах к массе нетто продукта.
Среднюю пробу продукта в противнях (лотках или другой упаковке массой нетто более 1 кг) помещают на ровную поверхность (без уклона). Содержимое противня разрезают посредине вдоль и поперек на четыре равные части. Если разрез приходится на позвоночную кость (рыба с твердой костью) допускается сместить его выше или ниже середины противня, но не более, чем на 1 см.
На чистую предварительно взвешенную тарелку выкладывают из противня лопаточкой или широким ножом без острия любую четвертую часть. Выкладывание начинают от середины противня, двигая лопаточкой к его краям.
Отобранную пробу взвешивают вместе с тарелкой и по разности определяют массу пробы.
Отделяют рыбу от заливки, соуса, пользуясь ложкой или тупой стороной ножа для удаления остатков соуса, пряностей или других специй с кусков рыбы.
Взвешивают тарелку с соусом и по разности между массой пробы и массой тарелки определяют массу его.
Массу рыбы находят по разности между массой пробы и массой соуса.
Соотношение массы рыбы и жидкой части вычисляют в процентах и распространяют на всю упаковку.
4.5.6. Определение соотношения рыбы и плотной части в мучных изделиях с начинками из рыбы или рыбных фаршей и других морепродуктов
500 г сырых размороженных или жареных изделий взвешивают на технических весах с абсолютной погрешностью не более 1 г, отделяют фарш от теста и взвешивают фарш (при массе изделия более 500 г отбирают 1 экз.).
4.5.7. Массовую долю фарша () в % к массе изделий вычисляют по формуле
![]() ,
,
где - масса изделий, г;
- масса фарша, г.
4.6. Определение общей кислотности - по ГОСТ 27082-89 (титриметрическим методом).
4.7. Определение крахмала - по ГОСТ 10574-91 (количественный метод).
5. МЕТОДЫ АНАЛИЗА ИКРЫ
5.1. Подготовка средней пробы к анализу - по п.2.3.
5.2. Определение величины вакуума в банках с икрой
5.2.1. Сущность метода
Метод основан на определении величины вакуума вакуумметром.
5.2.2. Аппаратура
Вакуумметр по ГОСТ 2405-88.
5.2.3. Проведение анализа
Банку, предназначенную для анализа, моют и тщательно протирают сухой тряпкой.
Для проверки величины вакуума полой иглой, навинченной на штуцер вакуумметра, прокалывают крышку банки. При этом эластичная резиновая пробка, в которую вставлен запиленный по конусу и отточенный конец иглы, уплотняется, предотвращая потерю вакуума при анализе.
Крышку банки прокалывают так, чтобы конец иглы не попадал на кольцо жесткости или маркировочные знаки.
По отклонению стрелки вакуумметра определяют величину вакуума в банке.
5.3. Определение воды - по п.3.3.1.
5.4. Определение хлористого натрия (поваренной соли) - по пп.3.5.1; 3.5.2.
5.5. Методы определения азота летучих оснований в икре рыб
5.5.1. Определение азота летучих оснований титриметрическим методом
5.5.1.1. Сущность метода - по п.3.2.1.1.
Свободные и связанные летучие основания отгоняют с паром.
Образующийся аммиак взаимодействует с серной кислотой, избыток которой оттитровывают щелочью.
5.5.1.2. Аппаратура, материалы и реактивы - по п.3.2.1.2 со следующими дополнениями:
Пипетки по ГОСТ 29227-91, вместимостью 100 см.
Аппарат для встряхивания.
Колбы мерные с пришлифованной пробкой по ГОСТ 1770-74, вместимостью 200 см.
Воронка стеклянная по ГОСТ 25336-82, диаметром 70-80 мм.
Марля медицинская по ГОСТ 9412-93.
Вода дистиллированная по ГОСТ 6709-72.
5.5.1.3. Подготовка вытяжки икры
От 9 до 10 г тонкоизмельченной икры взвешивают с абсолютной погрешностью не более 0,1 г, переносят в мерную колбу с пришлифованной пробкой вместимостью 200 см и заливают дистиллированной водой до
объема.
Смесь взбалтывают в течение 30 мин на аппарате для встряхивания, доводят объем дистиллированной водой до 200 см, после чего фильтруют через марлю, уложенную на воронку.
Профильтрованную вытяжку используют для отгонки летучих оснований.
5.5.1.4. Проведение анализа
В отгонную колбу вместимостью 700-1000 см вносят 100 см
профильтрованной вытяжки, прибавляют 1 г окиси магния и во избежание вспенивания, кусочек чистого парафина. Колбу быстро закрывают пробкой, соединенной с парообразователем с интенсивно кипящей водой и с каплеуловителем.
Параллельно с рабочим проводят контрольный анализ (отгоняют 100 см дистиллированной воды и 1 г окиси магния).
Отгонку проводят в течение 30 мин с момента закипания жидкости в колбе. Дистиллят собирают в коническую колбу вместимостью 300-500 см с 15-20 см
раствора серной кислоты 0,05 моль/дм
, погружая в нее конец холодильника.
По окончании отгонки конец холодильника обмывают дистиллированной водой в приемную колбу, избыток серной кислоты в нем оттитровывают раствором гидроксида натрия 0,1 моль/дм с индикатором метиловым красным
.
5.5.1.5. Обработка результатов
Массовую долю азота летучих оснований () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на титрование серной кислоты в контрольном анализе, см
;
- объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на титрование серной кислоты в рабочем анализе, см
;
- коэффициент пересчета рабочего раствора гидроксида натрия на точный раствор 0,1 моль/дм
(0,1 н);
0,0014 - количество азота, эквивалентное 1 см точного раствора гидроксида натрия 0,1 моль/дм
, г;
- общий объем приготовленной вытяжки из икры, см
;
- объем профильтрованной вытяжки, взятый для отгонки, см
;
- масса икры, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,005%.
Вычисление проводят до третьего десятично
го знака.
5.5.2. Определение азота летучих оснований колориметрическим методом - по п.3.2.2, кроме приготовления стандартного раствора хлористого аммония.
Стандартный раствор хлористого аммония
0,9548 г дважды перекристаллизованного и высушенного при комнатной температуре до постоянной массы хлористого аммония, отвешенных с абсолютной погрешностью не более 0,0001 г, растворяют в мерной колбе вместимостью 0,5 дм. Приготовленный основной стандартный раствор, содержащий 0,5 мг азота в 1 см
, сохраняется в течение нескольких дней в темном прохладном месте. Перед началом работы 5 см
раствора отбирают пипеткой в мерную колбу вместимостью 500 см
и доводят объем дистиллированной водой до метки. Это рабочий стандартный раствор, содержащий 0,005 мг азота в 1 см
.
5.6. Методы определения уротропина
5.6.1. Определение уротропина (гексаметилентетрамина) титрованием
5.6.1.1. Сущность метода
Метод основан на разложении уротропина в кислой среде до формальдегида, окислении его йодом в муравьиную кислоту в щелочной среде с последующим титрованием избытка йода тиосульфатом натрия.
5.6.1.2. Аппаратура, материалы и реактивы
Микроизмельчитель тканей.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83.
Штативы с кольцами и зажимами.
Колба плоскодонная (для парообразования) по ГОСТ 25336-82, вместимостью 2000 см.
Холодильник стеклянный лабораторный с прямой трубкой по ГОСТ 25336-82.
Трубка соединительная стеклянная по ГОСТ 25336-82, диаметром 10 мм, высотой 1200 мм.
Каплеуловитель стеклянный лабораторный по ГОСТ 25336-82.
Колба Кьельдаля по ГОСТ 25336-82, вместимостью 500-750 см.
Колба коническая с притертой пробкой (колба Эрленмейера) по ГОСТ 25336-82, вместимостью 300-500 см.
Колба мерная по ГОСТ 1770-74, вместимостью 200 см.
Пипетки по ГОСТ 29227-91, вместимостью 1, 10, 20 см.
Часы.
Бюретка по ГОСТ 29251-91, вместимостью 25 см с ценой деления 0,1 см
.
Фуксин основной для приготовления фуксинсернистой кислоты.
Кислота соляная по ГОСТ 3118-77, концентрированная.
Кислота ортофосфорная по ГОСТ 6552-80, раствор 250 г/дм (25%-ный).
Кислота серная по ГОСТ 4204-77, концентрированная и раствор 5 моль/дм (10 н).
Йод по ГОСТ 4159-79, раствор 0,05 моль/дм (0,1 н) в йодистом калии.
Калий йодистый по ГОСТ 4232-74.
Натрий сернистокислый (сульфит) по ГОСТ 195-77 или натрий сернистокислый кислый (бисульфит натрия).
Натрий серноватистокислый (тиосульфат), раствор 0,1 моль/дм (0,1 н).
Крахмал растворимый по ГОСТ 10163-76, раствор 10 г/дм (1%-ный).
Натрия гидроксид по ГОСТ 4328-77, раствор 1 моль/дм (1 н).
Вода дистиллированная по ГОСТ 6
709-72.
5.6.1.3. Приготовление раствора фуксинсернистой кислоты
0,2 г фуксина (основного) растворяют при нагревании в 120 см горячей воды.
После охлаждения прибавляют в раствор 2 г безводного сульфита натрия в 20 см воды, 2 см
концентрированной соляной кислоты и доводят объем до 200 см
. Реактив выдерживают в течение суток в темном месте.
5.6.1.4. Проведение анализа
Перед определением воду в колбе-парообразователе отгонного аппарата доводят до интенсивного кипения, которое поддерживается до окончания анализа.
К холодильнику отгонного аппарата присоединяют приемную коническую колбу со шлифом, в которую предварительно наливают 5 см дистиллированной воды.
В колбу Кьельдаля помещают 5-6 г тщательно измельченной икры, взвешенной с абсолютной погрешностью не более 0,01 г, добавляют 200 см дистиллированной воды и энергично перемешивают. Через 30 мин в колбу с навеской прибавляют 10 см
раствора фосфорной кислоты 250 г/дм
, перемешивают содержимое и немедленно закрывают тщательно пригнанной пробкой, соединяющей колбу с парообразователем с одной стороны, с другой - холодильником через каплеуловитель (во избежание переброса кислоты).
Образовавшийся в кислой среде формальдегид отгоняют с водяным паром через холодильник и собирают в приемной колбе со шлифом. Отгонку проводят до получения 100-200 см дистиллята.
После получения 100 см дистиллята проводят пробу на полноту отгонки формальдегида. Для этого 5 см
дистиллята смешивают с 1 см
концентрированной серной кислоты и прибавляют 5 см
раствора фуксинсернистой кислоты. В присутствии формальдегида жидкость окрашивается в фиолетовый цвет.
Отгонку прекращают после получения отрицательной реакции на формальдегид.
К дистилляту в приемной колбе прибавляют 20 см раствора йода в йодистом калии 0,05 моль/дм
, 10 см
раствора гидроксида натрия 1 моль/дм
; колбу, плотно закрыв пробкой, взбалтывают и оставляют на 15 мин.
По истечении указанного времени содержимое колбы подкисляют 11 см раствора серной кислоты 0,5 моль/дм
, избыток йода оттитровывают раствором серноватистокислого натрия (тиосульфата) 0,1 моль/дм
в присутствии раствора крахмала 10 г/дм
.
Параллельно проводят контрольный анализ со свежеперегнанной дистиллирова
нной водой.
5.6.1.5. Обработка результатов
Массовую долю уротропина () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора серноватистокислого натрия 0,1 моль/дм
, израсходованный на титрование йода в контрольном анализе, см
;
- объем раствора серноватистокислого натрия 0,1 моль/дм
, израсходованный на титрование йода, в рабочем анализе, см
;
- коэффициент пересчета на точный раствор серноватистокислого натрия 0,1 моль/дм
;
0,00117 - количество уротропина, эквивалентное 1 см точного раствора серноватистокислого натрия 0,1 моль/дм
, г;
- навеска икры, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,01%.
Вычисление проводят до второго десятичного з
нака.
5.6.2. Определение уротропина (гексаметилентетрамина) колориметрическим методом
5.6.2.1. Сущность метода
Метод основан на способности формальдегида, образующегося при разложении уротропина в кислой среде, давать окрашенный комплекс с реактивом Нэша.
5.6.2.2. Аппаратура, материалы и реактивы
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83.
Баня водяная.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Ступка фарфоровая по ГОСТ 9147-80.
Чашка фарфоровая по ГОСТ 9147-80, вместимостью 50-100 см.
Колбы мерные по ГОСТ 1770-74, вместимостью 25, 100, 200 и 1000 см.
Колбы плоскодонные конические по ГОСТ 25336-82, вместимостью 250, 500, 1000-2000 см.
Насадка Вюрца по ГОСТ 25336-82.
Аллонж по ГОСТ 25336-82.
Холодильник Либиха по ГОСТ 25336-82.
Трубка стеклянная диаметром 10 мм, длиной 1200 мм.
Бюретка по ГОСТ 29251-91, вместимостью 25 см.
Пипетка по ГОСТ 29227-91, вместимостью 10 см.
Магний сернокислый по ГОСТ 4523-77, ч.д.а.
Кислота лимонная моногидрат и безводная по ГОСТ 3652-69, х.ч.
Уротропин (гексаметилентетрамин) по Госфармакопее СССР с массовой долей основного вещества не менее 990 г/дм (99%-ный).
Натрий двууглекислый по ГОСТ 4201-79, х.ч., раствор 1 моль/дм (1 н).
Кислота серная по ГОСТ 4204-77, х.ч., раствор 810 г/дм (81%-ный).
Динатриевая соль хромотроповой кислоты, раствор 5 г/дм (0,5%-ный) в дистиллированной воде (раствор в склянке с притертой пробкой хранится не более 5 сут).
Вода дистиллированная по ГОСТ 6709-72.
Аммоний уксуснокислый по ГОСТ 3117-78, ч.д.а.
Кислота уксусная по ГОСТ 61-75, х.ч.
Ацетилацетон по ГОСТ 10259-78, ч.д.а.
Бумага индикаторная "Рифан" или "Универсальна
я".
5.6.2.3. Приготовление растворов
Ацетилацетоновый реактив (реактив Нэша)
В мерной колбе вместимостью 1 дм растворяют в дистиллированной воде 150 г уксуснокислого аммония, добавляют 3 см
ледяной уксусной кислоты, объем в колбе доводят водой до метки и перемешивают. Приготовленный раствор уксуснокислого аммония в склянке с притертой пробкой сохраняется 2-3 мес.
Для приготовления реактива Нэша к 100 см раствора уксуснокислого аммония добавляют 1 см
ацетилацетона и тщательно перемешивают. Реактив Нэша годен для использования в течение 2 сут.
5.6.2.4. Проведение анализа
В фарфоровую ступку отвешивают с абсолютной погрешностью не более 0,1 г от 7 до 8 г хорошо измельченной средней пробы икры и тщательно растирают с 2 г лимонной кислоты и 30 г сульфата магния.
Смесь количественно переносят в термостойкую колбу 2 вместимостью 500 см (черт.6). Остатки из ступки смывают 10 см
дистиллированной воды и отбирают небольшими кусочками чистой ваты или фильтровальной бумаги, которые помещают в ту же колбу.
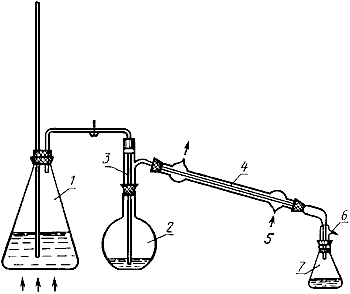
1 - парообразователь; 2 - отгонная колба; 3 - насадка Вюрца; 4 - водяной холодильник; 5 - аллонж с отводом; 6 - регулятор давления; 7 - приемная колба
Черт.6
Колбу 2 немедленно соединяют через насадку Вюрца 3 с колбой-парообразователем 1, в котором интенсивно кипит вода, и с холодильником 4.
Образующийся формальдегид отгоняют с водяным паром, собирая дистиллят в приемную колбу 7, соединенную с холодильником при помощи аллонжа 5 с обязательным отводом 6 для регулирования давления в системе.
После получения 100 см дистиллята проводят пробу на полноту отгонки формальдегида. Для этого 0,5 см
дистиллята собирают в пробирку, смешивают с 0,5 см
раствора хромотроповой кислоты 5 г/дм
и добавляют 4 см
раствора серной кислоты 810 г/дм
. В присутствии формальдегида смесь окрашивается в розовый или фиолетовый цвет. При отрицательной реакции на формальдегид отгонку прекращают.
Дистиллят нейтрализуют раствором двууглекислого натрия 1 моль/дм до рН 7 (по индикаторной бумаге) и количественно переносят дистиллированной водой в мерную колбу вместимостью 200 см
, объем в мерной колбе доводят до метки и тщательно перемешивают.
Для проведения цветной реакции отбирают пипеткой 1 см нейтрализованного дистиллята в мерную колбу вместимостью 25 см
и добавляют 1-2 см
реактива Нэша. Обмывают горло колбы 3-5 см
дистиллированной воды, перемешивают содержимое и термостатируют 15 мин в водяной бане температурой 50 °С. По истечении указанного времени содержимое колбы охлаждают проточной водопроводной водой, доводят объем дистиллированной водой до метки и тщательно перемешивают.
Оптическую плотность окрашенных растворов измеряют в кюветах с рабочей длиной 10 мм фотоэлектроколориметром или спектрофотометром при длине волны 412 нм по отношению к контрольной пробе из 1 см дистиллированной воды и 1 см
реактива Нэша, термостатированной 15 мин при 50 °С. Содержание уротропина, соответствующее определенной оптической плотности, рассчитывают по градуировочному
графику.
5.6.2.5. Построение градуировочного графика
Готовят основной стандартный раствор уротропина. Для этого 1 г гексаметилентетрамина, взвешенного с абсолютной погрешностью не более 0,0001 г, количественно переносят дистиллированной водой в мерную колбу вместимостью 100 см. После растворения уротропина объем в колбе доводят до метки дистиллированной водой и тщательно перемешивают. Отбирают пипеткой 10 см
приготовленного раствора гексаметилентетрамина, вносят в колбу отгонного аппарата и после добавления 2 г лимонной кислоты и 30 г сульфата магния отгоняют формальдегид с водяным паром по п.5.6.1.4.
Нейтрализованный и разведенный до 200 см дистиллят является основным стандартным раствором (с массовой концентрацией 0,5 г уротропина в 1 см
) и служит для приготовления серии рабочих стандартных растворов с различной концентрацией уротропина.
В 5 мерных колб вместимостью 100 см последовательно наливают из бюретки 2, 4, 6, 8 и 10 см
основного стандартного раствора. Колбы доливают дистиллированной водой до метки и тщательно перемешивают. 1 см
каждого из приготовленных растворов содержит 0,010; 0,020; 0,030; 0,040; 0,050 мг уротропина.
Из каждого из пяти растворов берут пипеткой по 1 см в мерные колбы вместимостью 25 см
и приливают по 1 см
реактива Нэша.
Все дальнейшие операции проводят по п.5.6.2.4.
Подготовку серии стандартных растворов проводят три раза, начиная каждый раз с приготовления основного раствора гексаметилентетрамина. Для каждого раствора одной и той же концентрации берут среднее значение оптической плотности из трех измерений.
По полученным данным строят градуировочный график, откладывая на оси абсцисс количество уротропина, а на оси ординат - соответствующие оптические плотно
сти.
5.6.2.6. Обработка результатов
Массовую долю уротропина в икре () в процентах вычисляют по формуле
![]() ,
,
где - содержание уротропина, найденное по градуировочному графику, мг;
- объем дистиллята после нейтрализации и разведения, см
;
- объем разведенного дистиллята, взятый для цветной реакции, см
;
- масса исследуемого образца, г;
1000 - коэффициент пересчета миллиграммов в граммы.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,01%.
Вычисление проводят до второго десятичного знака.
5.7. Определение сорбиновой кислоты колориметрическим методом
5.7.1. Сущность метода
Метод основан на способности малонового альдегида, в который окисляется сорбиновая кислота в кислой среде, образовывать окрашенный комплекс с тиобарбитуровой кислотой.
5.7.2. Средства измерений, оборудование, реактивы, материалы
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Колбы мерные по ГОСТ 1770-74, вместимостью 500 и 100 см.
Стаканы по ГОСТ 25336-82, вместимостью 150 см.
Цилиндры мерные по ГОСТ 1770-74.
Воронки химические по ГОСТ 25336-82, диаметром 6-8 см.
Бюретки по ГОСТ 29251-91, вместимостью 25 см.
Пипетки по ГОСТ 29227-91, вместимостью 5, 10, 15 см.
Палочки стеклянные.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Ареометр общего назначения по ГОСТ 18481-81 для определения плотности жидкости (=1000-1100 кг/м
).
Вода дистиллированная по ГОСТ 6709-72.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Кислота уксусная по ГОСТ 61-75.
Калий двухромовокислый по ГОСТ 4220-75, раствор 0,02 моль/дм.
Кислота трихлоруксусная, водный раствор 200 г/дм (20%-ный).
Кислота сорбиновая (2,4 гексадиеновая) с массовой долей основного вещества не менее 99%.
Кислота тиобарбитуровая, свежеприготовленный раствор 0,02 моль/д м.
Допускается применение аналогичных по метрологическим характеристикам средств измерений, аналогичного по техническим характеристикам оборудования и материалов с требованиями безопасности не ниже, указанных в настоящем стандарте.
(Измененная редакция, Изм. N 1).
5.7.3. Приготовление реактивов
Раствор тиобарбитуровой кислоты
0,2880 г тиобарбитуровой кислоты переносят в мерную колбу вместимостью 100 см 90%-ной уксусной кислотой (
=1066 кг/м
). Колбу заполняют на 2/3 объема, содержимое слегка нагревают на водяной бане и взбалтывают. После растворения тиобарбитуровой кислоты объем жидкости в колбе доливают до метки уксусной кислотой и перемешивают. Приготовленный раствор тиобарбитуровой кислоты годен в течение одних суток.
Стандартный раствор сорбиновой кислоты
Навеску 0,1 г свежевозогнанной или свежеперекристаллизованной из спирта сорбиновой кислоты, взвешенную с абсолютной погрешностью не более 0,0001 г, количественно переносят дистиллированной водой в мерную колбу вместимостью 1000 см, заполняют ее на
объема и энергично перемешивают. После полного растворения сорбиновой кислоты колбу доливают водой до метки и перемешивают содержимое.
1 см основного стандартного раствора содержит 0,1 мг сорбиновой кислоты.
5.7.4. Приготовление вытяжки из пробы продукции
В высокий стакан вместимостью 150 см помещают 1 г тщательно измельченной пробы исследуемой продукции (пробы с низким содержанием влаги необходимо измельчать до размера частиц не более 1 мм), взвешенной с абсолютной погрешностью не более 0,01 г, и растирают ее стеклянной палочкой с резиновым наконечником, постепенно приливая небольшими порциями 25 см
дистиллированной воды. Полученную смесь выдерживают 25 мин при периодическом перемешивании.
В стакан добавляют 10 см водного раствора трихлоруксусной кислоты 200 г/дм
и осторожно, во избежание вспенивания, нагревают содержимое до кипения при непрерывном помешивании. После 10 мин спокойного кипения горячую смесь фильтруют через смоченный водой бумажный фильтр в мерную колбу вместимостью 500 см
. При фильтровании применяют воронку с обогревом или поддерживают температуру фильтруемой смеси, помещая стакан в горячую водяную баню. Стакан и осадок на фильтре промывают 6-7 раз кипящей дистиллированной водой, собирая промывные воды в ту же мерную колбу.
Фильтрат проверяют на полноту осаждения белков, добавляя несколько капель 200 г/дм трихлоруксусной кислоты. При помутнении раствора проводят повторное осаждение белков трихлоруксусной кислотой и горячее фильтрование.
Жидкость должна занимать не более объема колбы.
(Измененная редакция, Изм. N 1).
5.7.5. Проведение анализа
В колбу с фильтратом приливают 10 см раствора двухромовокислого калия 0,02 моль/дм
и 15 см
раствора тиобарбитуровой кислоты 0,02 моль/дм
.
После тщательного перемешивания колбу помещают в кипящую водяную баню и выдерживают 30 мин. Уровень воды в бане должен быть выше уровня жидкости в колбах. По истечении указанного времени колбу охлаждают на воздухе или в проточной воде, объем раствора доводят дистиллированной водой до метки, жидкость перемешивают. Одновременно проводят контрольный анализ, используя все реактивы без вытяжки из пробы продукции.
Оптическую плотность окрашенного раствора измеряют спектрофотометром или фотоэлектроколориметром при длине волны 532 нм в кюветах с рабочей длиной 10 мм по отношению к контрольному раствору. Содержание сорбиновой кислоты, соответствующее определенной оптической плотности, рассчитывают по градуировочному графику.
Окраска раствора устойчива и сохраняется в течение нескольких часов.
(Измененная редакция, Изм. N 1).
5.7.6. Построение градуировочного графика
В мерные колбы вместимостью 500 см последовательно вносят из бюретки основной стандартный раствор сорбиновой кислоты в количествах, указанных в табл.5.
Таблица 5
Номер колбы | Количество основного стандартного раствора, см | Количество сорбиновой кислоты, мг |
1 | 5 | 0,5 |
2 | 10 | 1,0 |
3 | 15 | 1,5 |
4 | 20 | 2,0 |
5 | 25 | 2,5 |
Контрольный анализ | 0 | 0 |
В каждую колбу приливают по 10 см раствора трихлоруксусной кислоты 200 г/дм
, разбавляют содержимое дистиллированной водой до
объема колбы и проводят все дальнейшие операции в соответствии с п.5.7.5.
Оптическую плотность окрашенных растворов измеряют спектрофотометром или фотоэлектроколориметром при длине волны 532 нм.
Подготовку серии растворов для построения градуировочного графика проводят три раза, начиная каждый раз с приготовления основного раствора сорбиновой кислоты. Для каждого раствора одинаковой концентрации берут среднее значение оптической плотности трех измерений.
По полученным данным строят градуировочный график, откладывая на оси абсцисс количество сорбиновой кислоты, на оси ординат - соответствующие оптические плотности.
5.7.7. Обработка результатов
Массовую долю сорбиновой кислоты в продукте () в процентах вычисляют по формуле
![]() ,
,
где - масса пробы продукции, взятая для приготовления вытяжки, г;
- содержание сорбиновой кислоты, найденное по градуировочному графику, мг;
1000 - коэффициент пересчета миллиграммов в граммы.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,02%.
Вычисление проводят до второго десятичного знака.
(Измененная редакция, Изм. N 1).
5.8. Определение тяжелых металлов (олова - по ГОСТ 26935-86, свинца - по ГОСТ 26932-86).
5.9. Определение наличия песка
5.9.1. Сущность метода
Метод основан на термохимической минерализации (сжигание, обработка соляной кислотой) навески икры и весовом определении песка.
5.9.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83.
Баня водяная.
Шкаф сушильный.
Электропечь сопротивления лабораторная.
Эксикатор по ГОСТ 25336-82.
Ступка фарфоровая по ГОСТ 9147-80.
Чашка фарфоровая по ГОСТ 9147-80.
Тигель фарфоровый по ГОСТ 9147-80.
Колба коническая по ГОСТ 25336-82, вместимостью 250 см.
Воронка стеклянная по ГОСТ 25336-82.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Фильтр обеззоленный.
Кислота соляная по ГОСТ 3118-77, раствор 100 г/дм (10%-ный).
5.9.3. Проведение анализа
20-50 г измельченной икры, взвешенных с абсолютной погрешностью не более 0,01 г, подсушивают в фарфоровой чашке или большом тигле в сушильном шкафу, затем обугливают на плитке или в муфельной печи. Уголь выщелачивают горячей водой и фильтруют. Фильтр с осадком озоляют. Золу обрабатывают раствором соляной кислоты 100 г/дм в течение 30 мин на кипящей водяной бане и фильтруют через обеззоленный фильтр. Осадок на фильтре промывают горячей водой до исчезновения реакции на хлор (проба с раствором азотнокислого серебра). Фильтр вместе с осадком сжигают и прокаливают в предварительно взвешенном фарфоровом тигле. Тигель охлаждают в эксикаторе и взвешивают с абсолютной погрешностью не более 0,001 г.
5.9.4. Обработка результатов
Массовую долю песка () в процентах вычисляют по формуле
![]() ,
,
где - масса икры, г;
- масса тигля, г;
- масса тигля с прокаленным осадком, г.
Вычисление проводят до второго десятичного знака.
6. МЕТОДЫ АНАЛИЗА КОНЦЕНТРАТОВ, БЕЛКОВОЙ МАССЫ, БУЛЬОНОВ, ГИДРОЛИЗАТОВ И ПАСТ
6.1. Подготовка средней пробы к анализу - по п.2.4.
6.2. Определение воды - по п.3.3.1.
6.3. Определение жира - по пп.3.7.1; 3.7.2; 3.7.4.
Для жидких продуктов навеску массой 3-5 г, взвешенную с абсолютной погрешностью не более 0,001 г, предварительно высушенную с песком до постоянной массы, переносят в фарфоровую ступку и растирают до получения тонкого порошка, который количественно переносят в пакет для дальнейшей экстракции эфиром.
6.4. Определение общего азота - по пп.8.9.1; 8.9.2; 8.9.4.
Пересчет на белковые вещества не проводят.
6.5. Методы определения аминного азота
6.5.1. Определение аминного азота осаждением фосфатом меди
6.5.1.1. Сущность метода
Метод основан на осаждении белка фосфатом меди и титриметрическом определении аминного азота в фильтрате.
6.5.1.2. Аппаратура, реактивы и материалы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Колбы мерные по ГОСТ 1770-74, вместимостью 50, 100, 1000 см.
Пипетки градуированные по ГОСТ 29227-91, вместимостью 2, 5, 10, 20 см.
Бюретка по ГОСТ 29251-91, вместимостью 50 см.
Воронки по ГОСТ 25336-82.
Колбы конические по ГОСТ 25336-82, вместимостью 100 см.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 50 и 250 см.
Капельница лабораторная по ГОСТ 25336-82.
Фильтры бумажные с синей лентой.
Кислота соляная, х.ч., по ГОСТ 3118-77, плотностью 1119 кг/м.
Кислота уксусная, х.ч., по ГОСТ 61-75 ледяная.
Потенциометр постоянного тока по ГОСТ 9245-79.
Калий йодистый, х.ч., по ГОСТ 4232-74, раствор 100 г/дм (10%-ный).
Крахмал растворимый по ГОСТ 10163-76, раствор 10 г/дм (1%-ный).
Натрий серноватистокислый (тиосульфат), раствор 0,01 моль/дм (0,01 н).
Натрий тетраборнокислый (бура), х.ч., по ГОСТ 4199-76.
Натрия гидроксид, ч.д.а., по ГОСТ 4328-77, раствор 0,5 моль/дм (0,5 н).
Натрий фосфорнокислый трехзамещенный, ч.д.а., по ГОСТ 201-76.
Натрий фосфорнокислый двузамещенный 12-водный по ГОСТ 4172-76.
Медь двухлористая 2-водная, ч.д.а., по ГОСТ 4167-74.
Вода дистиллированная по ГОСТ 6709-72.
Тимолфталеин, раствор 2,5 г/дм (0,25%-ный) в 50%-ном этиловом сп
ирте.
6.5.1.3. Приготовление растворов
Раствор хлорной меди
27,3 г хлорной меди растворяют в 500 см дистиллированной воды, перемешивают и доводят объем до 1000 см
дистиллированной водой.
Раствор фосфата натрия
68,5 г трехзамещенного фосфорнокислого натрия растворяют в 500 см дистиллированной воды, перемешивают и доводят объем до 1000 см
или 64,5 г двузамещенного 12-водного фосфорнокислого натрия растворяют в 500 см
дистиллированной воды, освобожденной кипячением от углекислого газа, добавляют 7,2 г гидроксида натрия и доводят объем до 1000 см
дистиллированной водой.
Боратный буферный раствор
28,6 г тетрабората натрия растворяют в 750 см дистиллированной воды, добавляют 50 см
раствора соляной кислоты 1 моль/дм
(1 н) и доводят объем дистиллированной водой до 1000 см
(рН 8,8).
Суспензия фосфата меди
Смешивают один объем раствора хлорида меди с двумя объемами раствора фосфата натрия и приливают два объема боратного буфера.
Суспензия готовится перед началом работы.
6.5.1.4. Проведение анализа
В мерную колбу вместимостью 50 см помещают 0,2-0,3 г упаренного или 0,10-0,15 г сухого гидролизата, отвешенного с абсолютной погрешностью не более 0,001 г, добавляют 4 см
воды, 5 капель тимолфталеина и по каплям раствор 0,5 моль/дм
гидроокиси натрия до слабо-голубого окрашивания (рН 10,2). После этого в колбу добавляют 20 см
суспензии фосфата меди, хорошо перемешивают. При исчезновении осадка добавляют еще 5 см
суспензии, объем в колбе доводят до метки дистиллированной водой, энергично перемешивают и фильтруют через плотный фильтр. Фильтрат должен быть абсолютно прозрачным.
В две колбы отмеряют по 10 см фильтрата, подкисляют 0,4 см
ледяной уксусной кислоты, добавляют 7,5 см
раствора йодида калия 100 г/дм
и титруют выделившийся йод раствором 0,01 моль/дм
тиосульфата натрия. В процессе титрования, когда раствор примет соломенно-желтую окраску, добавляют 1,5 см
крахмала (появляется синяя окраска) и продолжают титрование до исчезновения синей окраски.
Одновременно проводят контрольное определение без исследуемого раст
вора.
6.5.1.5. Обработка результатов
Массовую долю аминного азота () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора тиосульфата натрия 0,01 моль/дм
, израсходованный на титрование исследуемого образца, см
;
- объем раствора тиосульфата натрия 0,01 моль/дм
, израсходованный на титрование контрольного образца, см
;
- коэффициент пересчета на точный раствор 0,01 моль/дм
тиосульфата натрия;
- объем приготовленного раствора, см
;
- объем фильтрата, взятый на титрование, см
;
0,28 - количество аминного азота, соответствующее 1 см точного раствора 0,01 моль/дм
тиосульфата натрия, мг;
- масса исследуемого образца, г;
1000 - коэффициент пересчета мг в г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до третьего десятично
го знака.
6.5.2. Определение аминного азота формольным титрованием
6.5.2.1. Сущность метода
Метод основан на связывании аминных групп с формалином и косвенном определении их количества по результатам титрования карбоксильных групп.
6.5.2.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Потенциометр постоянного тока измерительный с пределами измерений значений рН от 0 до 14 по ГОСТ 9245-79.
Стаканы стеклянные лабораторные по ГОСТ 25336-82, вместимостью 50, 100 см.
Пипетки по ГОСТ 29227-91, вместимостью 2, 20 см.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 10 см.
Колба мерная по ГОСТ 1770-74, вместимостью 100 см.
Бюретка по ГОСТ 29251-91, вместимостью 25 см.
Мешалка магнитная.
Вода дистиллированная по ГОСТ 6709-72.
Натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм (0,1 н) и 0,02 моль/дм
(0,02 н).
Формалин по ГОСТ 1625-89, раствор 400 г/дм (40%-ный).
Кислота серная по ГОСТ 4204-77, раствор 0,05 моль/дм (0,1 н).
Спирт этиловый ректификованный по ГОСТ 5962-67.
Индикатор смешанный (Таширо).
6.5.2.3. Приготовление реактивов
Нейтрализованный формалин
Формалин нейтрализуется в день анализа раствором 0,1 моль/дм (0,1 н) гидроксида натрия до рН=7.
Индикатор смешанный
0,12 г метилового красного и 0,08 г метиленового синего растворяют отдельно в небольшом количестве спирта 900 г/дм (90%-ного). Растворы сливают в мерную колбу вместимостью 100 см
, объем доводят спиртом до метки.
6.5.2.4. Проведение анализа
Сначала проводят контрольное титрование.
В стеклянный стакан наливают 20 см дистиллированной воды, помещают магнит. В стакан помещают электроды, приливают 10 см
нейтрализованного формалина, включают мешалку и титруют жидкость раствором 0,02 моль/дм
гидроксида натрия до рН=9.
При проведении рабочего анализа в стакан вместимостью 50 см помещают 0,2-0,3 г исследуемого образца, взвешенного с абсолютной погрешностью не более 0,001 г, и доводят объем дистиллированной водой до 20 см
. В стакан опускают магнит, электроды.
Раствор нейтрализуют 0,1 моль/дм гидроксида натрия до рН 6,8-7,0.
При работе с очень кислым гидролизатом (рН 1,0) начинать нейтрализацию удобнее раствором 1 моль/дм (1 н) гидроксида натрия, постепенно переходя к концентрации 0,1 моль/дм
и 0,02 моль/дм
.
По окончании нейтрализации в гидролизат добавляют 10 см формалина (рН 4,5-5,5) и титруют раствором 0,02 моль/дм
(0,02 н) гидроксида натрия до
рН 9.
6.5.2.5. Обработка результатов
Массовую долю аминного азота () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида натрия 0,02 моль/дм
, израсходованный на титрование исследуемого образца, см
;
- объем раствора гидроксида натрия 0,02 моль/дм
, израсходованный на титрование контрольного образца, см
;
- коэффициент пересчета на точный раствор 0,02 моль/дм
гидроксида натрия;
0,28 - количество аминного азота, соответствующее 1 см раствора 0,02 моль/дм
гидроксида натрия, мг;
1000 - коэффициент пересчета мг в г;
- навеска исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до третьего десятичного з
нака.
6.6. Определение хлористого натрия (поваренной соли) - по п.3.5.1.
Навеску образца гидролизата 0,2-0,3 г отвешивают с абсолютной погрешностью не более 0,001 г и дальше проводят определение по п.3.5.1.3.
6.7. Определение золы - по п.11.6.
6.8. Определение перекиси водорода в белковой массе
6.8.1. Сущность метода
Метод основан на извлечении перекиси водорода из навески белковой массы водой и количественном определении ее титрованием.
6.8.2. Аппаратура, материалы и реактивы
Колбы конические по ГОСТ 25336-82, вместимостью 100 и 250 см по ГОСТ 1770-74.
Колба мерная по ГОСТ 1770-74, вместимостью 100 см.
Колбы конические по ГОСТ 25336-82, вместимостью 100 и 250 см.
Пипетка по ГОСТ 29227-91, вместимостью 5 и 10 см.
Бюретка по ГОСТ 29251-91, вместимостью 25 см.
Воронка стеклянная по ГОСТ 25336-82, диаметром 50-70 мм.
Калий марганцовокислый по ГОСТ 20490-75, раствор 0,01 моль/дм (0,05 н).
Кислота серная по ГОСТ 4204-77, раствор 100-150 г/дм (10-15%-ный).
Вода дистиллированная по ГОСТ 6709-72.
(Измененная редакция, Изм. N 1).
6.8.3. Проведение анализа
Навеску анализируемого продукта массой от 9 до 10 г, взвешенную с абсолютной погрешностью не более 0,1 г, количественно переносят дистиллированной водой через воронку в мерную колбу вместимостью 100 см. Количество воды должно быть не более
объема колбы. Содержимое колбы перемешивают и настаивают в течение 10 мин, периодически взбалтывая. В колбу доливают дистиллированную воду до метки, содержимое перемешивают и фильтруют через бумажный фильтр в чистую сухую колбу. Отбирают пипеткой в коническую колбу 10 см
фильтрата, приливают 5 см
серной кислоты 100-150 г/дм
и титруют раствором 0,01 моль/дм
перманганата калия до появления слабо-розовой окраски.
Одновременно проводят контрольный анализ с навеской исследуемого продукта до обработки его перекисью водорода.
6.8.4. Обработка результатов
Массовую долю перекиси водорода в белковой массе () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора марганцовокислого калия 0,01 моль/дм
, израсходованный на титрование 10 см
рабочего образца, см
;
- объем раствора марганцовокислого калия 0,01 моль/дм
, израсходованный на титрование 10 см
контрольного образца, см
;
- объем вытяжки, см
;
- объем фильтрата, взятый для титрования, см
;
- коэффициент пересчета на точный раствор 0,01 моль/дм
перманганата калия;
0,00085 - количество перекиси водорода, соответствующее 1 см 0,01 моль/дм
раствора перманганата калия, г;
- навеска исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,05%.
Вычисление проводят до второго десятич
ного знака.
6.9. Определение растворимости белка в воде
6.9.1. Сущность метода
Метод основан на извлечении из белка водорастворимой фракции, обезвоживании нерастворимой части центрифугированием, выпариванием, высушиванием и весовом определении ее.
6.9.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Центрифуга.
Шкаф сушильный.
Эксикатор по ГОСТ 25336-82.
Стаканчики для взвешивания (бюксы) с притертой крышкой по ГОСТ 25336-82.
Колба мерная по ГОСТ 1770-74, вместимостью 250 см.
Пипетка по ГОСТ 29227-91, вместимостью 20 см.
Вода дистиллированная по ГОСТ 6709-72.
6.9.3. Проведение анализа
Навеску рыбного белка (с известной массовой долей воды) массой от 4,5 до 5 г растирают в ступке с небольшим количеством дистиллированной воды в течение 3-5 мин и количественно переносят в мерную колбу вместимостью 250 см. Колбу доливают водой до метки и, закрыв пробкой, содержимое взбалтывают, переворачивая колбу 10-15 раз. Содержимое колбы центрифугируют в течение 20 мин со скоростью 1000 об/мин. Пипеткой отбирают 20 см
центрифугата и переносят в предварительно высушенную и тарированную бюксу.
Бюксу с центрифугатом помещают в сушильный шкаф и выпаривают жидкость при температуре не выше 70 °С.
Остаток в бюксе сушат еще 2 ч при температуре 100-105 °С, затем охлаждают в эксикаторе и взвешивают.
6.9.4. Обработка результатов
Растворимость рыбного белка () в процентах вычисляют по формуле
![]() ,
,
где - массовая доля воды в рыбном белке, %;
- масса сухого остатка после выпаривания 20 см
центрифугата, г;
- масса рыбного белка, г;
- объем раствора в мерной колбе, см
;
- объем центрифугата, отобранный для высушивания, см
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 1%.
Вычисления проводят до единиц
ы.
6.10. Определение прозрачности и растворимости гидролизата
6.10.1. Сущность метода
Метод основан на визуальном определении прозрачности гидролизата в проходящем свете и растворимости.
6.10.2. Проведение анализа
5 г гидролизата, отвешенных с абсолютной погрешностью не более 0,01 г, растворяют, помешивая, в 250 см дистиллированной воды температурой 18-20 °С.
Прозрачность раствора и отсутствие нерастворимого осадка определяют визуально.
6.11. Определение растворимости концентрата (бульонных таблеток)
Таблетку концентрата растворяют в 200 см горячей воды температурой 70-80 °С, перемешивая стеклянной палочкой.
Отсутствие нерастворимого осадка определяют визуально.
6.12. Определение азота летучих оснований - по пп.3.2.1; 3.2.2.
6.13. Определение общей кислотности - по ГОСТ 27082-89.
6.14. Определение активной кислотности гидролизата (рН).
Навеску гидролизата разводят дистиллированной водой в соотношении 1:10 и определяют активную кислотность по ГОСТ 28972-91.
6.15. Определение тяжелых металлов - ГОСТ 26927-86-ГОСТ 26928-86, ГОСТ 26929-94, ГОСТ 26930-86-ГОСТ 26932-86, ГОСТ 26934-86, ГОСТ 26935-86.
7. МЕТОДЫ АНАЛИЗА ЖИРОВ, КРИСТАЛЛИЧЕСКОГО СПЕРМАЦЕТА, ЖИДКИХ ВИТАМИННЫХ ПРЕПАРАТОВ И СЫРЬЯ ДЛЯ ИХ ПРОИЗВОДСТВА
7.1. Подготовка средней пробы к анализу - по пп.2.5 и 2.12.
7.2. Методы определения цвета жира
7.2.1. Определение цвета жира визуально
7.2.1.1. Сущность метода
Метод основан на визуальном определении цвета жира в проходящем дневном свете.
7.2.1.2. Аппаратура, материалы
Стакан химический бесцветного стекла по ГОСТ 25336-82, диаметром 50-100 мм.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Электроплитка бытовая по ГОСТ 14919-83.
7.2.1.3. Проведение анализа.
Часть профильтрованной средней пробы или жидкого витаминного препарата наливают в стакан диаметром 50-100 мм из бесцветного стекла (слабоокрашенный жир или препарат наливают в стакан диаметром 100 мм).
Жир или жидкий витаминный препарат нагревают до температуры, при которой проводится определение прозрачности, и рассматривают в проходящем дневном свете, определяя цвет и оттенок.
7.2.2. Определение цвета жира цветомером ВНИИЖ-16
7.2.2.1. Сущность метода
Метод основан на сравнении цвета жира с цветом стандартных стеклянных фильтров.
7.2.2.2. Цвет жира определяют цветомером ВНИИЖ-16* и выражают числом красных единиц при 35 желтых. При этом цвет одной половины поля зрения, освещенной световым потоком, прошедшим через слой жира, сравнивают с цветом другой половины поля зрения, которая может быть освещена световым потоком переменного цвета. Изменения цвета последнего достигают при помощи набора светофильтров, выделяющих световые потоки определенной величины и цвета. Величину светового потока и его цвет измеряют в условных цветовых единицах.
________________
* Цвет всех видов жиров допускается определять на других приборах, дающих сопоставимые результаты.
7.2.2.3. Аппаратура, реактивы
Цветомер ВНИИЖ-16 (черт.7).
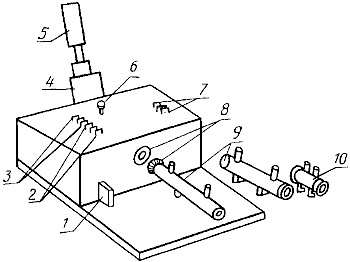
1 - кювета для масла толщиной 10 мм; 2 - набор желтых светофильтров; 3 - набор красных светофильтров; 4 - кожух призмы; 5 - окуляр; 6 - ручка перемещения клина; 7 - набор нейтральных светофильтров; 8 - верхнее и нижнее окна прибора; 9 - кювета для масла толщиной 130 мм; 10 - кювета для масла толщиной 50 мм
Черт.7
Растворитель.
Цветомер ВНИИЖ-16 состоит из корпуса с вмонтированной оптической системой цветных фильтров, осветителя, основными частями которого являются кожух с белой, рассеивающей свет, поверхностью экрана, двух ламп накаливания, набора кювет различной длины.
Прибор предназначен для измерения цвета жира, выражаемого числом красных единиц в интервале от 2 до 70 при 20-125 желтых.
В световой поток, прошедший через верхнее окно прибора, могут быть введены:
- желтые фильтры, выделяющие потоки, равные 20, 35 и 70 единицам желтого цвета;
- красные фильтры, выделяющие потоки, равные 10, 20 и 30 единицам красного цвета;
- двойной клин, позволяющий получать переменные световые потоки в интервале 1-10 единиц красного цвета.
Правая половина поля зрения освещается током, прошедшим через слой жира, левая - через систему цветных фильтров.
Прибор должен быть установлен перед осветителем так, чтобы освещенность полей зрения окуляра была одинаковой. При правильной установке граница между полями должна быть отчетливо видна.
7.2.2.4. Подготовка пробы
Пробу анализируемого жира нагревают до температуры, при которой предусмотрено определение его прозрачности. При заполнении кюветы жиром не должно быть воздушных пузырьков.
7.2.2.5. Проведение анализа
Заполненную жиром кювету с толщиной слоя 50 мм ставят перед нижним окном прибора и, введя в поток желтый фильтр, уравнивают окраску обеих половин поля зрения перемещением клина. Измерения проводят с таким желтым фильтром (или суммой фильтров), с которым наилучшим образом выравнивается окраска.
Если перемещением клина не достигается равенство окраски, вводят последовательно дополнительные красные фильтры 10, 20 и 30 единиц красного цвета. В этом случае к отсчету, полученному по шкале красного клина, добавляют соответствующее число красных единиц. Значение цвета получают как среднеарифметическое 3-5 измерений.
Перед заполнением кюветы новой порцией жира и после окончания измерений кювету промывают растворителем или теплой водой с мылом и высушивают при слабом нагревании.
Разборка цветомера ВНИИЖ-16 не допускается, так как при этом нарушается юстировка прибора и градуировка его шкалы.
7.2.3. Определение цвета жира спектрофотометром или фотоэлектроколориметром (кроме жира лососевых и окуневых рыб)
7.2.3.1. Сущность метода
Метод основан на определении величины оптической плотности жира.
7.2.3.2. Аппаратура, материалы и реактивы
Фотоэлектроколориметр или спектрофотометр с пределами измерений оптической плотности от 0 до 1,3.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Эфир медицинский по Госфармакопее СССР.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Колба плоскодонная по ГОСТ 25336-82, вместимостью 100 см.
Воронка стеклянная по ГОСТ 25336-82.
Термометр стеклянный технический по ГОСТ 28498-90 с пределом измерений температуры от 0 до 150 °С.
7.2.3.3. Проведение анализа
Пробу анализируемого жира при необходимости нагревают до температуры, при которой предусмотрено определение его прозрачности, и фильтруют через бумажный фильтр. Профильтрованный жир наливают в кювету с рабочей длиной 3-10 мм (в зависимости от цвета жира) и определяют оптическую плотность при длине волны 440 нм по сравнению с пустой кюветой. Если после фильтрации жир остается непрозрачным, его нагревают до 40 °С.
Перед заполнением кюветы новой порцией жира и после окончания измерения его оптической плотности кювету промывают спирто-эфирной смесью (1:2) и высушивают.
7.2.3.4. Обработка результатов
Оптическую плотность () в относительных единицах, характеризующую цвет жира, вычисляют по формуле
![]() ,
,
где - оптическая плотность исследуемого жира;
- рабочая длина кюветы, мм.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,015 относительных единиц.
Вычисление проводят до третьего десятичного знака.
Цвет жира, соответствующий полученному значению оптической плотности, устанавливают по табл.6.
Таблица 6
Цвет жира | Величина оптической плотности ( |
Светло-желтый | До 0,6 |
Желтый | Св. 0,6 " 0,8 |
Темно-желтый (или светло-коричневый) | " 0,8 " 2,0 |
Коричневый | " 2,0 " 3,0 |
Темный | " 3,0 |
7.3. Определение прозрачности жира
7.3.1. Сущность метода
Метод основан на визуальном определении прозрачности жира, нагретого до требуемой температуры, в проходящем и отраженном свете.
7.3.2. Аппаратура
Баня водяная.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 100 см.
Стакан химический по ГОСТ 25336-82, вместимостью 200-300 см.
Термостат.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Палочка стеклянная.
7.3.3. Проведение анализа
Перед анализом жир, помещенный в химический стакан, осторожно нагревают на водяной бане до температуры 60-70 °С (медицинский рыбий жир должен иметь температуру 5 °С).
100 см жира переносят в цилиндр, медленно охлаждают при непрерывном перемешивании стеклянной палочкой до температуры, предусмотренной для определения прозрачности, и выдерживают в течение 3 ч при установленной температуре, периодически помешивая через каждые 15 мин.
По истечении указанного времени жир рассматривают в проходящем и отраженном свете на белом фоне.
Жир считается прозрачным, если в нем нет мути или взвешенных частиц.
7.4. Определение относительной плотности жира пикнометром
7.4.1. Сущность метода
Метод основан на определении отношения массы жира к массе воды при установленных для них температурах.
7.4.2. Аппаратура, материалы и реактивы.
Пикнометры стеклянные по ГОСТ 22524-77, градуированные при температуре 20 °С.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Пипетки по ГОСТ 29227-91.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Вода дистиллированная по ГОСТ 6709-72.
7.4.3. Проведение анализа
Пикнометр тщательно промывают хромовой смесью, водой, спиртом, просушивают и взвешивают на аналитических весах.
Для определения массы воды (водного числа) взвешенный пикнометр наполняют выше метки дистиллированной водой. Температуру воды доводят до 20 °С погружением пикнометра в водяную баню данной температуры на 20-30 мин, после чего доводят уровень воды в пикнометре до метки удалением излишка ее с помощью полосок фильтровальной бумаги.
Пикнометр вынимают из бани, тщательно вытирают снаружи и взвешивают.
После удаления воды пикнометр высушивают, вновь взвешивают, наполняют профильтрованным жиром (с некоторым избытком), доводят до температуры 20 °С (или указанной в стандарте на данный вид жира) погружением в водяную баню, имеющую ту же температуру, и выдерживают в ней пикнометр до тех пор, пока уровень мениска не перестанет изменяться. Избыток жира отбирают фильтровальной бумагой, свернутой в тонкую трубочку. После этого пикнометр с жиром тщательно вытирают и взвешивают.
7.4.4. Обработка результатов
Относительную плотность жира () вычисляют по формуле
![]() ,
,
где - масса пустого пикнометра, г;
- масса пикнометра с водой, г;
- масса пикнометра с жиром, г.
Относительную плотность вычисляют по формуле
![]() ,
,
где - относительная плотность жира, определенная при 20 °С;
0,9982 - плотность воды при 20 °С.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,001 условных единиц.
Вычисление проводят до третьего десятичного знака.
7.5. Определение примесей нежирового характера (отстоя) объемным методом в жире-полуфабрикате
7.5.1. Сущность метода
Метод основан на осаждении из жира в период отстаивания веществ нежирового характера, имеющих большую плотность, и определении их количества измерением объема.
7.5.2. Проведение анализа
После тщательного перемешивания пробы отбирают 100 см жира и вносят его в стеклянный мерный цилиндр вместимостью 100 см
, оставляют на 24 ч, после чего измеряют объем, занимаемый отстоем.
Определение проводят при температуре, установленной стандартом на исследуемый жир. Количество отстоя выражают в объемных единицах.
7.6. Определение примесей нежирового характера, исключая воду
7.6.1. Сущность метода
Метод основан на растворении жировой фракции липидов органическим растворителем, отделении веществ нежирового характера фильтрованием, обезвоживании их высушиванием и определении массы взвешиванием.
7.6.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Шкаф сушильный.
Колбы конические по ГОСТ 25336-82 вместимостью 250-300 см.
Секундомер механический.
Эксикатор по ГОСТ 25336-82.
Бюксы по ГОСТ 25336-82.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Палочка стеклянная.
Воронка стеклянная по ГОСТ 25336-82, диаметром 100 мм.
Воронка для горячего фильтрования.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Эфир петролейный, температурой кипения 40-60 °С или бензин-растворитель для резиновой промышленности по ТУ 38.401-67-108-92 температурой кипения до 90 °С.
7.6.3. Проведение анализа
Из разогретой и тщательно перемешанной пробы жира в коническую колбу вместимостью 250-300 см отбирают с абсолютной погрешностью не более 0,01 г навеску массой 50 г и растворяют в равном объеме петролейного эфира или бензина. Полученный раствор фильтруют через предварительно доведенный до постоянной массы при 100-105 °С фильтр. Колбу тщательно промывают эфиром или бензином, который фильтруют через тот же фильтр. Остаток на фильтре промывают небольшими порциями эфира или бензина до полного удаления жира, после чего фильтр с остатком в той же бюксе высушивают до постоянной массы при 100-105 °С.
При отсутствии бензина или достаточного количества петролейного эфира навеску жира фильтруют через воронку для горячего фильтрования при температуре не выше 60 °С, остаток на фильтре промывают петролейным или серным эфиром, после чего фильтр с остатком доводят до постоянной массы при 100-105 °С.
7.6.4. Обработка результатов
Массовую долю примесей в жире () в процентах вычисляют по формуле
![]() ,
,
где - навеска жира, г;
- масса высушенного бумажного фильтра с бюксой, г;
- масса бумажного фильтра с высушенным остатком и бюксой, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,02%.
Вычисление проводят до второго десятичного знака.
7.7. Определение воды - по п.3.3.3.
7.8. Определение температуры плавления спермацета
7.8.1. Сущность метода
Метод основан на визуальном определении момента начала движения жира в капилляре под давлением столба воды.
7.8.2. Аппаратура
Баня водяная.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 °С по ГОСТ 28498-90.
Стакан химический по ГОСТ 25336-82.
Трубка капиллярная стеклянная с внутренним диаметром 1,0-1,5 мм и длиной 100-120 мм.
Секундомер механический.
7.8.3. Проведение анализа
Для наполнения спермацетом тонкостенной капиллярной трубки с внутренним диаметром 1,0-1,5 мм и длиной 100-120 мм (предварительно хорошо вымытой и высушенной) опускают конец капилляра в расплавленный на водяной бане спермацет и заполняют его на 10-20 мм по высоте.
Заполненный капилляр охлаждают холодной водой. Капилляр с застывшим спермацетом прикрепляют к термометру так, чтобы столбик спермацета находился против шарика, и погружают термометр с капилляром в химический стакан с водой, предварительно нагретой до температуры 25 °С.
Ртутный резервуар термометра помещают ниже поверхности воды на 4 см и осторожно, перемешивая воду мешалкой, нагревают ее.
Показания термометра в момент, когда спермацет под напором столба воды поднимается в капилляре вверх, принимают за температуру плавления.
Температуру водяной бани поднимают со скоростью не более 1 °С в мин. Около точки перехода спермацета в жидкое состояние скорость повышения температуры должна быть не более 0,5-0,75 °С в мин.
7.9. Определение кислотного числа.
7.9.1. Сущность метода
Метод основан на взаимодействии свободных жирных кислот, содержащихся в 1 г жира, с гидроксидом калия.
7.9.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Секундомер механический.
Баня водяная.
Колбы конические с притертой пробкой по ГОСТ 25336-82, вместимостью 250 см.
Колба коническая с боковой отводной трубкой.
Капельница по ГОСТ 25336-82.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 50 см.
Холодильник стеклянный лабораторный (обратный) по ГОСТ 25336-82.
Бюретки по ГОСТ 29251-91, вместимостью 25 и 50 см.
Фенолфталеин (1%-ный).
Калия гидроксид по ГОСТ 24363-80, раствор 0,1 моль/дм (0,1 н) или натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм
(0,1 н).
Эфир медицинский по Госфармакопее СССР.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Алкалиблау 6Б.
Тимолфталеин, спиртовой раствор 10 г/дм (1%-ный).
7.9.3. Проведение анализа
В коническую колбу вместимостью 250 см отвешивают с абсолютной погрешностью не более 0,01 г (в зависимости от ожидаемого кислотного числа) 2-10 г профильтрованного жира или 1-2 г препарата "Витамин А в жире", или концентрата витамина А, приливают 30-50 см
нейтрализованной (по тому индикатору, с которым проводится титрование) смеси спирта с этиловым эфиром (1:2) и перемешивают взбалтыванием. Если при этом жир не растворяется, содержимое колбы слегка нагревают на водяной бане с обратным холодильником, а затем охлаждают до температуры 15-20 °С. В полученный раствор прибавляют 1 см
спиртового раствора фенолфталеина 10 г/дм
и при постоянном взбалтывании быстро титруют раствором гидроксида калия 0,1 моль/дм
или натрия до появления слабо-розовой окраски, устойчивой в течение 30 с.
Во избежание гидролиза раствора мыла количество спирта в нейтрализованной смеси должно превышать количество гидроксида натрия или калия, пошедшее на титрование, в 5 раз.
При исследовании темно-окрашенных жиров титрование лучше вести в конической колбе с боковой отводной трубкой с добавлением 2 см спиртового раствора тимолфталеина 10 г/дм
в качестве индикатора или 20 см
спиртового раствора 20 г/дм
алкалиблау 6Б.
Наблюдают за изменением окраски смеси во время титрования в тонком слое, находящемся в отводной трубке колбы.
Тимолфталеин в кислой среде бесцветен, а в щелочной дает голубое окрашивание; при титровании темно-окрашенных жиров он принимает голубовато-зеленый или грязно-зеленый цв
ет.
7.9.4. Обработка результатов
Кислотное число исследуемого жира () в мг КОН на 1 г жира вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида калия или гидроксида натрия 0,1 моль/дм
, израсходованный на титрование, см
;
- коэффициент пересчета на точный раствор 0,1 моль/дм
щелочи;
5,61 - количество гидроксида калия, соответствующее 1 см точного раствора 0,1 моль/дм
, мг;
- масса исследуемого жира, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1 мг KОН.
Вычисление проводят до второго десятичного знак
а.
7.10. Определение числа омыления
7.10.1. Сущность метода
Метод основан на взаимодействии свободных и связанных жирных кислот, содержащихся в 1 г жира, со щелочью и определении числа омыления обратным титрованием.
7.10.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Колбы конические по ГОСТ 25336-82, вместимостью 250 см со шлифом.
Холодильник стеклянный лабораторный (обратный) по ГОСТ 25336-82.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 50 см.
Бюретка по ГОСТ 29251-91, вместимостью 25-50 см.
Кислота соляная по ГОСТ 3118-77, раствор 0,5 моль/дм (0,5 н).
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный) или алкалиблау спиртовой раствор 20 г/дм
(2%-ный) (при анализе темноокрашенных жиров).
Спирт этиловый ректификованный по ГОСТ 5962-67.
Калия гидроксид по ГОСТ 24363-80, спиртовой раствор 0,5 моль/дм (0,5 н)
.
7.10.3. Приготовление спиртового раствора 0,5 моль/дм гидроксида калия
30 г гидроксида калия растворяют в 20 см воды в мерной колбе вместимостью 1 дм
и доливают до метки этиловым спиртом.
Раствор оставляют на сутки для отстаивания в закрытой колбе.
Прозрачный раствор осторожно декантируют в склянку из темного стекла.
7.10.4. Проведение анализа
В колбу для омыления отвешивают с абсолютной погрешностью не более 0,001 г 1,5-2 г хорошо перемешанного и профильтрованного жира и приливают из бюретки 25 см спиртового раствора гидроксида калия 0,5 моль/дм
. Колбу соединяют с обратным холодильником, глубоко опускают в сильно кипящую водяную баню и кипятят 1 ч при периодическом взбалтывании до полного омыления жира.
Об окончании реакции омыления судят по отсутствию помутнения пробы от нескольких капель воды.
К полученному горячему прозрачному мыльному раствору приливают 0,5 см раствора фенолфталеина 10 г/дм
или при анализе темноокрашенных жиров раствора алкалиблау 20 г/дм
и сразу титруют раствором соляной кислоты 0,5 моль/дм
до нейтральной реакции.
Одновременно проводят контрольный анализ без навески жира.
7.10.5. Обработка результатов
Число омыления исследуемого жира () в мг КОН на 1 г жира вычисляют по формуле
![]() ,
,
где 28,055 - количество гидроксида калия, соответствующее 1 см раствора соляной кислоты 0,5 моль/дм
, мг;
- коэффициент пересчета на точный раствор соляной кислоты 0,5 моль/дм
;
- объем раствора соляной кислоты 0,5 моль/дм
, израсходованный на титрование раствора в контрольном анализе, мл;
- объем раствора соляной кислоты 0,5 моль/дм
, израсходованный на титрование в рабочем анализе, см
;
- навеска жира, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 2,0 мг.
Вычисление проводят до един
ицы.
7.11. Методы определения йодного числа
7.11.1. Определение йодного числа титрованием с использованием йодно-ртутного раствора (применяется при разногласиях в оценке качества продукта)
7.11.1.1. Сущность метода
Метод основан на взаимодействии йода с непредельными жирными кислотами жира.
7.11.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Колбы с притертой пробкой по ГОСТ 25336-82, вместимостью 500 см.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 100 см.
Бюретки по ГОСТ 29251-91, вместимостью 50, 25 см с делениями на 0,1 см
.
Капельницы по ГОСТ 25336-82.
Ртуть двухлористая (сулема) по Госфармакопее СССР.
Йод по ГОСТ 4159-79.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Кислота соляная по ГОСТ 3118-77.
Натрий серноватистокислый (тиосульфат), водный раствор 0,1 моль/дм.
Калий йодистый по ГОСТ 4232-74, водный раствор 100 г/дм (10%-ный) (не содержащий йодноватокислой соли).
Хлороформ по Госфармакопее СССР, не содержащий перекисей, остающийся бесцветным после взбалтывания его с йодистым калием.
Крахмал растворимый по ГОСТ 10163-76, раствор 10 г/дм (1%-ный), к которому прибавляют кристалл для стойкости при хранении.
Вода дистиллированная по ГОСТ 6709-72
.
7.11.1.3. Приготовление йодно-ртутного раствора
Йодно-ртутный раствор (раствор Гюбля) готовят смешиванием отдельно приготовленных спиртовых растворов двухлористой ртути (сулемы) и йода.
25 г йода растворяют в 500 см спирта 950 г/дм
, а 30 г сулемы растворяют в 500 см
спирта 950 г/дм
и профильтровывают. Оба раствора хранят в отдельных склянках из темного стекла с притертыми пробками и смешивают в равных объемах за 48 ч до начала определения.
7.11.1.4. Определение испытания
В конической колбе с притертой пробкой взвешивают 0,2 г профильтрованного жира с абсолютной погрешностью не более 0,0002 г, приливают 15-20 см хлороформа, свободного от перекисей, и 25 см
йодно-ртутного раствора. Колбу закрывают притертой пробкой, смоченной раствором йодистого калия во избежание улетучивания йода. Содержимое колбы осторожно перемешивают. Колбу ставят в темное место при температуре 20 °С на 24 ч.
По окончании настаивания в колбу приливают 15 см раствора йодистого калия 100 г/дм
и 100 см
дистиллированной воды. Если при этом выпадает осадок йодистой ртути, не растворяющейся при взбалтывании, доставляют еще 5 см
раствора йодистого калия.
Смесь взбалтывают и титруют выделившийся йод раствором серноватистокислого натрия 0,1 моль/дм до светло-желтой окраски. После этого прибавляют 1 см
раствора крахмала 10 г/дм
и продолжают титрование до исчезновения синего окрашивания.
Одновременно проводят контрольное определение без навески жи
ра.
7.11.1.5. Обработка результатов
Йодное число () в г йода на 100 г жира вычисляют по формуле
![]() ,
,
где 0,01269 - количество йода, соответствующее 1 см раствора тиосульфата натрия 0,1 моль/дм
, г;
- коэффициент пересчета на точный раствор 0,1 моль/дм
тиосульфата натрия;
- объем раствора тиосульфата натрия 0,1 моль/дм
, израсходованный в контрольном анализе, см
;
- объем раствора тиосульфата натрия 0,1 моль/дм
, израсходованный в рабочем анализе, см
;
- навеска жира, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 1 г йода.
Вычисление проводят до еди
ницы.
7.11.2. Определение йодного числа титрованием с использованием солянокислого раствора хлористого йода
7.11.2.1. Сущность метода - по п.7.13.1.1.
7.11.2.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Колбы по ГОСТ 25336-82 с притертой пробкой вместимостью 350-500 см.
Колбы мерные по ГОСТ 1770-74, вместимостью 1 дм.
Пипетки стеклянные по ГОСТ 29227-91, вместимостью 1; 3 и 10 см.
Часы песочные на 5, 10 и 15 мин.
Воронка делительная по ГОСТ 25336-82, вместимостью 500 см.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 100 см с делениями через 1 см
.
Хлороформ по Госфармакопее СССР.
Кислота соляная по ГОСТ 3118-77.
Вода дистиллированная по ГОСТ 6709-72.
Эфир медицинский по Госфармакопее СССР, свободный от перекисей.
Калий йодистый по ГОСТ 4232-74, водный раствор 100 г/дм (10%-ный).
Натрий серноватистокислый (тиосульфат), водный раствор 0,1 моль/дм.
Крахмал растворимый по ГОСТ 10163-76, раствор 10 г/дм (1%-ный) свежеприготовленный.
Калий йодноватокислый по ГОСТ 4202-
75.
7.11.2.3. Приготовление раствора хлористого йода 0,2 моль/дм (0,2 н)
В склянку с притертой пробкой вносят 11,1 г йодистого калия, 7 г йодноватокислого калия, 50 см воды, 50 см
концентрированной соляной кислоты и взбалтывают до полного растворения йода. Приливают 20 см
хлороформа и добиваются фиолетовой окраски слоя хлороформа добавлением по каплям водного раствора йодноватокислого калия 10 г/дм
при энергичном взбалтывании.
После отстаивания водный слой сливают в мерную колбу и доводят объем водой до 1 дм. Реактив хранят в склянке темного стекла.
7.11.2.4. Проведение анализа
В колбу с притертой пробкой отвешивают с абсолютной погрешностью не более 0,0001 г 0,08-0,12 г профильтрованного жира.
Для растворения жира в колбу приливают 3 см этилового эфира, свободного от перекисей, и добавляют из бюретки 25 см
0,2 моль/дм
солянокислого раствора хлористого йода.
Колбу плотно закрывают пробкой, перемешивают и оставляют стоять 5-10 мин, затем вносят 10 см раствора йодистого калия 100 г/дм
, 50 см
дистиллированной воды и выделившийся йод титруют раствором тиосульфата натрия 0,1 моль/дм
до светло-желтой окраски.
После этого в колбу прибавляют 1 см свежеприготовленного раствора крахмала 10 г/дм
, 2-3 см
хлороформа, свободного от перекисей, и продолжают титрование до полного исчезновения синего окрашивания.
Одновременно проводят контрольное определение без навески ж
ира.
7.11.2.5. Обработка результатов
Йодное число () в г йода на 100 г жира вычисляют по формуле
![]() ,
,
где 0,01269 - количество йода, соответствующее 1 см раствора тиосульфата натрия 0,1 моль/дм
, г;
- коэффициент пересчета на точный раствор тиосульфата натрия 0,1 моль/дм
;
- объем раствора тиосульфата натрия 0,1 моль/дм
, израсходованный в контрольном анализе, см
;
- объем раствора тиосульфата натрия 0,1 моль/дм
, израсходованный в рабочем анализе, см
;
- навеска жира, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 3 г.
Вычисление проводят до еди
ницы.
7.12. Определение перекисного числа
7.12.1. Сущность метода
Метод основан на взаимодействии перекисей, содержащихся в жире, с йодистым калием в присутствии ледяной уксусной кислоты с выделением йода, который оттитровывают раствором тиосульфата натрия.
7.12.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Часы песочные на 2 мин или секундомер.
Колбы с притертой пробкой по ГОСТ 25336-82, вместимостью 250-300 см.
Микробюретка по ГОСТ 29251-91, вместимостью 10 см с делениями на 0,01-0,02 см
.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 50 и 100 см.
Пипетки по ГОСТ 29227-91, вместимостью 1 см.
Натрий серноватистокислый (тиосульфат), раствор 0,01 моль/дм.
Хлороформ по Госфармакопее СССР.
Кислота уксусная ледяная по ГОСТ 61-75.
Калий йодистый по ГОСТ 4232-74, насыщенный водный раствор.
Крахмал растворимый по ГОСТ 10163-76, водный раствор 10 г/дм (1%-ный).
Вода дистиллированная по ГОСТ 6709-72
.
7.12.3. Проведение анализа
В коническую колбу с притертой пробкой вносят навеску тщательно перемешанного и профильтрованного жира массой 1 г, взятую с абсолютной погрешностью не более 0,0001 г.
Китовые жиры предварительно осторожно подогревают на водяной бане до температуры 60-70 °С. Навеску жира в колбе растворяют в 30 см смеси, состоящей из 12 см
хлороформа и 18 см
ледяной уксусной кислоты. К раствору приливают 1 см
насыщенного на холоде раствора йодистого калия и смесь равномерно взбалтывают точно 2 мин.
В колбу добавляют 100 см свежепрокипяченной дистиллированной воды, 1 см
раствора крахмала 10 г/дм
и немедленно титруют в присутствии крахмала выделившийся йод раствором тиосульфата натрия 0,01 моль/дм
до исчезновения синего окрашивания. Одновременно проводят контрольный анализ без навески жир
а.
7.12.4. Обработка результатов
Перекисное число исследуемого жира () в процентах йода вычисляют по формуле
![]() ,
,
где - объем раствора тиосульфата натрия 0,01 моль/дм
, израсходованный на титрование в рабочем анализе, см
;
- объем раствора тиосульфата натрия 0,01 моль/дм
, израсходованный на титрование в контрольном анализе, см
;
- навеска жира, г;
- коэффициент пересчета на точный раствор тиосульфата натрия 0,01 моль/дм
(0,01 н);
0,001269 - количество йода, соответствующее 1 см точного раствора тиосульфата натрия 0,01 моль/дм
, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,02%.
Вычисление проводят до второго десятичного з
нака.
7.13. Определение неомыляемых веществ
7.13.1. Сущность метода
Метод основан на омылении триглицеридов щелочью, экстракции неомыляемых веществ этиловым спиртом, обезвоживании и определении их количества взвешиванием.
7.13.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня песочная.
Шкаф сушильный.
Колбы конические со шлифом по ГОСТ 25336-82, вместимостью 250-300 см.
Холодильник стеклянный лабораторный (обратный) по ГОСТ 25336-82 со шлифом.
Воронка делительная по ГОСТ 25336-82, вместимостью 500 см.
Колбы с притертой пробкой по ГОСТ 25336-82, вместимостью 250-350 см.
Термометр стеклянный технический по ГОСТ 28498-90 с пределом измерений температуры от 0 до 150 °С.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 25 и 100 см.
Воронка по ГОСТ 25336-82, диаметром 50 мм.
Холодильник стеклянный лабораторный (водяной) по ГОСТ 25336-82.
Аллонж по ГОСТ 25336-82.
Колба коническая по ГОСТ 25336-82, вместимостью 250-350 см.
Капельница по ГОСТ 25336-82.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Калия гидроксид по ГОСТ 24363-80, спиртовой раствор 2 моль/дм (2 н).
Эфир медицинский по Госфармакопее СССР.
Натрий сернокислый безводный по ГОСТ 4166-76.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный).
Вода дистиллированная по ГОСТ 6709-72
.
7.13.3. Проведение анализа
В коническую колбу взвешивают 2-3 г с абсолютной погрешностью не более 0,001 г тщательно перемешанного и профильтрованного жира. Китовые жиры предварительно подогревают на водяной бане до температуры 60-79 °С. К навеске жира приливают 20 см свежеприготовленного спиртового раствора гидроксида калия 2 моль/дм
и, отметив уровень раствора, нагревают колбу на песочной бане с обратным холодильником в течение 2 ч (1 ч для рыбьих жиров). Уровень раствора доводят этиловым спиртом до первоначального и прибавляют 80 см
дистиллированной воды.
После этого содержимое колбы нагревают еще 30 мин. Полученный раствор должен быть прозрачным. После охлаждения раствор количественно переносят в делительную воронку и экстрагируют неомыляемые вещества этиловым эфиром. При образовании эмульсии к эфирному раствору добавляют несколько капель спирта для расслоения. Экстракцию проводят в три приема, используя для первой экстракции 50 см эфира, для последующих - по 25 см
. Собранные вместе в делительной воронке эфирные вытяжки промывают водой до полного удаления раствора мыла, о чем свидетельствует исчезновение окраски промывных вод при реакции с фенолфталеином.
Эфирный раствор количественно переносят через воронку с фильтром и безводным сернокислым натрием в предварительно высушенную и взвешенную колбу.
Эфир отгоняют на водяной бане с холодильником, остатки эфира удаляют на водяной бане без холодильника. Колбу с неомыляемыми веществами высушивают при температуре 100-105 °С до постоянной массы.
7.13.4. Обработка результатов
Массовую долю неомыляемых веществ () в процентах вычисляют по формуле
![]() ,
,
где - навеска жира, г;
- масса пустой колбы, г;
- масса колбы с остатком после высушивания, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до первого десятичного знака.
7.14. Методы определения витаминов A, D и Е в тканях и органах (печень и др.) рыб, жирах рыб, морских млекопитающих и в жидких витаминных препаратах
7.14.1. Определение витаминов А и D в жирах рыб и морских млекопитающих
Витамин А в препарате и витамины А и D в медицинском жире определяют по ФС 42-965-75.
Концентраты витамина А в жире, полученные методом молекулярной дистилляции и содержащие свыше 5000 ME витамина в 1 г, омылению не подвергают. Навеску концентрата в количестве 0,1-0,2 г непосредственно растворяют в хлороформе в мерной колбе вместимостью 25 см с пришлифованной пробкой. Колориметрирование и расчет проводят по ФС 42-965-75.
7.14.2. Определение витамина А в тканях и органах (печень и др.) рыб, морских млекопитающих, морских беспозвоночных и продуктах их переработки колориметрическим методом
7.14.2.1. Сущность метода
Метод основан на взаимодействии витамина А с треххлористой сурьмой с образованием окрашенного комплекса.
7.14.2.2. Аппаратура, материалы и реактивы
Холодильник стеклянный лабораторный по ГОСТ 25336-82.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Термометр ртутный стеклянный лабораторный по ГОСТ 28498-90 с пределом измерений температуры от 0 до 150 °С.
Секундомер механический.
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Воронки химические по ГОСТ 25336-82, диаметром 35-50 мм.
Колбы конические с притертой пробкой по ГОСТ 25336-82, вместимостью 100 см.
Пипетка по ГОСТ 29227-91, вместимостью 1 и 5 см.
Воронки делительные по ГОСТ 25336-82, вместимостью 300-500 см.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 100 см.
Баня водяная.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Калия гидроксид по ГОСТ 24363-80, х.ч.
Натрия гидроксид по ГОСТ 4328-77, х.ч.
Натрий сернокислый безводный по ГОСТ 4166-76, х.ч.
Бензидин-основание.
Азот газообразный по ГОСТ 9293-74.
Ангидрид уксусный по ГОСТ 5815-77, ч.д.а.
Кислота серная по ГОСТ 4204-77, ч.д.а.
Медь сернокислая 5-водная по ГОСТ 4165-78, ч.д.а.
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный).
Калий йодистый по ГОСТ 4232-74, ч.д.а.
Калий марганцовокислый по ГОСТ 20490-75, х.ч.
Пирогаллол А по ТУ 6-09-5319-86, ч.д.а.
Спирт этиловый ректификованный по ГОСТ 5962-67.
Сурьма треххлористая.
Хлороформ по Госфармакопее СССР.
Эфир медицинский по Госфармакопее СССР.
Вода дистиллированная по ГОСТ 6709-72.
7.14.2.3. Подготовка и очистка реактивов
Азот
Для очистки от примесей азот пропускают последовательно через поглотители: щелочный раствор пирогаллола А (5 г пирогаллола на 100 см раствора КОН 500 г/дм
), концентрированную серную кислоту, стеклянную вату и безводный хлористый кальций.
Натрий сернокислый (безводный)
Безводный сернокислый натрий подсушивают при температуре 105-110 °С в течение 2-3 ч.
Спирт этиловый
Этиловый ректификованный спирт освобождают от альдегидов перегонкой над твердой* гидроксидом калия или натрия или раствором 10 г на 1 дм спирта.
________________
* Текст соответствует оригиналу. - .
Начальную и конечную партии отгона отбрасывают.
Эфир этиловый
В склянку с корковой пробкой помещают 500 см эфира, 50 см
раствора KМnО
40 г/дм
, 5 см
раствора NaOH 400 г/дм
или КОН, взбалтывают содержимое и оставляют на сутки в темноте. Смесь (верхний слой) переносят в делительную воронку (нижний слой сливают), высушивают в течение суток сернокислым натрием, перегоняют и сохраняют в темноте.
Эфир необходимо проверять на отсутствие перекисей по качественной реакции с йодистым калием.
Хлороформ
Хлороформ промывают 5-6 раз дистиллированной водой (2:1), высушивают безводным сернокислым натрием, перегоняют в темную склянку и хранят в прохладном месте в течение 5-6 недель.
Хлороформ необходимо проверять на отсутствие свободного хлора, соляной кислоты и фосгена. Наличие фосгена и соляной кислоты устанавливают с бензидином следующим образом: несколько кристаллов бензидина растворяют в 10 см хлороформа (в склянке с притертой пробкой) и оставляют на ночь. При наличии соляной кислоты и фосгена раствор становится мутным.
Треххлористая сурьма
Треххлористую сурьму промывают хлороформом до тех пор, пока хлороформ, используемый для промывки, не будет прозрачным и бесцветным. Высушивают промытую хлороформом треххлористую сурьму в эксикаторе над серной кислотой в течение 1-2 сут в темноте.
Для приготовления раствора 230 г треххлористой сурьмы растворяют в колбе в 1 дм хлороформа для наркоза при нагревании на теплой водяной бане. Колбу закрывают притертой пробкой и оставляют на ночь. Прозрачную часть раствора сливают в мерный цилиндр вместимостью 1 дм
, прибавляют 2% (по объему) уксусного ангидрида, переносят в темную склянку, перемешивают и закрывают притертой пробкой. Раствор годен для анализа через сутки в течение 5-6 недель.
7.14.2.4. Проведение анализа
К навеске исследуемой пробы массой 2-3 г, взятой с абсолютной погрешностью не более 0,001 г, добавляют 1 см водного раствора КОН 600 г/дм
и 10-20 см
этилового спирта. Смесь нагревают на водяной бане при температуре 85-90 °С с обратным холодильником до полного растворения исследуемого образца, но не менее 2 ч. Если полного растворения не происходит, добавляют еще 10-20 см
спирта и продолжают нагревание. После омыления жира к раствору добавляют 10-20 см
спирта и двойное (по отношению к общему объему спирта) количество воды. Содержимое колбы количественно переносят в делительную воронку.
Неомыляемую фракцию трижды извлекают эфиром. Первый раз добавляют 50 см эфира, второй и третий раз - по 30 см
. Во избежание образования стойкой эмульсии экстракцию эфиром проводят в хорошо охлажденном растворе и при осторожном перемешивании содержимого воронки.
Объединенные эфирные вытяжки промывают дистиллированной водой, добавляя по 30-40 см до исчезновения щелочной реакции промывных вод (проба с фенолфталеином). Промытые эфирные вытяжки медленно фильтруют в колбу для отгона через бумажный фильтр, на который предварительно насыпают 8 г безводного сульфата натрия.
Фильтр с сульфатом натрия три раза промывают эфиром по 10 см, собирая фильтрат в ту же колбу.
Эфир отгоняют в токе азота при температуре не выше 40 °С.
Остаток вначале растворяют в небольшом объеме хлороформа для наркоза, затем разбавляют тем же хлороформом так, чтобы в 1 см раствора содержалось около 30 ME витамина А.
0,4 см полученного хлороформного раствора переносят в кювету фотоэлектроколориметра с рабочей длиной 10 мм, быстро прибавляют 4 см
хлороформного раствора хлорида сурьмы, содержащего 20 г/дм
уксусного ангидрида, и измеряют оптическую плотность раствора фотоэлектроколориметром при длине волны 620 нм. Показания прибора отмечают не позднее, чем через 5 с после добавления в кювету хлороформного раствора хлорида сурьмы. Содержание витамина А, соответствующее определенной оптической плотности, рассчитывают по градуировочному г
рафику.
7.14.2.5. Построение градуировочного графика
Навеску раствора ретинола ацетата в масле (для инъекции), содержащего 100000 ME в 1 см (фактическое содержание витамина А в 1 см
раствора устанавливают как указано в ФС 42-101-72) массой 0,1 г, взвешенную с абсолютной погрешностью не более 0,0001 г, разводят в таком количестве хлороформа для наркоза, чтобы получить раствор, содержащий 100 ME витамина А в 1 см
.
Приготовленный раствор используют для получения ряда разведений с содержанием витамина А от 5 до 50 ME в 1 см с интервалом в 5 ME.
Из каждого разведения отбирают по 0,4 см раствора, проводят цветную реакцию в кювете с рабочей длиной 10 мм так же, как при анализе исследуемого раствора и определяют оптическую плотность при длине волны 620 нм.
По полученным данным строят градуировочный график, откладывая на оси абсцисс количества витамина А, на оси ординат - соответствующие оптические плотности.
Градуировочный график строят каждый раз при смене реактивов, особенно треххлористой сурьмы.
7.14.2.6. Обработка результатов
Массовую долю витамина А () в 1 грамме исследуемого продукта (фарша) в международных единицах (ME) вычисляют по формуле
,
где - содержание витамина А в 1 см
раствора, найденное по градуировочному графику в международных единицах (ME);
- разведение, см
;
- навеска образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 10%.
Вычисление проводят до единицы.
7.14.3. Определение витамина Е в жире - по ГОСТ 9393-82.
7.15. Определение ионола в ветеринарном жире - по ГОСТ 9393-82.
8. МЕТОДЫ АНАЛИЗА КОРМОВОЙ МУКИ ИЗ РЫБЫ, МОРСКИХ МЛЕКОПИТАЮЩИХ И РАКООБРАЗНЫХ
8.1. Подготовка средней пробы - по п.2.6.
8.2. Определение внешнего вида муки
8.2.1. Сущность метода
Метод основан на визуальном определении внешнего вида муки.
8.2.2. Проведение испытания
Среднюю пробу муки, рассыпанную тонким слоем на пергаментной бумаге, полимерной пленке или другом материале, внимательно просматривают на присутствие комков, плесени, величину гранул.
8.3. Определение крупности помола
8.3.1. Сущность метода
Метод основан на фракционировании частиц муки просеиванием ее через сито и определении массы фракции взвешиванием.
8.3.2. Аппаратура и материалы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Сито диаметром 150 мм с сеткой проволочной стальной тканой для мукомольной промышленности со стороной отверстия 3,2 мм.
Стакан химический по ГОСТ 25336-82, вместимостью 200 см.
8.3.3. Проведение анализа
Навеску муки массой 100 г, взвешенную с абсолютной погрешностью не более 0,5 г, просеивают через сито с квадратными отверстиями, со стороной равной 3,2 мм.
Остаток крупных частиц на сите переносят в тарированный стакан и взвешивают. Масса остатка в граммах, выраженная в процентах от общей массы пробы, характеризует крупность помола.
8.4. Определение содержания металлопримесей в кормовой рыбной муке
8.4.1. Сущность метода
Метод основан на извлечении ферромагнитных частиц магнитом и определении их массы взвешиванием.
8.4.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Лист стекла или плексигласа размером 500x500 мм.
Магнит.
Стакан химический по ГОСТ 25336-82, вместимостью 100 см.
Натрия гидроксид по ГОСТ 4328-77, раствор 100 г/дм(10%-ный).
Бумага фильтровальная по ГОСТ 12026-76.
Сита из сетки проволочной стальной тканой для мукомольной промышленности с размерами сторон 0,5 и 1,0 мм.
8.4.3. Проведение анализа
Навеску муки массой 250 г, взятую из средней пробы без просеивания через сито и измельчения в ступке, рассыпают на стекле, плексигласе или мраморной доске слоем толщиной 5 мм и извлекают металлические частицы подковообразным магнитом. Во избежание потерь при снятии металлических частиц с магнита, его полюсы предварительно обертывают папиросной бумагой, через которую собирают металлопримеси. Для этого проводят магнитом продольные и поперечные бороздки по всей поверхности муки, чтобы покрыть ими весь образец муки без промежутков.
Снимают с магнита выбранные частицы железа, осторожно снимают бумагу с полюсов магнита и ссыпают металлические частицы на чистый лист белой бумаги. Образец муки заравнивают и повторяют обработку магнитом. Эту операцию проводят несколько раз до тех пор, пока на магните не будут больше собираться частицы железа.
После этого, держа за края бумагу, на которой собрано железо, водят ее по полюсам магнита и осторожно сдувают примешанную муку так, чтобы металлические частицы во время сдувания удерживались магнитом. Магнит следует укрепить в деревянном штативе полюсами вверх.
Очищенные от муки металлические частицы переносят в стакан вместимостью 100 см и заливают 25 см
раствора гидроксида натрия 100 г/дм
или калия и кипятят 30 мин на слабом огне; при этом происходит разрушение остатков частиц муки гидроксидом натрия. Содержимое стакана разбавляют тройным количеством воды и фильтруют через бумажный фильтр.
Фильтр промывают один-два раза водой и подсушивают в сушильном шкафу при 100 °С в течение 30 мин.
Металлические частицы собирают с фильтра магнитом через бумагу, переносят на тарированное часовое стекло и взвешивают с абсолютной погрешностью не более 0,01 г.
8.4.4. Обработка результатов
Содержание металлопримесей () в млн
(мг/кг) рассчитывают по формуле
![]() ,
,
где - масса исследуемого продукта, г;
- масса часового стекла, г;
- масса часового стекла с частицами железа, г;
10 - коэффициент пересчета г в мг и в кг.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, расхождения между которыми не должны превышать 0,03 млн (мг/кг).
Вычисление проводят до второго десятичного знака.
8.5. Определение размера металлопримесей
8.5.1. Сущность метода - по п.8.3.1.
8.5.2. Аппаратура и материалы - по п.8.3.2.
8.5.3. Проведение анализа
Взвешенные металлические частицы просеивают через два металлических сита, верхнее - с диаметром отверстий 2 мм и нижнее - с отверстиями диаметром 0,5 мм. Металлические частицы, задержанные на том и другом сите, взвешивают отдельно и выражают их количество в миллиграммах на килограмм муки (или в граммах на тонну). Металлические частицы, задержанные на первом сите, считают крупнее 2 мм, на втором сите - крупнее 0,5 мм.
8.6. Определение воды - по ГОСТ 13496.3-92.
(метод применяют при разногласиях в оценке качества продукции)
8.7. Определение хлористого натрия - по пп.3.5.1 и 3.5.3.
8.8. Определение жира - по пп.3.7.1; 3.7.2; 3.7.4.
8.9. Методы определения белковых веществ (сырого протеина)
8.9.1. Определение массовой доли белковых веществ макрометодом
8.9.1.1. Сущность метода
Метод основан на окислении органического вещества при сжигании его в серной кислоте в присутствии катализатора, отгонке образующегося аммиака паром, улавливании его раствором серной кислоты и определении содержания азота методом титрования.
Белковые вещества определяют, умножая количества общего азота на коэффициент 6,25. Метод применяют при разногласиях в оценке качества продукции.
8.9.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83 или газовые горелки.
Холодильник шариковый по ГОСТ 25336-82.
Воронка капельная по ГОСТ 25336-82.
Бюретка по ГОСТ 29251-91, вместимостью 50 см.
Каплеуловитель по ГОСТ 25336-82.
Колбы для сжигания по ГОСТ 25336-82, вместимостью 100 см.
Колбы плоскодонные или круглодонные по ГОСТ 25336-82, вместимостью 500-700 см.
Колбы плоскодонные по ГОСТ 25336-82, вместимостью 250-300 см.
Капельница по ГОСТ 25336-82.
Вода дистиллированная по ГОСТ 6709-72.
Кислота серная по ГОСТ 4204-77, концентрированная и раствор 0,05 моль/дм (0,1 н).
Натрия гидроксид по ГОСТ 4328-77, раствор 330 г/дм (33%-ный) прокипяченный и раствор 0,1 моль/дм
(0,1 н).
Медь сернокислая 5-водная по ГОСТ 4165-78.
Калий сернокислый по ГОСТ 4145-74.
Метиловый красный спиртовой раствор 0,2 г/дм (0,02%-ный).
Индикатор смешанный (Таширо
).
8.9.1.3. Приготовление раствора смешанного индикатора - по п.6.5.2.3.
8.9.1.4. Проведение анализа
Навеску муки массой 0,2-0,3 г взвешивают с абсолютной погрешностью не более 0,0005 г (для рыбы 0,6-1,0 г, для тузлука до 5 г) в закрытую с одной стороны трубочку из фильтровальной бумаги или из станиоля, помещают в колбу для сжигания вместимостью 100 см, добавляют несколько мелких кристаллов медного купороса (0,2-0,3 г) и приливают 10-20 см
серной кислоты плотностью 1840 кг/м
.
Колбу с содержимым осторожно нагревают в вытяжном шкафу, не допуская разбрызгивания жидкости. Когда содержимое колбы станет однородным, прекращают нагревание, дают остыть, добавляют 0,5 г сернокислого калия и продолжают нагревание до тех пор, пока жидкость в колбе не станет прозрачной, зеленовато-голубой окраски без бурого оттенка. Внутренние стенки колбы должны быть совершенно чистыми. Это достигается осторожным взбалтыванием содержимого колбы для смывания со стенок темных, обугленных частиц муки.
По окончании сжигания содержимое колбы охлаждают и количественно переносят в отгонную колбу вместимостью 500-750 см (черт.8).
Колбу для сжигания тщательно ополаскивают, проверяя полноту смывания добавлением 1-2 капель раствора метилового красного.
Общий объем раствора в отгонной колбе должен быть не более 250-300 см.
Приемником служит коническая колба вместимостью 250-300 см, в которую из бюретки налито 25-30 см
раствора серной кислоты 0,05 моль/дм
. Конец трубки холодильника должен быть погружен в раствор серной кислоты.
В отгонную колбу осторожно, по стенкам, избегая смешивания жидкостей, приливают 50-70 см раствора гидроксида натрия 330 г/дм
, бросают кусочек лакмусовой бумаги и быстро закрывают ее пробкой, соединенной посредством каплеуловителя с холодильником, осторожно перемешивают содержимое и нагревают. Реакция жидкости в колбе должна быть резко щелочной.
После закипания жидкости в колбе приемник опускают так, чтобы конец трубки холодильника находился на некотором расстоянии от поверхности раствора и продолжают отгонку до тех пор, пока не отгонится не менее жидкости.
Конец отгонки определяют по лакмусовой бумаге. Если отгонка закончена, капля дистиллята не должна вызывать посинения красной лакмусовой бумаги. При появлении в конце отгонки при кипении толчков отгонку прекращают.
По окончании отгонки конец трубки холодильника обмывают водой в приемную колбу и содержащийся в ней избыток серной кислоты оттитровывают раствором гидроксида натрия 0,1 моль/дм в присутствии метилового красного или двойного индикатора.
Одновременно проводят контрольный анализ без навески исследуемого об
разца.
8.9.1.5. Обработка результатов
Массовую долю белковых веществ () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на титрование серной кислоты в контрольном анализе, см
;
- объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на титрование избытка серной кислоты в рабочем анализе, см
;
- коэффициент пересчета на точный раствор гидроксида натрия 0,1 моль/дм
, г;
0,0014 - количество азота, эквивалентное 1 см раствора гидроксида натрия, 0,1 моль/дм
, г;
6,25 - коэффициент пересчета количества азота на белковые вещества;
- навеска муки, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5% для кормовой муки и 0,2% для остальной продукции.
Вычисление проводят до второго десятичного з
нака.
8.9.2. Определение массовой доли белковых веществ макрометодом с селеновой смесью - по п.8.9.1, но в качестве катализатора используют 2 г селеновой смеси, состоящей из 1,9 весовых частей надсернокислого калия по ГОСТ 4146-74 и 0,1 части селенистокислой меди (0,1 часть селенистокислой меди можно заменить смесью из 0,05 частей сернокислой меди и 0,05 частей селена элементарного).
8.9.3. Определение массовой доли белковых веществ макрометодом с перекисью водорода без отгонки
8.9.3.1. Сущность метода
Метод основан на сжигании навески исследуемого образца в серной кислоте в присутствии перекиси водорода и определении общего азота кипячением минерализованной пробы с 20 см раствора гидроокиси натрия 0,1 моль/дм
.
8.9.3.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83 или горелки газовые.
Колбы мерные по ГОСТ 1770-74, вместимостью 500 и 200 см.
Бюретки по ГОСТ 29251-91, вместимостью 25 и 50 см.
Капельницы по ГОСТ 25336-82.
Часы механические по ГОСТ 10733-98.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 100 и 50 см.
Колбы конические по ГОСТ 25336-82, вместимостью 250 и 500 см.
Колбы круглодонные длинногорлые (Кьельдаля) по ГОСТ 25336-82, вместимостью 100-250 см.
Кислота серная по ГОСТ 4204-77, плотностью 1840 кг/м и раствор 0,05 моль/дм
(0,1 н).
Водорода перекись по ГОСТ 10929-76, раствор 300 г/дм (30%-ный).
Натрия гидроксид по ГОСТ 4328-77, раствор 150 г/дм (15%-ный).
Вода дистиллированная по ГОСТ 6709-72.
Смешанный индикатор: смешивают равные объемы спиртовых растворов метилового красного 2 г/дм (0,2%-ного) и метиленового синего 1 г/дм
(0,1%-н
ого).
8.9.3.3. Проведение анализа
Предварительно определяют количество гидроксида натрия в 20 смраствора 0,1 моль/дм
после кипячения по отношению к серной кислоте 0,05 моль/дм
и к добавляемым реактивам. Это количество условно называют "рабочим титром щелочи" и обозначают
.
Установка "рабочего титра" 20 см 0,1 моль/дм
раствора гидроокиси натрия и проверка реактивов
В коническую колбу вместимостью 500 см наливают 100 см
дистиллированной воды, добавляют 1,5 см
концентрированной серной кислоты и 5 капель смешанного индикатора.
Содержимое колбы осторожно нейтрализуют раствором гидроксида натрия 150 г/дм до переходной окраски и подкисляют несколькими см
раствора серной кислоты 0,05 моль/дм
; вновь точно нейтрализуют раствором гидроксида натрия 0,1 моль/дм
.
В колбу приливают из бюретки 20 см раствора гидроксида натрия 0,1 моль/дм
, ставят на горящую плитку и кипятят 15 мин, считая с момента закипания, быстро охлаждают под краном или погружают в холодную воду.
Содержимое колбы нейтрализуют раствором серной кислоты 0,05 моль/дм и добавляют избыток ее 2-3 см
для растворения карбонатов. Отмечают объем добавленного раствора серной кислоты.
Добавляют 2-3 капли смешанного индикатора и проводят обратное титрование раствором гидроксида натрия 0,1 моль/дм до появления такой же окраски, какая была перед кипячением. "Рабочий титр" (
) 20 см
раствора гидроксида натрия 0,1 моль/дм
по отношению к раствору серной кислоты 0,05 моль/дм
вычисляют по формуле
![]() ,
,
где - объем раствора серной кислоты 0,05 моль/дм
, прилитый в колбу после кипячения, см
;
- поправочный коэффициент на точный раствор 0,05 моль/дм
серной кислоты;
- объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на обратное титрование, см
;
- поправочный коэффициент на точный раствор 0,1 моль/дм
гидроксида натрия.
Пример.
Перед кипячением в колбу прилито 20 см раствора гидроксида натрия (
=1). После кипячения добавлено 22 см
раствора серной кислоты
=1. На обратное титрование пошло 2,5 см
раствора гидроксида натрия (
=1).
=22·1-2,5·1=19,5.
Это свидетельствует о том, что при кипячении произошло снижение "рабочего титра" раствора гидроксида натрия вследствие возможного присутствия солей аммония в реактивах или под влиянием других факторов.
Исходя из установленного "рабочего титра" раствора щелочи, можно в дальнейшем определять количество азота по снижению "рабочего титра" щелочи, так как каждый см раствора ее эквивалентен 1,4 мг азота. При последующих определениях необходимо пользоваться одними и теми же реактивами. Необходимо ежемесячно и при смене титрованных растворов проверять "рабочий титр" раствора щелочи.
0,8-1,4 г рыбной муки (для рыбы и продуктов с высоким содержанием воды навеска 1,8-2,0 г), отвешенных с абсолютной погрешностью не более 0,0005 г, помещают в колбу для сжигания вместимостью 100-250 см, добавляют соответственно 20 или 10-12 см
серной кислоты плотностью 1840 кг/м
и 5 см
перекиси водорода. Происходит саморазогревание смеси и выделение газов.
По окончании выделения газов колбу нагревают в колбонагревателе до закипания серной кислоты, немного охлаждают, осторожно, по стенке колбы, добавляют 2-3 см перекиси водорода и продолжают нагревание. Добавление перекиси водорода с последующим нагреванием проводят несколько раз, постепенно уменьшая ее количество. Процесс минерализации продолжается 35-45 мин.
По окончании минерализации (жидкость в колбе должна быть полностью обесцвечена) и охлаждения колбы содержимое ее количественно переносят в мерную колбу вместимостью 500 см при исследовании рыбной муки и 200 см
при исследовании рыбы и рыбных продуктов с высоким содержанием воды. Объем жидкости в колбе доводят до метки дистиллированной водой и тщательно перемешивают содержимое.
Пипеткой отбирают 50 см приготовленного раствора, переносят в коническую колбу вместимостью 500 см
, добавляют 50 см
дистиллированной воды, 5 капель смешанного индикатора.
Содержимое колбы осторожно нейтрализуют раствором гидроксида натрия 150 г/дм до переходной окраски, подкисляют несколькими см
раствора серной кислоты 0,05 моль/дм
, а затем вновь точно нейтрализуют раствором гидроксида натрия 0,1 моль/дм
.
После этого в колбу приливают из бюретки 20 см раствора гидроксида натрия 0,1 моль/дм
и ставят на горячую плитку. Содержимое колбы интенсивно кипятят точно 15 мин, считая с момента закипания, быстро охлаждают (под краном или погружают в холодную воду), нейтрализуют раствором серной кислоты 0,05 моль/дм
и добавляют избыток ее 2-3 см
для растворения карбонатов, отмечая количество добавленного раствора серной кислоты.
Добавляют 2-3 капли смешанного индикатора и проводят обратное титрование раствором гидроксида натрия 0,1 моль/дм до появления такой же окраски, какая была пер
ед кипячением.
8.9.3.4. Обработка результатов
Массовую долю белковых веществ () в процентах вычисляют по формуле
![]() ,
,
где - "рабочий титр" 20 см
раствора гидроксида натрия 0,1 моль/дм
(0,1 н);
- объем, в котором растворена минерализованная навеска, см
;
- объем раствора серной кислоты 0,05 моль/дм
, прилитый для нейтрализации и подкисления жидкости после ее кипячения, см
;
- объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на обратное титрование избытка серной кислоты, см
;
- объем раствора, взятый для кипячения, см
;
- коэффициент пересчета на точный 0,05 моль/дм
раствор серной кислоты;
- коэффициент пересчета на точный раствор 0,1 моль/дм
гидроксида натрия;
0,0014 - количество азота, эквивалентное 1 см раствора гидроксида натрия 0,1 моль/дм
, г;
6,25 - коэффициент пересчета азота на белковые вещества;
- навеска муки, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,5%.
Вычисление проводят до второго деся
тичного знака.
8.9.4. Определение массовой доли белковых веществ полумикрометодом
8.9.4.1. Аппаратура, материалы и реактивы по п.8.9.1.2 со следующими дополнениями:
Колба мерная по ГОСТ 1770-74, вместимостью 100 см.
Кислота серная по ГОСТ 4204-77, раствор 0,01 моль/дм (0,02 н).
Натрия гидроксид по ГОСТ 4328-77, раствор 0,02 моль/дм (0,02 н).
8.9.4.2. Проведение анализа
0,2-0,3 г муки (при исследовании рыбы и продуктов рыбного промысла с повышенным содержанием воды навеску увеличивают до 3 г, тузлука - до 5 г) взвешивают с абсолютной погрешностью не более 0,0005 г в закрытую с одной стороны трубочку из фильтровальной бумаги или станиоля, помещают в колбу для сжигания вместимостью 100 см, добавляют несколько мелких кристаллов сернокислой меди и приливают 10-20 см
серной кислоты плотностью 1840 кг/м
.
Колбу с содержимым осторожно нагревают в вытяжном шкафу, не допуская разбрызгивания жидкости. Когда содержимое колбы станет однородным, прекращают нагревание, дают остыть, прибавляют 0,5 г сернокислого калия и продолжают нагревание до тех пор, пока жидкость в колбе не станет прозрачной зеленовато-голубой без бурого оттенка. Внутренние стенки колбы должны быть совершенно чистыми. Это достигается осторожным взбалтыванием содержимого колбы для смывания со стенок темных, обугленных частиц муки.
По окончании сжигания содержимое колбы охлаждают и количественно переносят в мерную колбу вместимостью 100 см. Из полученного раствора отбирают пипеткой 10 см
жидкости и через воронку вносят в колбу прибора для отгонки аммиака, смывая остаток с воронки дистиллированной водой.
Отгонку аммиака водяным паром ведут в приборе, приведенном на черт.8.
1 - парообразователь; 2 - предохранительный сосуд; 3 - воронка; 4 - колба-смеситель; 5 - холодильник; 6 - колба-приемник
Черт.8
В коническую колбу-приемник 6 наливают из бюретки 25-30 см 0,01 моль/дм
раствора серной кислоты и опускают в нее конец холодильника 5. Закрывают нижнее отверстие предохранительного сосуда 2. Через воронку 3 наливают в колбу-смеситель 4 5-6 см
раствора гидроокиси натрия 330 г/дм
; воронку 3 заменяют стеклянной палочкой или применяют зажим Мора и затем пускают пар.
Отгонку с водяным паром продолжают до тех пор, пока в приемную колбу не отгонится 50-60 см жидкости. По окончании отгонки конец холодильника обмывают дистиллированной водой, собирают воду в приемник и титруют избыток кислоты раствором 0,02 моль/дм
гидроксида натрия в присутствии двойного индикатора.
8.9.4.3. Обработка результатов
Массовую долю белковых веществ () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида натрия 0,02 моль/дм
, израсходованный на титрование серной кислоты в контрольном анализе, см
;
- объем раствора гидроксида натрия 0,02 моль/дм
, израсходованный на титрование избытка серной кислоты в рабочем анализе, см
;
0,00028 - количество азота, эквивалентное 1 см 0,02 моль/дм
раствора гидроксида натрия, г;
- коэффициент пересчета на точный раствор 0,02 моль/дм
гидроксида натрия;
6,25 - коэффициент пересчета азота на белковые вещества;
- объем, в котором растворена сожженная навеска, см
;
- объем раствора, взятый для отгонки, см
;
- навеска исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать для кормовой муки 0,5%, для остальной продукции 0,2%.
Вычисление проводят до второго десятично
го знака.
8.9.5. Содержание белковых веществ (сырого протеина) в кормовой рыбной муке, направляемой на производство комбикормов - по ГОСТ 13496.4-93*.
________________
* На территории Российской Федерации в части приложения действует ГОСТ Р 51417-99.
8.10. Методы определения антиокислителя-ионола
8.10.1. Определение массовой доли ионола колориметрическим методом
8.10.1.1. Сущность метода
Метод основан на способности ионола давать розовое окрашивание с хлорным железом и ,
-дипиридилом.
Метод применяют при разногласиях в оценке качества продукции.
8.10.1.2. Аппаратура, материалы и реактивы
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Холодильник стеклянный лабораторный по ГОСТ 25336-82.
Парообразователь.
Электроплитка бытовая по ГОСТ 14919-83.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Колбы мерные по ГОСТ 1770-74, вместимостью 25, 100, 200 см.
Пробирки стеклянные по ГОСТ 25336-82.
Пипетки градуированные по ГОСТ 29227-91.
Часы механические по ГОСТ 10733-98.
Колбы конические со шлифом по ГОСТ 25336-82, вместимостью 25 и 50 см.
Трубки соединительные стеклянные по ГОСТ 25336-82.
Компаратор.
Кальций хлористый кристаллический.
Железо треххлористое 6-водное по ГОСТ 4147-74, свежеприготовленный раствор 2 г/дм (0,2%-ный).
,
-дипиридил (4-метил-2,6-дитретичного бутилфенола).
Спирт этиловый ректификованный по ГОСТ 5962-67, растворы 960 г/дм (96%-ный) и 500 г/дм
(50%-ный).
Ионол технический перекристаллизованный из этилового спирта 960 г/дм (96%-ного).
Вода дистиллированная по ГОСТ 6709-7
2.
8.10.1.3. Подготовка к анализу
8.10.1.3.1. Приготовление раствора ,
-дипиридила.
200 мг ,
-дипиридила растворяют в 1 см
этилового спирта 960 г/дм
и доводят объем дистиллированной водой до метки в мерной колбе вместимостью 100 см
.
8.10.1.3.2. Приготовление основного стандартного раствора ионола.
0,04 г перекристаллизованного ионола растворяют в этиловом спирте 960 г/дм в мерной колбе вместимостью 200 см
, доводят объем до метки спиртом и перемешивают. Раствор содержит 0,2 мг ионола в 1 см
.
Основной стандартный раствор ионола годен к употреблению в течение месяца при хранении его в холодном темном месте.
8.10.1.3.3. Приготовление рабочего стандартного раствора ионола.
Раствор готовят непосредственно перед определением.
2,5 см основного стандартного раствора ионола разбавляют этиловым спиртом 500 г/дм
в мерной колбе вместимостью 25 см
. Раствор содержит 0,02 мг ионола в 1 см
.
8.10.1.4. Проведение анализа
В колбу для отгонки, соединенную с холодильником, помещают 3 г кристаллического хлористого кальция и 40 см дистиллированной воды. Содержимое колбы охлаждают до комнатной температуры. Отвешивают 2 г исследуемой рыбной муки с абсолютной погрешностью не более 0,001 г и помещают в ту же колбу. Колбу закрывают пробкой, в которую вставлены две стеклянные трубки, одна - для ввода в колбу пара из парообразователя, другая - для отвода дистиллята через холодильник в приемную колбу и нагревают, отгоняя ионол паром.
Дистиллят собирают в мерную колбу вместимостью 200 см. Интенсивность отгонки должна быть такой, чтобы 100 см
дистиллята были отогнаны за 20-30 мин. Когда в приемнике соберется 100 см
дистиллята, отгонку прекращают. Форштос холодильника смывают небольшими порциями подогретого до 70 °С этилового спирта 960 г/дм
, сливая его в мерную колбу с дистиллятом. Содержимое колбы охлаждают, доводят объем до метки этиловым спиртом, закрывают пробкой и перемешивают.
Для проведения цветной реакции в одну коническую колбу с притертой пробкой вместимостью 25-50 см вносят пипеткой 4 см
дистиллята и 4 см
спирта 500 г/дм
, в другую - 8 см
этилового спирта 500 г/дм
(контрольный опыт). В каждую колбу приливают по 2 см
раствора
,
-дипиридила, перемешивают, добавляют по 2 см
раствора хлорного железа, снова перемешивают и ставят на 30 мин в темное место.
По истечении указанного времени измеряют оптическую плотность окрашенных растворов фотоэлектроколориметром в кювете с рабочей длиной 10 мм при длине волны 530 нм. Содержание ионола, соответствующее определенной оптической плотности, рассчитывают по градуировочному
графику.
8.10.1.5. Построение градуировочного графика
В 8 конических колб с притертыми пробками вместимостью 25-50 см вносят от 0 до 2,5 см
стандартного рабочего раствора, содержащего 0,02 мг ионола в 1 см
(см. табл.7) и доводят объем в каждой колбе до 8 см
спиртом 500 г/дм
.
Таблица 7
Номер колбы | Количество рабочего стандартного раствора, см | Количество добавленного спирта 500 г/дм | Количество ионола в колбе, мг |
1 | 0,00 | 8,00 | 0,000 |
2 | 0,25 | 7,75 | 0,005 |
3 | 0,50 | 7,50 | 0,010 |
4 | 1,00 | 7,00 | 0,020 |
5 | 1,25 | 6,75 | 0,025 |
6 | 1,50 | 6,50 | 0,030 |
7 | 2,00 | 6,00 | 0,040 |
8 | 2,50 | 5,50 | 0,050 |
В каждую колбу приливают по 2 см
,
-дипиридила, 2 см
свежеприготовленного раствора хлорного железа 2 г/дм
, выдерживают 35 мин в темноте и определяют оптическую плотность фотоэлектроколориметром по отношению к этиловому спирту 500 г/дм
в кювете с рабочей длиной 10 мм, начиная с контрольного опыта. Добавление
,
-дипиридила и хлорного железа в колбы проводят с таким интервалом, чтобы колориметрирование содержимого очередной колбы началось ровно через 35 мин.
Величину оптической плотности контрольного опыта вычитают из величин оптической плотности, полученных при измерении серии стандартных растворов ионола (колбы 2-8).
По полученным данным строят градуировочный график, откладывая на оси абсцисс содержание ионола, на оси ординат - разность оптической плотности стандартного и контрольного растворо
в.
8.10.1.6. Обработка результатов
Массовую долю ионола () в процентах вычисляют по формуле
![]() ,
,
где - содержание ионола, найденное по градуировочному графику, мг;
- навеска муки, г;
- объем дистиллята, взятый для цветной реакции, см
;
- объем дистиллята в мерной колбе, см
;
1000 - коэффициент пересчета мг в г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений (каждое вычислено по трем измерениям), допускаемые расхождения между которыми не должны превышать 0,01%.
Вычисление проводят до второго десятичного знака.
8.10.2. Определение массовой доли ионола визуально-колориметрическим методом
8.10.2.1. Сущность метода - по п.8.10.1.1.
8.10.2.2. Аппаратура, реактивы и материалы - по п.8.10.1.2.
8.10.2.3. Подготовка к анализу
Приготовление шкалы стандартных растворов ионола.
В двенадцать пробирок (по 3 пробирки для каждой концентрации) наливают пипеткой рабочий стандартный раствор ионола и раствор этилового спирта 500 г/дм в количествах, указанных в табл.8.
Таблица 8
Номер пробирки | Объем, см | Количество ионола в пробирке, мг | Количество ионола в муке, % | |
рабочего стандартного раствора ионола | этилового спирта | |||
1 | 0,4 | 7,6 | 0,008 | 0,02 |
2 | 1,2 | 6,8 | 0,024 | 0,06 |
3 | 2,0 | 6,0 | 0,040 | 0,10 |
4 | 4,0 | 4,0 | 0,080 | 0,20 |
8.10.2.4. Проведение анализа
Дистиллят готовят по п.8.10.1.4.
4 см полученного дистиллята вносят в чистую, сухую пробирку (анализ ведут параллельно с приготовлением шкалы), добавляют 4 см
этилового спирта, 500 г/дм
(50%-ного), 2 см
,
-дипиридила и перемешивают. Добавляют 2 см
хлорного железа, снова перемешивают и ставят на 35 мин в темное место.
По истечении указанного времени сравнивают (в компараторе или без него) окраску исследуемой пробы со шкалой стандартных растворов.
Если интенсивность окраски исследуемого раствора находится в интервале между двумя окрасками шкалы, указывают пределы массовой доли ионола в процентах
.
8.10.2.5. Обработка результатов
Массовую долю ионола () в процентах вычисляют по формуле
![]() ,
,
где - массовая доля ионола в пробирке стандартной шкалы, окраска которой соответствует окраске исследуемого образца, мг;
- объем дистиллята в мерной колбе, см
;
- объем дистиллята, взятый для колориметрирования, см
;
- навеска муки, г;
1000 - коэффициент для пересчета мг в г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,01%.
Вычисление проводят до второго десятичного знака.
8.11. Методы определения кальция
8.11.1. Определение массовой доли кальция объемным методом
8.11.1.1. Сущность метода
Метод основан на осаждении кальция щавелевокислым аммонием, растворении осадка в серной кислоте и количественном определении выделившейся при этом щавелевой кислоты титрованием.
Метод применяют при разногласиях в оценке качества продукции.
8.11.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электропечь сопротивления лабораторная.
Баня водяная.
Колбы конические по ГОСТ 25336-82.
Тигли фарфоровые по ГОСТ 9147-80.
Колбы мерные по ГОСТ 1770-74, вместимостью 100, 1000 см.
Воронки по ГОСТ 25336-82, диаметром 50-70 мм.
Пипетки по ГОСТ 29227-91, вместимостью 1, 5, 10, 25 см.
Эксикатор по ГОСТ 25336-82.
Бюретка по ГОСТ 29251-91, вместимостью 25 см.
Палочки стеклянные.
Цилиндры мерные по ГОСТ 1770-74, вместимостью 10, 25 см.
Бумага универсальная индикаторная.
Фильтры обеззоленные с белой лентой диаметром 70-100 мм.
Кислота соляная по ГОСТ 3118-77, раствор 200 г/дм (20%-ный).
Аммиак водный по ГОСТ 3760-79, раствор 100 г/дм (10%-ный) и 10 г/дм
(1%-ный).
Кислота серная по ГОСТ 4204-77, раствор 50 г/дм (5%-ный) и разведенная 1:8.
Кальций хлористый, раствор 100 г/дм (10%-ный).
Термометр стеклянный технический по ГОСТ 28498-90 с пределом измерений температуры от 0 до 500 °С.
Аммоний щавелевокислый по ГОСТ 5712-78, насыщенный раствор.
Метиловый красный, раствор 2 г/дм (0,2%-ный) спиртовой раствор.
Калий марганцовокислый по ГОСТ 20490-75, х.ч., раствор 0,02 моль/дм (0,1 н.) (3,165 г марганцовокислого калия растворяют в 1000 см
дистиллированной воды).
Кислота щавелевая, х.ч., по ГОСТ 22180-76.
Вода дистиллированная по ГОСТ 67
09-72.
8.11.1.3. Проведение анализа
Навеску муки массой 1,0-1,5 г (3,0-4,0 сырья) взвешивают с абсолютной погрешностью не более 0,001 г в предварительно прокаленном до постоянной массы фарфоровом тигле и обугливают в муфельной печи при 200-300 °С, затем повышают температуру до 550 °С и озоляют. По окончании озоления тигель охлаждают 25-30 мин в эксикаторе и взвешивают. Прокаливание проводят до постоянной массы.
К полученной золе добавляют 2-3 капли воды, 2 см раствора соляной кислоты 200 г/дм
, подогревают до полного растворения золы и переносят раствор в мерную колбу вместимостью 100 см
, тщательно смывая воронку и тигель. Объем раствора в колбе доводят до метки дистиллированной водой.
25 см солянокислой вытяжки помещают в колбу вместимостью 250 см
, добавляют 10 см
насыщенного раствора щавелевокислого аммония, затем, добавляя по каплям раствор аммиака 100 г/дм
, нейтрализуют соляную кислоту по метилроту до перехода розового окрашивания в желтое и появления слабого запаха аммиака. Колбу помещают на плитку, нагревают и оставляют для осаждения на 2-3 ч. Осадок отфильтровывают через бумажный фильтр, колбу и осадок на фильтре промывают раствором аммиака 10 г/дм
до полного удаления щавелевокислого аммония (полноту удаления проверяют добавлением нескольких капель раствора хлористого кальция 100 г/дм
к последней порции промывной жидкости). В случае появления мути промывку осадка продолжают.
Фильтр с осадком переносят в колбу, в которой проводили осаждение, добавляют 50 см раствора серной кислоты 50 г/дм
, нагревают до температуры 90 °С для растворения осадка. Горячую смесь титруют раствором марганцовокислого калия 0,02 моль/дм
до слабого розового окрашивания, не исчезающего в течение 1 мин. Параллельно титруют контрольную пробу - 50 см
серной кислоты 50 г/дм
с фильтром, нагретую д
о 90 °С.
8.11.1.4. Обработка результатов
Массовую долю кальция () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора марганцовокислого калия 0,02 моль/дм
, израсходованный на титрование рабочей пробы, см
;
- объем раствора марганцовокислого калия 0,02 моль/дм
, израсходованный на титрование контрольной пробы, см
;
- коэффициент пересчета на точный раствор 0,02 моль/дм
марганцовокислого калия;
0,002 - количество кальция, соответствующее 1 см раствора 0,02 моль/дм
марганцовокислого калия, г;
- объем солянокислой вытяжки, взятый для определения кальция, см
;
- навеска исследуемого образца, г;
- объем солянокислой вытяжки, см
.
Коэффициент пересчета на точный 0,02 моль/дм (0,1 н) раствор марганцовокислого калия устанавливают следующим образом. Навеску перекристаллизованной щавелевой кислоты 2-3 г, отвешенную с абсолютной погрешностью не более 0,001 г, растворяют в мерной колбе вместимостью 250 см
. К 20 см
приготовленного раствора приливают 3 см
серной кислоты, разведенной 1:8, нагревают на водяной бане до 90 °С и медленно титруют раствором марганцовокислого калия до появления розового окрашивания.
Коэффициент нормальности вычисляют по формуле
![]() ,
,
где - объем раствора щавелевой кислоты, израсходованный на титрование, см
;
- объем раствора марганцовокислого калия, израсходованный на титрование, см
;
- практический титр раствора щавелевой кислоты, который рассчитывается следующим образом:
![]() ;
;
- теоретический титр раствора 0,05 моль/дм
(0,1 н) щавелевой кислоты, который рассчитывают следующим образом:
![]() .
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до второго десятичного знака.
8.11.2. Определение содержания кальция трилонометрическим методом по ГОСТ 26570-95.
8.12. Методы определения фосфора
8.12.1. Определение фосфора объемным методом по ГОСТ 26657-97.
8.12.2. Колориметрический метод определения фосфора
8.12.2.1. Сущность метода
Метод основан на способности фосфатов давать с молибденовокислым аммонием комплексные соединения, которые восстанавливаются до окрашенного в голубой цвет молибденового окисла, называемого молибденовой синью.
8.12.2.2. Аппаратура, материалы и реактивы
Фотоэлектроколориметр или спектрофотометр с пределами измерения оптической плотности от 0 до 1,3.
Термостат.
Колбонагреватель или электроплитка бытовая по ГОСТ 14919-83.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Бумага масштабно-координатная по ГОСТ 334-73.
Колбы для сжигания по ГОСТ 25336-82, вместимостью 50-100 см.
Колбы мерные по ГОСТ 1770-74, вместимостью 100 см.
Пробирки с делениями по ГОСТ 25336-82, вместимостью 10 см.
Пипетки по ГОСТ 29227-91, вместимостью 1, 2, 5 и 10 см.
Вода дистиллированная по ГОСТ 6709-72.
Кислота серная по ГОСТ 4204-77, концентрированная, 0,05 (0,1 н) и 2,5 моль/дм (5 н) растворы.
Натрий сернистокислый 7-водный.
Аммоний молибденовокислый по ГОСТ 3765-78, х.ч., раствор 25 г/дм (2,5%-ный).
Водорода пероксид по ГОСТ 10929-76.
Натрий бисульфит технический водный раствор по ГОСТ 902-76.
Калий фосфорнокислый однозамещенный по ГОСТ 4198-75.
Эйконоген (1-амино-2-нафтол-4-сульфоновая кислота).
8.12.2.3. Подготовка к анализу
8.12.2.3.1. Приготовление раствора 1-амино-2-нафтол-4-сульфоновой кислоты.
15 г бисульфита натрия и 0,5 г сернистокислого натрия растворяют в теплой дистиллированной воде в мерной колбе вместимостью 100 см, добавляют 0,25 г эйконогена. После растворения последнего (при слабом нагревании на водяной бане) раствору дают охладиться и доводят объем до метки. Полученный основной раствор годен в течение 15 сут и должен храниться в темном прохладном месте (в случае выпадения кристаллов раствор подогревают на водяной бане до их растворения); перед употреблением его разводят дистиллированной водой в соотношении 1:4.
8.12.2.3.2. Приготовление стандартного раствора фосфорнокислого однозамещенного калия.
В мерную колбу вместимостью 1000 см отвешивают с абсолютной погрешностью не более 0,0001 г 350,9 мг фосфорнокислого калия, приливают 20 см
серной кислоты 0,05 моль/дм
и доводят объем дистиллированной водой до метки. 1 см
раствора содержит 0,08 мг фосфора.
8.12.2.4. Проведение анализа
Навеску образца массой (0,35-0,45 г - для рыбной и китовой граксовой муки; 0,8-1,0 г - для китовой сальной, мясной и муки из водорослей; 1,0-1,5 г - для рыбного и китового сырья), взвешенную с абсолютной погрешностью не более 0,0001 г, помещают в колбу для сжигания, приливают 10 см концентрированной серной кислоты. Колбу нагревают сначала осторожно, во избежание сильного вспенивания. Через 1,5-2,0 ч, дав колбе остыть, приливают несколько капель пероксидом водорода (непосредственно в жидкость, а не по стенкам колбы) и продолжают нагревание на сильном огне. Эту операцию повторяют через 20-30 мин до полного обесцвечивания раствора и после 5-10 мин кипения дают колбе остыть.
Параллельно проводят контрольный анализ с пероксидом водорода.
Содержимое остывшей колбы переносят многократным смыванием небольшими порциями дистиллированной воды в мерную колбу вместимостью 100 см, после охлаждения объем жидкости доводят до метки и тщательно перемешивают.
Для проведения цветной реакции пипеткой отбирают из колбы в две мерные пробирки вместимостью 10 см определенное количество анализируемого и контрольного растворов: при массовой доле фосфора в образце от 1 до 2% - 2 см
, от 2% и более - 0,5-1,0 см
и добавляют соответствующее количество раствора серной кислоты 2,5 моль/дм
, указанное в табл.9.
Таблица 9
Анализируемый раствор, см | 2,5 моль/дм |
0,5 | 1,25 |
1,0 | 1,00 |
2,0 | 0,50 |
После этого в пробирки приливают по 1,25 см молибденовокислого аммония и 0,5 см
раствора эйконогена, объем смеси доводят дистиллированной водой до метки, энергично встряхивают пробирку и помещают ее в термостат на 25 мин при 37 °С.
Оптическую плотность определяют фотоэлектроколориметром в кюветах с рабочей длиной 3 мм при длине волны 690 нм против контроля. Содержание фосфора, соответствующее определенной оптической плотности, рассчитывают по градуировочному графику.
8.12.2.5. Построение градуировочного графика
В мерные пробирки вместимостью 10 см вносят из микробюретки стандартный раствор однозамещенного фосфорнокислого калия в количествах, указанных в табл.10.
Таблица 10
Номер пробирки | Количество стандартного раствора однозамещенного фосфорнокислого калия, см | Массовая доля фосфора, мг |
1 | 0,5 | 0,04 |
2 | 1,0 | 0,08 |
3 | 1,5 | 0,12 |
4 | 2,0 | 0,16 |
5 | 2,5 | 0,20 |
В каждую пробирку приливают по 1,25 см раствора серной кислоты 2,5 моль/дм
, 1,25 см
молибденовокислого аммония и 0,5 см
раствора эйконогена, объем смеси доводят дистиллированной водой до метки и далее поступают так же, как с пробой.
По полученным данным строят градуировочный график, откладывая на оси абсцисс - массовую долю фосфора, соответствующую определенному разведению, по оси ординат - оптические плотности.
8.12.2.6. Обработка результатов
Массовую долю фосфора () в процентах вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- количество фосфора, найденное по градуировочному графику, мг;
- объем, до которого доводят минерализованную пробу, см
;
- объем исследуемого раствора, взятый для проведения цветной реакции, см
.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисления проводят до первого десятичного знака.
8.13. Определение песка
8.13.1. Сущность метода
Метод основан на разрушении органических веществ муки соляной кислотой, отмывании песка водой и весовом определении его.
8.13.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электропечь сопротивления.
Эксикатор по ГОСТ 25336-82.
Стакан химический по ГОСТ 25336-82, вместимостью 150-200 см.
Стекло часовое диаметром 70-80 мм.
Палочка стеклянная по ГОСТ 25336-82.
Пипетка Мора вместимостью 5, 10 см.
Фильтры обеззоленные.
Тигли фарфоровые по ГОСТ 9147-80.
Кислота соляная по ГОСТ 3118-77, разведенная водой в соотношении 1:1.
8.13.3. Проведение анализа
Навеску муки массой от 18 до 20 г, взятую из средней пробы без просеивания через сито и измельчения в ступке, взвешенную с абсолютной погрешностью не более 0,1 г, помещают в стакан вместимостью 150 см, наливают 40-50 см
соляной кислоты (1:1) и нагревают до кипения, непрерывно помешивая, пока масса в стакане не перестанет вспучиваться. Накрывают стакан часовым стеклом и оставляют кипеть 15 мин.
Прекратив нагревание, стакан доливают водой почти доверху, энергично размешивают содержимое стеклянной палочкой и оставляют в покое на 3-5 мин, после чего приступают к отмыванию песка.
К водопроводному крану или большой бутыли с водой присоединяют стеклянную трубку с шаровидным расширением посредине и оттянутым концом с диаметром отверстия 1-2 мм (удобно пользоваться пипеткой Мора вместимостью 5-10 см). В расширение трубки вкладывают кусочек ваты в качестве фильтра и устанавливают подачу воды 150-170 см
/мин.
Отрегулировав подачу воды, подставляют под струю стакан с гидролизатом, погружая трубку до половины высоты стакана (4-5 см от его дна). Слив воды происходит через край стакана.
Продолжительность отделения разрушенных органических веществ - 20 мин. По окончании отмывания на дне стакана остается песок и небольшое количество крупных частиц муки. Для удаления этих частиц осадок заливают 25-30 см насыщенного раствора поваренной соли, перемешивают и, дав песку осесть на дно, осторожно сливают жидкость вместе со взвешенными частицами муки. Обработку раствором соли повторяют 3-4 раза до прекращения всплывания частиц муки.
Осадок в стакане промывают 2-3 раза декантацией и количественно переносят на обеззоленный фильтр, прокаливают в предварительно взвешенном тигле в муфельной печи при температуре 500-550 °С в течение 15 мин, охлаждают и взвешивают.
8.13.4. Обработка результатов
Массовую долю песка () в процентах вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- масса пустого тигля, г;
- масса тигля с песком после прокаливания, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
8.14. Определение посторонних примесей (стекла)
8.14.1. Сущность метода
Метод основан на извлечении из муки неорганических примесей водой, фильтровании их, высушивании и определении компонентов с помощью оптических приборов.
8.14.2. Аппаратура, материалы и реактивы - по п.8.13.2 со следующими дополнениями:
лупа по ГОСТ 25706-83 или микроскоп по ГОСТ 8074-82.
8.14.3. Проведение анализа
Навеску муки массой от 18 до 20 г, взятую из средней пробы без просеивания через сито и измельчения в ступке, обрабатывают по п.8.13.3 (без прокаливания).
Полученный осадок подсушивают фильтровальной бумагой и исследуют под лупой или микроскопом при увеличении 20-50. Частицы стекла отличаются от песка по прозрачности и по острым очертаниям граней.
При обнаружении частиц, похожих на стекло, весь осадок передают в соответствующий институт или лабораторию для получения окончательного заключения после исследования в поляризованном свете.
При наличии в лаборатории поляризационной лупы исследование выделенного из муки песка проводят следующим образом. Песок распределяют тонким слоем на предметном стекле, помещают на столик лупы (между поляризатором и анализатором) и исследуют в простом свете при увеличении 10. Обнаруженные частицы, похожие на стекло, наблюдают в поляризованном свете, после чего медленно вращают анализатор до затемнения поля, сосредоточив внимание на подозрительных частицах. В поляризованном свете кварцевый песок выступает в виде ярких кристалликов на темном фоне, а частицы стекла исчезают вовсе или принимают вид тусклых кусочков со светлыми очертаниями. Для получения более отчетливой картины рекомендуется смочить подозрительные частицы каплей воды, накрыть покровным стеклом и еще раз просмотреть в поляризованном свете. В капле воды стекло принимает вид равномерно темных кусочков.
Наличие стекла в муке не допускается.
8.15. Определение углекислого кальция
8.15.1. Сущность метода
Метод основан на взаимодействии соляной кислоты с углекислым кальцием и количественном определении его титрованием избытка кислоты.
8.15.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электроплитка бытовая по ГОСТ 14919-83.
Стаканы стеклянные по ГОСТ 25336-82.
Колбы мерные по ГОСТ 1770-74.
Бюретка по ГОСТ 29251-91.
Кислота соляная по ГОСТ 3118-77, раствор 1 моль/дм (1 н).
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный).
Натрия гидроксид по ГОСТ 4328-77, раствор 1 моль/дм (1 н).
Вода дистиллированная по ГОСТ 6709-72.
8.15.3. Проведение анализа
Навеску муки массой 3-5 г, взвешенную с абсолютной погрешностью не более 0,001 г, помещают в стеклянный стакан вместимостью 500 см, добавляют 100-150 см
горячей воды и 100 см
раствора соляной кислоты 1 моль/дм
. После прекращения выделения пузырьков углекислоты содержимое стакана подогревают и выдерживают при слабом кипении 25-30 мин. После охлаждения к раствору добавляют 3-4 капли спиртового раствора фенолфталеина 10 г/дм
и титруют раствором гидроксида натрия 1 моль/дм
до появления розового окрашивания.
8.15.4. Обработка результатов
Массовую долю углекислого кальция () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора соляной кислоты 1 моль/дм
, израсходованный для растворения навески, см
;
- объем раствора гидроксида натрия 1 моль/дм
, израсходованный для титрования избытка кислоты, см
;
- коэффициент пересчета на точный раствор соляной кислоты 1 моль/дм
;
- коэффициент пересчета на точный раствор 1 моль/дм
щелочи;
0,05 - количество углекислого кальция, соответствующее 1 см точного раствора соляной кислоты 1 моль/дм
, г;
- навеска исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
9. МЕТОДЫ АНАЛИЗА КЛЕЙДАЮЩЕГО СЫРЬЯ, КЛЕЯ И КЛЕЕВЫХ БУЛЬОНОВ
9.1. Подготовка средней пробы - по п.2.7.
9.2. Определение клеющей способности - по ГОСТ 2067-93.
9.3. Определение сухого остатка
9.3.1. Сущность метода
Метод основан на обезвоживании навески высушиванием и определении сухих веществ взвешиванием.
9.3.2. Аппаратура и материалы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Шкаф сушильный.
Термометр стеклянный технический по ГОСТ 28498-90 с пределом измерений температуры от 0 до 150 °С.
Палочки стеклянные.
Стаканчики для взвешивания (бюксы) стеклянные по ГОСТ 25336-82.
Эксикатор по ГОСТ 25336-82.
9.3.3. Проведение анализа
Навеску галерты массой от 1,5 до 2 г, взвешенную с абсолютной погрешностью не более 0,001 г, помещают тонким слоем в предварительно высушенную бюксу со стеклянной палочкой и высушивают в шкафу, периодически перемешивая, сначала при 60-80 °С, а затем при 100-105 °С до постоянной массы.
9.3.4. Обработка результатов
Массовую долю сухого остатка () в процентах вычисляют по формуле
![]() ,
,
где - масса галерты, г;
- масса пустой бюксы, г;
- масса бюксы с галертой после сушки, г.
За окончательный результат принимают среднее арифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до первого десятичного знака.
9.4. Определение жира - по п.3.7.1; 3.7.2 (метод применяют при разногласиях в оценке качества продукции).
9.4.1. Определение жира экстракционным методом в аппарате Сокслета
Навеску галерты массой 6-8 г смешивают до загустения с тщательно промытым прокаленным песком и высушивают в шкафу при температуре 100-105 °С до постоянной массы.
Высушенную навеску галерты растирают в фарфоровой ступке, помещают в пакет или патрон из фильтровальной бумаги и переносят в эксикатор аппарата Сокслета. Дальнейшее определение проводят по п.3.7.1.3.
9.5. Определение золы - по п.11.6.
9.6. Определение хлористого натрия - по п.3.5.
9.7. Определение муравьиной кислоты
9.7.1. Сущность метода
Метод основан на взаимодействии кислоты с раствором щелочи и определении количества кислоты титрованием.
9.7.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Колба коническая по ГОСТ 25336-82, вместимостью 200-300 см.
Натрия гидроксид по ГОСТ 4328-77, раствор 0,1 моль/дм (0,1 н).
Фенолфталеин, спиртовой раствор 10 г/дм (1%-ный).
Вода дистиллированная по ГОСТ 6709-72.
9.7.3. Проведение анализа
Навеску жидкого клея массой от 1,5 до 2 г, взвешенную с абсолютной погрешностью не более 0,001 г, помещают в колбу вместимостью 200-300 см, растворяют, подогревая на водяной бане, в 100 см
дистиллированной воды, добавляют несколько капель фенолфталеина и титруют раствором гидроксида натрия 0,1 моль/дм
до появления розового окрашивания.
9.7.4. Обработка результатов
Массовую долю муравьиной кислоты () в процентах вычисляют по формуле
![]() ,
,
где - объем раствора гидроксида натрия 0,1 моль/дм
, израсходованный на титрование, см
;
- коэффициент пересчета на точный раствор 0,1 моль/дм
гидроокиси натрия;
0,0046 - количество муравьиной кислоты, соответствующее 1 см раствора 0,1 моль/дм
гидроокиси натрия, г;
- навеска клея, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,05%.
Вычисление проводят до второго десятичного знак
а.
9.8. Определение условной вязкости по Энглеру
9.8.1. Сущность метода
Метод основан на определении вискозиметром времени истечения раствора клея стандартной концентрации по сравнению с водой.
9.8.2. Аппаратура и материалы
Вискозиметр для определения условной вязкости по ГОСТ 1532-81.
Секундомер механический.
Баня водяная.
Термометр ртутный стеклянный лабораторный с пределами измерений от 0 до 200 и от 0 до 50 °С по ГОСТ 28498-90.
Колба мерная по ГОСТ 1770-74, вместимостью 200 см.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Бензин авиационный марки Б-70 по ГОСТ 1012-72.
Марля медицинская по ГОСТ 9412-93.
Вода дистиллированная по ГОСТ 6709-72.
9.8.3. Подготовка к анализу
Прибор устанавливают так, чтобы три штифта во внутреннем измерительном сосуде находились в горизонтальной плоскости, для чего регулируют установочные винты треножника, пока все три острия указателей уровня не будут едва заметны над поверхностью воды, налитой во внутренний сосуд. Объем воды регулируется удалением или добавлением жидкости при помощи пипетки.
Установленный треножник не следует сдвигать с места. Нагревательную баню вискозиметра необходимо каждый раз устанавливать на треножнике в одном и том же положении.
Определение водного числа вискозиметра Энглера
Водным числом вискозиметра называется время истечения 200 см дистиллированной воды при 20 °С.
Время, необходимое для истечения указанного количества воды, должно быть не менее 50 и не более 52 с. Среднее для данного прибора время, принимаемое за водное число прибора, должно быть определено не менее 6 раз подряд.
Во внутренний сосуд немного выше, чем острия штифтов, наливают дистиллированную воду температурой 20 °С, поддерживая ее на том же уровне в течение 10-15 мин с помощью водяной бани.
Слегка приподнимают штепсель и выпускают немного воды из сосуда, чтобы вся сточная трубка также заполнялась водой. Штепсель должен быть новый, деревянный, без зазубрин и повреждений на конце.
Из внутреннего сосуда отбирают пипеткой излишек воды, чтобы ее поверхность находилась точно на уровне остриев штифтов.
Под сточное отверстие вискозиметра ставят чистую сухую измерительную колбу.
Установив прибор, закрывают его крышкой, слегка придерживая рукой штепсель, запирающий сточное отверстие.
Убедившись, что температура воды во внутреннем сосуде равна 20 °С, быстро (не двигая прибора) приподнимают штепсель и одновременно другой рукой пускают в ход секундомер.
Штепсель кладут рядом на стекло. Внимательно наблюдая за колбой, останавливают секундомер в момент, когда колба наполнится до метки 200 см. Счет ведут по нижнему мениску; луч зрения должен быть перпендикулярен оси колбы.
9.8.4. Проведение анализа
Перед каждым определением внутренний сосуд вискозиметра и его сточную трубку тщательно промывают чистым бензином и просушивают воздухом (вытирать внутренний сосуд не допускается), капли с внутреннего сосуда могут быть сняты кусочком фильтровальной бумаги с ровно обрезанными краями.
Раствор клея 18%-ной концентрации по товарно-сухому клею или 15%-ной - по беззольному и безводному клею фильтруют сначала через металлическое, а затем через шелковое сито (или марлю), подогревают до 30 °С и наполняют им внутренний сосуд вискозиметра. Необходимо следить за тем, чтобы не образовывалось пузырьков воздуха. Уровень залитого раствора клея должен немного превышать острия штифта. Деревянный штепсель должен плотно закрывать сточное отверстие.
Во внешний сосуд прибора наливают воду температурой несколько выше 80 °С. Чтобы во время опыта температура исследуемого клея была одинаковой (точно 30 °С), температуру клея, залитого во внутренний сосуд, доводят точно до 30 °С и выдерживают ее в течение 5 мин. Температуру бани устанавливают на 0,2 °С выше температуры клея и поддерживают ее на этом уровне на протяжении опыта, перемешивая содержимое бани мешалкой.
Подняв немного деревянный штепсель, дают стечь излишку клея, чтобы уровень его совпадал с верхними точками остриев. Если клея вытечет больше, чем нужно, следует добавить его по каплям, погружая острия, следя за тем, чтобы в клее не оставалось пузырьков воздуха.
Установив прибор, его закрывают крышкой и под сточное отверстие ставят чистую сухую измерительную колбу. Клей непрерывно перемешивают термометром, осторожно вращая вокруг штепселя крышку прибора, в которую вставлен термометр.
Когда термометр будет показывать точно 30 °С, быстро вынимают штепсель и одновременно нажимают кнопку секундомера. Как только уровень раствора клея в измерительной колбе дойдет точно до метки 200 см, секундомер останавливают и отсчитывают время.
9.8.5. Обработка результатов
Вязкость клея (в градусах Энглера) при 30 °С вычисляют по формуле
![]() ,
,
где - время истечения из вискозиметра 200 см
исследуемого раствора при температуре 30 °С, с;
- время истечения из вискозиметра 200 см
дистиллированной воды при температуре 20 °С (водное число вискозиметра), с.
При определении водного числа вискозиметра Энглера расхождение между двумя параллельными определениями не должно превышать 0,5 с.
При определении вязкости по Энглеру расхождение между двумя параллельными определениями не должно превышать 1 с.
9.9. Проба на загнивание
9.9.1. Сущность метода
Метод основан на органолептическом определении запаха и визуальном осмотре термостатированной пробы.
В результате жизнедеятельности микроорганизмов органические вещества клея разрушаются. Появляются признаки его загнивания (следы плесени, гнилостный запах).
9.9.2. Аппаратура
Шкаф сушильный.
Термостат.
Чашки Петри по ГОСТ 25336-82.
9.9.3. Проведение анализа
В чашку Петри, предварительно простерилизованную в сушильном шкафу при 160 °С в течение 2 ч или в автоклаве обычным способом и охлажденную до комнатной температуры, наливают 25 см раствора клея стандартной концентрации (18% по товарно-сухому клею или 15% по беззольному и безводному клею).
Чашку накрывают крышкой, помещают в термостат и выдерживают при температуре 25 °С в течение 3 сут, по истечении которых клей не должен иметь признаков загнивания (гнилостного запаха), а также следов плесени.
9.10. Методы определения кислотности
9.10.1. Определение общей (титруемой) кислотности - по ГОСТ 27082-89.
9.10.2. Определение активной кислотности (рН) потенциометрическим методом - по ГОСТ 28972-91.
9.10.3. Определение активной кислотности колориметрическим методом - по п.10.5.2.
9.11. Определение азота летучих оснований - по пп.3.2.1; 3.2.2.
9.12. Определение величины адгезии
9.12.1. Сущность метода
Метод основан на определении величины усилия, необходимого для расслаивания двух склеенных полос по месту склейки.
9.12.2. Аппаратура и материалы
Разрывная машина РМ-30.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Ткань-кирза двухслойная по ГОСТ 19196-93, артикул 6882.
9.12.3. Подготовка к анализу
Ткань-кирзу нарезают на полоски шириной 75 мм и длиной 130 мм. Навеску клея для определения адгезии рассчитывают, исходя из содержания в клее сухих веществ так, чтобы в навеске содержалось 0,75 г сухих веществ.
В табл.11 указаны навески клея, которые нужно отбирать в зависимости от содержания сухих веществ в исследуемых образцах.
Таблица 11
Массовая доля сухих веществ в исследуемом образце клея, % | Навеска клея, г, наносимая на | |
одну полоску ткани | две полоски ткани | |
35 | 1,07 | 2,14 |
36 | 1,04 | 2,08 |
37 | 1,02 | 2,04 |
38 | 0,98 | 1,96 |
39 | 0,96 | 1,92 |
40 | 0,94 | 1,88 |
41 | 0,91 | 1,82 |
42 | 0,90 | 1,80 |
43 | 0,87 | 1,74 |
44 | 0,85 | 1,70 |
45 | 0,83 | 1,66 |
Две полоски ткани взвешивают каждую отдельно с абсолютной погрешностью не более 0,01 г.
Навеску клея равномерно размазывают по рабочей поверхности полосок стеклянной лопаточкой. 10 мм по длине полоски с одной стороны и 20 мм с другой остаются свободными от клея.
Полоски склеивают (получают "склейки") проглаживанием пальцами по пять раз с каждой стороны, оставляют для высушивания на воздухе 5-6 ч при температуре 20-25 °С.
В подсушенных "склейках" определяют остаточное содержание воды, взвешивая их через 5-6 ч. Высушивание прекращают после того, как массовая доля воды достигнет 10-20%.
Высушенную "склейку" разрезают ножницами по длине на три равные части и получают три параллельные "склейки" размером 25х130 мм каждая.
9.12.4. Проведение анализа
Расслаивание "склеек" проводят на разрывной машине, например, РМ-30 по шкале до 10 кг, при свободном положении ограничителя маятника и расстоянии между зажимами для полосок ткани не более 15 мм.
Для расслаивания большие концы "склейки", не намазанные клеем, закрепляют в зажимах машины, после чего включают пуск и с помощью самописца проводят запись диаграммы, по которой устанавливают расслаивающее усилие.
Скорость движения нижнего зажима при испытании проб клея должна быть равна 200 мм/мин.
Во время испытания маятник разрывной машины при опускании необходимо слегка поддерживать рукой, чтобы опускание происходило плавно и сила инерции не оказывала влияния на показания расслаивающего усилия.
Если отсутствует самописец, показания маятника машины (максимальные и минимальные значения расслаивающего усилия) записывают в журнал. Запись показаний проводят через равные промежутки расслаиваемой длины "склейки" (через 5-10 мм).
Из минимальных и максимальных значений подсчитывают среднее значение расслаивающего усилия.
9.12.5. Обработка результатов
Величину расслаивающего усилия () в кг/см, затраченного на расслаивание погонного сантиметра склейки, вычисляют по формуле
![]() ,
,
где - средняя величина разрывного усилия, выведенная из трех параллельных определений, кг;
2,5 - ширина склейки, см.
10. МЕТОДЫ АНАЛИЗА КОРМОВЫХ ПРОДУКТОВ, КОНСЕРВИРОВАННЫХ ПИРОСУЛЬФИТОМ НАТРИЯ И КИСЛОТАМИ
10.1. Подготовка средней пробы к анализу - по п.2.8.
10.2. Определение воды - по п.3.3.1.
10.3. Определение хлористого натрия - по п.3.5.1.
10.4. Определение свободной сернистой кислоты в пересчете на сернистый ангидрид - по ГОСТ 27001-86.
10.5. Методы определения активной кислотности (рН)
10.5.1. Определение рН потенциометрическим методом - по ГОСТ 28972-91
10.5.2. Определение рН колориметрическим методом
10.5.2.1. Сущность метода
Метод основан на изменении окраски индикатора в водной вытяжке (фильтрате) анализируемого продукта.
10.5.2.2. Аппаратура, материалы
Шкала стандартных растворов.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Ступка фарфоровая с пестиком по ГОСТ 9147-80.
Воронки стеклянные по ГОСТ 25336-82.
Колбы плоскодонные по ГОСТ 25336-82.
Чашка фарфоровая по ГОСТ 9147-80.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Вода дистиллированная по ГОСТ 6709-72.
10.5.2.3. Проведение анализа
От 9 до 10 г анализируемого продукта, отвешенных с абсолютной погрешностью не более 0,01 г, количественно переносят в ступку 100 см дистиллированной воды. Смесь тщательно растирают и фильтруют через складчатый фильтр.
Водную вытяжку наливают в пробирку, входящую в комплект шкалы, или в пробирку, аналогичную по цвету и диаметру пробиркам шкалы, добавляют индикатор согласно инструкции, прилагаемой к шкале стандартных растворов, и сравнивают полученную окраску анализируемого раствора с окраской стандартных растворов шкалы.
11. МЕТОДЫ АНАЛИЗА МОРСКИХ БЕСПОЗВОНОЧНЫХ И ПРОДУКТОВ ИХ ПЕРЕРАБОТКИ
11.1. Подготовка средней пробы к анализу - по п.2.9.
11.2. Определение воды - по п.3.3.1.
11.3. Определение хлористого натрия - по пп.3.5.1; 3.5.2.
11.4. Определение белковых веществ - по пп.8.9.1; 8.9.2; 8.9.4.
11.5. Определение жира - по пп.3.7.1; 3.7.2; 3.7.4.
11.6. Определение золы
11.6.1. Сущность метода
Метод основан на удалении органических веществ из навески анализируемого продукта сжиганием и определении золы взвешиванием.
11.6.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электропечь сопротивления лабораторная.
Тигли фарфоровые по ГОСТ 9147-80.
Эксикатор по ГОСТ 25336-82.
Шкаф сушильный.
Электроплитка бытовая по ГОСТ 14919-83.
11.6.3. Проведение анализа
Навеску сухой массы от 1,5 до 2 г (сырой массы около 4 г), взвешенную с абсолютной погрешностью не более 0,001 г, помещают в предварительно прокаленный, доведенный до постоянной массы, фарфоровый тигель, осторожно обугливают на плитке до прекращения выделения дыма, а затем озоляют в муфельной печи при температуре 500 °С. После взвешивания остывшего в эксикаторе тигля с золой его повторно прокаливают в течение 1 ч до постоянной массы, охлаждают и взвешивают с абсолютной погрешностью не более 0,001 г.
Цвет золы может быть белым, желтым, серым, оранжевым и другим, но зола не должна содержать черных вкраплений.
11.6.4. Обработка результатов
Массовую долю золы () в процентах вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- масса пустого тигля, г;
- масса тигля с золой, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, расхождение между которыми не должны превышать 0,01%.
Вычисление проводят до второго десятичного знака.
11.7. Определение песка
11.7.1. Сущность метода
Метод основан на сжигании органических веществ навески, обработке золы раствором соляной кислоты и определении количества песка взвешиванием.
11.7.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Электропечь сопротивления лабораторная.
Электроплитка бытовая по ГОСТ 14919-83.
Баня водяная.
Эксикатор по ГОСТ 25336-82.
Чашка фарфоровая по ГОСТ 9147-80, диаметром 70-90 мм.
Тигель фарфоровый по ГОСТ 9147-80.
Воронка стеклянная по ГОСТ 25336-82, диаметром 50-70 мм.
Стакан химический по ГОСТ 25336-82, вместимостью 200-300 см.
Палочка стеклянная.
Фильтры обеззоленные.
Кислота соляная по ГОСТ 3118-77, раствор 100 г/дм (10%-ный) плотностью 1050 кг/м
.
11.7.3. Проведение анализа
Навеску измельченного продукта 20-30 г, взвешенную с абсолютной погрешностью не более 0,1 г, подсушивают в фарфоровой чашке или в большом тигле в сушильном шкафу, осторожно обугливают на слабом огне, затем прокаливают в муфельной печи до получения белой или слегка сероватой золы.
При исследовании сушеных беспозвоночных навеску продукта для озоления уменьшают в 2-3 раза.
Золу в тигле обрабатывают раствором соляной кислоты 100 г/дм в течение 20-30 мин на кипящей водяной бане. Для ускорения растворения золу перемешивают стеклянной палочкой. Раствор фильтруют через предварительно взвешенный беззольный фильтр, избегая попадания на фильтр нерастворившегося остатка. Обработку золы соляной кислотой повторяют несколько раз. Нерастворившийся остаток в тигле количественно переносят на фильтр и промывают горячей водой до исчезновения реакции на ион хлора (проба с раствором азотнокислого серебра). Фильтр с осадком сжигают и прокаливают в предварительно взвешенном фарфоровом тигле. Тигель охлаждают в эксикаторе и взвешивают с абсолютной погрешностью не более 0,001 г.
11.7.4. Обработка результатов
Массовую долю песка () в процентах вычисляют по формуле
![]() ,
,
где - масса тигля с прокаленным остатком, г;
- масса пустого тигля, г;
- навеска исследуемого образца, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,02%.
Вычисление проводят до второго десятичного знака.
11.8. Определение минеральных примесей (песка, известковых образований "жемчуг")
11.8.1. Сущность метода
Метод основан на разрушении тканей мидий щелочью, отмывании механических неорганических включений дистиллированной водой и определении их количества взвешиванием.
11.8.2. Аппаратура, материалы и реактивы
Шкаф сушильный.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Стаканы химические по ГОСТ 25336-82, вместимостью 500, 1000 см.
Колба коническая по ГОСТ 25336-82, вместимостью 250 см.
Воронки химические по ГОСТ 25336-82, диаметром 70-80 мм.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Натрия гидроксид по ГОСТ 4328-77, ч.д.а., раствор 300 г/дм (30%-ный).
Вода дистиллированная по ГОСТ 6709-72 или вода питьевая по ГОСТ 2874-82*.
________________
* На территории Российской Федерации действует ГОСТ Р 51232-98.
Вата минеральная по ГОСТ 4640-93.
11.8.3. Проведение анализа
Навеску фарша массой от 45 до 50 г, взвешенную с абсолютной погрешностью не более 0,1 г, помещают в коническую колбу вместимостью 250 см и заливают 50-70 м
профильтрованного через вату раствора гидроксида натрия 300 г/дм
. После взбалтывания содержимого стенки колбы смывают небольшим количеством воды и помещают колбу в кипящую водяную баню на 40-60 мин (до полного гидролиза мяса).
Для равномерного прогревания колбу периодически взбалтывают, при этом песок, "жемчуг" и кусочки ракушек оседают на дно колбы.
По окончании гидролиза содержимое колбы разводят в полтора-два раза горячей водой и после отстаивания осадка осторожно сливают раствор. Обработку горячей водой повторяют 5-7 раз до полного отмывания осадка. Все смывные воды собирают в один стакан. При обнаружении в смывных водах песка или известковых образований им дают отстояться, жидкость сверху осторожно сливают, осадок промывают водой и количественно переносят в колбу с основным осадком. Осадок (песок, "жемчуг" и кусочки ракушек) количественно переносят дистиллированной водой на высушенный и взвешенный фильтр.
Фильтр с осадком высушивают при температуре 100-105 °С около 1 ч, затем выдерживают на воздухе около 0,5 ч и взвешивают с абсолютной погрешностью не более 0,001 г. После этого быстро удаляют с фильтра поочередно "жемчуг", кусочки ракушек (створки) и песок. Каждый раз после удаления очередной порции примесей фильтр взвешивают.
11.8.4. Обработка результатов
По разности масс между первым и вторым взвешиванием фильтра определяют количество "жемчуга", между вторым и третьим взвешиванием - количество ракушек, между третьим и четвертым взвешиванием - количество песка.
Количество примеси каждого вида выражают в процентах к массе мяса, взятой для анализа. Общее количество минеральных включений в мясе мидий () вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- масса фильтра, г;
- масса фильтра с осадком после высушивания, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений.
Вычисление проводят до второго десятичного знака.
11.9. Определение хитина в продуктах переработки беспозвоночных
11.9.1. Сущность метода
Метод основан на выделении частиц хитина и определении в них азота макрометодом по п.8.9.1.
11.9.2. Аппаратура, материалы и реактивы
Шкаф сушильный.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Центрифуга лабораторная.
Аппарат для встряхивания жидкостей.
Баня водяная.
Насос водоструйный лабораторный стеклянный по ГОСТ 25336-82.
Колба коническая по ГОСТ 25336-82, вместимостью 200-250 см.
Колба Бунзена, вместимостью 250-500 см.
Пробирки центрифужные.
Холодильник обратный по ГОСТ 25336-82.
Фильтр стеклянный (крупнопористый) N 1 по ГОСТ 25336-82.
Кислота муравьиная по ГОСТ 5848-73, 900 г/дм (90%-ная).
Ацетон по ГОСТ 2603-79, растворы 700, 960-980 г/дм (70, 96-98%-ный).
Натрия гидроксид по ГОСТ 4328-77, раствор 50 г/дм (5%-ный).
11.9.3. Проведение анализа
Навеску фарша массой от 9 до 10 г (или муки от 4,5 до 5 г), взвешенную с абсолютной погрешностью не более 0,1 г, помещают в колбу со шлифом вместимостью 200-250 см, добавляют 100 см
ацетона и нагревают на водяной бане с обратным холодильником в течение 45 мин.
После нагревания содержимое колбы количественно переносят ацетоном в центрифужную пробирку вместимостью 200 см и центрифугируют в течение 20 мин при 2000 об/мин.
Надосадочную жидкость отбрасывают.
К осадку добавляют 100 см раствора ацетона 700 г/дм
, перемешивают и вновь центрифугируют в указанном режиме. Полученную жидкую фракцию также сливают.
К осадку, содержащемуся в пробирке, приливают 100 см муравьиной кислоты 900 г/дм
, закрывают пробкой и встряхивают в течение 18 ч.
Содержимое вновь центрифугируют в течение 15 мин при 2000 об/мин и жидкую фракцию отбрасывают.
Полученный осадок промывают ацетоном, центрифугируют в течение 20 мин, сливают жидкую фазу и вторично промывают ацетоном.
К промытому осадку добавляют 100 см раствора гидроксида натрия 50 г/дм
и нагревают на водяной бане.
Полученную массу фильтруют через стеклянный крупнопористый фильтр N 1 и промывают остаток на фильтре кипящей водой. Фильтрат отбрасывают, а остаток высушивают при 100-105 °С до постоянной массы.
Сухой остаток количественно переносят дистиллированной водой в колбу Кьельдаля вместимостью 100-250 см, выпаривают воду до 5 см
и определяют массовую долю азота макрометодом по п.8
.9.1.
11.9.4. Обработка результатов
Массовую долю хитина () в процентах вычисляют по формуле
![]() ,
,
где - массовая доля азота хитина в исследуемом образце, в процентах, определенная макрометодом;
14,5 - коэффициент пересчета азота хитина в хитин.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,05%.
Вычисление проводят до второго десятичного знака.
11.10. Определение массовой доли частиц депротеинизированного панциря в мясе криля и продуктах из него
11.10.1. Сущность метода
Метод основан на выделении панциря из анализируемого продукта щелочным гидролизом и весовом определении его.
11.10.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Шкаф сушильный.
Эксикатор по ГОСТ 25336-82.
Колба коническая по ГОСТ 25336-82, вместимостью 200 см.
Колба для фильтрования под вакуумом по ГОСТ 25336-82.
Насос водоструйный лабораторный по ГОСТ 25336-82.
Фильтр стеклянный крупнопористый N 1 по ГОСТ 25336-82.
Натрия гидроксид по ГОСТ 4328-77, водный раствор 50 г/дм (5%-ный).
Вода дистиллированная по ГОСТ 6709-72.
Бумага лакмусовая красная.
11.10.3. Проведение анализа
Навеску фарша массой 10-20 г, отвешенную с абсолютной погрешностью не более 0,1 г, помещают в коническую колбу вместимостью 200 см, добавляют 6-кратное количество водного раствора гидроксида натрия 50 г/дм
и нагревают в сушильном шкафу в течение 2 ч при температуре 140 °С. Горячий гидролизат фильтруют под вакуумом (от 266 до 399 гПа) через предварительно высушенный до постоянной массы стеклянный фильтр N 1 (диаметром отверстий 100-120 мкм).
Остаток на фильтре промывают горячей дистиллированной водой до нейтральной реакции на лакмус и высушивают до постоянной массы.
11.10.4. Обработка результатов
Массовую долю частиц панциря () в процентах вычисляют по формуле
![]() ,
,
где - навеска исследуемого образца, г;
- масса фильтра, г;
- масса фильтра с частицами панциря, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,05%.
Вычисление проводят до второго десятичного знака.
12. МЕТОДЫ АНАЛИЗА НАТУРАЛЬНОЙ АМБРЫ
12.1. Подготовка средней пробы к анализу - по п.2.10.
12.2. Определение цвета
12.2.1. Цвет амбры определяют в целом куске при дневном освещении (визуально).
12.2.2. Если цвет амбры вызывает сомнение, кусок амбры разбирают на две равные части и просматривают цвет на разломе куска.
12.3. Определение запаха
12.3.1. Для определения запаха отбирают из нескольких мест каждой партии разные по цвету куски амбры.
12.3.2. Запах определяют на поверхности куска амбры и на разломе его.
12.4. Определение растворимости амбры
12.4.1. Сущность метода
Метод основан на растворении органических веществ амбры этиловым спиртом и определении их количества взвешиванием.
12.4.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Эксикатор по ГОСТ 25336-82.
Колбы конические по ГОСТ 25336-82, вместимостью 200, 300 см.
Воронки стеклянные по ГОСТ 25336-82, диаметром 100 мм.
Цилиндр мерный по ГОСТ 1770-74, вместимостью 100 см.
Фильтры бумажные с синей лентой.
Спирт этиловый ректификованный по ГОСТ 5962-67.
12.4.3. Проведение анализа
2-3 г измельченной амбры взвешивают с абсолютной погрешностью не более 0,0002 г в конической колбе вместимостью 200-300 см.
В колбу с навеской амбры приливают 100 см этилового спирта 960 г/дм
(96%-ного), помещают ее на водяную баню, нагретую до температуры 60-70 °С, где выдерживают до полного растворения амбры.
Полученный спиртовой раствор фильтруют через предварительно высушенный до постоянной массы бумажный фильтр.
Колбу 2-3 раза ополаскивают подогретым до 60-70 °С спиртом (по 10 см), сливая его на фильтр. Остаток на фильтре промывают подогретым на водяной бане до 60-70 °С спиртом до тех пор, пока фильтрат не станет совершенно бесцветным. Фильтр с остатком высушивают до постоянной массы в эксикаторе при температуре 20 °С.
12.4.4. Обработка результатов
Растворимость амбры () в процентах вычисляют по формуле
![]() ,
,
где - масса исследуемого образца, г;
- масса сухого фильтра, г;
- масса высушенного фильтра с остатком, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,3%.
Вычисление проводят до первого десятичного знака.
12.5. Определение воды - по п.3.3.3.
Навеску амбры от 45 до 50 г взвешивают с погрешностью не более 0,1 г.
12.6. Определение золы - по п.11.6.
13. МЕТОДЫ АНАЛИЗА ЧЕШУИ, ЖЕМЧУЖНОГО ПАТА И ПЕРЛАМУТРОВОГО ПРЕПАРАТА
13.1. Подготовка средней пробы к анализу - по п.2.11.
13.2. Определение золы - по п.11.6.
13.3. Методы определения сырого гуанина
13.3.1. Определение сырого гуанина спектрофотометрическим методом
13.3.1.1. Определение сырого гуанина в жемчужном пате
13.3.1.1.1. Сущность метода
Метод основан на выделении гуанина из жемчужного пата ацетоном, растворении в гидроксиде натрия и определении его количества по оптической плотности.
13.3.1.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Фотоэлектроколориметр с пределами измерений оптической плотности от 0 до 1,3 по ТУ 3-3.1766-82, ТУ 3.31860-85, ТУ 3-3.2164-89.
Центрифуга лабораторная по ТУ 27-32-26-77-86, =4000 об/мин.
Шкаф сушильный.
Колбы мерные по ГОСТ 1770-74, вместимостью 200 см.
Баня водяная.
Пипетки по ГОСТ 29227-91 с ценой деления 0,1 см.
Пробирки стеклянные по ГОСТ 25336-82.
Пробирки центрифужные.
Цилиндры мерные, вместимостью 10, 25 и 50 см.
Ацетон по ГОСТ 2603-79.
Бензин авиационный марки Б-70 по ГОСТ 1012-72 или эфир петролейный.
Вода дистиллированная по ГОСТ 6709-72.
Натрия гидроксид по ГОСТ 4328-77, раствор 1 моль/дм (1 н).
Спирт этиловый технический по ГОСТ 17299-78.
Хлороформ по Госфармакопее СССР.
13.3.1.1.3. Проведение анализа
Навеску жемчужного пата массой от 0,8 до 1,0 г, отвешенную с абсолютной погрешностью не более 0,0001 г, помещают в центрифужную пробирку и приливают 10-20 см ацетона. Содержимое пробирки тщательно размешивают. Суспензию центрифугируют 10 мин при 4000 об/мин. Обработку пата свежими порциями растворителя повторяют 2-3 раза до тех пор, пока капля центрифугата, нанесенная на фильтровальную бумагу, после испарения не будет оставлять следов нитролака. После этого растворитель сливают, а гуанин подсушивают на водяной бане для удаления ацетона.
Гуанин из пробирки количественно переносят в мерную колбу вместимостью 200 см небольшими порциями раствора 1 моль/дм
гидроксида натрия. Для лучшего растворения гуанина колбу подогревают на водяной бане при температуре 70 °С в течение 5-10 мин. Раствор охлаждают до комнатной температуры и доводят объем до метки раствором гидроксида натрия 1 моль/дм
.
0,2-0,3 см полученного раствора разбавляют раствором гидроксида натрия 1 моль/дм
до 10 см
, тщательно перемешивают, заливают в кювету с рабочей длиной 10 мм и определяют оптическую плотность при длине волны 275 нм по отношению к раствору гидроксида натрия 1 моль/дм
.
Содержание гуанина, соответствующее определенной оптической плотности, рассчитывают по градуировочному график
у.
13.3.1.1.4. Построение градуировочного графика
Гуанин предварительно обрабатывают следующим образом: 5-10 г сырого гуанина разбавляют бензином в соотношении 1:10 и центрифугируют. Центрифугат сливают, а обработку повторяют сначала бензином, а затем смесью бензина с хлороформом.
Для более полного разделения суспензии при центрифугировании смесь растворителей должна иметь плотность не более 1300 кг/м.
Очищенный гуанин подсушивают на водяной бане для удаления растворителей, а затем в сушильном шкафу до постоянной массы при температуре 100-105 °С. Хранят гуанин в склянке с притертой пробкой из темного стекла.
Для приготовления стандартных растворов навеску очищенного гуанина 25 мг, взвешенную с абсолютной погрешностью не более 0,001 г, помещают в мерную колбу вместимостью 200 см, приливают раствор гидроксида натрия 1 моль/дм
при подогревании на водяной бане температурой 70 °С.
После охлаждения содержимого до комнатной температуры объем в колбе доводят до метки раствором гидроксида натрия 1 моль/дм. Хранят раствор не более 3 сут в прохладном, защищенном от света месте.
1 см приготовленного раствора содержит 0,125 мг гуанина.
Для построения градуировочного графика готовят серию растворов с различным содержанием гуанина в 1 см. Соотношение стандартного раствора и гидроксида натрия приведено в табл.12.
Таблица 12
Количество гуанина в приготовленном растворе, мг/10 см | Количество стандартного раствора гуанина, см | Количество добавляемой щелочи, см |
0,012 | 0,1 | 9,9 |
0,025 | 0,2 | 9,8 |
0,050 | 0,4 | 9,6 |
0,075 | 0,6 | 9,4 |
0,100 | 0,8 | 9,2 |
0,125 | 1,0 | 9,0 |
0,150 | 1,2 | 8,8 |
0,175 | 1,4 | 8,6 |
0,200 | 1,6 | 8,4 |
0,225 | 1,8 | 8,2 |
0,250 | 2,0 | 8,0 |
Хранят серию приготовленных растворов не более 3 сут в темном прохладном месте.
Градуировочный график строят не реже двух раз в год.
Оптическую плотность серии растворов определяют против раствора гидроксида натрия 1 моль/дм в кювете с рабочей длиной 10 мм. По полученным данным строят градуировочный график, откладывая на оси абсцисс содержание гуанина, на оси ординат - соответствующие оптические плотности.
13.3.1.1.5. Обработка результатов
Массовую долю гуанина в жемчужном пате () в процентах вычисляют по формуле
![]() ,
,
где - содержание гуанина, найденное по градуировочному графику, мг;
- объем раствора гидроксида натрия 1 моль/дм
, израсходованного для растворения гуанина, см
;
10 - разведение раствора, см;
- объем исследуемого раствора, израсходованный для колориметрирования, см
;
- масса исследуемого образца, мг.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,2%.
Вычисление проводят до первого десятичного знак
а.
13.3.1.2. Определение гуанина в перламутровом препарате, приготовленном с добавлением спирта
Навеску препарата от 0,25 до 0,30 г помещают в предварительно высушенную до постоянной массы бюксу, подсушивают на водяной бане для удаления остатков спирта, а затем в сушильном шкафу до постоянной массы при температуре 105 °С.
Гуанин из бюксы количественно переносят в мерную колбу вместимостью 200 см небольшими порциями раствора 1 моль/дм
гидроксида натрия и далее поступают так, как указано в п.13.3.1.1.3 для гуанина, выделенного из жемчужного пата.
Обработка результатов - по п.13.3.1.1.5.
13.3.1.3. Определение гуанина в перламутровом препарате, приготовленном с добавлением касторового масла
Навеску препарата от 0,25 до 0,30 г помещают в центрифужную пробирку и приливают 10-20 см подогретого этилового спирта. Содержимое пробирки тщательно размешивают. Суспензию центрифугируют 10 мин при 4000 об/мин. Обработку препарата свежими порциями спирта повторяют 2-3 раза до тех пор, пока капля центрифугата, нанесенная на фильтровальную бумагу, после испарения не будет оставлять следов масла.
По окончании центрифугирования жидкость сливают, а гуанин подсушивают на водяной бане для удаления спирта, затем в сушильном шкафу до постоянной массы и далее поступают как указано в п.13.3.1.1.3.
13.3.1.4. Обработка результатов - по п.13.3.1.1.5.
13.3.2. Методы определения сырого гуанина в чешуе, жемчужном пате и перламутровом препарате взвешиванием
13.3.2.1. Определение сырого гуанина в жемчужном пате
13.3.2.1.1. Сущность метода
Метод основан на выделении (отмывании) гуанина из жемчужного пата ацетоном, центрифугировании (фильтровании) и определении массы гуанина взвешиванием.
13.3.2.1.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Шкаф сушильный лабораторный.
Баня водяная.
Центрифуга лабораторная, 4000 об/мин по ТУ 27-32-26-77-86.
Стаканчики для взвешивания (бюксы) по ГОСТ 25336-82.
Стаканы стеклянные лабораторные по ГОСТ 25336-82.
Ацетон по ГОСТ 2603-79.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
13.3.2.1.3. Проведение анализа
Навеску пата массой 3-5 г разбавляют ацетоном в соотношении 1:50, осторожно, тщательно перемешивают и центрифугируют. Центрифугат осторожно сливают. Обработку ацетоном повторяют до тех пор, пока капля центрифугата, нанесенная на фильтровальную бумагу, после испарения не будет оставлять следов лака.
Очищенный от лака гуанин количественно переносят в предварительно высушенную бюксу небольшими порциями ацетона. Бюксу помещают на водяную баню для удаления ацетона, а затем - в сушильный шкаф для высушивания гуанина до постоянной массы.
Вместо центрифугирования выделения гуанина из суспензии можно проводить фильтрованием через предварительно высушенный до постоянной массы фильтр. Осадок на фильтре промывают 5-6 раз ацетоном и высушивают до постоянной массы при 100-105 °С.
13.3.2.1.4. Обработка результатов
Массовую долю гуанина в жемчужном пате () в процентах вычисляют по формуле
![]() ,
,
где - масса бюксы с навеской после высушивания, г;
- масса бюксы, г;
- масса жемчужного пата, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
13.3.2.2. Определение сырого гуанина в перламутровом препарате
13.3.2.2.1. Сущность метода
Метод основан на определении разницы между общей массой плотных веществ и белковых примесей взвешиванием.
13.3.2.2.2. Определение плотных веществ в перламутровом препарате, приготовленном с применением этилового спирта
13.3.2.2.2.1. Сущность метода - по п.3.3.1.1.
13.3.2.2.2.2. Аппаратура, материалы и реактивы
Стаканчики для взвешивания (бюксы) по ГОСТ 25336-82.
Шкаф сушильный лабораторный.
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Эксикатор по ГОСТ 25336-82.
13.3.2.2.2.3. Проведение анализа
Навеску препарата от 0,8 до 1,0 г помещают в бюксу, предварительно высушенную до постоянной массы, взвешивают с абсолютной погрешностью не более 0,001 г и высушивают до постоянной массы при температуре 100-105 °С.
13.3.2.2.2.4. Обработка результатов
Массовую долю плотных веществ () в процентах вычисляют по формуле
![]() ,
,
где - масса бюксы с навеской после высушивания, г;
- масса бюксы, г;
- масса перламутрового препарата, г.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление доводят до первого десятичного знака.
13.3.2.2.3. Определение плотных веществ в перламутровом препарате, приготовленном с применением касторового масла
13.3.2.2.3.1. Сущность метода
Метод основан на растворении масла, содержащегося в препарате, этиловым спиртом, выделении плотного остатка ацетоном и определении его массы взвешиванием.
13.3.2.2.3.2. Аппаратура, реактивы и материалы - по п.13.3.2.2.2.2 со следующими дополнениями:
Центрифуга лабораторная по ТУ 27-32-26-77-86.
Пробирки центрифужные по ГОСТ 25336-82.
Баня водяная.
Спирт этиловый технический по ГОСТ 17299-78.
Ацетон по ГОСТ 2603-79.
Бензин авиационный марки Б-70 по ГОСТ 1012-72.
13.3.2.2.3.3. Проведение анализа
Навеску препарата массой от 0,8 до 1,0 г, взвешенную с абсолютной погрешностью не более 0,0001 г, помещают в центрифужную пробирку, приливают 10-20 см подогретого этилового спирта и тщательно перемешивают.
После центрифугирования в течение 10 мин при 4000 об/мин осторожно сливают жидкость. Обработку препарата спиртом повторяют до тех пор, пока капля центрифугата, нанесенная на фильтровальную бумагу, не будет оставлять следов масла. Осевший на дно центрифужной пробирки плотный остаток препарата количественно переносят небольшими порциями ацетона или бензина в предварительно высушенную до постоянной массы бюксу, помещают ее на водяную баню для удаления растворителя, а затем в сушильный шкаф для высушивания остатка до постоянной массы.
Массовую долю плотных веществ в перламутровом препарате определяют по п.13.3.2.2.2.3 и вычисляют по п.13.3.2.2.2.4.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
13.3.2.2.4. Определение белковых примесей в перламутровом препарате
13.3.2.2.4.1. Сущность метода
Метод основан на экстракции гуанина из перламутрового препарата раствором соляной кислоты, отделении примесей и определении их массы взвешиванием.
13.3.2.2.4.2. Аппаратура, материалы и реактивы
Весы лабораторные с пределом допускаемой абсолютной погрешности взвешивания не более ±0,01 г по ГОСТ OIML.R 76-1-2011 или по нормативным документам, действующим на территории государства, принявшего стандарт.
Баня водяная.
Шкаф сушильный лабораторный.
Стакан химический по ГОСТ 25336-82, вместимостью 200-300 см.
Воронка стеклянная по ГОСТ 25336-82.
Кислота соляная по ГОСТ 3118-77, раствор 50 г/дм (5%-ный).
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
13.3.2.2.4.3. Проведение анализа
Навеску препарата от 0,8 до 1,0 г, взвешенную с абсолютной погрешностью не более 0,0001 г, помещают в химический стакан вместимостью 200 см, заливают 50 см
раствора соляной кислоты 50 г/дм
и нагревают до кипения. Гуанин быстро растворяется, а примеси коагулируют и всплывают на поверхность раствора.
После кипячения в течение 1-2 мин содержимое стакана в горячем виде фильтруют через высушенный до постоянной массы фильтр. Осадок промывают два раза горячим раствором соляной кислоты 50 г/дм, а затем горячей водой, подкисленной соляной кислотой. По окончании фильтрования фильтр с осадком высушивают до постоянной массы и взвешивают.
13.3.2.2.4.4. Обработка результатов
Массовую долю белковых примесей () в процентах вычисляют по формуле
![]() ,
,
где - масса перламутрового препарата, г;
- масса фильтра с осадком после высушивания, г;
- масса фильтра, г.
13.3.2.2.5. Обработка результатов по определению гуанина в перламутровом препарате
Массовую долю сырого гуанина () в перламутровом препарате в процентах вычисляют по формуле
![]() ,
,
где - массовая доля плотных веществ в препарате в процентах;
- массовая доля белковых примесей в препарате в процентах.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
13.4. Определение плотного остатка в жемчужном пате
13.4.1. Сущность метода - по п.3.3.1.1.
13.4.2. Аппаратура, материалы и реактивы- по п.13.3.2.2.4.2.
13.4.3. Проведение анализа
Навеску пата массой 1-2 г, помещенную в предварительно высушенную бюксу, высушивают в сушильном шкафу в течение 2 ч при температуре 60-65 °С, а затем до постоянной массы при 100-105 °С.
13.4.4. Обработка результатов
Массовую долю плотного остатка в жемчужном пате определяют по п.13.3.2.2.2.4.
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,1%.
Вычисление проводят до первого десятичного знака.
Электронный текст документа
и сверен по:
Рыба и рыбные продукты. Методы анализа,
маркировка, упаковка: Сб. ГОСТов. -
, 2010
Редакция документа с учетом
изменений и дополнений подготовлена

{kind=link}